

Disruption of a metabolic pathway causes tolerance, high persistence, and MIC-shifted resistance to diverse antibiotics.
Abstract
A critical challenge for microbiology and medicine is how to cure infections by bacteria that survive antibiotic treatment by persistence or tolerance. Seeking mechanisms behind such high survival, we developed a forward-genetic method for efficient isolation of high-survival mutants in any culturable bacterial species. We found that perturbation of an essential biosynthetic pathway (arginine biosynthesis) in a mycobacterium generated three distinct forms of resistance to diverse antibiotics, each mediated by induction of WhiB7: high persistence and tolerance to kanamycin, high survival upon exposure to rifampicin, and minimum inhibitory concentration–shifted resistance to clarithromycin. As little as one base change in a gene that encodes, a metabolic pathway component conferred multiple forms of resistance to multiple antibiotics with different targets. This extraordinary resilience may help explain how substerilizing exposure to one antibiotic in a regimen can induce resistance to others and invites development of drugs targeting the mediator of multiform resistance, WhiB7.
INTRODUCTION
Antibiotics revolutionized the practice of medicine by offering a cure for many infectious diseases, enhancing the survival of premature newborns, improving the success of surgery, and permitting immunosuppressive treatments of malignancies and autoimmune conditions. However, less than a century after the widespread adoption of antibiotics, these gains are threatened by antimicrobial resistance (AMR)—a term used here to refer to the ability of microbial pathogens to avoid or delay death upon exposure to antimicrobial compounds that were expected to kill them (1). An estimated 700,000 people die each year from antimicrobial-resistant infections (2), and by 2050, deaths due to AMR are projected to reach a staggering 10.5 million per year—more than five times COVID-19’s global toll in the year 2020. A large proportion of AMR deaths—more than a quarter—are due to drug-resistant tuberculosis (TB). TB is caused primarily by Mycobacterium tuberculosis (Mtb) and was the leading cause of death due to a single infectious agent before being overtaken by COVID-19 in 2020. Drug-sensitive TB can be cured in 85% of cases by treatment with a combination of four drugs for 2 months followed by two of the four for an additional 4 months, but for drug-resistant TB, treatment options are generally much longer, more toxic, and less effective, with success rates of only 39 to 57% depending on the number of drugs to which the bacteria are resistant (3).
Antibiotic resistance on the part of Mtb and other major human pathogens can manifest in at least three different ways (Fig. 1). First, minimum inhibitory concentration (MIC)–shifted resistance occurs when the minimum concentration of antibiotic required to stop growth of a clonal bacterial population is increased. A second manifestation, tolerance, refers to a reduced rate of killing for an entire bacterial population upon antibiotic exposure. Last, a biphasic kill curve reflecting a reduced rate of killing for a subpopulation is the hallmark of persistence, which results from phenotypic heterogeneity within a clonal population. This heterogeneity can potentially be epigenetic (4), transcriptional (5), posttranscriptional (6), translational (7), posttranslational (8), or metabolic (9) and can also arise from biased partitioning of cellular components during division (10, 11). In unstressed conditions such as during logarithmic growth in vitro, most bacterial populations contain a small proportion of stochastically generated persister cells [for example, on the order of 10−6 for Staphylococcus pyogenes (12)]. Resistance relative to this basal level of persistence can therefore manifest as an increased proportion of persister cells, known as high persistence (13). A cell that survives antibiotic exposure through tolerance or persistence will give rise in the absence of the antibiotic to a population that will exhibit the same rate of killing or proportion of persisters upon reexposure to the same drug under the same conditions (Fig. 1), and neither tolerance nor persistence leads to an increase in the MIC for populations derived from the tolerant or persistent cells. MIC-shifted resistance, tolerance, and high persistence can each result from either acquisition of heritable mutations or stably retained genes (genetic resistance) or conditional induction of diverse, incompletely understood resistance mechanisms in bacteria that are genetically identical to those that are not resistant (phenotypic resistance).
Fig. 1. Manifestations of antibiotic resistance.
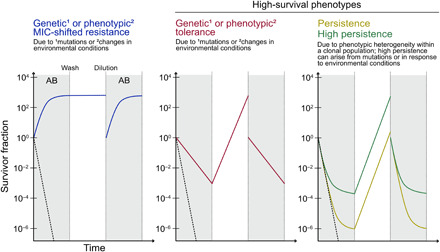
Schematics show the survivor fraction over time upon exposure of a bacterial population to an antibiotic (AB; gray background), removal of the antibiotic (wash) and outgrowth (white background), and dilution to the original OD followed by reexposure to the same antibiotic. The killing kinetics for a completely susceptible population are depicted by dashed black lines.
Standard clinical assays used to assess antibiotic resistance do not evaluate tolerance or high persistence. However, these types of resistance have major clinical consequences, such as long treatment times for TB (14, 15) and recurrence of disease after conclusion of antibiotic therapy for many infections (16, 17). In addition, pools of tolerant and persistent cells foster the emergence of genetic MIC-shifted resisters because of their prolonged survival under antibiotic selection (18), especially when there is an accompanying increase in mutation rates (19) or competence levels (20).
Exacerbating the problem, diverse conditions that bacteria face in the host can trigger phenotypic resistance (21) in the form of MIC-shifted resistance, tolerance, and/or high persistence. Triggers include stresses imposed by substerilizing host immunity, such as nutrient and micronutrient limitation, reactive oxygen and nitrogen species, hypoxia, and low pH. Equally challenging, infecting bacteria may develop phenotypic resistance when they encounter subinhibitory concentrations of an antibiotic due to its slow penetration or intermittently waning concentration between doses in certain biological niches. In the standard combination regimens for TB, the distinct pharmacokinetics of each antibiotic ensure that mycobacteria in pulmonary cavities are exposed to subinhibitory antibiotic concentrations of each member of the combination for hours each day (22). Such patterns of exposure can induce phenotypic resistance not only to the triggering drug but also to unrelated ones (23). This can reduce treatment efficacy, extend treatment duration, and foster emergence of genetic MIC-shifted resistance.
High-survival (HS) mutants (Fig. 1), which display tolerance (24, 25) and/or high persistence (13), have provided a powerful route to uncovering mechanisms of phenotypic resistance. In addition, such mutations have been identified in clinical isolates during or after antibiotic treatment (26, 27) and have been shown to precede mutations that lead to genetic MIC-shifted resistance both in vitro (18) and in patients (28). However, the systematic isolation of these mutants poses challenges. Here, using the model mycobacterium Mycobacterium smegmatis (Msm) and extending results to Mtb, we introduce a facile forward-genetic method—applicable to any culturable bacterial species, such as those flagged by the World Health Organization as top priorities for the development of new antibiotics—for isolation of mutants that have markedly increased tolerance and/or persistence to a given antibiotic.
We found that loss of function of ArgA or ArgD, dispensable members of the essential arginine biosynthesis pathway in Msm, confers both tolerance and high persistence to kanamycin through up-regulation of the gene encoding the transcriptional regulator WhiB7 and the downstream effector, Eis. Loss of ArgA or ArgD also elicits HS upon exposure to rifampicin and MIC-shifted resistance to the macrolide clarithromycin, both of which are also conferred by whiB7 up-regulation. Our results reveal that perturbation of an essential pathway can trigger multiple forms of resistance to antibiotics targeting other pathways.
These findings have major implications for antibiotic therapy in three respects. Regimens should be designed so that no component induces phenotypic resistance to the others. Targets that are poorly vulnerable should be avoided, as the partial inhibition typically achieved by an antibiotic is likely to trigger phenotypic resistance mechanisms rather than killing the pathogen. Last, WhiB7 emerges as a target whose inhibition might increase the efficiency and efficacy of antibiotic regimens for mycobacterial diseases.
RESULTS
A novel method allows in vitro isolation of mutants displaying HS without an increase in MIC
The systematic isolation of HS mutants without an increase in MIC, which comprise mutants that display tolerance and/or high persistence, is challenging because they are difficult to separate from MIC-shifted genetic resisters that emerge during antibiotic exposure. To overcome this problem, we developed a four-step in vitro method that allows for spatiotemporal separation of genetic MIC-shifted resisters from survivors without an increase in MIC, including both HS mutants and wild-type (WT) persisters, thereby permitting isolation and subsequent validation of HS mutants from a genetically heterogeneous bacterial population (Fig. 2A). In the first step, survivors of antibiotic exposure are isolated. A large number of bacteria (~107) are seeded onto a filter that is then placed onto an agar plate containing 10× the MIC of an antibiotic (fig. S1A). Bacteria are exposed to the antibiotic for a period of time predetermined to kill approximately 99.9999% of a WT population (fig. S1B). During the exposure period, genetic MIC-shifted resisters with ΔMIC > 10-fold grow and will form visible colonies on the filters given sufficient incubation time, while genetic MIC-shifted resisters with ΔMIC between 2- and 10-fold and non–MIC-shifted survivors cannot form visible colonies. Next, the filter is transferred to an agar plate containing 2× the MIC of the same antibiotic. Genetic MIC-shifted resisters with ΔMIC between 2- and 10-fold now grow and form visible colonies given sufficient incubation time. Last, the filter is transferred to an agar plate containing 0.4% charcoal for 24 hours to remove residual antibiotic without affecting cell viability (29) before final transfer to a plate of growth-permissive agar for recovery of candidate non–MIC-shifted survivors, which grow as spatially isolated colonies. Colonies generated from non–MIC-shifted survivors are distinguished temporally from those formed by genetic MIC-shifted resisters by their late emergence, i.e., only during the recovery phase.
Fig. 2. A forward-genetic, in vitro screen yields HS mutants in Msm.
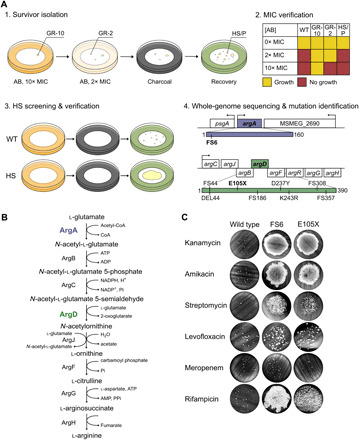
(A) HS mutants are isolated in vitro in four steps. (1) Survivor isolation. AB, antibiotic. GR-10, genetic MIC-shifted resister with ΔMIC > 10-fold. GR-2, genetic MIC-shifted resister with ΔMIC between 2- and 10-fold. HS, high-survival mutant. P, WT persister. (2) MIC verification. (3) HS screening and verification. (4) Whole-genome sequencing and mutation identification. We identified one Msm mutant with a mutation in argA (purple) and eight with mutations in argD (green), all isolated on kanamycin. Mutations annotated by their effect on the protein product. FS, frameshift. DEL, deletion (DEL44 is missing amino acids 44 to 51). (B) ArgA (purple) and ArgD (green) are members of the arginine biosynthesis pathway. CoA, coenzyme A. ATP, adenosine triphosphate. ADP, adenosine diphosphate. NADP+, nicotinamide adenine dinucleotide phosphate. NADPH, reduced form of NADP+. AMP, adenosine monophosphate. Pi, inorganic phosphate; PPi, inorganic pyrophosphate. (C) Survival of ArgA-FS6 and ArgD-E105X upon exposure to 10× the MIC of the aminoglycosides kanamycin (KAN), amikacin (AMK), and streptomycin (STR); levofloxacin (LVX); meropenem (MEM); or rifampicin (RIF). Representative of 2 to 36 independent experiments (FS6: KAN 13, AMK 2, STR 2, LVX 2, MEM 2, RIF 2; E105X: KAN 36, AMK 3, STR 3, LVX 2, MEM 2, RIF 2). See also fig. S1 and table S2.
