

Targeting carbonic anhydrase IX acidifies intracellular pH, disrupts redox homeostasis, and creates vulnerability to ferroptosis.
Abstract
The metabolic mechanisms involved in the survival of tumor cells within the hypoxic niche remain unclear. We carried out a synthetic lethal CRISPR screen to identify survival mechanisms governed by the tumor hypoxia–induced pH regulator carbonic anhydrase IX (CAIX). We identified a redox homeostasis network containing the iron-sulfur cluster enzyme, NFS1. Depletion of NFS1 or blocking cyst(e)ine availability by inhibiting xCT, while targeting CAIX, enhanced ferroptosis and significantly inhibited tumor growth. Suppression of CAIX activity acidified intracellular pH, increased cellular reactive oxygen species accumulation, and induced susceptibility to alterations in iron homeostasis. Mechanistically, inhibiting bicarbonate production by CAIX or sodium-driven bicarbonate transport, while targeting xCT, decreased adenosine 5′-monophosphate–activated protein kinase activation and increased acetyl–coenzyme A carboxylase 1 activation. Thus, an alkaline intracellular pH plays a critical role in suppressing ferroptosis, a finding that may lead to the development of innovative therapeutic strategies for solid tumors to overcome hypoxia- and acidosis-mediated tumor progression and therapeutic resistance.
INTRODUCTION
The microenvironment of solid tumors is heterogeneous in oxygen availability, and most solid tumor types contain regions of hypoxia (1, 2). Hypoxia initiates a global adaptive transcriptional response (3), and engagement of the hypoxia-inducible factor (HIF) transcriptional program triggers a metabolic shift that is critical for cell survival (3). The decreased availability of oxygen initiates a shift in carbon utilization to increase dependence on glycolysis for energy production. This leads to accumulation of acidic by-products and reliance upon pH regulatory enzymes and transporters to maintain an alkaline intracellular pH (pHi) to sustain proliferation and survival (4). As a consequence of the increased efflux of lactate and protons, the extracellular environment acidifies. Akin to hypoxia, acidic pH also leads to metabolic rearrangement to meet cellular macromolecular and bioenergetic demands and sustain survival (4). Identifying strategies to target these features is important for improving therapeutic efficacy in these microenvironments.
Carbonic anhydrases are a family of metalloenzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate and protons (5). Carbonic anhydrase IX and XII (CAIX/XII) are hypoxia-induced (6), cell surface, extracellular facing pH regulatory enzymes. CAIX and CAXII aid in buffering pHi through cooperation with bicarbonate transporters (7–9). CAIX, in particular, predicts poor prognosis in a number of solid tumor types (10, 11) and plays a critical role in several features of high-grade tumors including invasion (12, 13), metastasis (14, 15), and cancer stem cell survival (16). While inhibitors of CAIX are under clinical evaluation (17), identifying survival mechanisms governed by CAIX and combinatorial approaches to improving therapeutic response and limit resistance is of critical importance.
Regulated cell death is a critical determinant in the success of cancer therapy. Hypoxia selects for mutations in TP53 enriching for cells in these niches that are less sensitive to initiation of apoptosis (18). Fortunately, additional modes of regulated cell death have emerged that can be engaged to overcome resistance to apoptosis (19). Ferroptosis is a nonapoptotic form of iron-dependent cell death resulting from toxic accumulation of phospholipid peroxidation (20). Ferroptosis is intimately linked to the metabolic status of the cell being influenced by redox homeostasis, iron, amino acid and lipid metabolism, and mitochondrial activity (21). Rewired cellular metabolism in response to hypoxia and acidosis influences many of the same pathways (4, 22, 23). The HIF pathway has even been implicated in both protecting from and inducing ferroptosis (24, 25). However, the molecular determinants dictating its role and ways to target these features are unclear.
Here, we identified cellular axes compensating for CA9 loss following an unbiased, genome-wide synthetic lethal CRISPR screen in hypoxic triple-negative breast cancer (TNBC) cells. We uncovered a pattern indicative of a vulnerability to perturbations in redox homeostasis, including the gene encoding the cysteine desulfurase, NFS1, an enzyme critical for iron-sulfur cluster biogenesis. During our investigation into the link between CA9 and NFS1, we found that CAIX influenced cell death by ferroptosis. Suppression of CAIX expression or CAIX/XII activities increased cellular reactive oxygen species (ROS) levels, creating a vulnerability to increases in cellular iron levels associated with NFS1 depletion, addition of exogenous iron, or transit in the bloodstream. Furthermore, the CAIX-generated bicarbonate was important in preventing this. In summary, the data described here demonstrate that the role of CAIX in maintaining an alkaline pHi is critical to suppress ferroptosis.
RESULTS
Identification of a synthetic lethal interaction between CA9 and NFS1
To identify synthetic lethal interactions with CA9, we knocked the gene out in SUM159PT TNBC cells using CRISPR-Cas9 (fig. S1, A to C). We then screened the genome-wide GeCKO lentiCRISPRv2 library A consisting of 65,383 single-guide RNAs (sgRNAs) targeting 19,050 genes in a dropout screen to identify synthetic lethal interactions (Fig. 1A). Control cells expressing a nontargeting sgRNA (NTC) and CA9 (carbonic anhydrase IX) knockout (KO) (CA9KO) cells were maintained in 1% O2 following transduction with the lentiviral library and passaged for 20 generations to allow adequate gene dropout time. Changes in sgRNA abundance corresponding to each gene between the initial (T0) and the final time point (T11; representing 20 generations) were determined by next-generation sequencing. We identified 762 statistically significant hits that we classified by biological function using gene ontology (Fig. 1B and table S1). Among the significantly enriched processes affected by loss of CA9 were RNA metabolism, nonsense-mediated decay, ribosomal RNA processing, selenocysteine synthesis, selenoamino acid metabolism, tricarboxylic acid (TCA) cycle, and electron transport pathways (Fig. 1B and fig. S1D). Further refining the CA9 synthetic lethal interactions into a network of functional pathways exposed vulnerable gene networks associated with cytoskeleton, cell cycle and mitosis, ribosome biogenesis, RNA processing, mitochondrial organization, DNA damage repair (DDR) and nucleic acid (NA) metabolism, and redox homeostasis (Fig. 1C). The redox homeostasis network is particularly interesting, as CAIX function has not been previously linked to redox homeostasis. Among the genes we identified involved in redox homeostasis were thioredoxin (TXN), glutathione S-transferases (GSTM2 and GSTM5), glutathione reductase (GSR), and the molybdenum cofactor (MOCS3), all genes playing a critical role in antioxidant defense (26). In addition, a set of genes involved in iron-sulfur cluster biogenesis including the cysteine desulfurase (NFS1), iron-sulfur cluster scaffold protein (ISCU), iron-sulfur cluster adapter protein (ISCA2), adenosine 5′-triphosphate–binding cassette subfamily B member 7 (ABCB7), and glutaredoxin 5 (GLRX5) were identified (27). Ranking hits according to their fitness score, one of the top synthetic lethal genes in the screen was NFS1 (Fig. 1D). NFS1 catalyzes the first step in the generation of iron-sulfur clusters, cofactors for proteins involved in a number of cellular functions, including the TCA cycle and electron transport chain, by removing the thiol group from cysteine generating alanine and a persulfide group on the scaffold protein ISCU (28, 29). Moreover, NFS1 was recently implicated in protection from the iron-dependent, oxidative form of cell death, ferroptosis (30). Given the evidence demonstrating that CAIX affects cellular metabolism and the abundance of enzymes involved in cell metabolism requiring iron-sulfur clusters as cofactors, we decided to pursue this synthetic lethal interaction (29, 31–35).
Fig. 1. Identification of a synthetic lethal interaction between CA9 and NFS1.
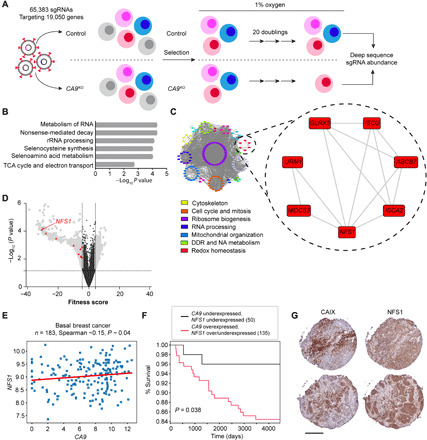
(A) Diagram outlining scheme of screen. (B) Gene ontology identification of pathways most significantly affected by CA9 loss during growth in hypoxia. rRNA, ribosomal RNA. (C) Cytoscape visualization of gene networks identified color-coded according to biological pathway. DDR, DNA damage repair; NA, nucleic acid. (D) Volcano plot depicting synthetic lethal hit ranking according to fitness score. Red triangles denote redox homeostasis network genes from (C). (E) Relationship of NFS1 and CA9 expression in the TCGA invasive breast cancer dataset in basal cases. n = 183 cases. (F) Kaplan-Meier curve visualizing survival fractions of patients from the entire invasive breast cancer dataset stratified according to CA9 and NFS1 expression. (G) Representative sequential cores depicting CAIX and NFS1 expression on a TNBC tissue microarray by immunohistochemistry; n = 28. Scale bar, 200 μm. Statistical significance was assessed using (E) Spearman rank correlation or (F) Mantel-Cox log-rank test.
With little being known about the importance of NFS1 expression across various cancers, we interrogated datasets from The Cancer Genome Atlas (TCGA) for NFS1 expression. We identified a number of solid cancers where NFS1 expression is elevated relative to normal tissue expression, including breast cancer (fig. S1E). Since the screen was carried out in a TNBC cell line, we took a closer look at the TCGA invasive breast cancer cohort after stratifying the patient population according to molecular subtype and identified CA9 and NFS1 expression that were correlated in the basal subtype (Fig. 1E). Furthermore, patients with elevated CA9 and NFS1 mRNA expression in their tumors in this cohort survive for shorter periods (Fig. 1F). To confirm the transcription data from the TCGA datasets extended to expression of the two proteins, we stained a small breast tumor tissue microarray consisting of triple-negative cases for CAIX and NFS1 (Fig. 1G). We and others have previously shown that CAIX is highly expressed and is a marker of worse patient prognosis in breast cancer (11, 14). CAIX was expressed in ~65% of the TNBC cases in this array, whereas NFS1 was expressed in nearly all cases (fig. S1F). These data suggest that both CAIX and NFS1 are expressed in a subset of patients with breast cancer where cotargeting may be beneficial.
