

Lamin B1 controls 53BP1 recruitment to DNA damage.
Abstract
Double-strand breaks (DSBs) are harmful lesions and a major cause of genome instability. Studies have suggested a link between the nuclear envelope and the DNA damage response. Here, we show that lamin B1, a major component of the nuclear envelope, interacts directly with 53BP1 protein, which plays a pivotal role in the DSB repair. This interaction is dissociated after DNA damage. Lamin B1 overexpression impedes 53BP1 recruitment to DNA damage sites and leads to a persistence of DNA damage, a defect in nonhomologous end joining and an increased sensitivity to DSBs. The identification of interactions domains between lamin B1 and 53BP1 allows us to demonstrate that the defect of 53BP1 recruitment and the DSB persistence upon lamin B1 overexpression are due to sequestration of 53BP1 by lamin B1. This study highlights lamin B1 as a factor controlling the recruitment of 53BP1 to DNA damage sites upon injury.
INTRODUCTION
Genome stability is ensured by the coordination of cell cycle control, DNA repair, apoptosis, and senescence. Double-strand breaks (DSBs) are very harmful lesions that can be generated by endogenous and exogenous stresses (1). DSB repair defects lead to cellular death, senescence, and genome instability. In mammalian cells, nonhomologous end joining (NHEJ) plays a pivotal role in DSB repair, and consistently, its defect leads to high sensitivity to DSB-inducing agents and genome rearrangements. However, while NHEJ is essential for the maintenance of genome stability, it can also generate genetic instability through chromosomal sequence capture or translocations (2–4). Moreover, NHEJ has been suggested to contribute to triggering chromothripsis observed in tumors (5).
The tumor suppressor p53-binding protein (53BP1) protein, in association with telomere-associated protein RIF1 (RIF1), PAX-interacting protein 1 (PTIP), Bloom syndrome protein (BLM) proteins, favors NHEJ DSB repair by protecting DNA extremities from resection through the 53BP1-RIF1–dependent recruitment of the shielding complex (6–10), a required step for competitor pathways, such as homologous recombination or alternative end-joining (11–16). 53BP1 deficiency leads to radiosensitivity, and 53BP1-deficient mice are prone to tumor development (17). However, uncontrolled 53BP1 recruitment increases genetic instability in mitosis (18, 19). Together, these observations indicate the need to tightly control NHEJ and 53BP1 recruitment to DNA damage. Various sophisticated mechanisms controlling 53BP1 binding to damaged chromatin have been reported. Several modifications of chromatin and of associated proteins that surround DSBs modulate the accessibility of 53BP1 to DNA damage sites. In addition, post-translational modifications of 53BP1 itself tightly control its recruitment to damaged DNA and its subsequent role in DNA damage response (DDR) (18–22).
Different studies have suggested a link between nuclear envelope proteins and the DDR. In mammalian cells, the integrity and plasticity of the nuclear envelope are ensured by the two types of nuclear lamins, A-type and B-type, and their associated proteins (23). While lamin B1, a major component of the nuclear lamina, encoded by the LMNB1 gene, is present throughout the organism’s lifespan, lamin A encoded by the LMNA gene is mainly expressed in differentiated cells (23). Various degenerative pathologies are associated with LMNA gene mutations, among which the severe premature aging Hutchinson-Gilford progeria syndrome (HGPS). In this case, a deficient processing of the precursor pre–lamin A leads to the expression of a mutated, immature, and farnesylated lamin A protein named progerin (24, 25). Beyond the impact of lamins on nuclear integrity, on gene expression regulation and possibly on DNA replication, a role for lamins, especially for lamin A, in DNA damage signaling and repair has been also reported. The persistence of DNA damage, as visualized by the presence of the DSB marker γH2AX foci, has been observed in progeria cells, after pre–lamin A overexpression and in a mouse model with defective ZMPSTE24 protease, which is involved in pre–lamin A processing to generate mature lamin A (26–29). The persistence of DNA damage has been proposed to participate in the aging phenotypes of HGPS (26, 27). Moreover, a decrease in 53BP1 stability was reported in mouse embryonic fibroblasts lacking lamin A gene expression (30). It has been also shown that lamin A can coimmunoprecipate with 53BP1 (31) and that pre–lamin A overexpression leads to a mislocalization of 53BP1 into the cytoplasm (32). Lamin B1 decrease, a situation that can be observed upon senescence induction, leads to misregulation of DNA repair factors expression, especially those involved in DSB repair, and may lead to DNA repair defect (33, 34).