In the second step of the method, colonies that emerged during recovery are picked and outgrown in liquid medium, and their susceptibility to the antibiotic is tested to confirm the absence of an increase in MIC compared to the WT. Cultures confirmed to have arisen from non–MIC-shifted survivors can be WT persisters or harbor HS mutations. These two cases are distinguished in the third step by screening cultures for an increase in survival relative to the WT. Equal volumes of cultures diluted to the same optical density (OD) are seeded onto separate filters, and filters are placed onto an agar plate containing 10× the MIC of the antibiotic as in the first step of the method. After the exposure period, filters are transferred to agar plates with charcoal for 24 hours and then to growth-permissive agar plates for recovery. After recovery, the number of surviving bacteria is compared to that of a WT control (which typically has a number of survivors on the order of 100 to 101; see fig. S1B). Candidates with an increased number of survivors in at least two independent filter experiments qualify as HS mutants. In the final step, the genomes of confirmed HS mutants are sequenced to identify candidate HS mutations.
Loss-of-function mutations in ArgA and ArgD lead to three types of resistance to antibiotics with distinct targets in Msm
We used this method to isolate HS mutants from a WT population of Msm, which we assumed would contain spontaneous mutants that may display an HS phenotype; details of the screen are shown in table S1. Using the aminoglycoside kanamycin, which inhibits protein synthesis by binding to the 30S ribosomal subunit, as the exposure antibiotic, we recovered nine mutants with mutations in genes belonging to the arginine biosynthesis pathway: one in argA (MSMEG_2691) and eight in argD (MSMEG_3773) (Fig. 2, A and B, and table S2). In addition to a large increase in survival upon exposure to kanamycin, the mutants had increased survival when exposed to additional aminoglycosides, including amikacin and streptomycin, and to rifampicin, which targets the RNA polymerase RpoB (Fig. 2C shows ArgD-E105X as a representative argD mutant; the seven other argD mutants are shown in fig. S1C). In contrast, these mutants did not show increased survival when exposed to levofloxacin or meropenem, which respectively target DNA gyrase and penicillin binding proteins (Fig. 2C and fig. S1C). We confirmed that the MIC of all antibiotics for which the mutants had an HS phenotype was unchanged compared to the WT (fig. S2A). A quantitative kill curve performed in liquid medium containing kanamycin showed that the mutants exhibited both a slowed kill rate for the bulk population (genetic tolerance) and a proportion of persister cells five to six orders of magnitude higher than the WT (high persistence) after 12 hours of antibiotic exposure, which is when the WT kill curve starts to reflect the slow kill rate of the persister population (Fig. 3A). The mutants displayed a third type of resistance—genetic MIC-shifted resistance—to the macrolide clarithromycin (Fig. 3B), which binds to the 50S ribosomal subunit to inhibit protein synthesis.
Fig. 3. HS and MIC-shifted resistance phenotypes are caused by loss of ArgA or ArgD function and are complemented by provision of l-arginine.
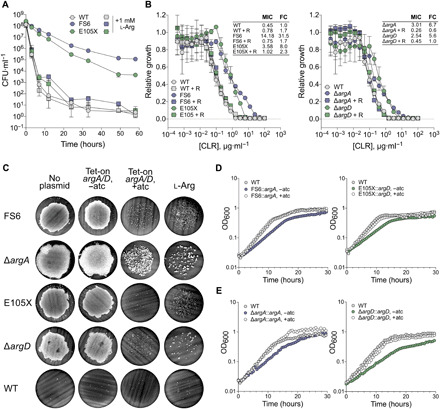
Error bars: ±SD. (A) Colony-forming unit (CFU) counts during exposure of ArgA-FS6 and ArgD-E105X to kanamycin (3.5 μg·ml−1) (5× MIC). Symbols, means of three replicates. Representative of four independent experiments. (B) Clarithromycin (CLR) MIC (μg·ml−1) for ArgA-FS6, ArgD-E105X, ΔargA, and ΔargD with or without provision of 1 mM l-arginine (R). Growth relative to a no-antibiotic control. Symbols, means of three replicates. Values were calculated as described in Materials and Methods. Fold change (FC) relative to WT (same in both graphs). Representative of two independent experiments. (C) Kanamycin (10× MIC) survival of argA and argD mutants complemented with an anhydrotetracycline (atc)–inducible (tet-on) copy of WT argA (ArgA-FS6, ΔargA) or argD (ArgD-E105X, ΔargD, WT) or grown with provision of 1 mM l-arginine (l-Arg). Representative of 5 to 55 independent experiments [FS6: 13, genetic complementation (comp) 8, l-Arg comp 5; ΔargA: 44, genetic comp 9, l-Arg comp 13; E105X: 36, genetic comp 8, l-Arg comp 23; ΔargD: 55, genetic comp 17, l-Arg comp 19]. (D and E) Growth of tet-on strains from (C) with or without atc. Symbols, mean OD600 of two to three replicates. Representative of 5 to 10 independent experiments (FS6, 5; E105X, 10; ΔargA, 9; ΔargD, 10). See also fig. S2.
Because many of the mutations were frameshifts, we suspected that all of them led to loss of function. To test this, we transformed the argA and argD mutants with anhydrotetracycline (atc)–inducible (tet-on) copies of WT argA and argD, respectively. Growing the tet-on strains in the presence of atc complemented the increase in survival upon exposure to kanamycin (Fig. 3C), genetic MIC-shifted resistance to clarithromycin (fig. S2B), and the growth defect exhibited by the FS6 and E105X mutants (Fig. 3D), whose respective generation times were 1.51 and 1.62 times longer than that of the WT when grown in 7H9 medium (fig. S2C). To further test whether loss of ArgA or ArgD function was responsible for the resistance phenotypes of the argA and argD mutants, we constructed a genetic knockout strain for each gene (fig. S2D) and showed that these strains recapitulated the HS (Fig. 3C and fig. S2, A and E) and genetic MIC-shifted resistance phenotypes (Fig. 3B and fig. S2F) as well as the growth defect (Fig. 3E and fig. S2C) of the mutants in an ArgA- or ArgD-complementable manner. In addition to clarithromycin, loss of ArgA or ArgD function also conferred MIC-shifted resistance to the macrolides azithromycin and erythromycin (fig. S2, G and H).
Provision of l-arginine complements the resistance phenotypes of the argA and argD mutants
Because ArgA and ArgD are involved in arginine biosynthesis, we measured l-arginine levels in the ΔargA and ΔargD mutants to determine whether arginine deficiency might play a role in the resistance phenotypes. Neither strain had an arginine level significantly different from that of the WT (fig. S3A), most likely due to recruitment of other enzymes to replace the functions of ArgA and ArgD. Such recruitment could reflect and/or result in transcriptional and other changes that could lead to the resistance phenotypes observed. Provision of exogenous l-arginine might forestall those changes, presumably by shutting down the arginine biosynthesis pathway, and complement the resistance phenotypes. Genetic tolerance and high persistence to kanamycin were complemented by growing the mutants in medium supplemented with 1 mM l-arginine (Fig. 3, A and C), but not d-arginine or l-lysine (fig. S3B). Provision of l-arginine also reversed genetic MIC-shifted resistance to clarithromycin (Fig. 3B) and HS upon exposure to rifampicin (fig. S2E) and restored a WT growth rate (fig. S3C). To understand the kinetics of complementation of the HS phenotype by l-arginine, we grew the ArgA-FS6 mutant in medium supplemented with 1 mM l-arginine for varying lengths of time before seeding filters and exposing them to kanamycin. We found that between 14 and 24 hours of growth in medium supplemented with l-arginine was required for maximum complementation of the HS phenotype (fig. S3D). In contrast, the growth phenotype was complemented within 1 hour of placing bacteria into medium supplemented with 1 mM l-arginine (fig. S3C). This result suggested a dissociation between the HS and growth phenotypes.
HS upon exposure to kanamycin is not a consequence of slow growth or a general decrease in metabolic activity
We used the ArgD-E105X::tet-on argD strain to further investigate whether the HS and growth phenotypes could be dissociated. We grew this strain in medium with varied concentrations of atc and tested the survival upon exposure to kanamycin and the growth rate at each concentration. A low concentration of atc (0.8 ng/μl) restored growth rate to that of the WT but did not complement the HS phenotype (Fig. 4A). In addition, the ArgD-E105X mutant grew at the same rate as the WT in Sauton’s, a defined minimal medium (Fig. 4B), but still displayed HS upon exposure to kanamycin (Fig. 4C). Further, in contrast to reports that observed an inverse relationship between growth rate and phenotypic resistance to antibiotics (30–32), WT Msm grown in Sauton’s had a generation time 1.42 times longer than that of the WT grown in 7H9 (Fig. 4B and fig. S2C) and comparable to the generation times of the FS6 and ΔargA mutants in 7H9 (fig. S2C) but did not display increased phenotypic resistance to kanamycin (Fig. 4C). Together, these results demonstrated that slow growth is neither necessary nor sufficient for HS upon exposure to kanamycin in mycobacteria.
Fig. 4. HS upon exposure to kanamycin is not a consequence of slow growth or generally decreased metabolic activity.
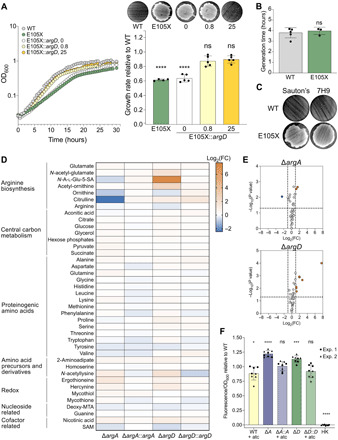
ns, not significant; *P < 0.05; ***P < 0.001; ****P <0.0001. Error bars: ±SD. (A) ArgD-E105X with an atc-inducible copy of WT argD (E105X::argD) was grown in 0, 0.8, or 25 ng/ml of atc. Left: Growth curves. Symbols, mean OD600 of two to three replicates. Right: Growth rate relative to WT mean [bars, means of four to five replicate cultures (symbols) pooled from two independent experiments] and kanamycin survival (filters above bars). Generation times compared to WT by one-way analysis of variance (ANOVA). Growth curves and filters representative of two independent experiments. (B) Generation times of WT versus ArgD-E105X in Sauton’s (unpaired two-tailed t test). Bars, means of N independent experiments (symbols; N = 5 for WT and 3 for E105X) with three to four replicates each. (C) Kanamycin survival of WT and ArgD-E105X in Sauton’s. Representative of four to five independent experiments (E105X in Sauton’s, 4; WT in Sauton’s versus 7H9, 5). (D and E) Metabolites from ΔargA, ΔargA::argA, ΔargD, and ΔargD::argD provisionally identified by accurate mass and retention time. (D) Colors reflect mean log2 FC relative to WT from three independent experiments (three replicates each). N-A-l-Glu-5-SA, N-acetyl-l-glutamate-5-semialdehyde; deoxy-MTA, deoxy-methylthioadenosine; SAM, S-adenosylmethionine. (E) Mean log2 FC compared to a theoretical mean of 0 (two-tailed one-sample t test). Dotted lines, cutoffs (significance, P = 0.05; FC, ±2). (F) Strains from (D) were incubated with resazurin. Fluorescence (excitation 544 nm, emission 590 nm) was normalized to OD600 before addition of resazurin and then to WT mean. Bars, means of eight replicate cultures (symbols) pooled from two independent experiments (exp.). Means compared to a theoretical mean of 1 (two-tailed one-sample t test). HK, heat-killed. See also data S1.