Combined loss of CA9 and NFS1 increases cellular iron pools and lipid peroxidation
To validate NFS1 as a hit from the screen, we took the top two performing sgRNAs targeting NFS1 from the library pool and generated NTC and CA9KO cells expressing each sgRNA. We then transduced each line with Cas9 to create double (CA9KO) or single (NTC) NFS1 KOs and assessed viability (fig. S2, A and B). In agreement with the pooled library screen, double KO cell lines had two- (sgNFS1_1) and threefold (sgNFS1_2) more cell death than the single KOs over a 7-day growth period (fig. S2, A and B). As an orthogonal approach, we used small interfering RNA (siRNA)–mediated suppression of NFS1 in SUM159PT CA9KO cells (Fig. 2, A and B, and fig. S2C). Depletion of NFS1 in CA9KO cells with two independent siRNAs led to 3.5- and fourfold increases in cell death relative to NTC cells (Fig. 2, A and B), thus providing additional, independent confirmation of this synthetic lethal interaction. With NFS1 critically involved in iron-sulfur cluster biogenesis, we assessed whether mitochondrial iron levels were affected upon NFS1 depletion in CA9KO cells using rhodamine B-[1,10-phenanthrolin-5-yl)aminocarbonyl]benzyl ester (RPA), a dye quenched in the presence of Fe2+ (36). To evaluate iron levels before the induction of cell death following NFS1 depletion, we assessed these cells after a 72-hour expression of the siRNA versus the 96-hour time point where cell death was observed. We observed a decrease in RPA fluorescence in NFS1-depleted CA9KO cells indicative of increased iron levels (Fig. 2, C and D). Increased cellular iron levels can react with H2O2 via the Fenton reaction to produce ROS, leading to lipid peroxidation, a hallmark of ferroptosis (37). To address whether the observed increase in mitochondrial iron levels affected lipid peroxidation, we stained cells with BODIPY C11. In agreement with the increased iron levels observed in the NFS1-depleted CA9KO cells, we observed a concomitant increase in lipid peroxidation (Fig. 2, E to G).
Fig. 2. Combined loss of CA9 and NFS1 increases cellular iron pools and lipid peroxidation.
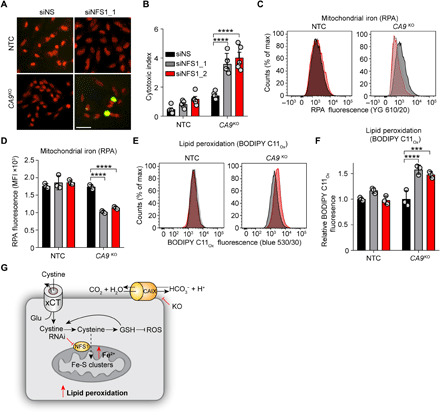
(A and B) Cell viability of the indicated cell lines following NFS1 depletion with siRNA over 96 hours in hypoxia. siNS, nonsilencing control siRNA. (A) Representative IncuCyte images. Green, cytotoxicity; red, nuclei. (B) Quantification of data in (A). Cells in (A) and (B) assessed for mitochondrial iron (RPA fluorescence) (C and D) and lipid peroxidation (BODIPY C11Ox fluorescence) (E and F) by flow cytometry. Flow plots (C and E) are colored according to (B); siNS, black; siNFS1_1, gray; siNFS1_2, red. MFI, mean fluorescence intensity. (G) Model depicting increase in mitochondrial iron and lipid peroxidation following suppression of NFS1 and CA9 expression. GSH, glutathione. Bars indicate means ± SEM. Shown are representative experiments from experiments performed at least twice. ***P < 0.001 and ****P < 0.0001. Statistical significance was assessed by two-way analysis of variance (ANOVA).
NFS1 suppression combined with CAIX/XII inhibitors triggers ferroptosis
To assess whether pharmacological inhibition of CAIX could recapitulate the phenotype associated with genetic loss of CA9, we suppressed NFS1 expression with siRNA in SUM159PT cells and administered concentrations of SLC-0111, a clinically evaluated small molecule inhibitor of CAIX/XII (17), that effectively inhibits CAIX activity in cells (Fig. 3, A and B, and fig. S2D) (32, 34). In agreement with previous findings, a reduction in NFS1 levels alone resulted in very little cell death in these hypoxic cultured cells (Fig. 3, A and B, and fig. S2E) (30). However, combining NFS1 depletion with SLC-0111 triggered a dose-dependent increase in cell death (Fig. 3, A and B). Treating this combination with the radical trapping antioxidant and ferroptosis inhibitor, ferrostatin-1 (Fer-1) (20), completely blocked the increased cell death (Fig. 3, A and B). We confirmed these findings in additional models of TNBC [MDA-MB-231 LM2-4 (LM2-4) and 4T1] as well as in models of bladder (UC-16) and colon cancer (HT-29) (Fig. 3C and fig. S2, F to H). Moreover, the combination of NFS1 suppression and SLC-0111 resulted in increased lipid peroxidation (Fig. 3, D and E, and fig. S2, I and J). To demonstrate that this effect was not unique to SLC-0111, we used an additional ureidosulfonamide inhibitor of CAIX/XII, compound 13, which also effectively inhibits CAIX/XII activity in cells (fig. S2K) (38). Combining compound 13 treatment with NFS1 depletion in SUM159PT (Fig. 3F) and LM2-4 (Fig. 3G) resulted in increased cell death in both cell lines in a dose-dependent manner, thus providing evidence that, in addition to genetic loss of CA9, inhibition of CAIX/XII activity results in increased cellular dependence on NFS1 for survival and that combined loss of both results in ferroptosis.
Fig. 3. NFS1 suppression combined with CAIX/XII inhibitors triggers ferroptosis.
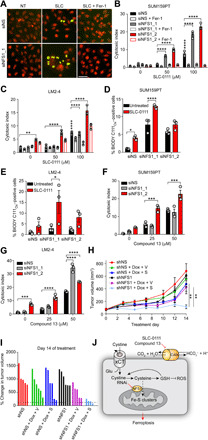
(A to C) Cell viability data of the indicated cell lines following NFS1 depletion in combination with SLC-0111 in the presence or absence of Fer-1 (2 μM) for 72 hours in hypoxia. (A) Representative IncuCyte images of the indicated treatments. Green, cytotoxicity; red, nuclei. (B and C) Quantitation of IncuCyte data for the indicated cell lines treated and cultured in hypoxia for 72 hours. (D and E) Lipid peroxidation (BODIPY C11Ox fluorescence) of the indicated cell lines treated as in (B) and (C) by flow cytometry. (F and G) Cell viability of the indicated cell lines following NFS1 depletion and treatment with compound 13 for 72 hours in hypoxia. (H) Tumor growth curve of orthotopic SUM159PT control (shNS) or NFS1-depleted (shNFS1) tumors treated with vehicle or SLC-0111 following induction of shRNA with Dox. n = 4 to 6 per group. V, vehicle; S, SLC-0111. (I) Waterfall plot depicting percent change in individual tumor volume of the groups color coded as in (H) following 14 days of treatment. (J) Model depicting CAIX inhibition combined with NFS1 depletion triggering ferroptosis. Bars represent means ± SEM (B to H). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was assessed using two-way ANOVA followed by Tukey’s multiple comparison test post hoc (B to G) or by ANOVA followed by Bonferroni correction (H). Shown are representative data from experiments performed at least twice. Scale bar, 50 μm.
To evaluate this relationship in vivo, we generated SUM159PT cell lines expressing doxycycline (Dox)–inducible short hairpin–mediated RNA (shRNA) to NFS1 (fig. S2L) and verified that inducible depletion of NFS1 in combination with CAIX inhibition resulted in enhanced cell death (fig. S2M). Cells were implanted orthotopically, and tumors were allowed to form before initiation of treatment. Tumors expressing control shRNA (shNS) or NFS1 shRNA (shNFS1) in the absence of Dox grew at similar rates (Fig. 3H and fig. S2N). CAIX/XII inhibition (shNS + Dox + SLC-0111) or NFS1 depletion (shNFS1 + Dox + vehicle) alone had little impact on tumor growth in this model. However, combining both treatments (shNFS1 + Dox + SLC-0111) caused significant suppression of tumor growth (Fig. 3, H and I). Together, these data suggest that targeting both CAIX and NFS1 is an effective therapeutic strategy to overcome resistance to CAIX/XII inhibition in solid tumors (Fig. 3J).
CAIX suppresses ROS production
We next sought to determine the mechanistic details behind the enhanced ferroptosis resulting from cotargeting CAIX/XII and NFS1. Since the combination increases lipid peroxidation and can be blocked in the presence of Fer-1 (Fig. 3, A to C), we next assessed whether cellular ROS levels were affected in the absence of CAIX expression or activity. We identified increased ROS in CA9KO and SLC-0111–treated and CAIX-depleted cells (Fig. 4, A to C, and fig. S3, A to C). Restoring CAIX expression in CAIX-depleted cells returned ROS to basal, hypoxic levels (Fig. 4C). We then evaluated whether NFS1 depletion would further affect ROS production (fig. S3, D and E). Depletion of NFS1 did not significantly affect cellular production of ROS, whereas inhibition of CAIX/XII did irrespective of NFS1 levels. Moreover, as expected, mitochondrial iron increased following NFS1 depletion, inducing an iron starvation response increasing transferrin receptor levels, but blocking CAIX/XII activity had no further impact (Fig. 4D and fig. S3, F and G). To address whether the increased ROS was responsible for the increased cell death observed upon cotargeting CAIX and NFS1, we combined NFS1 knockdown with SLC-0111 in the presence or absence of the antioxidant Trolox (Fig. 4E). Similar to what we observed with Fer-1, treatment with Trolox suppressed the increased cell death accompanying this combination. These findings are in agreement with those previously published, revealing that NFS1 does not affect cellular ROS levels, yet NFS1-depleted cells were more sensitive to treatments inducing accumulation of ROS (30). Furthermore, treatment with the iron chelator deferoxamine (Fig. 4E) also significantly reduced the efficacy of this combination, suggesting that the increased ROS accompanying CAIX/XII inhibition creates a cellular vulnerability to the increased iron following NFS1 suppression, thereby triggering ferroptosis (Fig. 3, A to C). To test this directly, we treated SUM159PT cells with physiological concentrations of iron in the form of ferric citrate (Fig. 4F). Notably, ferric citrate alone did not affect cell viability. However, combining ferric citrate with CAIX/XII inhibition led to robust cell death at a concentration of SLC-0111 capable of increasing ROS levels. Furthermore, in addition to SLC-0111, combining compound 13 or compound 11 with ferric citrate led to significant increases in cell death that could be reversed by treatment with Fer-1 or Trolox in both SUM159PT and LM2-4 cells (Fig. 4, G and H).