However, to date, no genetic disease associated with a decrease of lamin B1 level has been reported. Recently, it has been reported identification of de novo mutations in LMNB1 that result in a dominant and damaging effect on nuclear envelope formation and that cause microcephaly in humans (35, 36). In contrast, various pathological situations have been associated with lamin B1 overexpression. A duplication of the LMNB1 gene, resulting in lamin B1 overexpression, is responsible for adult-onset autosomal dominant leukodystrophy (ADLD) (37). Overexpression of lamin B1 was also reported in two repair syndromes, namely, ataxia telangiectasia (A-T) and Werner syndromes, caused respectively by mutations in ATM and WRN genes, and which are associated with genetic instability, cancer predisposition, and premature aging (38, 39). We have previously identified lamin B1 accumulation in A-T cells. We showed that normalizing lamin B1 rescued the nuclear shape alteration (NSA) and decreased the premature senescence observed in these cells (38). Last, NSA is one of the characteristics of tumor cells, and for decades, it has been used by cytologists to classify tumors in relation with their aggressiveness. In several tumors, a high level of lamin B1 has been found (40–44). However, the impact of NSA and especially of lamin B1 accumulation on genome stability or on DSB repair has not been studied yet.
Here, we determine the effect of lamin B1 overexpression on DSB repair. In contrast to what has been reported for lamin A deficiency or mutant pre–lamin A accumulation (30–32), we find that neither the 53BP1 level nor its nuclear localization is altered upon lamin B1 overexpression. However, lamin B1 overexpression impedes 53BP1 recruitment to DNA damage. We identify a direct interaction between endogenous lamin B1 and 53BP1 proteins. In cells, this interaction is dissociated after exposure to ionizing radiation (IR), allowing rapid 53BP1 recruitment to DNA damage. Together, these data highlight a previously unindentified mechanism that modulates 53BP1 recruitment after DNA injury and unveil another link between the nuclear envelope, DSB repair, and genome stability.
RESULTS
Lamin B1 overexpression leads to DSB accumulation and to a higher sensitivity to genotoxic stress
Lamin B1 was overexpressed in human transformed fibroblasts, and 48 hours after, cell survival was measured upon exposure to genotoxic stress. Cloning efficiency assay, performed after irradiation exposure, highlights an elevated sensitivity of lamin B1–overexpressing cells compared to control cells (Fig. 1A). Consistently, upon IR, the metaphases of lamin B1–overexpressing cells show a significantly higher level of chromosome abnormalities including chromatid breaks (Fig. 1B). In unchallenged lamin B1–overexpressing cells, the presence of an elevated number of chromosomal DSBs (Fig. 1B) was confirmed by the quantification of γH2AX foci (Fig. 1C). The γH2AX foci increased upon a two- to fivefold lamin B1 overexpression, a level comparable to what is observed in ADLD or in tumor cells (Fig. 1C). An increase of γH2AX level was also detected and quantified by Western blotting (Fig. 1C). A slower kinetics of IR-induced γH2AX foci disappearance in lamin B1–overexpressing cells versus control cells is consistent with a DSB repair defect. Three hours after irradiation, 65 and 80% of γH2AX foci persist in lamin B1–overexpressing-cells (for a 2- to 5-fold overexpression and for >5-fold overexpression, respectively), while less than 25% of γH2AX foci persist in control cells (Fig. 1D). Quantification of breaks on metaphases (in unchallenged cells and upon IR) and analysis of γH2AX foci 3 hours after irradiation showed that lamin A overexpression also triggers DNA damage persistence (fig. S1, A and B). The MDC1 protein, which is involved in the first steps of the DDR, binds to DSBs through γH2AX and is required for 53BP1 recruitment (45, 46). A decrease in the disappearance of IR-induced MDC1 foci was also observed in lamin B1–overexpressing cells compared to control cells (fig. S2). These data suggest a DSB repair defect triggered by lamin B1 dysregulation downstream of γH2AX and MDC1 recruitments.