Given reports that metabolic inactivity can protect bacteria from antibiotic lethality (33), we next wondered whether a generalized decrease in metabolic activity could underlie the resistance phenotypes in our mutants. We extracted metabolites from WT Msm, the ΔargA and ΔargD mutants, and their respective complemented strains and compared the levels of 40 metabolites (provisionally identified based on accurate mass and retention time) that represented diverse metabolic categories, including the arginine biosynthesis pathway, central carbon metabolism, proteinogenic amino acids, amino acid precursors and derivatives, redox molecules, nucleoside-related molecules, and cofactor-related molecules (Fig. 4D). Outside of molecules in the arginine biosynthesis pathway, the levels of no metabolites were decreased more than twofold compared to the WT in either mutant (Fig. 4, D and E, and data S1). Metabolic blocks could lead to accumulation of metabolites in blocked pathways; the levels of only two metabolites outside of the arginine biosynthesis pathway in the ΔargA mutant and three in the ΔargD mutant were increased more than twofold compared to the WT (Fig. 4, D and E, and data S1). To complement our metabolomics findings, we measured respiratory activity using a resazurin assay, in which the nonfluorescent molecule resazurin is converted to the fluorescent product resorufin during bacterial respiration (34). Compared to WT Msm, the ΔargA and ΔargD mutants respectively displayed 1.22- and 1.13-fold higher metabolism of resazurin (Fig. 4F), indicative of increased rather than decreased respiration. Together, these results showed that the resistance phenotypes in the ΔargA and ΔargD mutants were not associated with a generalized decrease in metabolic activity.
ΔargA and ΔargD mutants overexpress genes encoding the transcriptional regulator WhiB7 and members of its regulon, including the aminoglycoside-modifying enzyme Eis
To obtain mechanistic insight into the resistance phenotypes, we analyzed the transcriptomes of the ΔargA and ΔargD mutants and a WT Msm control grown without or with provision of 1 mM l-arginine, corresponding respectively to a condition in which the mutants displayed resistance phenotypes and a condition in which the phenotypes were complemented (fig. S4A). In the WT control, the expression levels of only three genes differed by more than fourfold with versus without provision of l-arginine (fig. S4B and data S2). In contrast, expression levels of 83 genes in the ΔargA mutant and 104 genes in the ΔargD mutant differed by more than fourfold between conditions (Fig. 5A and data S2). Among genes whose expression was increased more than fourfold with provision of l-arginine, the two mutants had only seven in common, including three in common with the WT (Fig. 5B). In contrast, among genes whose expression was increased more than fourfold without provision of l-arginine, the mutants had 46 in common (Fig. 5B). This suggested that genes expressed in response to loss of either ArgA or ArgD could underlie the resistance phenotypes.
Fig. 5. ΔargA and ΔargD mutants overexpress genes encoding the transcriptional regulator WhiB7 and members of its regulon, including the aminoglycoside-modifying enzyme Eis.
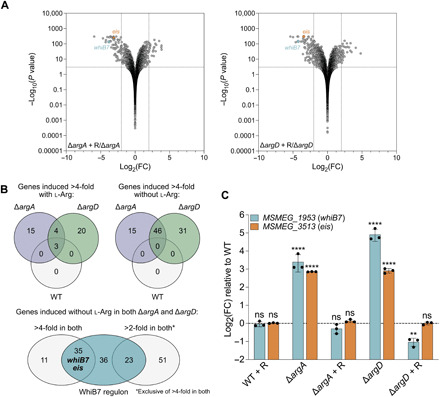
RNA sequencing was performed on RNA extracted from ΔargA, ΔargD, and WT strains of Msm grown ± provision of 1 mM l-arginine (R). (A) −Log10 P value versus log2 FC for each gene (symbols) in the ΔargA (left) and ΔargD (right) mutants grown with versus without provision of l-arginine. Dotted lines, cutoffs (FC ± 4, P value 0.05). (B) Top: Intersection of transcripts induced >4-fold with (left) and without (right) provision of l-arginine in the ΔargA, ΔargD, and WT strains. Bottom: Intersection of transcripts induced >4-fold and >2-fold without provision of l-arginine in both the ΔargA and ΔargD strains with the Msm WhiB7 regulon reported in (35). The >2-fold category is exclusive of transcripts induced >4-fold in both mutants. (C) whiB7 and eis transcript levels in the ΔargA, ΔargD, and WT strains grown ± provision of 1 mM l-arginine. Bars, mean log2 fold change relative to WT of three replicates (symbols). Error bars, ±SD. ΔCp values compared to WT ΔCp (one-way ANOVA). **P < 0.01; ****P < 0.0001. Representative of two independent experiments. See also fig. S4 and data S2.
Three other genes in the arginine biosynthesis pathway—argC, argB, and argJ—were among the 46 highly expressed in both mutants without provision of l-arginine, suggesting activation of the pathway. Genes encoding the transcriptional regulator WhiB7 (MSMEG_1953) and members of its regulon were also highly expressed; 35 of the 46 genes, plus 23 additional genes up-regulated at least twofold in both mutants, are part of the Msm WhiB7 regulon (Fig. 5B and data S2) (35). WhiB7 is a member of the WhiB-like family of transcriptional regulators, which are unique to actinomycetes and contain four conserved cysteine residues involved in the binding of iron-sulfur clusters (36, 37). WhiB7 has an AT (adenine-thymine)-hook domain that enables it to bind AT-rich DNA sequences in the promoters of the genes in its regulon (36, 37). The WhiB7 regulon includes WhiB7 itself in addition to genes encoding efflux pumps and enzymes that modify antibiotics or their targets; deletion of whiB7 renders mycobacteria more susceptible to ribosome-targeting antibiotics (35, 36, 38). We therefore hypothesized that overexpression of WhiB7 mediated the resistance phenotypes for aminoglycosides, rifampicin, and clarithromycin observed in our mutants. Among WhiB7 regulon members up-regulated in our mutants, we hypothesized that the aminoglycoside-modifying enzyme Eis (MSMEG_3513) was responsible for mediating HS upon exposure to aminoglycosides. We confirmed increased expression of whiB7 and eis in the ΔargA and ΔargD mutants and complementation of expression levels of both genes upon provision of l-arginine by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Fig. 5C).
Mutations upstream of whiB7 and/or eis confer HS upon exposure to aminoglycosides or rifampicin and MIC-shifted resistance to clarithromycin
Our hypothesis that whiB7 and/or eis overexpression mediated the resistance phenotypes to aminoglycosides, rifampicin, and clarithromycin was supported by an evolution experiment designed to select for mutants in the ΔargA and ΔargD backgrounds for which HS upon exposure to kanamycin was no longer complemented by provision of l-arginine (Fig. 6A). We grew the mutants in medium containing l-arginine such that they would have a WT level of survival when exposed to kanamycin. Next, we subjected them to cycles of kanamycin exposure until survival increased, indicating emergence of a mutation that prevented complementation of the HS phenotype by l-arginine or conferred HS de novo. The genomic sequences of four mutants isolated from cultures with an HS phenotype despite provision of l-arginine (Fig. 6B) contained mutations in either the 5′ untranslated region (5′UTR) of whiB7 (39) or the putative promoter region of eis (Fig. 6C and table S2), which contains a 5–base pair (bp) AT-rich region starting 45 bp before the annotated start codon that may constitute the WhiB7 binding site (Fig. 6C). Moreover, two additional HS mutants that we isolated on kanamycin along with the argA and argD mutants in our initial screen also had mutations in the 5′UTR of whiB7 (Fig. 6C and table S2). We dubbed these HS mutants ups_w-m1 and ups_w-m2 for upstream whiB7 mutant. Similar to the ΔargA and ΔargD mutants, both ups_w-m1 and ups_w-m2 displayed HS upon exposure to kanamycin, amikacin, streptomycin, and rifampicin (Fig. 6D and fig. S5A), for which their MICs were within 2.25-fold that of the WT (fig. S5B), well below the concentration used in the plates for the HS assay. They also exhibited genetic MIC-shifted resistance to clarithromycin, azithromycin, and erythromycin (Fig. 6E and fig. S5, C and D).
Fig. 6. Overexpression of whiB7 confers HS upon exposure to kanamycin or rifampicin and MIC-shifted resistance to CLR.
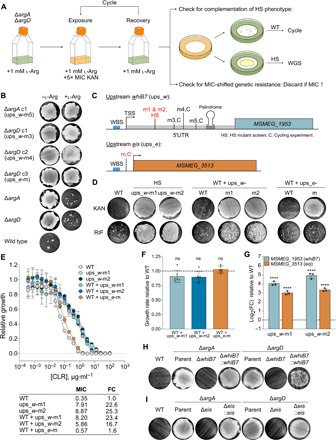
Error bars: ±SD. (A) Cycling experiment (see main text). l-Arg, l-arginine. KAN, kanamycin. WGS, whole-genome sequencing. (B) KAN survival of mutants from (A) grown ± provision of 1 mM l-Arg. (C) Schematics of whiB7 (MSMEG_1953) and eis (MSMEG_3513) showing mutants in (B) and two HS mutants (red; used in subsequent experiments). TSS, transcription start site. WBS, WhiB7 binding site. (D) KAN or rifampicin (RIF) survival of ups_w-m1 and ups_w-m2 mutants and WT containing 485 bp upstream + MSMEG_1953 from WT, ups_w-m1, or ups_w-m2 or 236 bp upstream + MSMEG_3513 from WT or ups_e-m. Representative of two to eight independent experiments (KAN, RIF; ups_w-m: 8, 4; ups_w-m2: 6, 3; WT + ups_w-wt: 3, 2; ups_w-m1: 5, 2; ups_w-m2: 3, 2; ups_e-wt: 3, 2; ups_e-m: 4, 2). (E) CLR MIC (μg·ml−1) for strains in (D). Growth relative to no-antibiotic control. Values calculated as described in Materials and Methods. FC relative to WT. Symbols, means of three replicates. Representative of two independent experiments. (F) Growth rates relative to WT of WT + ups_w-m1, ups_w-m2, and ups_e-m. Bars, means of six independent experiments (symbols) with three to five replicates each. Generation times compared to WT (one-way ANOVA). (G) whiB7 and eis transcript levels in ups_w-m1 and ups_w-m2 mutants. Bars, mean log2 FC relative to WT of three replicates (symbols). ΔCp values compared to WT ΔCp (one-way ANOVA). ****P < 0.0001. Representative of two independent experiments. (H and I) KAN survival of ΔargAΔwhiB7, ΔargAΔeis, ΔargDΔwhiB7, and ΔargDΔeis complemented with the ups_e-WT construct in (C) or a longer ups_w-WT construct containing 800 bp upstream of whiB7. Representative of three to seven independent experiments (ΔwhiB7: ΔargA, 7; ΔargD, 4; Δeis: 4). See also fig. S5 and table S2.
We hypothesized that the mutations in the four cycling mutants and two additional HS mutants resulted in constitutive overexpression of the downstream gene to confer resistance. We cloned whiB7 from the ups_w-m1 and ups_w-m2 HS mutants as representatives of the whiB7 5′UTR-mutated strains along with the 485-bp region upstream, which contains the WhiB7 binding site and complete 5′UTR [Fig. 6C, as described in (39)]. We also cloned eis along with the 236 bp, preceding it from the evolution experiment mutant with a mutation upstream of eis (ups_e-m for upstream eis mutant). Upon transforming these constructs into WT Msm, all transformants displayed HS upon exposure to kanamycin (Fig. 6D) and amikacin (fig. S5A). In addition, the ups_w transformants displayed HS upon exposure to rifampicin (Fig. 6D) and genetic MIC-shifted resistance to clarithromycin (Fig. 6E). MICs for the three strains were within 2.25-fold of the WT MICs for the antibiotics upon exposure to which the strains displayed HS (fig. S5B), again well below the concentrations used in the HS assay. The strains all had a WT growth rate (Fig. 6F), further supporting our evidence that slow growth is not required for the whiB7- and/or eis-mediated HS phenotypes. We confirmed overexpression of whiB7 and eis for the two HS mutants with mutations upstream of whiB7 by qRT-PCR (Fig. 6G). These results demonstrated that mutations upstream of whiB7 or eis confer HS upon exposure to aminoglycosides and that mutations upstream of whiB7 additionally confer HS upon exposure to rifampicin and genetic MIC-shifted resistance to macrolides, most likely by mediating constitutive overexpression of other members of the WhiB7 regulon, such as efflux pumps. Differences in expression level of the specific direct effectors or in efficiency of drug neutralization by these effectors likely underlie differences in the types and strengths of resistance phenotype observed for each drug.