Fig. 4. CAIX suppresses ROS production.
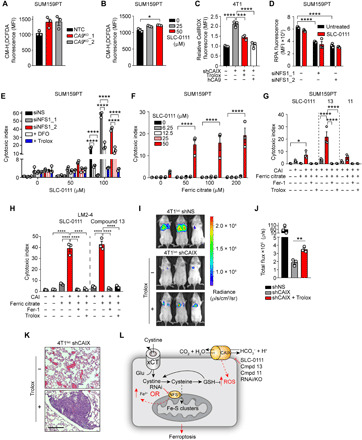
Cellular ROS levels in hypoxic SUM159PT CA9KO (A) SLC-0111–treated (B) and CAIX-depleted 4T1luc cells (C) following 72 hours of exposure to hypoxia detected with CM-H2DCFDA (A and B) and CellROX Deep Red (C). (D) Mitochondrial iron (RPA fluorescence) levels in hypoxic NFS1-depleted SUM159PT cells treated with SLC-0111 for 72 hours in hypoxia. (E) Cell viability of SUM159PT cells treated with NFS1 siRNA and the indicated concentrations of SLC-0111 in the presence or absence of deferoxamine (DFO; 10 μM) or Trolox (100 μM) for 72 hours in hypoxia. (F) Cell viability of SUM159PT cells treated with the indicated concentrations of ferric citrate in combination with SLC-0111 for 72 hours in hypoxia. (G) Cell viability of SUM159PT or (H) LM2-4 cells treated with ferric citrate together with SLC-0111, compound 13 (13), or compound 11 (11) in the presence or absence of Fer-1 (2 μM) or Trolox (100 μM) for 72 hours in hypoxia. CAI, carbonic anhydrase inhibitor. (I) Bioluminescent images depicting metastatic burden of intravenously injected 4T1luc shNS, 4T1luc shCAIX, and 4T1luc shCAIX cells pretreated with Trolox. n = 3 per group. (J) Quantitation of total chest bioluminescence of the groups in (H). (K) Representative hematoxylin and eosin–stained lung sections from shCAIX groups in (H). Scale bar, 200 μm. (L) Model depicting increased ROS resulting from CAIX/XII inhibition inducing vulnerability to perturbations in iron levels. Bars represent means ± SEM. Shown are representative experiments from experiments performed at least twice. *P < 0.05, **P < 0.01, and ****P < 0.0001. Statistical significance was assessed using one-way (B, C, G, and H) or two-way (D to F) ANOVA followed by Tukey’s multiple comparisons test post hoc or by t test (I).
We previously demonstrated reduced metastatic capacity of breast cancer cells with suppressed CAIX expression (14). Oxidative stress has been identified as a hurdle to systemic metastatic spread (39). Furthermore, ferroptosis was recently found to limit hematogenous metastatic spread of cells with high cellular ROS levels (40). Given the vulnerability to physiological iron concentrations following CAIX/XII inhibition, we addressed whether the increased ROS associated with CAIX depletion impeded the hematogenous metastatic capacity of these cells (Fig. 4, I to K). In agreement with our previous findings, depletion of CAIX affected the metastatic capability of 4T1 cells. The pretreatment of shCAIX cells with Trolox significantly reduced cellular ROS levels (Fig. 4C) and enhanced their metastatic capacity (Fig. 4, I to K). Collectively, the data suggest that CAIX protects cells from ferroptosis by limiting ROS production and cotargeting CAIX with chemical agents that perturb iron or antioxidant homeostasis and may be an effective therapeutic strategy (Fig. 4L).
CAIX/XII inhibition potentiates ferroptosis induced by xCT inhibition
We next sought to assess whether combining CAIX/XII inhibition with limiting cyst(e)ine availability, the substrate of NFS1, through inhibition of the cystine glutamate antiporter xCT, would lead to a similar enhancement of ferroptosis. To target xCT, cells were treated with the small molecule inhibitor erastin (20). Notably, at doses higher than 0.5 μM, erastin is lethal on its own in SUM159PT cells (fig. S4A). To identify whether CAIX/XII inhibition could enhance erastin-induced cell death, we treated cells with SLC-0111 and a concentration of erastin that did not induce cell death following a 72-hour treatment (Fig. 5A and fig. S4A). Notably, SLC-0111 also causes very little cell death on its own in this model. However, the combination treatment resulted in a significant increase in cell death that could be reversed in the presence of Fer-1, Trolox, and 2-mercaptoethanol (2-ME) but not inhibitors of necroptosis (Nec1s) or apoptosis (ZVAD-FMK) (Fig. 5, A to C), suggesting that ferroptosis is the primary mode of cell death induced by the combination and restoring cysteine uptake or buffering ROS alleviates this. Moreover, the SLC-0111–erastin combination resulted in marked accumulation of lipid peroxidation (Fig. 5D). To assess the range at which this combination was effective, we treated SUM159PT with multiple doses of SLC-0111 and erastin (Fig. 5E). CAIX/XII inhibition potentiated erastin-induced ferroptosis in a dose-dependent manner. We extended this finding to additional models of TNBC, pancreatic, and bladder cancer (Fig. 5, F and G, and fig. S4, B and C). In addition, this finding was not unique to the SLC-0111 compound, as cells treated with the same titration of erastin in the presence of compound 13 or compound 11 evoked similar dose-dependent increases in cell death (Fig. 5, H and I). Alternatively, to assess whether this effect was unique to erastin, we carried out similar combination experiments with SLC-0111 and an additional inhibitor of xCT, sulfasalazine (Fig. 5J) (41). As sulfasalazine is a much less potent inhibitor of xCT than erastin, much higher doses were needed before sulfasalazine caused any cell death on its own (Fig. 5J). Nevertheless, the addition of SLC-0111 was able to enhance sulfasalazine-induced cell death in a dose-dependent manner across multiple TNBC models (Fig. 5J and fig. S4, D and E). To further confirm the on-target nature of the CAIX/XII inhibitors, we depleted CAIX, CAXII, or both simultaneously and treated with erastin or sulfasalazine (fig. S4, F to H). In support of the findings from the pharmacological inhibition studies, suppressing CAIX or CAXII expression increased the ferroptosis resulting from xCT inhibition.
Fig. 5. CAIX/XII inhibition potentiates ferroptosis induced by xCT inhibition.
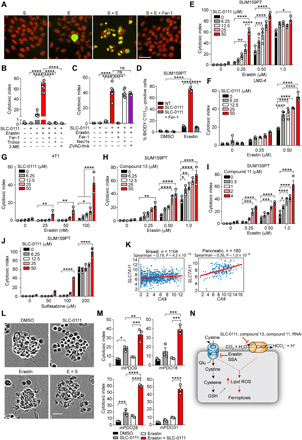
(A) IncuCyte images of cell viability following treatment of SUM159PT cells with SLC-0111 (S) (50 μM) and erastin (E) (0.5 μM) for 72 hours in hypoxia in the presence or absence of Fer-1 (2 μM). Green, cytotoxicity; red, nuclei. Cell viability of SUM159PT cells treated with SLC-0111 and erastin as in (A) in the presence of (B) Fer-1 (2 μM), Trolox (100 μM), 2-ME (50 μM), (C) Nec1s (50 μM), or ZVAD-FMK (25 μM) for 72 hours in hypoxia. ns, not significant. (D) Lipid peroxidation (BODIPY C11Ox fluorescence) in SUM159PT cells treated as in (A). Cell viability of (E, H, I, and J) SUM159PT, (F) LM2-4luc, and (G) 4T1luc cells treated with the indicated concentrations of erastin together with SLC-0111 (A to G), compound 13 (H), compound 11 (I), or sulfasalazine (SSA) together with SLC-0111 (J) for 72 hours in hypoxia. (K) Analysis of CA9 and SLC7A11 expression in the indicated TCGA datasets. (L and M) Cell viability of PDAC patient–derived organoids treated with erastin (10 μM) and SLC-0111 (50 μM) for 72 hours. (L) Representative images of the indicated combinations in mPDO28. (M) Cell viability data of organoids from four separate patients following treatment with the indicated compounds. (N) Model depicting CAIX/XII inhibition in combination with blocking cystine uptake increases ferroptosis. Bars represent means ± SEM. Shown are representative experiments from experiments performed at least twice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was assessed using one-way (J) or two-way ANOVA (B to G) followed by Tukey’s multiple comparisons test post hoc or (H) Spearman rank correlation. Scale bars, 50 μm.
To evaluate the clinical relevance of cotargeting CAIX and xCT, we assessed the expression of CA9 and SLC7A11, the gene encoding xCT, across human cancers in the TCGA. Positive correlations were observed between the two genes in cohorts of patients with breast and pancreatic ductal adenocarcinoma (PDAC) (Fig. 5K). To cotarget CAIX/XII and xCT in a clinically relevant model, we treated CAIX expressing organoids derived from liver metastases of patients with metastatic PDAC with erastin and SLC-0111 (Fig. 5, L and M, and fig. S4I). Similar to our findings in cell line models, this combination led to substantive increases in cell death in organoids from multiple patients outperforming treatment with either compound alone (Fig. 5, L and M). Therefore, cotargeting CAIX/XII and xCT enhances ferroptosis and is an effective strategy to pharmacologically exploit the synthetic lethal interaction between CA9 and NFS1 (Fig. 5N).