Fig. 1. Enhanced genotoxic stress sensitivity and increased DSB formation in lamin B1–overexpressing cells.

(A) Clonogenic survival curves of control (Ctrl) and lamin B1–overexpressing cells (LMNB1) after γ-irradiation. Forty-eight hours after transfection, cells are plated (100, 500, 1000, or 2000 cells per well of six-well plate) and irradiated (Cs137, 0.5, 1, or 2 Gy at 1.77 Gy/min; iBL637 irradiator). Eight to 11 days later, cell colonies were stained by gentian violet and counted. Multiple t test P values *P < 0.05; **P < 0.005. (B) Chromosomal aberrations were scored in metaphase spreads prepared from lamin B1–overexpressing cells or control cells, under basal conditions (NI) or irradiation (2 Gy) 24 hours before cell harvest. Histogram shows the mean number of chromatid breaks scored per metaphase [n = 89 (Ctrl, NI) n = 98; (LMNB1, NI); n = 94 (Ctrl, IR); and n = 94 (LMNB1, IR)]. Example of chromosomes with chromatid breaks found in lamin B1–overexpressing cells are shown in the left. (C) Analysis of basal γH2AX foci and protein levels in control and lamin B1–overexpressing cells 48 hours after transfection. γH2AX protein levels were analyzed by Western blot (left, top) and quantified from three independent experiments with ImageJ (right, top). Representative images and quantification of γH2AX foci by immunofluorescent staining are shown in the left and right bottom panels, respectively. Scale bar, 10 μm. (D) Analysis of IR-induced γH2AX foci kinetics after irradiation (0.5 Gy), in control (Ctrl) and lamin B1–overexpressing (LMNB1) cells. γH2AX foci were counted with Metamorph software from four independent experiments. For (C) and (D), lamin B1–overexpressing cells were divided into two categories: cells expressing moderate (up to fivefold) or higher (more than fivefold) levels of lamin B1. For (B), (C), and (D), unpaired t test P values *P < 0.05; **P < 0.005; ***P < 0.0001. Error bars, SEM.
Lamin B1 overexpression alters the NHEJ efficiency
We then determined whether the sensitivity to genotoxic agents and the persistence of DSBs after irradiation could result from a defect in DSB repair by NHEJ. For this purpose, cells containing an intrachromosomal substrate, which allows the measurement of NHEJ events, were cotransfected with vectors encoding the meganuclease I-SceI and lamin B1 (Fig. 2A). The measurement of CD4 expression by flow cytometry monitors the repair frequency of I-SceI–induced DSBs by NHEJ, as previously described (2, 16, 47–49). Lamin B1 overexpression resulted in a twofold decrease in the frequency of CD4-positive cells following I-SceI expression (Fig. 2B). The I-SceI level after lamin B1 overexpression is comparable with level found in cells transfected with an empty vector (Fig. 2A, right). These data show that lamin B1 overexpression represses NHEJ. As lamins may have a role in gene expression (23), the expression levels of the essential proteins of the NHEJ pathway (KU70, KU80, DNA-PKcs, XRCC4, and ligase IV) were examined by Western blot after lamin B1 overexpression. No changes in the levels of these proteins were observed in lamin B1–overexpressing cells (fig. S3A).
Fig. 2. Lamin B1 overexpression decreases NHEJ efficiency.