Deleting whiB7 or eis in the ΔargA and ΔargD backgrounds abolishes HS upon exposure to kanamycin
To further test whether whiB7 and/or eis overexpression were mediators of HS upon exposure to kanamycin in the ΔargA and ΔargD mutants and evaluate whether there were additional contributors, we deleted either whiB7 or eis in the ΔargA and ΔargD backgrounds to create ΔargAΔwhiB7, ΔargAΔeis, ΔargDΔwhiB7, and ΔargDΔeis double-deletion strains (fig. S5E). HS upon exposure to kanamycin was abolished with the deletion of either whiB7 (Fig. 6H) or eis (Fig. 6I). We complemented the Δeis strains with ups_e-wt but were unable to complement the ΔwhiB7 strains with ups_w-wt containing 485 bp upstream of whiB7 (fig. S5F), suggesting that this construct may be missing uncharacterized regulatory elements important for up-regulation of whiB7 in the context of the ΔargA and ΔargD mutants. We achieved complementation with a longer construct beginning 800 bp upstream of the whiB7 start codon (ups800_w-wt). However, complementation was not seen in every experiment and was more complete in the ΔargAΔwhiB7 mutant compared to the ΔargDΔwhiB7 mutant, suggesting the existence of additional regulatory factors at the promoter level or at posttranscriptional or posttranslational levels that may differ among experiments and between the two strains. Neither the ups800_w-wt (fig. S5G) nor the ups_e-wt construct (Fig. 6D) conferred an HS phenotype on the WT. Together, these results demonstrated that eis was the sole direct mediator of HS upon exposure to kanamycin in the ΔargA and ΔargD mutants.
An ΔargD mutant in Mtb displays HS upon exposure to gentamicin
To investigate whether disruption of arginine biosynthesis would confer HS upon exposure to aminoglycosides in Mtb, we constructed an ΔargD (rv1655) Mtb mutant (fig. S6A). We chose to construct the argD mutant since we had obtained a larger diversity of mutations in this gene in Msm compared to argA, which suggested that spontaneous mutations may arise more frequently in this gene, making it potentially more relevant to HS in physiological contexts. Unlike the Msm ΔargD mutant, the Mtb ΔargD mutant was an arginine auxotroph (Fig. 7A), and this auxotrophy was bactericidal (Fig. 7B), consistent with what has been reported for Mtb ΔargB and ΔargF mutants (40). To minimize killing due to auxotrophy in experiments to test the survival of the mutant upon exposure to antibiotics, we assessed the killing kinetics of a panel of aminoglycosides for which the Msm E105X mutant displayed HS (fig. S6B) to determine which produced the fastest killing of WT Mtb (fig. S6C). On the basis of the results, we chose exposure to gentamicin at a concentration equivalent to ~15× the MIC (fig. S6D) for 14 days. To test survival of the ΔargD mutant upon exposure to gentamicin, we washed cultures of the mutant grown with provision of 1 mM l-arginine, resuspended in medium without l-arginine supplementation, and seeded filters after 4 days of arginine starvation, a window within which we did not observe death from arginine auxotrophy (Fig. 7B). The Mtb ΔargD mutant displayed a statistically significant increase in survival upon exposure to gentamicin that was complemented by constitutively expressed argD, although the effect size was smaller than for the Msm ΔargA and ΔargD mutants (Fig. 7, C and D). The increase in survival was complemented by growth in 1 mM l-arginine in two of three experiments. Over an exposure period of 14 days, the viable Mtb ΔargD mutant population would be expected to decrease by two log10 from auxotrophy alone (Fig. 7B); thus, the ΔargD mutant displayed increased survival despite substantial loss of viability from arginine deprivation. Similar to the Msm ΔargA and ΔargD mutants, the Mtb ΔargD mutant also overexpressed both whiB7 (rv3197A) and eis (rv2196) (Fig. 7E), demonstrating that perturbation of arginine biosynthesis leads to whiB7 up-regulation in Mtb and suggesting that whiB7 and eis could also be the mediators of the HS phenotype in Mtb.
Fig. 7. An Mtb ΔargD mutant displays HS upon exposure to gentamicin.
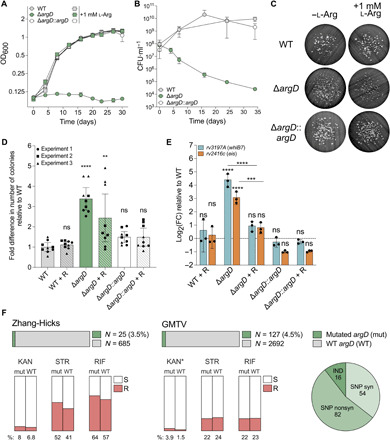
**P < 0.01; ***P < 0.001; ****P < 0.0001. Error bars: ±SD. (A) Growth of ΔargD and ΔargD::argD (constitutive expression) ± provision of 1 mM l-arginine. Symbols, mean OD600 of three replicates. Representative of three to four independent experiments. (B) CFU counts during arginine starvation of ΔargD and ΔargD::argD. Symbols, means of three replicates. Representative of three independent experiments. (C and D) Gentamicin survivors (15× MIC) for ΔargD and ΔargD::argD grown ± provision of 1 mM l-arginine (R). (D) Bars, mean fold difference relative to WT for replicates (symbols) pooled from three experiments [three replicates each; representative example in (C)]. Normalized counts compared to WT (one-way ANOVA). (E) whiB7 and eis transcript levels in strains from (C) and (D). Bars, mean log2 FC relative to WT of three replicates (symbols). ΔCp values compared to all other ΔCp values (one-way ANOVA). Indicators above bars, comparison to WT. Representative of two independent experiments. (F) Horizontal bars, argD mutations (green) among 710 Mtb clinical isolates from references (41, 42) and 2819 from the Genome-based Mycobacterium tuberculosis Variation (GMTV) Database (43). Circle graph, argD mutation types; IND, indel; SNP, single-nucleotide polymorphism; syn, synonymous; nonsyn, nonsynonymous. Vertical bars, MIC-shifted resistance (red) to kanamycin (KAN), streptomycin (STR), and/or rifampicin (RIF) for argD mutated (mut) and argD WT strains. R, resistant; S, sensitive. Asterisk (*) indicates significant difference between mut and WT. See also fig. S6 and tables S3 to S5.
Mutations in argD are present among Mtb clinical isolates
Given the potential clinical relevance of increased survival upon exposure to aminoglycosides, which are used to treat Mtb infections resistant to first-line antibiotics, we investigated whether clinical isolates harbor mutations in argD. Among 710 clinical isolates from patients in China (41, 42), 25 strains (3.5%) had missense (13 strains) or synonymous (12 strains) mutations in argD (Fig. 7F and table S3). Although synonymous mutations would not be expected to affect protein function, they could affect expression, e.g., due to introduction of rare codons. Since HS mutations may precede the emergence of genetic MIC-shifted resistance (18, 28), we quantified the proportion of strains displaying genetic MIC-shifted resistance to kanamycin, streptomycin, and/or rifampicin, three antibiotics for which we observed a strong HS phenotype in our Msm mutants, among strains with and without mutations in argD. A greater proportion of strains with mutations in argD had genetic MIC-shifted resistance to kanamycin (8% versus 6.8%), streptomycin (52% versus 41%), and/or rifampicin (64% versus 57%) (Fig. 7F), suggesting trends that could potentially achieve statistical significance with larger sample sizes.
To this end, we searched the Genome-based Mycobacterium tuberculosis Variation Database (43), which contains 2819 genomic sequences of Mtb isolates from around the world, the majority are from Russia (38%) or Malawi (35%). We found a total of 111 single-nucleotide polymorphisms (SNPs), including 68 missense and 43 synonymous mutations (Fig. 7F and table S4), and 16 indels in argD (Fig. 7F and table S5). Assuming that all are in unique strains, this represents 4.5% of the total number of strains in the database, a similar proportion to that in the 710 clinical isolates from China. We found a statistically significant enrichment for genetic MIC-shifted resistance to kanamycin (3.9% versus 1.5%; P = 0.036) among strains with mutations in argD compared to strains without mutations (Fig. 7F). Given our results showing that ArgD function is essential in vitro, the SNPs in these clinical isolates may represent mutations that do not completely abrogate the aminotransferase function of ArgD. Alternatively, some of these mutations may abolish ArgD function, as the existence of indels suggests that Mtb can scavenge arginine from the human host despite its apparent inability to do so in mice (40); if so, then the amount it scavenges could be sufficient to prevent death from auxotrophy but insufficient to complement HS induced by loss of ArgD function. Overall, these findings demonstrate that argD mutations are present in diverse Mtb clinical isolates and that strains bearing them are enriched for genetic MIC-shifted resistance, suggesting that these mutations may have clinical relevance.
DISCUSSION
We developed a powerful method for the isolation of HS mutants and used it to uncover mutants in mycobacteria with defects in the arginine biosynthesis pathway that display three forms of resistance to antibiotics targeting other pathways, a finding that has implications for developing antibiotics and using them in combination. The predominant approach for isolation of HS mutants relies on cyclic exposure of mutagenized bacterial populations to one or more antibiotics in liquid culture with outgrowth periods in antibiotic-free medium between cycles and monitoring of the survival fraction via separate liquid exposure assays at various intervals. After an increase in survival fraction is detected, individual mutants are isolated as colonies by plating on solid agar, grown in liquid medium, and individually tested for an increase in survival upon antibiotic exposure (13, 44–47). Although this approach has facilitated identification of HS mutants in several species, including Mtb (44), its limitations have likely hindered the discovery of HS mutants on a larger scale. If only one antibiotic is used for the exposure cycles, then genetic MIC-shifted resisters can emerge and overwhelm a liquid culture. A combination of antibiotics is often used to prevent genetic MIC-shifted resistance (44, 45), but this precludes discovery of mutants with HS specific to a single antibiotic, which could have clinical relevance. In addition, mutants with fitness defects in antibiotic-free conditions may be lost during outgrowth, and this approach requires an additional step on solid agar after cycling in liquid to isolate individual mutants. Moreover, testing HS candidates for an increase in survival by liquid exposure assay is labor intensive, requiring removal of the antibiotic via several rounds of washing, plating of serial dilutions on solid agar, and enumeration of colony-forming units (CFUs). The method presented here circumvents these problems. Spatiotemporal separation of cells that survive antibiotic exposure on the filter surface means that growth of genetic MIC-shifted resisters does not affect detection of non–MIC-shifted survivors, including HS mutants. Outgrowth on the filter as spatially separated colonies also prevents loss of mutants with fitness defects, allowing recovery of a wider variety of mutants, and eliminates the need for an extra step to isolate them. After recovery, the survival phenotypes of individual candidates can be assessed in a few quick steps—filter seeding followed by two plate transfers—permitting rapid screening of large numbers of candidates. These advantages coupled with flexibility in the size and format of the input bacterial population, drug concentration, and exposure time suit the method to a variety of applications with any culturable microorganism.