Cotargeting CAIX/XII and xCT blunts signaling through adenosine 5′-monophosphate–activated protein kinase to enhance ferroptosis in a pH-dependent manner
To determine the mechanistic details surrounding the cell death occurring as a result of combined CAIX/XII and xCT inhibition, we first assessed the impact of CAIX activity on pHi (Fig. 6A). Since CAIX is involved in regulation of pHi through the production of bicarbonate (7), we assessed pHi in hypoxic SUM159PT cells following treatment with SLC-0111, compound 11, and compound 13 (Fig. 6A). As expected, we identified that all three inhibitors led to the acidification of pHi. Moreover, the bicarbonate produced by CAIX/XII can be imported by bicarbonate transporters, and CAIX has been found to associate with the sodium bicarbonate cotransporter NBCn1 (12). Therefore, we blocked sodium bicarbonate cotransporters with S0859 (42), reducing pHi (Fig. 6B), and evaluated whether the bicarbonate generated was important in protecting cells from ferroptosis (Fig. 6, C and D, and fig. S4J). Treating hypoxic SUM159PT (Fig. 6C) and LM2-4 (Fig. 6D) cells with S0859 in combination with erastin induced a dose-dependent increase in cell death to levels similar to those achieved by cotargeting CAIX/XII and xCT, suggesting that CAIX/XII-driven pHi regulation is critical for protection against ferroptosis. To further implicate pH in influencing ferroptosis downstream of cotargeting CAIX/XII or sodium bicarbonate cotransporters with xCT, we added NH4Cl to the combinations to raise pHi (Fig. 6, B, E, and F). Combining NH4Cl with SLC-0111 and erastin (Fig. 6E) or with S0859 and erastin (Fig. 6F) reduced ferroptosis in a dose-dependent manner, providing additional support that acidic pHi potentiates ferroptosis.
Fig. 6. Cotargeting CAIX/XII and xCT blunts signaling through AMPK to enhance ferroptosis in a pH-dependent manner.
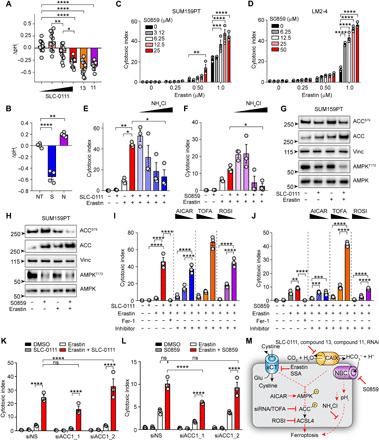
(A and B) pHi measurements in hypoxic SUM159PT cells following the indicated treatments for 72 hours. (A) SLC-0111 (0, 25, 50, and 100 μM,), compound 11 (2 μM), and compound 13 (50 μM); (B) NT, untreated; S, S0859 (25 μM); N, NH4Cl (2.5 mM). Cell viability of hypoxic (C) SUM159PT and (D) LM2-4 treated with the indicated concentrations of S0859 and erastin for 72 hours. Cell viability of SUM159PT cells treated with SLC-0111 (50 μM) and erastin (0.5 μM) (E) or S0859 (50 μM) and erastin (F) in the presence of NH4Cl (1, 2.5, 5, and 10 mM) for 72 hours in hypoxia. Indicated protein levels of SUM159PT cells treated with (G) SLC-0111 (12.5 μM) and erastin (0.25 μM) or (H) S0859 (12.5 μM) and erastin (0.25 μM) for 72 hours in hypoxia. Cell viability data of SUM159PT treated with erastin and SLC-0111 (I) or S0859 (J) together with the indicated compounds for 72 hours in hypoxia. AICAR (0.25, 0.5, and 1.0 μM), TOFA (12.5, 25, and 50 μM), and rosiglitazone (ROSI; 1.0, 5.0, and 10 μM). Cell viability data of SUM159PT treated with siRNA to ACC1 together with SLC-0111 (K) or S0859 (L) and erastin for 72 hours in hypoxia. (M) Model depicting role of CAIX and sodium-driven bicarbonate cotransporters in pH homeostasis and suppression of ferroptosis. Bars indicate means ± SEM. Shown are representative experiments from experiments performed at least twice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was assessed using t test with Welch’s correction (F) or one-way (A, B, E, I, and J) or two-way ANOVA (C, D, K, and L) followed by Tukey’s multiple comparisons test post hoc.
We next sought to identify the impact on cell signaling following treatment with SLC-0111 and erastin (Fig. 6G). CAIX has been shown to affect cellular metabolism (31–35), and adenosine 5′-monophosphate–activated protein kinase (AMPK) signaling is central to maintaining metabolic homeostasis. Activated in response to energy stress, ROS, and hypoxia (43), AMPK signaling has been implicated in protecting cells from ferroptosis (44). The combination of SLC-0111 and erastin at sublethal concentrations led to inactivation of AMPK following reduced phosphorylation of Thr172 (Fig. 6G). This inactivation was echoed by a reduction in phosphorylation of the AMPK target acetyl–coenzyme A (CoA) carboxylase 1 (ACC1) on Ser79, suggesting that the combination was activating ACC1 (Fig. 6G). We then assessed whether the combination of S0859 and erastin also affected AMPK activity (Fig. 6H). Similar to SLC-0111 treatment, combination of S0859 and erastin decreased phosphorylation of AMPK on Thr172 (Fig. 6H). This reduction in AMPK phosphorylation was mirrored by a reduction in phosphorylation of ACC1 on Ser79. To evaluate whether these changes in AMPK/ACC1 activation were relevant to the increased ferroptosis resulting from the SLC-0111–erastin and S0859-erastin combinations, we simultaneously activated AMPK with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (Fig. 6, I and J). Activation of AMPK reduced the enhanced cytotoxicity of the SLC-0111–erastin and S0859-erastin combinations in a dose-dependent manner. In parallel, we inhibited ACC1 with 5-(tetradecyloxy)-2-furoic acid (TOFA) (Fig. 6, I and J). Inhibition of ACC1 decreased cell death of both erastin combinations in a dose-dependent manner, suggesting that the signal transmitted from AMPK to ACC1 is biologically relevant to the ferroptosis induced by both the SLC-0111–erastin and S0859-erastin combinations. Furthermore, we inhibited acyl-CoA synthetase long-chain family member 4 (ACSL4), which is downstream of ACC1, with rosiglitazone (Fig. 6, I and J). ACSL4 expression is predictive of susceptibility to ferroptosis (45–47), and its activity is responsible for the conversion of polyunsaturated fatty acids into polyunsaturated fatty acyl-CoAs for incorporation into phospholipids (48, 49). We found that rosiglitazone treatment also reduced the cytotoxicity of the combinations (Fig. 6, I and J). To provide further confirmation of the importance of ACC1 in potentiating ferroptosis downstream of CAIX/XII or sodium bicarbonate cotransporter and xCT inhibition, we suppressed ACC1 expression with siRNA and treated cells with the SLC-0111–erastin and S0859-erastin combinations (Fig. 6, K and L, and fig. S4K). Reducing ACC1 expression partially suppressed the level of cytotoxicity induced by the SLC-0111–erastin (Fig. 6K) and S0859-erastin (Fig. 6L) combinations dependent on the extent of knockdown by the siRNAs. Thus, these cytotoxicity studies suggest that the observed reduction in AMPK activity following cotargeting CAIX/XII and xCT is responsible for the enhanced ferroptosis from this combination by activating ACC1. Together, these data suggest that pHi modulation is important in regulating ferroptosis. Furthermore, our data also implicate the importance of the AMPK/ACC1/ACSL4 axis downstream of cotargeting hypoxia-induced CAIX and xCT (Fig. 6M).
DISCUSSION
Advances in genetic manipulation and screening strategies have renewed interest in synthetic lethality to exploit vulnerabilities for cancer therapy and overcome therapeutic resistance (50). With inhibitors of CAIX/XII entering clinical evaluation, identifying cellular networks that become essential when CAIX/XII function is suppressed is critical to enhancing therapeutic benefit (17). In this study, we used an unbiased genetic screen and identified a novel synthetic lethal interaction between CA9 and NFS1. Moreover, we identified inhibition of xCT, a source of cellular cyst(e)ine upstream of NFS1, together with CAIX/XII inhibition to be a very effective alternative approach to exploiting this synthetic lethal interaction pharmacologically. Finding this interaction allowed us to uncover an important role for CAIX in redox homeostasis and protection from ferroptosis through pH regulation. Mechanistically, we found that CAIX guards against ROS accumulation and that cotargeting CAIX/XII and xCT blocked AMPK activity and activated downstream target ACC1. Together, this study demonstrates the importance of pHi regulation in protecting against ferroptosis.
Our study highlights an important role for NFS1, iron-sulfur cluster biogenesis, and regulation of iron levels in the survival of hypoxic cancer cells and tumors. NFS1 loss triggers the iron starvation response and increases cellular labile iron levels (30). In line with this, NFS1 has been previously shown to protect cells from ferroptotic cell death in high oxygen environments, with little effect on hypoxic cells and tumors (30). However, here, by triggering cellular accumulation of ROS following suppression of CAIX activity or expression, we identified increased cellular dependence on NFS1 and iron-sulfur cluster biogenesis in hypoxic cells and tumors. NFS1 has previously been shown to protect against ROS-inducing treatments (30), providing an explanation for this synthetic lethal interaction. Cellular adaptation to hypoxia typically involves a reduction in electron transport chain activity to reduce ROS accumulation (3). However, recent findings demonstrate that hypoxia leads to an increase in superoxide production from the mitochondria (51), creating a vulnerability to alterations in antioxidant buffering capacity and therapeutic strategies triggering iron accumulation.