(A) The intrachromosomal substrate (left): The reporter genes encode for the membrane antigens H2Kd and CD4. Before expression of the meganuclease I-SceI, CD4 is too far away from CMV promoter to be expressed. I-SceI sites flank a fragment containing the H2Kd gene. After cleavage by I-SceI meganuclease, rejoining of the DNA ends can lead to direct ligation of the CMV promoter with the CD4 fragment leading to the expression of CD4, monitored by flow cytometry. Western blot analysis (right) shows lamin B1 overexpression and the equivalent level of I-SceI expression between I-SceI and I-SceI + LMNB1 conditions. (B) NHEJ event frequency in control and lamin B1–overexpressing cells. An example of CD4-positive cell fluorescence-activated cell sorting quantification (NHEJ event) is shown in control and lamin B1–overexpressing cells (left). Frequencies of NHEJ events (CD4+) in lamin B1–overexpressing cells (I-SceI + LMNB1) relative to control cells (I-SceI) are shown in the right (two tailed Mann-Whitney test, **P < 0.005). The values correspond to 14 independent experiments. FITC, fluorescein isothiocyanate; PE: phycoerythrin.
Lamin B1 overexpression alters 53BP1 recruitment to the damage sites
The 53BP1 protein plays an important role in controlling DSB repair and favors NHEJ by repressing resection at the double-strand DNA ends. Lamin B1 overexpression triggers a decrease in 53BP1 IR-induced foci (IRIF) formation in a dose-dependent manner (Fig. 3A). The 53BP1-dependent recruitment to DNA damage of the downstream RIF1 protein, which plays a key role with shelterin complex to protect DSB from resection, was analyzed. Consistent with a defect of 53BP1 recruitment, Fig. 3B shows a defect in RIF1 foci formation after irradiation.
Fig. 3. Lamin B1 overexpression impedes 53BP1 recruitment to DNA damage sites.

(A) Postirradiation kinetics of 53BP1 foci in transfected cells with empty (Ctrl) or lamin B1–expressing vectors. Representative images of 53BP1 foci at 30 min after irradiation (2 Gy), visualized by immunofluorescence staining, are shown in the top panel. 53BP1 foci were counted from three independent experiments with Metamorph software in control cells (Ctrl) and in lamin B1–overexpressing cells and were divided into two categories: those with moderate staining (up to fivefold lamin B1 level) or those with higher staining (more than fivefold) lamin B1 level (bottom). DAPI, 4′,6-diamidino-2-phenylindole. (B) Postirradiation formation of RIF1 foci in transfected cells with empty (Ctrl) or lamin B1–expressing vectors. Representative images of RIF1 foci at 1 hour after irradiation (5 Gy), visualized by immunofluorescence staining, are shown in the top. RIF1 foci were counted with Cell Profiler and KNIME software from three independent experiments in control cells (Ctrl) and in lamin B1–overexpressing cells with two- to fivefold lamin B1 level (bottom) (C) Recruitment kinetic of 53BP1 after laser microirradiation in U2OS cells transfected with either a 53BP1-GFP–expressing vector only (Ctrl) or both 53BP1-GFP–expressing and Ds-Red–lamin B1 expressing vectors (LMNB1). Cells were microirradiated with a 405-nm laser, and images were collected during a 75-min period to follow 53BP1 recruitment (top). GFP intensity at the breaks was then measured using NIS-Elements software (bottom). The values correspond to three independent experiments. Red line: region of irradiation; white line: scale bar. Unpaired t test P values *P < 0.05; ***P < 0.0001. Error bars, SEM. Scale bars, 10 μm.