Our screen for HS mutants in mycobacteria revealed that overexpression of whiB7, either through loss of ArgA or ArgD function or through mutation in the whiB7 5′UTR, led to genetic tolerance, high persistence, and genetic MIC-shifted resistance to antibiotics with distinct targets. Antibiotic tolerance and persistence are often associated with slowed or halted replication and/or reduced metabolic activity (16, 30, 32, 33, 48–54). This is sometimes interpreted as a mechanistic explanation for reduced lethality of the antibiotic, the inference being that processes targeted by the antibiotic are less active during slow growth or inactive when replication is arrested and are thus not susceptible to inhibition. However, actively replicating cells, including mycobacteria, can exhibit antibiotic persistence (7, 55–57). Here, we provide further compelling evidence that neither halted replication, slowed replication, nor slowed metabolism is required for HS by tolerance or persistence. The slower growth imposed by loss of ArgD function was not related to HS upon exposure to kanamycin, and neither the ΔargA nor the ΔargD mutant showed a generalized decrease in metabolic activity. Moreover, strains overexpressing whiB7 and/or eis due to upstream mutations displayed HS upon exposure to aminoglycosides and rifampicin but had a WT growth rate. HS in these strains and the ΔargA and ΔargD mutants relied on overexpression of WhiB7 and Eis, a mechanism that does not depend on nonreplication or slowed metabolism. Thus, our results support a growing recognition that specific alterations in cellular metabolism, rather than general slowing or cessation of metabolic activities, can mediate mechanisms of tolerance and persistence in mycobacteria that may be clinically relevant. Identification of specific mechanisms that confer tolerance and persistence, such as the one described here, could help guide discovery of compounds that prevent these types of resistance.
Our findings both support and extend the recent evidence from Lopatkin et al. (58) that “metabolic adaptation may represent a class of resistance mechanisms.” In that study, mutations in Escherichia coli genes serving central carbon metabolism conferred MIC-shifted resistance to antibiotics of diverse classes. In the one case studied in depth, this was associated with slowed metabolism (58). In our studies, mutations in mycobacteria in a metabolic pathway not only conferred resistance to antibiotics of multiple classes but also conferred three different forms of resistance—MIC-shifted resistance, genetic tolerance, and high persistence—and this was not attributable to slowed growth or decreased metabolism.
An open question raised by our work is how whiB7 expression is triggered by loss of ArgA or ArgD. Expression of whiB7 has been linked to other amino acid biosynthetic enzymes: Constitutive expression of MSMEG_4060, a putative aspartate aminotransferase, prevents antitermination in the 5′UTR of whiB7 in Msm, thereby repressing whiB7 expression (59). Further, MSMEG_0688, a member of the whiB7 regulon that is likely to be an alanine aminotransferase (60), amplifies the expression of whiB7, and MSMEG_6017, likely an alanine-valine aminotransferase (60), represses it (61). The mechanisms behind these interactions remain to be elucidated, but, combined with the results presented here, they point to a general link between amino acid biosynthetic pathways and regulation of whiB7 expression, likely through induction of metabolic shifts. WhiB7 activity is enhanced in reducing, but not oxidizing, conditions (39). Perturbations in amino acid biosynthetic pathways might invoke redox changes that could activate low levels of WhiB7 protein constitutively present in the cell, leading to amplification of whiB7 expression via autoinduction. In support of this, deleting argB or argF in Mtb results in both oxidative stress and up-regulation of whiB7 (40), although this appears to be opposite to the conditions that have been shown to favor WhiB7 activity (39). In addition, the arginine repressor ArgR, which might function as an activator of the arginine biosynthesis pathway (62), could regulate expression of whiB7; in support of this hypothesis, ArgR from Streptomyces coelicolor has been reported to bind to ARG boxes present upstream of whiB (63, 64), the ortholog of mycobacterial whiB2 (37). Future work will focus on elucidating the mechanism of whiB7 activation following loss of ArgA or ArgD.
Previous reports in Msm and Mtb have shown that ΔwhiB7 mutants are more susceptible to ribosome-targeting antibiotics (36, 38, 39). Overexpression of WhiB7 and/or Eis in Mtb due to upstream mutations has been associated with genetic MIC-shifted resistance to aminoglycosides (65, 66), in Msm, and induction of whiB7 expression via exposure to subinhibitory concentrations of macrolides or the amino acid analog acivicin results in MIC-shifted resistance to macrolides, including clarithromycin (39), as we observed in our mutants. Here, we show that WhiB7 can additionally mediate tolerance and high persistence to aminoglycosides. We further link WhiB7 to resistance to rifampicin in the form of non–MIC-shifted HS, demonstrating that WhiB7 affords protection against non–ribosome-targeting antibiotics. In addition to strong WhiB7-mediated HS phenotypes for ΔargA and ΔargD mutants in Msm, we showed a smaller but significant phenotype for the ΔargD mutant in Mtb upon exposure to the aminoglycoside gentamicin despite loss of viable cells to arginine auxotrophy, and we documented whiB7 up-regulation in this strain. Further, we found argD mutations in the genomic sequences of Mtb clinical isolates. We speculate that some of these mutations might partially disrupt function of ArgD in a way that allows synthesis of sufficient arginine to avoid auxotrophy while activating whiB7 expression to elicit an HS phenotype.
The lethality of starvation for l-arginine combined with the apparent inability of Mtb to scavenge the amino acid from murine hosts (40) has prompted interest in the l-arginine biosynthesis pathway as a target for new TB drugs (40, 67). The work we present here suggests that caution should be taken in developing inhibitors of this pathway for therapeutic use. Partial inhibition of enzymes in the arginine biosynthesis pathway, as would be expected for pharmaceutical interventions, might lead not to death of the bacilli but rather to induction of phenotypic resistance in the form of HS upon exposure to rifampicin and aminoglycosides, important components of first- and second-line therapy for TB, respectively. Along those lines, whiB7 overexpression and the resulting resistance phenotypes we showed here could be induced in many clinical contexts given the broad range of activators that have been identified, including a wide variety of structurally and mechanistically diverse compounds, subinhibitory concentrations of antibiotics, the transition from exponential to stationary phase, low iron, heat shock, hypoxia, and the macrophage-like intracellular environment (38, 39, 68–73). As suggested by our argA and argD mutants and by a study in Msm that documented up-regulation of whiB7 in response to mutations affecting ribosome assembly (74), it is also possible that mutations in a multitude of pathways could conduce to whiB7 overexpression. Thus, our results suggest that WhiB7 could be an important driver of diverse forms of resistance that could lead to treatment failure in TB and other mycobacterial infections. Development of drugs to target WhiB7 could block these forms of resistance and enhance treatment efficiency and efficacy.
Our results demonstrate a mechanism by which perturbation of an essential metabolic pathway elicits antibiotic resistance. Although metabolic perturbation was achieved by means of genetic mutation in our studies, subinhibitory concentrations of antibiotics could accomplish the same effect, leading to increased phenotypic resistance by WhiB7-mediated or other mechanisms. Standard treatment for drug-sensitive TB consists of 2 months of treatment with four drugs followed by 4 months of treatment with two of them. The requirement for such a prolonged course of antibiotics may be due, in part, to induction of phenotypic resistance by exposure to subinhibitory concentrations of one or more of the drugs in certain biological niches or during certain phases of treatment. Most current assays to evaluate drug interactions use growth inhibition as the output metric. Although these assays can capture phenotypic MIC-shifted resistance, they are blind to phenotypic tolerance and high persistence. This underscores the need for new assays such as the filter method introduced here to evaluate the bactericidal potential of drug combinations. Such assays will aid in investigating the extent to which induction of phenotypic resistance occurs with current and proposed treatment regimens for TB. Formulations that minimize induction of phenotypic resistance or inclusion of an agent that blocks this induction might greatly shorten the course of treatment.
MATERIALS AND METHODS
Bacterial strains and culture conditions
Strains used in this study are listed in table S6. Msm strains were cultured at 37°C with shaking in Middlebrook 7H9 medium supplemented with 0.2% glycerol, 0.05% tyloxapol, 0.5% bovine serum albumin fraction V, 0.2% dextrose, and 0.085% NaCl. For strains bearing chromosomally integrated antibiotic resistance cassettes, appropriate antibiotics [hygromycin (50 μg·ml−1), zeocin (25 μg·ml−1), and streptomycin (25 μg·ml−1)] were included during strain generation but not in cultures grown for experiments. Mtb strains were cultured in the same way but without shaking. For the Msm experiments conducted in Sauton’s minimal medium, the medium contained 0.05% potassium phosphate monobasic, 0.05% magnesium sulfate heptahydrate, 0.2% citric acid, 0.005% ferric ammonium citrate, 0.2% glycerol, 0.05% ammonium sulfate, 0.01% zinc sulfate, and 0.05% tyloxapol and was adjusted to pH 7.4 (75).
Plasmid construction
All plasmids were constructed using Invitrogen Gateway recombination cloning technology. Plasmids are indicated in parenthesis after the strain name in table S6. T10M-P1 is an atc-inducible mycobacterial promoter (76, 77), Ptb38 is a constitutive mycobacterial promoter (76), and pGMCS refers to the pDE43-MCS destination vector in Gateway cloning system, which confers streptomycin resistance (77). Primers used in cloning are listed in table S7.
Filter seeding
The bottom portion of a Swinnex Filter Holder sized for either 25- or 47-mm filters (MilliporeSigma, SX0002500 or SX0004700) was mounted on a 1200-cm3 Medi-Vac Guardian suction canister (Cardinal Health, 65651-212). Bacterial cells were deposited on Durapore hydrophilic polyvinylidene difluoride (PVDF) membrane filters with a pore size of 0.22 μm and a diameter of either 25 or 47 mm (MilliporeSigma, GVWP02500 or GVWP04700) to a final density of approximately 6 × 103 CFU/mm2. To achieve this density, 1 ml of bacterial culture diluted to OD = 0.03 was seeded onto 25-mm filters, and 5 ml of bacterial culture diluted to an OD of 0.02 was seeded onto 47-mm filters under the assumption that an OD of 1 is equivalent to a concentration of ~108 CFU/ml. See fig. S1A for a schematic representation of the filter seeding process.
HS mutant screening and verification
Survivor isolation
To isolate non–MIC-shifted survivors of antibiotic exposure, seeded filters were incubated at 37°C on 7H11 agar plates supplemented with 0.5% glycerol and containing 10× the MIC of an antibiotic for the predetermined exposure time for that antibiotic (table S8). This phase allowed killing of susceptible cells and emergence of colonies formed by MIC-shifted resisters with ΔMIC > 10-fold. Next, filters were transferred to plates containing 2× the MIC of the antibiotic and incubated for 5 days at 37°C. This step was to allow for emergence of colonies formed by MIC-shifted resisters with ΔMIC between 2- and 10-fold, and an exposure time of 5 days was chosen to allow sufficient time for formation of visible colonies. Filters were then transferred to plates containing 0.4% charcoal and incubated at 37°C for ~24 hours to remove residual antibiotic. Last, filters were transferred to 7H11 agar plates supplemented with 0.5% glycerol to allow for growth of any non–MIC-shifted survivors. To document the emergence of MIC-shifted and non–MIC-shifted survivors, photographs of filters were taken after both exposure phases and after recovery. Non–MIC-shifted candidates were considered to be those colonies that had emerged during the final incubation phase. Isolated colonies were picked from the filters and grown in 7H9 for further processing.
MIC verification
The absence of an increase in MIC for the antibiotic used in the survivor isolation step for cultures grown from colonies that emerged during recovery was confirmed using an abbreviated MIC assay. Two microliters of bacterial culture was inoculated in 200 μl of 7H9 supplemented with no antibiotic or 7H9 supplemented with 1× the MIC, 2× the MIC, 4× the MIC, or 10× the MIC of the antibiotic, and OD600 was read after 2 days of incubation at 37°C. Candidates that displayed growth at higher concentrations of the antibiotic than the WT were considered to be genetic MIC-shifted resisters and were excluded from further analysis, while candidates displaying WT susceptibility to growth inhibition were considered to have arisen from non–MIC-shifted survivors.