Metastasis is a very inefficient process that requires cells to survive immense selective pressure to seed distant organs (52). Oxidative stress is a critical hurdle for hematogenous spread (39). We have previously described a role for CAIX in multiple steps in the metastatic cascade (12, 14, 15). While we had demonstrated a requirement for CAIX to extravasate in the lungs, it was unclear as to why its expression was required in an environment that had such a high oxygen tension. Our findings here allow us to extend our previous observations by demonstrating that CAIX protects against ROS accumulation, which unabated becomes lethal upon encountering the increased iron concentration in the bloodstream (40). In line with recent findings, overcoming this oxidative stress with antioxidants permits survival in the blood and avoidance of ferroptosis, increasing metastatic capability (39, 40). Furthermore, the altered redox homeostasis accompanying CAIX/XII inhibition creates opportunities for future combinations with therapies altering cellular iron homeostasis (53).
Our work supports the role of the HIF pathway in protection of cancer cells against ferroptosis (25). Ferroptosis is an iron-dependent form of nonapoptotic cell death driven by toxic accumulation of lipid ROS (20). The lipid ROS arise from reactions of iron with lipid peroxides (54). An increased membrane lipid profile with a higher composition of polyunsaturated fatty acids induces susceptibility to ferroptosis (45, 46, 55). Hypoxia, through HIF-1, leads to increased expression of fatty acid synthase and stearoyl CoA desaturase 1 (SCD1), leading to increased production of polyunsaturated fatty acids (22). Here, we found that targeting the HIF-1 target CAIX together with xCT increased accumulation of lipid ROS and enhanced cell death by ferroptosis. Our investigation into downstream signaling revealed an activation of ACC1 following AMPK inhibition by SLC-0111 and erastin, and the function of ACSL4 was required downstream because treatment with rosiglitazone, an inhibitor of ACSL4, blocked the cytotoxicity of the combination. AMPK is activated in response to energy stress, including that induced by hypoxia. AMPK was recently shown to protect against ferroptosis in response to energy stress induced by glucose deprivation, leading to decreased polyunsaturated fatty acid levels (44). Loss of AMPK led to increased production of polyunsaturated fatty acids and sensitivity to erastin-induced ferroptosis in an ACSL4-dependent manner (44), which supports our findings. However, since ACC1 suppression did not completely restore viability following the SLC-0111–erastin combination, it is also possible that signaling through GPX4 (glutathione peroxidase 4) is also involved since TOFA can prevent its degradation (56). Energy stress is an important feature of the microenvironment of solid tumors, and AMPK has been implicated in playing a supportive role in tumor growth (57–59). Whether this has implications in resistance to ferroptosis in solid tumors remains to be determined, but targeting CAIX can be used as a strategy to overcome this.
Hypoxia-induced metabolic rewiring creates therapeutic opportunities in cancer (1, 3). Increased glycolytic flux puts pressure and added dependence on the pH regulatory machinery in the cell to maintain a pHi to maintain survival and sustain proliferation (4). Here, in agreement with our work in other models (31, 32, 34), we demonstrate that CAIX/XII inhibition acidifies pHi. In addition, we demonstrate the importance of the bicarbonate generated by CAIX/XII by phenocopying the enhanced ferroptosis upon cotargeting xCT by blocking sodium-driven bicarbonate transport. Acidosis leads to a number of metabolic changes that would increase susceptibility to ferroptosis including increased fatty acid synthesis, glutaminolysis, and autophagy and decreased mammalian target of rapamycin (mTOR) activity and suspension of the circadian clock (4, 60, 61). Oncogenic activation of mTOR through phosphatidylinositol 3-kinase–Akt has been implicated in the suppression of ferroptosis (62). Induction of acidosis may be an effective strategy to overcome this level of resistance. Since we have previously demonstrated that CAIX influences mTORC1 activity (16), it will be interesting to see whether inhibitors targeting CAIX could be used to overcome ferroptosis resistance in tumors with activation of this oncogenic axis. In summary, our data demonstrate new mechanisms for tumor hypoxia–induced CAIX in redox homeostasis and suppression of ferroptosis-mediated cell death, offering innovative strategies to enhance the activity of ferroptosis-inducing compounds against solid tumors.
MATERIALS AND METHODS
Cell lines
The SUM159PT cell line has been previously described (63) and was a gift from S. Gorski (BC Cancer Research Institute). MDA-MB-231 LM2-4luc+ cells were a gift from R. Kerbel (Sunnybrook Research Institute, Toronto) (64). 4T1luc+ cells have been previously described (65). UC-16 cells were a gift from P. Black (Vancouver Prostate Centre). HT-29 cells were a gift from A. Minchinton (BC Cancer Research Institute). Human embryonic kidney (HEK) 293T cells were a gift from S. Aparicio (BC Cancer Research Institute). All cells lines were validated by short tandem repeat analysis (Genetica) and mycoplasma testing using the LookOut Mycoplasma test. All cell lines were maintained in a humidified incubator at 37°C in an atmosphere containing 5% CO2. Cell lines used in hypoxia (1% O2) were routinely monitored for hypoxia-induced up-regulation of CAIX.
Organoid culture
Tumor tissue was collected from consented patients. The study was approved by the University of British Columbia research ethics board (REB) (H16-00291 and H18-03701). Patient-derived organoids were generated as previously described (66). Briefly, fresh liver metastatic biopsies from patients with PDAC were isolated and placed in RPMI 1640 medium supplemented with fetal bovine serum (FBS). The biopsy material was dissected and minced with scissors in the 10% FBS-supplemented RPMI 1640 medium. The resulting tissue chunks were washed, spun, and digested with collagenase/dispase (1 mg/ml). The collagenase/dispase was deactivated with 1% bovine serum albumin (BSA)–supplemented Dulbecco’s modified Eagle’s medium (DMEM); the tissue was washed and spun again and then digested with Accutase (500 U/ml). The digestion was again deactivated with 1% BSA-supplemented DMEM, and the resulting cell suspension was filtered through a 20-μm filter. The resulting cell suspension was spun and resuspended in complete organoid medium before being plated on 12-well plates coated with Matrigel. The organoids were grown at 37°C with 5% CO2 and passaged every 2 weeks.
Cell culture
SUM159PT cells are cultured in DMEM containing 10% FBS, 1× nonessential amino acids, hydrocortisone (1 μg/ml), and insulin (5 μg/ml). The MDA-MB-231 LM2-4 and PK-8 cells were cultured in RPMI 1640 containing 10% FBS. 4T1 cells were cultured in DMEM containing 10% FBS and 1× nonessential amino acids. HT-29 cells were cultured in DMEM containing 10% FBS. UC-16 cells were cultured in minimum essential medium containing 10% FBS, 1× nonessential amino acids, and 1 mM sodium pyruvate.
Chemicals/reagents/antibodies
Erastin (Selleckchem, S7242), Fer-1 (Cayman Chemical, 17729), Nec-1s (BioVision, 2263), Z-VAD-FMK (Selleckchem, S7023), sulfasalazine (Selleckchem, S1576), deferoxamine mesylate (Sigma-Aldrich, #D9533), Trolox (Sigma-Aldrich, #238813), ferric citrate (Sigma-Aldrich, #F3388), AICAR (Selleckchem, S1802), TOFA (Selleckchem, S6690), rosiglitazone (Selleckchem, S2556), S0859 (Cayman Chemicals, 18497), SLC-0111, compound 11, and compound 13 have been previously described (32, 38); RPA (Squarix Biotechnology, ME043), BODIPY 581/591 undecanoic acid (Thermo Fisher Scientific, #D3861), CM-H2DCFDA (Thermo Fisher Scientific, #C6827), CellROX Deep Red (Thermo Fisher Scientific, #C10422), SYTOX Green nucleic acid stain (Thermo Fisher Scientific, #S7020), HCS NuclearMask red stain (Thermo Fisher Scientific, #H10326), nigericin (Sigma-Aldrich, #N7143), and NH4Cl (Sigma-Aldrich, #A9434). Antibodies to AMPK (cs2532; 1:1000), AMPKT172 (cs2535; 1:500), ACC (cs3662; 1:500), and ACCS79 (cs3661; 1:500) were purchased from Cell Signaling Technology. Antibodies to CAIX were purchased from R&D Systems (AF2188; 1:1000) and BioScience Slovakia (M75; 1:1000). Antibodies to CA12 (ab195233; 1:1000) and ferrochelatase (ab137042; 1:1000) were purchased from Abcam. Antibodies to NFS1 (sc365308; 1:500) and CD71/TFRC (Transferrin receptor 1) (sc-32272; 1:500) were purchased from Santa Cruz Biotechnology. Antibody to vinculin (MAB3574; 1:1000) was purchased from Millipore.
Generation of CA9KO cell line
The procedure for generation of the CA9KO cell line is similar to what has been previously described for the generation of the MDA-MB-231 CA9KO using CRISPR-Cas9, and the same procedures were followed with the SUM159PT cell line (12). Briefly, cells were transfected with CG01 plasmid (GeneCopoeia) expressing Cas9 and sgRNA to CA9 using Lipofectamine 2000 as per the manufacturer’s suggestions and then expanded for 5 days. Cells were sorted for red fluorescent protein expression by flow cytometry using an AriaFusion and then cloned out from single cells in 96-well plates. Western blotting for CAIX expression following 72 hours of incubation in 1% O2 provided first pass filtering for positive KO clones. Genomic DNA was then extracted from these lines using QuickExtract solution (Lucigen, QE09050) and the CA9 locus around the sgRNA-targeted region amplified by polymerase chain reaction (PCR). Amplicons were sequenced by Sanger sequencing to screen for null mutations.
Lentiviral production
HEK293T cells were seeded at 5 × 104 cells/cm2 on 15-cm plates and transfected the following day with TransIT-LT1 (Mirus Bio, 2300) with 20 μg of human GeCKOv2 lentiCRISPRv2 KO pooled plasmid library A (a gift from F. Zhang; Addgene #1000000048), 10 μg of pVSVg, and 15 μg of psPAX2. After an overnight incubation, the medium was changed, and 30 ml of DMEM containing 10% FBS and 1% BSA (Sigma-Aldrich) was added to each 15-cm plate. After 60 hours, the medium was collected and centrifuged at 3000 rpm at 4°C for 5 min. The supernatant was then filtered through a 0.45-μm filter and concentrated 300× by centrifugation at 25,000 rpm at 4°C for 90 min.