To decipher whether lamin B1 overexpression might affect 53BP1 expression or stability, we measured the 53BP1 protein expression level by Western blot. Lamin B1 overexpression did not affect the protein level of 53BP1 (fig. S3B); thus, the defects in DSB repair and in 53BP1 foci formation observed here are not due to a defect in 53BP1 protein expression or stability, in contrast to what is observed in LMNA−/− cells (30, 31). Nevertheless, to exclude a potential alteration of lamin A level, we measured the level of lamin A/C after lamin B1 dysregulation and we did not observe any difference (fig. S3C). These data show that the impaired 53BP1 recruitment to DNA damage induced by lamin B1 is not associated with a change of lamin A and lamin C proteins. Differences in 53BP1 nuclear import that account for defective recruitment to DNA damage sites and for a NHEJ defect have been reported after depletion of the nuclear pore protein NUP153 (50). However, no difference in the cellular localization of 53BP1 was observed after lamin B1 dysregulation (fig. S3D), in contrast to what was recently reported for pre–lamin A overexpression (32). Together, these observations show that lamin B1 overexpression leads to a defect in 53BP1 foci formation, which is neither due to a defect in 53BP1 protein expression/stability nor to a defect in its import into the nucleus.
We next analyzed 53BP1 recruitment to damaged DNA by time-lapse video microscopy after microirradiation. Green fluorescent protein (GFP)–53BP1 and DsRed–lamin B1 were coexpressed in U2OS cells. Forty-eight hours later, the cells were microirradiated and monitored using live microscopy. Then, GFP-53BP1 recruitment to the laser track was analyzed. A lower GFP level was measured in DsRed–lamin B1–positive cells compared to control cells (Fig. 3C), confirming that lamin B1 overexpression induces a defect in 53BP1 recruitment to the DNA damage sites. Note here that neither lamin B1 staining/presence was shown to the laser track, suggesting that lamin B1 is not recruited to DNA damage after DNA damage.
Together, these observations demonstrate that lamin B1 overexpression leads to a defect in 53BP1 recruitment to damage sites. However, in contrast to data published for expression of pre–lamin A, the alteration of 53BP1 recruitment to DNA damage triggered by lamin B1 expression is not due to a defect in 53BP1 protein expression/stability or import into the nucleus, suggesting that the involved molecular mechanism is different from the one induced by lamin A dysregulation.
Lamin B1 interacts with the 53BP1 protein
Lamins have been shown to modulate the activity of proteins, such as transcription factors, by sequestering them (51). Since no defect in 53BP1 expression or nuclear import was observed upon lamin B1 overexpression, we hypothesized that lamin B1 might interact with 53BP1 and thus regulate its activity/recruitment after DNA injury. Immunoprecipitation assay showed that endogenous lamin B1 coprecipitates with endogenous 53BP1 (Fig. 4A). This interaction was not affected (Fig. 4A and fig. S4A) by a benzonase treatment (whose efficiency was verified; fig. S4B), thus indicating that the lamin B1 and 53BP1 proteins did not coprecipitate through DNA bridging. The lamin B1–53BP1 interaction was further confirmed by proximity ligation assay (PLA), which allows the visualization and quantification of in situ interactions (between proteins that are in close vicinity). The red dots in Fig. 4B correspond to in situ endogenous interactions between lamin B1 and 53BP1. The extinction of 53BP1 or lamin B1 by small interfering RNAs (siRNAs) strongly affects the frequency of PLA dots attesting to the specificity of the detected signal (Fig. 4B and fig. S4C). In contrast, after lamin B1 overexpression, a higher number of PLA dots was observed, reflecting an increase in interactions (Fig. 4C). Next, the localization of the interaction was investigated. Figure 4D and fig. S5A show that PLA dots are localized both in nucleus interior and at the nuclear periphery. However, according to the level of lamin B1 overexpression, the number of PLA dots is significantly increased at the periphery (50 to 65% compared to 30% without overexpression). This suggests that 53BP1 (at least a part) could be sequestered at nuclear envelop (NE) upon lamin B1 overexpression.
Fig. 4. 53BP1 interacts with lamin B1.