HS candidate screening and verification
To distinguish candidates that arose from WT persisters from those bearing HS mutations, we grew each candidate plus a WT control to mid-log phase in 7H9 and seeded them individually onto filters as described in the “Filter seeding” section. Filters were transferred to 7H11 agar plates supplemented with 0.5% glycerol and 10× the MIC of the antibiotic on which each candidate had been isolated and then incubated at 37°C for the appropriate exposure time (table S8). Filters were then transferred to 7H11 plates supplemented with 0.5% glycerol and 0.4% charcoal (charcoal plates) for approximately 24 hours at 37°C to remove residual antibiotic before final transfer to 7H11 plates supplemented with 0.5% glycerol (recovery plates). Candidates with a >10-fold increase in survivors relative to the WT control were tested again to confirm that the increase was reproducible. Candidates with a level of survival reproducibly >10-fold higher than that of the WT were considered to be HS mutants.
Filter assay for assessment of survival upon exposure to an antibiotic
For Msm strains, bacterial cultures were grown to mid-log phase in 7H9 supplemented with any compounds for which the effect on survival upon antibiotic exposure was to be tested or in Sauton’s minimal medium. Filters were seeded and processed as described in the “HS candidate screening and verification” section. Photographs of filters were taken after 3 days on recovery plates. To more easily visualize bacterial growth, raw images were changed to gray scale, and the brightness and contrast were adjusted. For each filter image, the same adjustments were applied to the entire image, and all adjustments were linear.
For Mtb strains, the procedure was the same except charcoal and recovery plates were supplemented with 1 mM l-arginine to support the auxotrophic ΔargD strain used in the experiments, and recovery plates were additionally supplemented with 10% OADC (oleic acid–albumin–dextrose–catalase). Photographs of filters were taken after about 20 days on recovery plates.
Determination of MICs
Varied concentrations of each antibiotic were prepared in the appropriate solvent (table S8) by performing twofold serial dilutions. Two microliters of each concentration and the solvent alone were deposited into three rows of a 96-well plate per strain to be tested. Bacterial cultures were grown to log phase in 7H9. For Msm, cultures were diluted to an OD of 0.001 or 0.025, and 198-μL aliquots were dispensed into wells prefilled with antibiotic. Plates were incubated at 37°C with shaking at 100 rpm for 2 to 3 days. For Mtb, cultures were diluted to an OD of 0.04 and dispensed into wells with antibiotic. Plates were incubated at 37°C without shaking for 9 days. Wells were resuspended before reading the OD600. MIC values were calculated in GraphPad Prism using an open-source file (available at www.graphpad.com/support/faq/fitting-bacterial-growth-data-to-determine-the-mic-and-nic/) that calculates the MIC based on a nonlinear regression model developed by Lambert and Pearson (78). In this method, the MIC is defined as the intersection of the horizontal line projected from the bottom plateau of the curve with a line projected from the inflection point with slope equivalent to the slope of the curve at that point.
Growth curves
For Msm, strains were grown in 7H9 or Sauton’s minimal medium to mid-log phase and then diluted to an OD of 0.05 in 7H9 supplemented with any compound for which the effect on growth was to be tested or in Sauton’s. Two to five replicates consisting of 100 μl of diluted culture each were added to a 96-well plate. A breathable membrane allowing for gas exchange was applied to the plate, which was inserted into a Spark multimode microplate reader (Tecan) set to 37°C. OD600 readings were taken every 30 min with orbital shaking at 180 rpm between reads.
For Mtb, strains were grown in 7H9 (+1 mM l-arginine for the ΔargD strain) and then diluted to an OD of 0.05 in 7H9 ± 1 mM l-arginine (ΔargD strain was washed before diluting). Three replicates consisting of 200 μl of diluted culture each were added to a 96-well plate. The plate was incubated at 37°C without shaking. To monitor growth, periodic OD600 readings were taken in a plate reader after resuspending the contents of each well.
Kanamycin kill curves
Msm strains were grown to mid-log phase in 7H9 with or without 1 mM l-arginine. Cultures were diluted in triplicate to an OD of 0.1 in 10 ml of 7H9 with or without 1 mM l-arginine. An aliquot from each culture was taken and serially diluted, and 10 μl of three dilutions was plated on 7H11 agar supplemented with 0.5% glycerol and 0.4% charcoal to enumerate input CFUs. Kanamycin was added to the 10-ml cultures to a final concentration of 3.5 μg·ml−1 (5× the MIC), and cultures were incubated at 37°C with shaking at 100 rpm. CFUs were enumerated at seven additional time points (~4, 8, 12, 24, 36, 48, and 60 hours) as described for the input CFUs; addition of charcoal in the plates for CFU enumeration eliminated the need for washing the cultures to remove kanamycin before plating. At later time points, to increase the limit of detection, cultures were concentrated 10- or 100-fold by spinning down 100 μl or 1 ml of culture, resuspending the pellet in 20 μl of 7H9, and plating.
Metabolomics
Msm strains were grown to mid-log phase in 7H9 or 7H9 + atc (250 ng/ml) for complemented strains. Three replicate 25-mm PVDF filters (MilliporeSigma, GVWP02500) were seeded with the equivalent of 1 ml of culture at OD600 = 1 and placed onto 7H11 agar plates supplemented with 0.5% glycerol and atc (250 ng/ml) for complemented strains. Filters were incubated at 37°C for 16 hours before being placed into tubes containing 800 μl of 40% acetonitrile, 40% methanol, and 20% H2O solution and silica beads. Bacteria were lysed by bead beating (two bead beating cycles separated by 5 min rest; each cycle consisted of three rounds of beating for 50 s at 6500 rpm followed by 30 s rest) in a Precellys 24 tissue homogenizer cooled with a Cryolys cooling unit (Bertin Instruments) to obtain metabolite extracts. Extracts were analyzed using a Diamond Hydride type C column (Cogent) on an Agilent 1200 Infinity liquid chromatography system coupled to an Agilent 6230 time-of-flight spectrometer (79). Metabolite peaks were identified on the basis of accurate mass and retention time and integrated in Agilent MassHunter Qualitative Analysis software. Peak integrations were normalized to residual protein concentration of the metabolite extracts quantified by the Bio-Rad DC Protein Assay kit.
Resazurin assay
Msm strains were grown to mid-log phase in 7H9 or 7H9 + atc (250 ng/ml). Cultures were diluted, split into four replicate wells of a 24-well plate, and incubated for 2 to 3 hours at 37°C with shaking at 100 rpm. Heat-killed cells were prepared by heating an aliquot of WT Msm culture to 90°C for 40 min. After incubation, OD600 was measured, and 100 μl of the alamarBlue Cell Viability Reagent (Invitrogen, DAL1100) was added to 800 μl of culture. Cultures were incubated for 30 min at 37°C with shaking at 100 rpm, then 200 μl from each culture was transferred to a clear-bottom black 96-well plate, and fluorescence at an excitation of 544 nm and emission of 590 nm was measured in a plate reader. Fluorescence readings were normalized to the OD600 of the cultures immediately preceding addition of alamarBlue.
RNA extraction
Total RNA was extracted from 10 to 20 ml of bacterial culture by washing the cells in guanidinium thiocyanate buffer, bead beating washed pellets into TRIzol, extracting RNA in chloroform, and purifying extracted RNA with the Direct-zol RNA miniprep kit (Zymo Research, R2052). After elution, DNA was removed from total RNA samples by incubation with TURBO DNase (Ambion, AM2238) at 37°C for 1 hour. Total RNA samples were then repurified using the RNA Clean and Concentrator-25 kit (Zymo Research, R1017) for RNA sequencing (RNA-seq) samples or the Direct-zol RNA miniprep kit for qRT-PCR samples.
Transcriptomics
Samples for RNA-seq were split into two groups of 12: WT Msm (WT-D) and ΔargD were cultured in triplicate in 7H9 or 7H9 supplemented with 1 mM l-arginine (R), and WT Msm (WT-A) and ΔargA were cultured the same way. Total RNA was extracted from 20 ml of culture grown to mid-log phase as described in the “RNA extraction” section. Ribosomal RNA (rRNA) was depleted from total RNA samples using the RiboMinus Transcriptome Isolation Kit for bacteria (Thermo Fisher Scientific, K155004), and rRNA-depleted RNA was concentrated using the RNA Clean and Concentrator-25 kit. Indexed RNA-seq libraries were prepared from rRNA-depleted RNA samples using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, E7760S) and NEBNext Multiplex Oligos for Illumina Index Primers Set 1 (New England Biolabs, E7335S; contains twelve unique index primers). RNA libraries were purified with AMPure XP Beads (Beckman Coulter, A63881), quality was checked on a Bioanalyzer, and the 12 uniquely indexed libraries in each group were pooled together. Sequencing of the two pools was performed on an Illumina HiSeq4000 instrument by the Genomics Core Facility of the Weill Cornell Medicine Core Laboratories Center. Pairwise differential expression analysis was performed by the Core on the eight conditions (WT-D, WT-D + R, ΔargD, ΔargD + R, WT-A, WT-A + R, ΔargA, ΔargA + R; each an average of results from three replicate libraries) to identify genes significantly differentially regulated between each condition.
Quantitative reverse transcription polymerase chain reaction
Primers and probes for qRT-PCR are listed in table S9. All were designed using the Biosearch Technologies RealTimeDesign qPCR Assay Design Software. Primers were ordered from Invitrogen, and probes were ordered from TIB Molbiol. Specificity of primer/probe sets was tested using a dilution series of genomic DNA from Msm or Mtb.
To measure transcript amounts, bacterial cultures (10 ml for Msm, 20 ml for Mtb) were grown in triplicate to mid-log phase in 7H9 with or without provision of 1 mM L-arginine. For the cultures without l-arginine supplementation, the auxotrophic Mtb ΔargD strain was grown to mid-log phase in medium with 1 mM l-arginine, washed, resuspended in medium without l-arginine, and incubated for 4 days. Total RNA was extracted as described in the “RNA extraction” section. cDNA was prepared using 500 ng of RNA as the template for M-MuLV reverse transcriptase (New England Biolabs, M0253L), and corresponding no-RT reactions were also prepared as a control for residual genomic DNA. Cycling with quantification was performed in a Roche LightCycler 480 instrument. Second-derivative analysis was performed in the LightCycler 480 Software (release 1.5.0 SP3) to obtain crossing points (Cp) for each sample for the test genes and an internal control (mysA, MSMEG_2758, for Msm and sigA, rv2703, for Mtb). Statistical analysis was performed on ΔCp values in GraphPad Prism using an ordinary one-way analysis of variance (ANOVA) test with multiple comparisons. For each test strain or condition, a ΔΔCp value was calculated by subtracting the average ΔCp of the three WT biological replicates from the average ΔCp of the three test strain/condition replicates. Log2 fold change values relative to the WT control were calculated as 2−ΔΔCp.
Knockout construction
In Msm, all genetic knockout strains (argD, MSMEG_3771; argA, MSMEG_2691; whiB7, MSMEG_1953; eis, MSMEG_3513) were produced by allelic exchange. Plasmids were constructed using the temperature-sensitive pXSTS plasmid backbone (80) containing a recombination fragment consisting of ~500 bp upstream of the gene to delete, a resistance cassette (either hygromycin or zeocin), and ~500 bp downstream of the gene to delete. Msm strains were transformed with a plasmid, plated on 7H11 plates supplemented with 0.5% glycerol and either hygromycin (50 μg·ml−1) or zeocin (25 μg·ml−1), and incubated for 4 days at 30°C. To select for single-crossover transformants, colonies were spotted with 5% pyrocatechol, and yellow colonies were grown at 37°C in 7H9 containing either hygromycin (50 μg·ml−1) or zeocin (25 μg·ml−1). To select for double-crossover transformants, an aliquot of each culture was spread onto 7H11 agar supplemented with 0.5% glycerol, hygromycin (50 μg·ml−1) or zeocin (25 μg·ml−1), and 10% sucrose, and plates were incubated at 40°C. Colonies were spotted with 5% pyrocatechol, and those that remained white were grown in 7H9 supplemented with hygromycin (50 μg·ml−1) or zeocin (25 μg·ml−1) and then validated by PCR. The PCR-verified strains chosen for use in subsequent experiments were further validated by whole-genome sequencing.