Screen
A total of 7 × 108 SUM15PT NTC or CA9KO cells were transduced at a multiplicity of infection of 0.3, representing 300× coverage of the library, in SUM159PT medium containing polybrene (800 μg/ml). The following day, the medium was removed, and the plates washed 2× with phosphate-buffered saline and fresh SUM (SUM159T) medium added containing puromycin (7 μg/ml). After 48 hours, the medium was removed, and the cells washed and trypsinized. Two aliquots of 2.5 × 107 cells were collected for T0 samples. A total of 2 × 107 cells were seeded onto T1000 HyCell multilayer flasks (Corning) in triplicate for culture in 1% O2 and passaged for 20 generations. Samples of 2 × 107 cells were collected every 72 hours. Genomic DNA was extracted using the QIAamp DNA Blood Kit (QIAGEN, #51194) as described by the manufacturer. Illumina adapters and barcodes were added to samples by PCR as previously described (67). Samples were sequenced on an Illumina HiSeq2500 by the Michael Smith Genome Sciences Centre, BC Cancer, Vancouver, Canada. Indexed reads were demultiplexed before analysis.
Computational and statistical analysis
Pooled sgRNA screening data were analyzed as previously described (68). Before analysis, the read counts from the demultiplexed sequencing files were generated using the MAGeCK software package (69). To calculate the differential cumulative change (DCC) between NTC and CA9KO, we normalized the read counts using cyclic loess normalization and sgRNAs with low read counts at the initial time point T0 were removed. The screen was performed as an end point assay with two time points (T0 and T11). Accordingly, the following formula was used
where is the normalized read counts at T11 and is the normalized read counts at T0 in replicates rep ∈ (1..2) for KO-B4 cells. Likewise, and are for NTC cells. To measure the gene level DCC, we used the two best sgRNAs with the maximum dropout for that gene. We used the Student’s t test in conjunction with the permutation test P value to classify sgRNA and its corresponding genes that were substantially different between the CA9KO and NTC cells. The permutation test was performed by estimating the frequency of randomized, shuffled DCC with maximum dropout compared to the observed gene level DCC value. Pathway enrichment analysis was performed on hits using the iDEP package (70). The gene network image was generated using Cytoscape software (71) for genes enriched in multiple pathways.
TCGA analysis
Comprehensive data analysis on gene expression from the TCGA database was performed by exploring the RNA sequencing expectation maximization–normalized expression data downloaded from https://portal.gdc.cancer.gov/ site. The normalized data were log-transformed (log2) to bring them to same scale. Spearman rank correlation was used to compute the correlation between CA9 and NFS1 or SLC7A11 in data of patient with breast and pancreatic cancer. Further subsetting the TCGA BRCA (breast cancer) dataset according to molecular subtype was performed using the PAM50 classifier (72). Statistical significance between normal and tumor samples for 24 different cancer types for NFS1 expression analysis was calculated using Mann-Whitney U test.
RNA interference
Dox-induced shRNA to NFS1 (V2THS_67142; ATGTCATTGACATCAAGTG) using the TRIPZ system (Dharmacon) was introduced by lentivirus. siRNA to NFS1 (GS9054), CA9 (GS768), CA12 (GS771), and ACACA (GS31) were purchased from QIAGEN. FlexiTube GeneSolution sets of four siRNAs were screened for each gene to identify two efficacious siRNAs to use in our studies. siRNAs were reverse-transfected using siLentFect (Bio-Rad, 1703360) or Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778075) as per the manufacturer’s instructions. The following day, the cells were split into experiments and cultured for an additional 72 hours. Protein levels were routinely monitored by Western blot.
Cell viability assays
Cells
Cells were plated in 96-well plates in the presence of treatment and medium containing SYTOX Green (125 ng/ml) and evaluated in the IncuCyte (Essen BioScience) for 72 hours in 1 or 21% O2. Following the 72-hour incubation period, HCS NuclearMask red dye was added to the wells to obtain an accurate number of cells remaining in each well. To assess cell viability, cytotoxic indeces were calculated as follows: (number of SYTOX Green–positive cells per mm2 ÷ number of HCS NuclearMask red cells per mm2) × 100. The following processing definition parameters were used in the analysis of the IncuCyte data: SUM159PT/UC-16/4T1: green, Top-Hat, radius of 100 μm, threshold [green calibrated unit (GCU)] of 8, and filters with an area of 10 to 1000 μm2; red, Top-Hat, radius of 100 μm, threshold (GCU) of 50, and filters with an area of 50 μm2 (minimum); LM2-4: green, Top-Hat, radius of 100 μm, threshold (GCU) of 8, and filters with an area of 10 to 1000 μm2; red, Top-Hat, radius of 100 μm, threshold (GCU) of 30, and filters with an area of 50 μm2 (minimum); PK-8: green, Top-Hat, radius of 100 μm; threshold (GCU) of 8, and filters with an area of 100 to 1600 μm2; red, Top-Hat, radius of 100 μm, threshold (GCU) of 12, and filters with an area of 100 μm2 (minimum); HT-29: green, Top-Hat, radius of 100 μm, threshold (GCU) of 10, filters with an area of 10 to 1000 μm2, and eccentricity of 0.8 (maximum); red, Top-Hat, radius of 100 μm, threshold (GCU) of 50, filters with an area of 50 μm2 (minimum), and eccentricity of 0.92 (maximum).
Organoids
For drug screening, Greiner 96-well plates were coated by diluting Matrigel 1:30 with organoid basal medium (66) and plating 100 μl per well. The Matrigel was allowed to polymerize for at least 1 hour before the remaining medium was removed and complete organoid medium containing 5000 cells per well was plated over. The organoids were allowed to grow for 96 hours before drugs were added in an additional 100 μl of organoid medium per well in triplicate for 72 hours. Organoids were stained with ethidium homodimer and Hoechst for 1 hour and scanned on an IN Cell Analyzer 2200 instrument. The resulting images were counted using IN Cell Analyzer 2200 software, and data were analyzed using the IN Cell Analyzer Workstation 3.7.3 software.
pHi measurements
pHi measurements were carried out as previously described (32). Briefly, cells were seeded into 96-well plates (3000 cells per well) in quadruplicates and then treated with SLC-0111, compound 13, compound 11, S0859, or NH4Cl and incubated in hypoxia for 72 hours. The pHi measurements were carried out using the Fluorometric Intracellular pH Assay Kit (Sigma-Aldrich, catalog no. MAK150) according to the manufacturer’s instructions. Briefly, growth medium was removed, and cells were loaded with 50 μl of BCFL-AM dye loading solution for 30 min at 37°C in hypoxia, followed by treatment with fresh inhibitor for 15 min. Ratiometric measurements were then carried out at λex = 505, λem = 535 and λex = 430, and λem = 535 using a SpectraMax i3x microplate reader (Molecular Devices). A calibration standard curve was prepared using pH calibration buffers (pH 6.0 to pH 8.0; increments of 0.5 pH units) containing 10 μM nigericin. Dye loading, drug treatment, and calibration were performed using Hanks’ balanced salt solution (HBSS) with 20 mM Hepes, without sodium bicarbonate, provided with the kit. A sigmoidal 4PL nonlinear regression model was used to fit the calibration curve and interpolate the experimental pHi values.
Western blotting
Cells were lysed in radioimmunoprecipitation assay buffer and quantified by bicinchoninic acid assay. Five micrograms of protein was separated on a 4 to 12% bis-tris gradient gel in 1× Mops buffer or a 3 to 8% tris-acetate gel in 1× tris-acetate SDS buffer. Gels were transferred to polyvinylidene difluoride for 1 hour at 100 V in ice-cold tris-glycine transfer buffer containing 20% methanol. Membranes were fixed in methanol for 20 s and dried before incubation with primary antibodies overnight in 1× TBST (Tris-buffered saline containing 0.05% Tween-20) (pH 7.6) containing 2% BSA. Horseradish peroxidase (HRP)–conjugated, species-specific, secondary antibodies (mouse, Cell Signaling Technology, #cs7076; rabbit, Jackson ImmunoResearch, #111-035-003; and goat, Jackson ImmunoResearch, #805-035-180) were added the following morning for 1 hour at room temperature and detected using enhanced chemiluminescence on a ChemiDoc (Bio-Rad).
Flow cytometry
Cells were seeded at 1 × 104 cells/cm2 in six-well plates, treated, and incubated in 1% O2 for 72 hours in respective growth medium. Following the 72-hour incubation, the cells were washed with 1× HBSS and stained with BODIPY 581/591 C11 (2 μM), CM-H2DCFDA (0.5 μM), or CellROX Deep Red (5 μM) for 15 min at 37°C or with RPA (0.5 μM) for 15 min at 37°C, followed by a 30-min incubation in HBSS at the same temperature. Cells were then washed and trypsinized. Collected cells were washed with HBSS + 2% FBS, resuspended in the same buffer, and analyzed on a BD LSRFortessa and collected with BD FACSDiva. Fluorescence following excitation with the 488-nm laser (BODIPY 581/591 C11 and CM-H2DCFDA), 568-nm laser (RPA), or 633-nm laser (CellROX Deep Red) was monitored in the blue 530/30 detector, yellow/green 610/20 detector, or red 670/14 detector, respectively. Data were analyzed in FlowJo v10 (FlowJo LLC). For BODIPY 581/591 C11 analyses, we report the percentage of the 10,000 events analyzed with increased green fluorescence (BODIPY C11Ox) signal above control samples. For CM-H2DCFDA, CellROX Deep Red, and RPA analyses, we report mean fluorescence intensity values for each respective fluorochrome.
“IN Cell” CAIX activity assay
IN Cell CAIX catalytic activity assays were carried out as described previously (12) with the following modifications: Assays were performed using SUM159PT cells following a 72-hour incubation in 1% O2 and were carried out on ice. For each sample, 5 × 105 cells were suspended in 100 μl of CO2-free isotonic buffer [20 mM Hepes, 130 mM NaCl, and 5 mM KCl (pH 8.0)] in a 2-ml flat bottom vial equipped with an 8-mm magnetic stir bar. Inhibitors (SLC-0111, compound 11, and compound 13) were added to the cell suspension to a final concentration of 50 μM or an equivalent volume of dimethyl sulfoxide (DMSO) vehicle added, and the sample was incubated at room temperature for 30 min. An additional 700 μl of ice-cold buffer was then added to the cell-inhibitor mixture, and a narrow pH electrode (Accumet, catalog no. 13-620-850) was immersed in the sample and equilibrated for 3 min. A total of 200 μl of CO2-saturated water was added to initiate the assay, and pH readings were recorded at 5-s intervals for up to 150 s. The change in pH was plotted as function of time. To determine the rate of spontaneous CO2 hydration, measurements were performed on cell-free (“buffer”) samples. The increase in hydration rate above the spontaneous rate is a measure of carbonic anhydrase activity. Three replicates were assayed for each condition evaluated in the assay.