(A) Coimmunoprecipitation of endogenous lamin B1 and 53BP1. Proteins from cellular lysates were subjected, before (−) or after (+) benzonase treatment, to immunoprecipitation with rabbit antibodies against lamin B1 or IgG as control. Coimmunoprecipitated proteins were revealed with mouse anti-53BP1 and rabbit anti–lamin B1 antibodies. Coimmunoprecipitation of lamin B1 and 53BP1 was confirmed in four independent experiments. (B) In situ interaction between endogenous 53BP1 and lamin B1. Cells were transfected with control siRNA (siCtrl), siRNA directed against lamin B1 mRNA specifically (siLMNB1) or against 53BP1 mRNA specifically (si53BP1,) and subjected to PLA using anti-53BP1 and lamin B1 antibodies. Example of an in situ interaction between endogenous 53BP1 and lamin B1 visualized as red fluorescent dots (left). Quantification of PLA dots per nucleus is shown (n > 100 nuclei analyzed per condition) (right). (C) In situ interaction between 53BP1 and lamin B1 proteins after lamin B1 overexpression. Cells transfected with empty vector (Ctrl) or with lamin B1–expressing vector (LMNB1) were subjected to PLA using anti-53BP1 and lamin B1 antibodies. Example of in situ interaction between 53BP1 and lamin B1 visualized as red fluorescent dots (left). Quantification of PLA dots per nucleus is shown (n > 400 nuclei analyzed per condition) (right). (D) In situ localization of the interaction between 53BP1 and lamin B1. Cells transfected with empty vector (Ctrl) or with lamin B1–expressing vector (LMNB1) were subjected to PLA using anti-53BP1 and lamin B1 antibodies. PLA dots at the nuclear membrane or in the nucleus interior were counted and expressed as a percentage of total PLA dots per cells from three to four independent experiments. Level of Lamin B1 overexpression was categorized in three classes as a fold increase compared to endogenous level: 1- to 2-fold, 2- to 5-fold, or 5- to 10-fold (respectively, intensity categories: 1 to 2, 2 to 5, or 5 to 10). Error bars, SEM. Scale bar, 10 μm. Unpaired t test P values *P < 0.05; ***P < 0.0001. ns, not significant.
Coimmunoprecipitation between lamin A and 53BP1 has been previously reported (31); however, there is no indication whether this interaction is direct. We confirmed that these proteins interact, at least in close vicinity, by PLA. This interaction between lamin A and 53BP1 is affected by lamin B1 depletion and is increased by lamin B1 overexpression (fig. S5, B and C), which may suggest a role for lamin B1 in this interaction. Localization of interaction between lamin A and 53BP1 was analyzed. The PLA dots localized either to nucleoplasm or nuclear periphery in endogenous conditions and are not relocalized to the periphery upon lamin A overexpression (at a level of overexpression comparable to that of lamin B1 (2- to 5-fold or >5-fold) (fig. S5D). Thus, in contrast with lamin B1 overexpression condition, 53BP1 does not seem to be relocalized or sequestered to the NE upon lamin A overexpression.
Lamin B1 and 53BP1 are dissociated after DNA damages
The interaction between endogenous lamin B1 and 53BP1 proteins was observed in unchallenged cells (Fig. 4, A and B), suggesting a physiological role for this interaction. We next studied the fate of this interaction in the presence of DNA damage. For this purpose, the interaction between lamin B1 and 53BP1 was monitored by PLA at various times after 2-Gy irradiation. A decrease in PLA dots was detected in control cells 30 min after irradiation (Fig. 5A, left). Notably, this timing corresponds to the time of maximal 53BP1 foci assembly in these cells (compare Figs. 5A and 3A). These data suggest that an interaction between lamin B1 and 53BP1 preexists under basal conditions but is disrupted after irradiation when the presence of 53BP1 is required at the damaged sites. Upon lamin B1 overexpression, dissociation of lamin B1–53BP1 interaction was no longer observed at 30 min (Fig. 5A, right). In addition, fig. S6A shows that there is no colocalization between (remaining) lamin B1–53BP1 PLA dots and 53BP1 foci, suggesting that the interaction between lamin B1 and 53BP1 is not colocalized at DNA breaks and the lamin B1 regulates 53BP1 recruitment at distance of damage. Thus, these data strongly suggest that the basal interaction between lamin B1 and 53BP1 must be inhibited after irradiation to enable 53BP1 to be recruited to sites of DNA damage. However, a higher level of lamin B1 impedes the dissociation of 53BP1 from lamin B1 and subsequently should impair the recruitment of 53BP1 to DNA damage sites. This hypothesis is highly consistent with the observed defects in the kinetics of 53BP1 foci formation and in the recruitment of 53BP1 to DNA damage sites as demonstrated in the above data (Fig. 3).