In Mtb, the ΔargD strain was constructed by recombinase-mediated recombination of a PCR product containing a hygromycin resistance cassette between sequences homologous to the ~500-bp regions upstream and downstream of the argD gene (Rv1655). Mtb H37Rv cells were first transformed with an episomal plasmid containing the phage-derived RecET recombinase (pNit-recET-sacB-kanR) (81) and grown to mid-log phase in 7H9 supplemented with kanamycin (25 μg·ml−1). RecET expression was induced by addition of isovaleronitrile to 1 μM for 24 hours and glycine to 0.2 M for 16 hours. Electrocompetent cells prepared from the induced culture were transformed with 1 μg of PCR product and allowed to recover for 24 hours in 7H9 supplemented with 1 mM l-arginine. Transformants were plated on 7H10 agar supplemented with 0.5% glycerol, 10% OADC, hygromycin (50 μg·ml−1), and 1 mM l-arginine. After 16 days of incubation at 37°C, colonies that had emerged were cultured in 7H9 supplemented with hygromycin (50 μg·ml−1) and 1 mM l-arginine and then validated by PCR. To select for clones that had lost the pNit-recET-sacB-kanR plasmid, each validated strain was streaked onto 7H10 agar supplemented with 0.5% glycerol, 10% OADC, 1 mM l-arginine, and 8.5% sucrose. Sucrose-resistant colonies were then patched first onto 7H10 agar supplemented with 0.5% glycerol, 10% OADC, 1 mM l-arginine, and kanamycin (25 μg·ml−1) and then onto the same agar without kanamycin. Cultures of kanamycin-sensitive strains were grown in 7H9 supplemented with hygromycin (50 μg·ml−1) and 1 mM l-arginine to obtain the final Mtb ΔargD strains. The strain chosen for use in subsequent experiments was further validated by whole-genome sequencing.
Whole-genome sequencing and SNP identification
Genomic DNA was isolated from stationary phase cultures by bead beating to break the cells followed by phenol-chloroform extraction and ethanol precipitation. Genomic DNA from HS mutants, cycling mutants, Msm ΔargA, Msm ΔargD, and Mtb ΔargD strains was sequenced by the Integrated Genomics Operation at the Memorial Sloan Kettering Cancer Center. Genomic DNA from Msm ΔargAΔwhiB7, Msm ΔargDΔwhiB7, Msm ΔargAΔeis, and Msm ΔargDΔeis strains was sequenced by BGI Genomics. Genetic knockouts were verified by visually confirming the absence of reads in the area of the expected knockout using the Integrative Genomics Viewer from The Broad Institute. SNPs were identified relative to the National Center for Biotechnology Information (NCBI) reference sequence NC_008596.1 for Msm and NZ_CM001515.1 for Mtb.
Clinical isolate search
Variant calling files for 710 Mtb clinical isolates associated with studies published by Zhang et al. (41) and Hicks et al. (42) were provided by the Fortune Lab (variants called based on NCBI reference sequence NC_000962.3). Variants in the Genome-based Mycobacterium tuberculosis Variation Database (43) were identified via the web-based interface (information was gathered on 25 September 2020).
Statistical analysis
All statistical tests were performed in GraphPad Prism except for the comparison of proportions of genetic antibiotic resistance between Mtb strains with and without argD mutations presented in Fig. 7F. Proportions were compared by calculating the test statistic for comparison of two population proportions
Where z is a value on the z distribution; and are the proportions with the characteristic of interest in samples 1 and 2, respectively; is the proportion with the characteristic of interest in the two samples combined; and n1 and n2 are the sizes of samples 1 and 2, respectively. The test was two-tailed.
Acknowledgments
We thank J. Bean (Sloan Kettering Institute), P. Zumbo [Weill Cornell Medicine (WCM)], L. Botella (Francis Crick Institute), S. Li (The Rockefeller University), N. Nahiyaan (WCM), T. Hartman (WCM), and S. Mishra (WCM) for assistance; D. Schnappinger (WCM) for advice; N. Hicks and S. Fortune (Harvard T.H. Chan School of Public Health) for providing variant calling files for 710 Mtb genomes; and R. Wigneshweraraj, J. Penadés, and D. Holden (MRC Centre for Molecular Bacteriology and Infection, Imperial College London) for critical reading of this manuscript. Funding: This work was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-Ph.D. Program (to S.M.S.), an F30 Predoctoral Fellowship from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number F30AI140623 (to S.M.S.), a National Defense Science and Engineering Graduate Fellowship from the U.S. Department of Defense (to S.M.S.), the Department of Infectious Disease at Imperial College London (to J.V.), the Potts Memorial Foundation (to J.V.), a European Commission Marie Skłodowska-Curie Actions Individual Fellowship (to H.B.), the Abby and Howard Milstein Program in Chemical Biology and Translational Medicine, and the William Randolph Hearst Trust. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Author contributions: Conceptualization: S.M.S., H.B., C.N., and J.V. Methodology: S.M.S. and J.V. Resources: S.E., K.R., and C.N. Visualization: S.M.S. and J.V. Investigation: S.M.S., H.B., R.J., and J.V. Formal analysis: S.M.S. and J.V. Project administration: S.M.S. and J.V. Validation: S.M.S. and J.V. Supervision: C.N. and J.V. Funding acquisition: S.M.S., C.N., and J.V. Writing—Original draft: S.M.S. and J.V. Writing—Review and editing: S.M.S., C.N., and J.V. Competing interests: The authors declare that they have no competing interests. C.N. serves on advisory boards for the Tres Cantos Open Lab Foundation, Tri-Institutional Therapeutics Discovery Institute, Bridge Medicines, Leap Therapeutics, and Pfizer’s External Sciences and Innovation. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All bacterial strains listed in this paper can be provided by C.N.’s or J.V.’s lab pending scientific review and a completed material transfer agreement. Requests should be directed to C.N. at cnathan@med.cornell.edu and J.V. at j.vaubourgeix@imperial.ac.uk.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabh2037/DC1
REFERENCES AND NOTES
- 1.Schrader S. M., Vaubourgeix J., Nathan C., Biology of antimicrobial resistance and approaches to combat it. Sci. Transl. Med. 12, eaaz6992 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.J. O’Neill, Review on AMR - Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations (2014).
- 3.World Health Organization, Global tuberculosis report 2019 (2019); http://apps.who.int/bookorders.
- 4.Adam M., Murali B., Glenn N. O., Potter S. S., Epigenetic inheritance based evolution of antibiotic resistance in bacteria. BMC Evol. Biol. 8, 52 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niki M., Niki M., Tateishi Y., Ozeki Y., Kirikae T., Lewin A., Inoue Y., Matsumoto M., Dahl J. L., Ogura H., Kobayashi K., Matsumoto S., A novel mechanism of growth phase-dependent tolerance to isoniazid in mycobacteria. J. Biol. Chem. 287, 27743–27752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romilly C., Lays C., Tomasini A., Caldelari I., Benito Y., Hammann P., Geissmann T., Boisset S., Romby P., Vandenesch F., A non-coding RNA promotes bacterial persistence and decreases virulence by regulating a regulator in Staphylococcus aureus. PLOS Pathog. 10, e1003979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su H.-W., Zhu J.-H., Li H., Cai R.-J., Ealand C., Wang X., Chen Y.-X., Kayani M. u. R., Zhu T. F., Moradigaravand D., Huang H., Kana B. D., Javid B., The essential mycobacterial amidotransferase GatCAB is a modulator of specific translational fidelity. Nat. Microbiol. 1, 16147 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Sakatos A., Babunovic G. H., Chase M. R., Dills A., Leszyk J., Rosebrock T., Bryson B., Fortune S. M., Posttranslational modification of a histone-like protein regulates phenotypic resistance to isoniazid in mycobacteria. Sci. Adv. 4, eaao1478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amato S. M., Fazen C. H., Henry T. C., Mok W. W. K., Orman M. A., Sandvik E. L., Volzing K. G., Brynildsen M. P., The role of metabolism in bacterial persistence. Front. Microbiol. 5, 70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergmiller T., Andersson A. M. C., Tomasek K., Balleza E., Kiviet D. J., Hauschild R., Tkačik G., Guet C. C., Biased partitioning of the multidrug efflux pump AcrAB-TolC underlies long-lived phenotypic heterogeneity. Science 356, 311–315 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Vaubourgeix J., Lin G., Dhar N., Chenouard N., Jiang X., Botella H., Lupoli T., Mariani O., Yang G., Ouerfelli O., Unser M., Schnappinger D., McKinney J., Nathan C., Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe 17, 178–190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bigger J. W., Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244, 497–500 (1944). [Google Scholar]
- 13.Moyed H. S., Bertrand K. P., hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155, 768–775 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Yew W. W., Barer M. R., Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 56, 2223–2230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connolly L. E., Edelstein P. H., Ramakrishnan L., Why is long-term therapy required to cure tuberculosis? PLOS Med. 4, e120 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gollan B., Grabe G., Michaux C., Helaine S., Bacterial persisters and infection: Past, present, and progressing. Annu. Rev. Microbiol. 73, 359–385 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Bakkeren E., Diard M., Hardt W. D., Evolutionary causes and consequences of bacterial antibiotic persistence. Nat. Rev. Microbiol. 18, 479–490 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Levin-Reisman I., Ronin I., Gefen O., Braniss I., Shoresh N., Balaban N. Q., Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Windels E. M., Michiels J. E., Fauvart M., Wenseleers T., Van den Bergh B., Michiels J., Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J. 13, 1239–1251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leung V., Lévesque C. M., A stress-inducible quorum-sensing peptide mediates the formation of persister cells with noninherited multidrug tolerance. J. Bacteriol. 194, 2265–2274 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabral D. J., Wurster J. I., Belenky P., Antibiotic persistence as a metabolic adaptation: Stress, metabolism, the host, and new directions. Pharmaceuticals 11, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strydom N., Gupta S. V., Fox W. S., Via L. E., Bang H., Lee M., Eum S., Shim T. S., Barry C. E., Zimmerman M., Dartois V., Savic R. M., Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: A mechanistic model and tool for regimen and dose optimization. PLOS Med. 16, e1002773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goossens S. N., Sampson S. L., Rie A. V., Mechanisms of drug-induced tolerance in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 34, e00141-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lederberg J., Zinder N., Concentration of biochemical mutants of bacteria with penicillin. J. Am. Chem. Soc. 70, 4267–4268 (1948). [DOI] [PubMed] [Google Scholar]
- 25.Tomasz A., Albino A., Zanati E., Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227, 138–140 (1970). [DOI] [PubMed] [Google Scholar]
- 26.Fisher R. A., Gollan B., Helaine S., Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15, 453–464 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bartell J. A., Cameron D. R., Mojsoska B., Haagensen J. A. J., Sommer L. M., Lewis K., Molin S., Johansen H. K., Bacterial persisters in long-term infection: Emergence and fitness in a complex host environment. PLOS Pathog. 6, e1009112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J., Gefen O., Ronin I., Bar-Meir M., Balaban N. Q., Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 367, 200–204 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Gold B., Roberts J., Ling Y., Quezada L. L., Glasheen J., Ballinger E., Somersan-Karakaya S., Warrier T., Warren J. D., Nathana C., Rapid, semiquantitative assay to discriminate among compounds with activity against replicating or nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 6521–6538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pontes M. H., Groisman E. A., Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci. Signal. 12, eaax3938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claudi B., Spröte P., Chirkova A., Personnic N., Zankl J., Schürmann N., Schmidt A., Bumann D., Phenotypic variation of salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158, 722–733 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Tuomanen E., Cozens R., Tosch W., Zak O., Tomasz A., The rate of killing of Escherichia coli by β-lactam antibiotics is strictly proportional to the rate of bacterial growth. J. Gen. Microbiol. 132, 1297–1304 (1986). [DOI] [PubMed] [Google Scholar]