Animal studies
All studies were performed in accordance with University of British Columbia and BC Cancer Research Institute Animal Care Committee guidelines under approved animal ethics protocol no. A18-0132.
Orthotopic xenograft
A total of 1 × 106 SUM159PT TRIPZ-shNS or TRIPZ-shNFS1 cells were injected into the left fourth mammary fat pad of 8- to 10-week-old female NSG (NOD scid gamma) mice in a solution consisting of 80% Matrigel/20% saline. Tumor growth was tracked by digital caliper, and volumes were calculated using the modified ellipsoid formula (l × w2 × π/6). Once average volume for the group reached ~80 mm3, mice were randomized to ensure equivalent average volumes, switched to Dox containing chow (625 mg/kg; Envigo), and dosed once daily by oral gavage with vehicle or SLC-0111 (100 mg/kg) (32). A cohort of mice bearing TRIPZ-shNS or TRIPZ-shNFS1 tumors was left untreated on regular chow as controls. Tumor volumes were statistically evaluated using TumGrowth (73).
Experimental metastasis
A total of 5 × 105 4T1luc shNS or shCAIX cells were injected into the lateral tail vein of female 7- to 9-week-old Balb/C mice (the Jackson Laboratory) in 100 μl of saline cell suspension (14). Before injection, cells in the Trolox-treated group were treated with 100 μM Trolox in suspension for 1 hour on ice (40). Mice were evaluated for the presence of metastasis weekly by bioluminescent imaging (IVIS Spectrum, PerkinElmer) following intraperitoneal injection of d-luciferin (PerkinElmer) at 150 mg/kg. Images were collected and analyzed using the Living Image software (PerkinElmer).
Breast tumor tissue microarray
This study was supported by the BC Cancer Tumor Tissue Repository, Victoria, BC, a member of the Canadian Tissue Repository Network in accordance with REB approval under human ethics protocol no. H18-03701. A 48-specimen array was constructed with 0.6-mm cores, spotted in duplicate, containing 28 triple-negative cases and 20 high-grade ductal cases. Arrays were stained manually with antibodies to CAIX (M-75; 1 μg/ml; BioScience Slovakia, AB1001) or NFS1 (20 μg/ml; Santa Cruz Biotechnology, sc365308). Following deparaffinization in xylene and gradual rehydration in graded ethanol baths, antigen retrieval was performed by microwave on high for 10 min in citrate buffer. Antibodies were added overnight at 4°C, and ImmPRESS HRP-conjugated, universal, secondary antibodies were added the following morning according to the manufacturer’s instructions (Vector Labs, #MP-7500), followed by DAB (3,3′-Diaminobenzidine) detection (Vector Labs, #SK-4100). Arrays were scored by two independent observers (S. Chafe and O. Nemirovsky). CAIX scoring was performed using a presence or absence approach as previously described (14), and NFS1 scoring was binned into high (2/3) and low (0/1) samples.
Statistics
Data were plotted using GraphPad Prism version 8.0. All statistical analyses were performed using GraphPad using a threshold of P < 0.05 for statistical significance.
Acknowledgments
Funding: This work was supported by a Foundation Scheme grant from the Canadian Institutes of Health Research (CIHR) (grant no. FDN-143318) and a grant from the Canadian Cancer Society Research Institute (CCSRI) (grant no. 703191) to S.D. F.J.V supported this project from operating grants from CIHR (PJT-156309) and Canada Foundation for Innovation (CFI-33364). F.S.V. is supported by funding from the College of Medicine, University of Saskatchewan. Author contributions: Conceptualization: S.C.C. and S.D. Methodology: S.C.C., W.S.B., P.C.M., L.H., S.K.M., and S.D. Software: F.S.V. Formal analysis: S.C.C., F.S.V., F.J.V., and S.D. Investigation: S.C.C., F.S.V., G.V., S.A., O.N., and A.M. Resources: F.S.V., F.C., C.T.S., J.M.K., A.M., D.F.S., D.J.R., and F.J.V. Writing (original draft): S.C.C. and S.D. Writing (review and editing): S.C.C., F.S.V., F.J.V., and S.D. Visualization: S.C.C. and F.S.V. Supervision: S.C.C., J.M.K., D.F.S., D.J.R., C.T.S., F.J.V., and S.D. Funding acquisition: F.J.V. and S.D. Competing interests: S.D., C.T.S., P.C.M., and F.C. are inventors on a patent related to this work filed by Welichem Biotech Inc., Burnaby, Canada, US 9,463,171 B2, filed: 9 January 2013, published: 11 October 2016. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The sequencing datasets generated for the genome-wide CRISPR screen used in this study are deposited to the Gene Expression Omnibus (GEO) database and can be obtained with the accession number GSE167481.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabj0364/DC1
REFERENCES AND NOTES
- 1.Wilson W. R., Hay M. P., Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 (2011). [DOI] [PubMed] [Google Scholar]
- 2.McKeown S. R., Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 87, 20130676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee P., Chandel N. S., Simon M. C., Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corbet C., Feron O., Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 17, 577–593 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Neri D., Supuran C. T., Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 10, 767–777 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Wykoff C. C., Beasley N., Watson P. H., Campo L., Chia S. K., English R., Pastorek J., Sly W. S., Ratcliffe P., Harris A. L., Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast. Am. J. Pathol. 158, 1011–1019 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swietach P., Wigfield S., Cobden P., Supuran C. T., Harris A. L., Vaughan-Jones R. D., Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J. Biol. Chem. 283, 20473–20483 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Švastová E., Hulíkova A., Rafajová M., Zat’ovičová M., Gibadulinová A., Casini A., Cecchi A., Scozzafava A., Supuran C. T., Pastorek J., Pastoreková S., Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 577, 439–445 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Chiche J., Ilc K., Laferriere J., Trottier E., Dayan F., Mazure N. M., Brahimi-Horn M. C., Pouyssegur J., Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 69, 358–368 (2009). [DOI] [PubMed] [Google Scholar]
- 10.McDonald P. C., Winum J. Y., Supuran C. T., Dedhar S., Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 3, 84–97 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chia S. K., Wykoff C. C., Watson P. H., Han C., Leek R. D., Pastorek J., Gatter K. C., Ratcliffe P., Harris A. L., Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J. Clin. Oncol. 19, 3660–3668 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Swayampakula M., McDonald P. C., Vallejo M., Coyaud E., Chafe S. C., Westerback A., Venkateswaran G., Shankar J., Gao G., Laurent E. M. N., Lou Y., Bennewith K. L., Supuran C. T., Nabi I. R., Raught B., Dedhar S., The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 36, 6244–6261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debreova M., Csaderova L., Burikova M., Lukacikova L., Kajanova I., Sedlakova O., Kery M., Kopacek J., Zatovicova M., Bizik J., Pastorekova S., Svastova E., CAIX regulates invadopodia formation through both a pH-dependent mechanism and interplay with actin regulatory proteins. Int. J. Mol. Sci. 20, 2745 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lou Y., McDonald P. C., Oloumi A., Chia S., Ostlund C., Ahmadi A., Kyle A., Auf dem Keller U., Leung S., Huntsman D., Clarke B., Sutherland B. W., Waterhouse D., Bally M., Roskelley C., Overall C. M., Minchinton A., Pacchiano F., Carta F., Scozzafava A., Touisni N., Winum J. Y., Supuran C. T., Dedhar S., Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 71, 3364–3376 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Chafe S. C., Lou Y., Sceneay J., Vallejo M., Hamilton M. J., McDonald P. C., Bennewith K. L., Moller A., Dedhar S., Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. 75, 996–1008 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Lock F. E., McDonald P. C., Lou Y., Serrano I., Chafe S. C., Ostlund C., Aparicio S., Winum J. Y., Supuran C. T., Dedhar S., Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 32, 5210–5219 (2013). [DOI] [PubMed] [Google Scholar]
- 17.McDonald P. C., Chia S., Bedard P. L., Chu Q., Lyle M., Tang L., Singh M., Zhang Z., Supuran C. T., Renouf D. J., Dedhar S., A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 43, 484–490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graeber T. G., Osmanian C., Jacks T., Housman D. E., Koch C. J., Lowe S. W., Giaccia A. J., Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379, 88–91 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Tang D., Kang R., Berghe T. V., Vandenabeele P., Kroemer G., The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X., Stockwell B. R., Conrad M., Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie H., Simon M. C., Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 292, 16825–16832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torti S. V., Torti F. M., Iron and cancer: More ore to be mined. Nat. Rev. Cancer 13, 342–355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou Y., Palte M. J., Deik A. A., Li H., Eaton J. K., Wang W., Tseng Y. Y., Deasy R., Kost-Alimova M., Dancik V., Leshchiner E. S., Viswanathan V. S., Signoretti S., Choueiri T. K., Boehm J. S., Wagner B. K., Doench J. G., Clish C. B., Clemons P. A., Schreiber S. L., A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang M., Chen P., Liu J., Zhu S., Kroemer G., Klionsky D. J., Lotze M. T., Zeh H. J., Kang R., Tang D., Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayes J. D., Dinkova-Kostova A. T., Tew K. D., Oxidative stress in cancer. Cancer Cell 38, 167–197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouault T. A., Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 16, 45–55 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Maio N., Jain A., Rouault T. A., Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 55, 34–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouault T. A., Maio N., Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 292, 12744–12753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez S. W., Sviderskiy V. O., Terzi E. M., Papagiannakopoulos T., Moreira A. L., Adams S., Sabatini D. M., Birsoy K., Possemato R., NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chafe S. C., McDonald P. C., Saberi S., Nemirovsky O., Venkateswaran G., Burugu S., Gao D., Delaidelli A., Kyle A. H., Baker J. H. E., Gillespie J. A., Bashashati A., Minchinton A. I., Zhou Y., Shah S. P., Dedhar S., Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol. Res. 7, 1064–1078 (2019). [DOI] [PubMed] [Google Scholar]