Fig. 5. 53BP1–lamin B1 interaction is dissociated after irradiation.

(A) Fate of the lamin B1–53BP1 interaction after DNA damage. Quantification of PLA kinetics of the endogenous interaction after irradiation (2 Gy) from three independent experiments (left). Quantification of PLA kinetics upon lamin B1 overexpression after irradiation (2 Gy) from three independent experiments (right). (B) Impact of ATM activity inhibition on 53BP1–Lamin B1 dissociation after IR induced DNA damages. Proteins from cellular lysates treated with ATM inhibitor (ATMi, +) or vehicle (−) were subjected to immunoprecipitation before (NIR) or 1 hour after irradiation (2 Gy) with rabbit antibodies against lamin B1 or IgG as control with DNAse treatment. Coimmunoprecipitated proteins were revealed by Western blot with mouse anti-53BP1 and rabbit anti–lamin B1 antibodies (left) and quantified from four independent experiments (right). (C) Impact of 53BP1 N-terminal phosphorylation on the dissociation between lamin B1 and 53BP1 after IR. 53BP1 knockout cells transfected with WT 53BP1 vector (53BP1 WT) or with 28A mutant 53BP1 vector (53BP1 28A) were subjected to PLA using anti-53BP1 and lamin B1 antibodies, before (NIR) or 1 hour after irradiation (2 Gy). Example of in situ interaction between 53BP1 constructs and lamin B1 visualized as red fluorescent dots (left). Quantification of PLA dots per nucleus from three independent experiments is shown (right). For (B) and (C), percentage of lamin B1–53BP1 association after irradiation was normalized to corresponding nonirradiated conditions. Error bars, SEM. Scale bar, 10 μm. Unpaired t test P values **P < 0.005; ***P < 0.0001.
Lamin A–53BP1 interaction also dissociates after damage, as it has been previously reported (31). However, the overexpression of lamin A does not impede its dissociation from 53BP1 (fig. S6B). This also suggests that lamin A overexpression impedes 53BP1 recruitment to DNA damage (fig. S6C) by different mechanism(s) than those triggered by lamin B1 overexpression.
Since interaction between lamin B1 and 53BP1 is decreased shortly after exposure to IR, this suggests that the DDR could be involved in the disruption of this interaction. Thus, we analyzed the impact of ATM activation on the dissociation between these proteins. Coimmunoprecipitation assays confirm a decrease of interaction between lamin B1 and 53BP1 upon irradiation (Fig. 5B and fig. S7A). Although a decrease was observed of 53BP1 coimmunoprecipitated with lamin B1 upon ATM inhibitor without irradiation (fig. S7A), the level of interaction between lamin B1 and 53BP1 in the presence of ATM inhibitor is still higher after IR (compared after radiation without ATM inhibitor). No difference of interaction was observed before and after radiation (Fig. 5B and fig. S7A) in the presence of ATM inhibitor, suggesting that ATM signaling may play a role in dissociation of the interaction.
Furthermore, 53BP1 is phosphorylated by ATM in response to DSB, and its N-terminal region contains 28 Ser/Thr-Gln (S/T-Q) residues that are known target sites for ATM (52, 53). Under unchallenged condition, 53BP1–lamin B1 interaction was confirmed in 53BP1-deficient cells expressing either a wild-type (WT) 53BP1 or a 28A unphosphorylable mutant 53BP1 (Fig. 3G). PLA experiments were performed allowing to analyze cells with comparable expression level of WT 53BP1 and 28A mutant 53BP1. The WT 53BP1–lamin B1 PLA dots are importantly decreased after IR, confirming the dissociation between lamin B1 and 53BP1 (Fig. 5C and fig. S7B). Unexpectedly, a decrease of 28A mutant 53BP1–lamin B1 PLA dots was observed without irradiation compared to WT 53BP1–lamin B1 (fig. S7B). However, after irradiation, the number of lamin B1 PLA dots with the 28A mutant 53BP1 is still more elevated than those obtained with WT 53BP1. No difference of interaction between lamin B1 with mutant 53BP1 was observed before or after radiation (Fig. 5C and fig. S7B). These data may suggest that 53BP1 phosphorylation could be required for its efficient dissociation after IR.