- 33.Lopatkin A. J., Stokes J. M., Zheng E. J., Yang J. H., Takahashi M. K., You L., Collins J. J., Bacterial metabolic state more accurately predicts antibiotic lethality than growth rate. Nat. Microbiol. 4, 2109–2117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campodónico V. L., Rifat D., Chuang Y.-M., Ioerger T. R., Karakousis P. C., Altered Mycobacterium tuberculosis cell wall metabolism and physiology associated with RpoB mutation H526D. Front. Microbiol. 9, 494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hurst-Hess K., Rudra P., Ghosh P., Mycobacterium abscessus WhiB7 regulates a species-specific repertoire of genes to confer extreme antibiotic resistance. Antimicrob. Agents Chemother. 61, e01347-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramón-García S., Ng C., Jensen P. R., Dosanjh M., Burian J., Morris R. P., Folcher M., Eltis L. D., Grzesiek S., Nguyen L., Thompson C. J., WhiB7, an Fe-S-dependent transcription factor that activates species-specific repertoires of drug resistance determinants in actinobacteria. J. Biol. Chem. 288, 34514–34528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bush M. J., The actinobacterial WhiB-like (Wbl) family of transcription factors. Mol. Microbiol. 110, 663–676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris R. P., Nguyen L., Gatfield J., Visconti K., Nguyen K., Schnappinger D., Ehrt S., Liu Y., Heifets L., Pieters J., Schoolnik G., Thompson C. J., Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 12200–12205 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burian J., Ramón-García S., Sweet G., Gómez-Velasco A., Av-Gay Y., Thompson C. J., The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J. Biol. Chem. 287, 299–310 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiwari S., Van Tonder A. J., Vilchèze C., Mendes V., Thomas S. E., Malek A., Chen B., Chen M., Kim J., Blundell T. L., Parkhill J., Weinrick B., Berney M., Jacobs W. R. Jr., Arginine-deprivation-induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 115, 9779–9784 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H., Li D., Zhao L., Fleming J., Lin N., Wang T., Liu Z., Li C., Galwey N., Deng J., Zhou Y., Zhu Y., Gao Y., Wang T., Wang S., Huang Y., Wang M., Zhong Q., Zhou L., Chen T., Zhou J., Yang R., Zhu G., Hang H., Zhang J., Li F., Wan K., Wang J., Zhang X. E., Bi L., Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 45, 1255–1260 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Hicks N. D., Yang J., Zhang X., Zhao B., Grad Y. H., Liu L., Ou X., Chang Z., Xia H., Zhou Y., Wang S., Dong J., Sun L., Zhu Y., Zhao Y., Jin Q., Fortune S. M., Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol. 3, 1032–1042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chernyaeva E. N., Shulgina M. V., Rotkevich M. S., Dobrynin P. V., Simonov S. A., Shitikov E. A., Ischenko D. S., Karpova I. Y., Kostryukova E. S., Ilina E. N., Govorun V. M., Zhuravlev V. Y., Manicheva O. A., Yablonsky P. K., Isaeva Y. D., Nosova E. Y., Mokrousov I. V., Vyazovaya A. A., Narvskaya O. V., Lapidus A. L., O’Brien S. J., Genome-wide Mycobacterium tuberculosis variation (GMTV) database: A new tool for integrating sequence variations and epidemiology. BMC Genomics 15, 308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torrey H. L., Keren I., Via L. E., Lee J. S., Lewis K., High persister mutants in Mycobacterium tuberculosis. PLOS ONE 11, e0155127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khare A., Tavazoie S., Extreme antibiotic persistence via heterogeneity-generating mutations targeting translation. mSystems 5, e00847-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slattery A., Victorsen A. H., Brown A., Hillman K., Phillips G. J., Isolation of highly persistent mutants of Salmonella enterica serovar typhimurium reveals a new toxin-antitoxin module. J. Bacteriol. 195, 647–657 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mechler L., Herbig A., Paprotka K., Fraunholz M., Nieselt K., Bertram R., A novel point mutation promotes growth phase-dependent daptomycin tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 5366–5376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shan Y., Brown Gandt A., Rowe S. E., Deisinger J. P., Conlon B. P., Lewis K., Formation A. P., Shan Y., Gandt A. B., Rowe S. E., Deisinger J. P., Conlon B. P., Lewis K., ATP-dependent persister formation in Escherichia coli. MBio 8, e02267-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conlon B. P., Rowe S. E., Gandt A. B., Nuxoll A. S., Donegan N. P., Zalis E. A., Clair G., Adkins J. N., Cheung A. L., Lewis K., Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 1, 16051 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Keren I., Minami S., Rubin E., Lewis K., Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2, e00100-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Helaine S., Cheverton A. M., Watson K. G., Faure L. M., Matthews S. A., Holden D. W., Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balaban N. Q., Merrin J., Chait R., Kowalik L., Leibler S., Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Stokes J. M., Lopatkin A. J., Lobritz M. A., Collins J. J., Bacterial metabolism and antibiotic efficacy. Cell Metab. 30, 251–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng E. J., Stokes J. M., Collins J. J., Eradicating bacterial persisters with combinations of strongly and weakly metabolism-dependent antibiotics. Cell Chem. Biol. 27, 1544–1552.e3 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Adams K. N., Takaki K., Connolly L. E., Wiedenhoft H., Winglee K., Humbert O., Edelstein P. H., Cosma C. L., Ramakrishnan L., Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wakamoto Y., Dhar N., Chait R., Schneider K., Signorino-Gelo F., Leibler S., McKinney J. D., Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Ueno H., Kato Y., Tabata K. V., Noji H., Revealing the metabolic activity of persisters in mycobacteria by single-cell D2O Raman imaging spectroscopy. Anal. Chem. 91, 15171–15178 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Lopatkin A. J., Bening S. C., Manson A. L., Stokes J. M., Kohanski M. A., Badran A. H., Earl A. M., Cheney N. J., Yang J. H., Collins J. J., Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 371, eaba0862 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burian J., Thompson C. J., Regulatory genes coordinating antibiotic-induced changes in promoter activity and early transcriptional termination of the mycobacterial intrinsic resistance gene whiB7. Mol. Microbiol. 107, 402–415 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Jansen R. S., Mandyoli L., Hughes R., Wakabayashi S., Pinkham J. T., Selbach B., Guinn K. M., Rubin E. J., Sacchettini J. C., Rhee K. Y., Aspartate aminotransferase Rv3722c governs aspartate-dependent nitrogen metabolism in Mycobacterium tuberculosis. Nat. Commun. 11, 1960 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.C. K. Lo Ng, thesis, University of British Columbia (2012). [Google Scholar]
- 62.Rustad T. R., Minch K. J., Ma S., Winkler J. K., Hobbs S., Hickey M., Brabant W., Turkarslan S., Price N. D., Baliga N. S., Sherman D. R., Mapping and manipulating the Mycobacterium tuberculosis transcriptome using a transcription factor overexpression-derived regulatory network. Genome Biol. 15, 502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Botas A., Pérez-Redondo R., Rodríguez-García A., Álvarez-álvarez R., Yagüe P., Manteca A., Liras P., ArgR of Streptomyces coelicolor is a pleiotropic transcriptional regulator: Effect on the transcriptome, antibiotic production, and differentiation in liquid cultures. Front. Microbiol. 9, 361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pérez-Redondo R., Rodríguez-García A., Botas A., Santamarta I., Martín J. F., Liras P., ArgR of Streptomyces coelicolor is a versatile regulator. PLoS ONE 7, e32697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reeves A. Z., Campbell P. J., Sultana R., Malik S., Murray M., Plikaytis B. B., Shinnick T. M., Posey J. E., Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob. Agents Chemother. 57, 1857–1865 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zaunbrecher M. A., Sikes R. D., Metchock B., Shinnick T. M., Posey J. E., Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 106, 20004–20009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mdluli K., Spigelman M., Novel targets for tuberculosis drug discovery. Curr. Opin. Pharmacol. 6, 459–467 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Geiman D. E., Raghunand T. R., Agarwal N., Bishai W. R., Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob. Agents Chemother. 50, 2836–2841 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larsson C., Luna B., Ammerman N. C., Maiga M., Agarwal N., Bishai W. R., Gene expression of Mycobacterium tuberculosis putative transcription factors whib1-7 in redox environments. PLOS ONE 7, e37516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin W., De Sessions P. F., Teoh G. H. K., Mohamed A. N. N., Zhu Y. O., Koh V. H. Q., Ang M. L. T., Dedon P. C., Hibberd M. L., Alonsoa S., Transcriptional profiling of Mycobacterium tuberculosis exposed to in vitro lysosomal stress. Infect. Immun. 84, 2505–2523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Homolka S., Niemann S., Russell D. G., Rohde K. H., Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: Delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLOS Pathog. 6, e1000988 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rohde K. H., Veiga D. F. T., Caldwell S., Balázsi G., Russell D. G., Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLOS Pathog. 8, e1002769 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pryjma M., Burian J., Kuchinski K., Thompson C. J., Antagonism between front-line antibiotics clarithromycin and amikacin in the treatment of Mycobacterium abscessus infections is mediated by the whiB7 gene. Antimicrob. Agents Chemother. 61, e01353-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gomez J. E., Kaufmann-Malaga B. B., Wivagg C. N., Kim P. B., Silvis M. R., Renedo N., Ioerger T. R., Ahmad R., Livny J., Fishbein S., Sacchettini J. C., Carr S. A., Hung D. T., Ribosomal mutations promote the evolution of antibiotic resistance in a multidrug environment. eLife 6, e20420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.B. W. Allen, Mycobacteria: General culture methodology and safety considerations, in Mycobacteria Protocols, T. Parish, N. G. Stoker, Eds. (Humana Press, 1998), pp. 15–30. [DOI] [PubMed] [Google Scholar]
- 76.Ehrt S., Guo X. V., Hickey C. M., Ryou M., Monteleone M., Riley L. W., Schnappinger D., Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33, e21 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schnappinger D., O’Brien K. M., Ehrt S., Construction of conditional knockdown mutants in mycobacteria. Methods Mol. Biol. 1285, 151–175 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Lambert R. J. W., Pearson J., Susceptibility testing: Accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J. Appl. Microbiol. 88, 784–790 (2000). [DOI] [PubMed] [Google Scholar]
- 79.M. Nandakumar, G. A. Prosser, L. P. S. de Carvalho, K. Rhee, in Mycobacteria Protocols, T. Parish, D. M. Roberts, Eds. (Springer New York, 2015), pp. 105–115. [Google Scholar]
- 80.D. Portevin, W. Malaga, C. Guilhot, in Mycobacteria Protocols: Second Edition, T. Parish, A. C. Brown, Eds. (Humana Press, 2009), pp. 229–243. [Google Scholar]
- 81.K. C. Murphy, K. Papavinasasundaram, C. M. Sassetti, in Mycobacteria Protocols, T. Parish, D. M. Roberts, Eds. (Springer New York, 2015), pp. 177–199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabh2037/DC1