- 32.McDonald P. C., Chafe S. C., Brown W. S., Saberi S., Swayampakula M., Venkateswaran G., Nemirovsky O., Gillespie J. A., Karasinska J. M., Kalloger S. E., Supuran C. T., Schaeffer D. F., Bashashati A., Shah S. P., Topham J. T., Yapp D. T., Li J., Renouf D. J., Stanger B. Z., Dedhar S., Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology 157, 823–837 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Gibadulinova A., Bullova P., Strnad H., Pohlodek K., Jurkovicova D., Takacova M., Pastorekova S., Svastova E., CAIX-mediated control of LIN28/let-7 axis contributes to metabolic adaptation of breast cancer cells to hypoxia. Int. J. Mol. Sci. 21, 4299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyd N. H., Walker K., Fried J., Hackney J. R., McDonald P. C., Benavides G. A., Spina R., Audia A., Scott S. E., Libby C. J., Tran A. N., Bevensee M. O., Griguer C., Nozell S., Gillespie G. Y., Nabors B., Bhat K. P., Bar E. E., Darley-Usmar V., Xu B., Gordon E., Cooper S. J., Dedhar S., Hjelmeland A. B., Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2, e92928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benej M., Svastova E., Banova R., Kopacek J., Gibadulinova A., Kery M., Arena S., Scaloni A., Vitale M., Zambrano N., Papandreou I., Denko N. C., Pastorekova S., CA IX stabilizes intracellular pH to maintain metabolic reprogramming and proliferation in hypoxia. Front. Oncol. 10, 1462 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petrat F., Weisheit D., Lensen M., de Groot H., Sustmann R., Rauen U., Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem. J. 362, 137–147 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conrad M., Pratt D. A., The chemical basis of ferroptosis. Nat. Chem. Biol. 15, 1137–1147 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Pacchiano F., Carta F., McDonald P. C., Lou Y., Vullo D., Scozzafava A., Dedhar S., Supuran C. T., Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 54, 1896–1902 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Piskounova E., Agathocleous M., Murphy M. M., Hu Z., Huddlestun S. E., Zhao Z., Leitch A. M., Johnson T. M., DeBerardinis R. J., Morrison S. J., Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ubellacker J. M., Tasdogan A., Ramesh V., Shen B., Mitchell E. C., Martin-Sandoval M. S., Gu Z., McCormick M. L., Durham A. B., Spitz D. R., Zhao Z., Mathews T. P., Morrison S. J., Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gout P. W., Buckley A. R., Simms C. R., Bruchovsky N., Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc– cystine transporter: A new action for an old drug. Leukemia 15, 1633–1640 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Ch’en F. F.-T., Villafuerte F. C., Swietach P., Cobden P. M., Vaughan-Jones R. D., S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br. J. Pharmacol. 153, 972–982 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herzig S., Shaw R. J., AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H., Zandkarimi F., Zhang Y., Meena J. K., Kim J., Zhuang L., Tyagi S., Ma L., Westbrook T. F., Steinberg G. R., Nakada D., Stockwell B. R., Gan B., Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doll S., Proneth B., Tyurina Y. Y., Panzilius E., Kobayashi S., Ingold I., Irmler M., Beckers J., Aichler M., Walch A., Prokisch H., Trumbach D., Mao G., Qu F., Bayir H., Fullekrug J., Scheel C. H., Wurst W., Schick J. A., Kagan V. E., Angeli J. P., Conrad M., ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kagan V. E., Mao G., Qu F., Angeli J. P., Doll S., Croix C. S., Dar H. H., Liu B., Tyurin V. A., Ritov V. B., Kapralov A. A., Amoscato A. A., Jiang J., Anthonymuthu T., Mohammadyani D., Yang Q., Proneth B., Klein-Seetharaman J., Watkins S., Bahar I., Greenberger J., Mallampalli R. K., Stockwell B. R., Tyurina Y. Y., Conrad M., Bayir H., Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon S. J., Winter G. E., Musavi L. S., Lee E. D., Snijder B., Rebsamen M., Superti-Furga G., Stockwell B. R., Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Küch E.-M., Vellaramkalayil R., Zhang I., Lehnen D., Brügger B., Sreemmel W., Ehehalt R., Poppelreuther M., Füllekrug J., Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 1841, 227–239 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Kang M. J., Fujino T., Sasano H., Minekura H., Yabuki N., Nagura H., Iijima H., Yamamoto T. T., A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. U.S.A. 94, 2880–2884 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Neil N. J., Bailey M. L., Hieter P., Synthetic lethality and cancer. Nat. Rev. Genet. 18, 613–623 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Hernansanz-Agustín P., Choya-Foces C., Carregal-Romero S., Ramos E., Oliva T., Villa-Piña T., Moreno L., Izquierdo-Álvarez A., Cabrera-García J. D., Cortés A., Lechuga-Vieco A. V., Jadiya P., Navarro E., Parada E., Palomino-Antolín A., Tello D., Acín-Pérez R., Rodríguez-Aguilera J. C., Navas P., Cogolludo Á., López-Montero I., Martínez-del-Pozo Á., Egea J., López M. G., Elrod J. W., Ruíz-Cabello J., Bogdanova A., Enríquez J. A., Martínez-Ruiz A., Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586, 287–291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanharanta S., Massague J., Origins of metastatic traits. Cancer Cell 24, 410–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S. E., Zhang L., Ma K., Riegman M., Chen F., Ingold I., Conrad M., Turker M. Z., Gao M., Jiang X., Monette S., Pauliah M., Gonen M., Zanzonico P., Quinn T., Wiesner U., Bradbury M. S., Overholtzer M., Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 11, 977–985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., Fulda S., Gascon S., Hatzios S. K., Kagan V. E., Noel K., Jiang X., Linkermann A., Murphy M. E., Overholtzer M., Oyagi A., Pagnussat G. C., Park J., Ran Q., Rosenfeld C. S., Salnikow K., Tang D., Torti F. M., Torti S. V., Toyokuni S., Woerpel K. A., Zhang D. D., Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R., Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.A. 113, E4966–E4975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimada K., Skouta R., Kaplan A., Yang W. S., Hayano M., Dixon S. J., Brown L. M., Valenzuela C. A., Wolpaw A. J., Stockwell B. R., Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeon S. M., Chandel N. S., Hay N., AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eichner L. J., Brun S. N., Herzig S., Young N. P., Curtis S. D., Shackelford D. B., Shokhirev M. N., Leblanc M., Vera L. I., Hutchins A., Ross D. S., Shaw R. J., Svensson R. U., Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab. 29, 285–302.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chhipa R. R., Fan Q., Anderson J., Muraleedharan R., Huang Y., Ciraolo G., Chen X., Waclaw R., Chow L. M., Khuchua Z., Kofron M., Weirauch M. T., Kendler A., McPherson C., Ratner N., Nakano I., Dasgupta N., Komurov K., Dasgupta B., AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 20, 823–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walton Z. E., Patel C. H., Brooks R. C., Yu Y., Ibrahim-Hashim A., Riddle M., Porcu A., Jiang T., Ecker B. L., Tameire F., Koumenis C., Weeraratna A. T., Welsh D. K., Gillies R., Alwine J. C., Zhang L., Powell J. D., Dang C. V., Acid suspends the circadian clock in hypoxia through inhibition of mTOR. Cell 174, 72–87.e32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wojtkowiak J. W., Rothberg J. M., Kumar V., Schramm K. J., Haller E., Proemsey J. B., Lloyd M. C., Sloane B. F., Gillies R. J., Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 72, 3938–3947 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yi J., Zhu J., Wu J., Thompson C. B., Jiang X., Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. U.S.A. 117, 31189–31197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flanagan L., Van Weelden K., Ammerman C., Ethier S. P., Welsh J., SUM-159PT cells: A novel estrogen independent human breast cancer model system. Breast Cancer Res. Treat. 58, 193–204 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Ebos J. M., Lee C. R., Cruz-Munoz W., Bjarnason G. A., Christensen J. G., Kerbel R. S., Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lou Y., Preobrazhenska O., auf dem Keller U., Sutcliffe M., Barclay L., McDonald P. C., Roskelley C., Overall C. M., Dedhar S., Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev Dyn 237, 2755–2768 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Huang L., Holtzinger A., Jagan I., BeGora M., Lohse I., Ngai N., Nostro C., Wang R., Muthuswamy L. B., Crawford H. C., Arrowsmith C., Kalloger S. E., Renouf D. J., Connor A. A., Cleary S., Schaeffer D. F., Roehrl M., Tsao M. S., Gallinger S., Keller G., Muthuswamy S. K., Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 21, 1364–1371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelson T., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paul J. M., Toosi B., Vizeacoumar F. S., Bhanumathy K. K., Li Y., Gerger C., El Zawily A., Freywald T., Anderson D. H., Mousseau D., Kanthan R., Zhang Z., Vizeacoumar F. J., Freywald A., Targeting synthetic lethality between the SRC kinase and the EPHB6 receptor may benefit cancer treatment. Oncotarget 7, 50027–50042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li W., Xu H., Xiao T., Cong L., Love M. I., Zhang F., Irizarry R. A., Liu J. S., Brown M., Liu X. S., MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge S. X., Son E. W., Yao R., iDEP: An integrated web application for differential expression and pathway analysis of RNA-seq data. BMC Bioinformatics 19, 534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopes C. T., Franz M., Kazi F., Donaldson S. L., Morris Q., Bader G. D., Cytoscape Web: An interactive web-based network browser. Bioinformatics 26, 2347–2348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Netanely D., Avraham A., Ben-Baruch A., Evron E., Shamir R., Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. 18, 74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enot D. P., Vacchelli E., Jacquelot N., Zitvogel L., Kroemer G., TumGrowth: An open-access web tool for the statistical analysis of tumor growth curves. Onco. Targets. Ther. 7, e1462431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabj0364/DC1