Lamin B1 interacts directly with the minimal region of 53BP1 required for its recruitment to DNA damage
As lamin B1 could affect the recruitment of 53BP1 to damaged chromatin through its interaction, we aimed at further characterizing this latter by first identifying the domain of 53BP1 involved in its interaction with lamin B1. Glutathione S-transferase (GST) pull-down assays, using different purified truncated 53BP1 constructions, showed that lamin B1 directly interacts with a 53BP1 fragment (ranging from amino acids 1222 to 1659) (Fig. 6A). This corresponds to the minimal region of 53BP1 required for its recruitment to DNA damage (the IRIF domain) (54), including the Tudor and ubiquitination-dependent recruitment (UDR) domains, key domains that allow 53BP1 to bind to histone modifications. At damaged chromatin, 53BP1 interacts with dimethylated lysine 20 of histone H4 (H4K20me2) and ubiquitinated lysine 15 of histone H2A (H2AK15ub) through its Tudor and UDR domains, respectively (46, 55, 56). Therefore, we expressed the IRIF domain of 53BP1 in human fibroblasts. Figure 6B shows that this domain coimmunoprecipitates with endogenous lamin B1. This interaction has also been detected in situ by PLA (Fig. 6C). The localization of PLA dots can be clearly visualized at nuclear periphery. In agreement with the GST pull-down assay and coimmunoprecipitation results, a significantly and statistically higher number of PLA dots per nucleus is observed with the IRIF domain of 53BP1 compared to other transfected 53BP1 fragments (Fig. 6D and fig. S8A). Together, these results validate an interaction between endogenous lamin B1 and the IRIF domain of 53BP1 in cells. Cells transfected with a 53BP1 fragment (ΔOD-IRIF) corresponding to the same region but lacking its oligomerization domain (deletion between residues 1222 to 1289) present an equivalent number of dots per nucleus, showing the specific involvement of the IRIF domain in the interaction, and not of another domain through dimerization with the endogenous 53BP1. Notably, the integrity of this IRIF region seems to be required for this interaction. This interaction is highly altered when only the MDC1 domain (residues 1288 to 1409) or the Tudor-UDR region (residues 1410 to 1659) is expressed in fibroblasts. Together, our data suggest that the sequestration of 53BP1 by lamin B1 requires the 53BP1 IRIF domain, which is involved in the recognition of the damaged chromatin.
Fig. 6. 53BP1 directly interacts with lamin B1.

(A) Scheme of GST-tagged 53BP1 constructs (left) and Coomassie-stained samples of purified recombinant proteins (GST-53BP1 N-term, GST-53BP1 IRIF; GST-53BP1 C-terminal; GST) used for interaction studies (middle). Interaction between Lamin B1 and IRIF domain of 53BP1 detected by GST pull-down experiment. Full-length lamin B1 was incubated with GST or GST-53BP1 fragments. The formed protein complexes were separated by 10% SDS–polyacryalmide gel electrophoresis (PAGE) and analyzed by Western blot using anti–lamin B1 antibody. The left lane corresponds to 100 ng of purified full-length lamin B1 used in these pull-down experiments (right). (B) Coimmunoprecipitation of 53BP1 IRIF and endogenous lamin B1 from SV40-fibroblasts transfected with HA-53BP1 IRIF. Top: Proteins from cellular lysates were subjected to immunoprecipitation with rabbit antibodies against lamin B1 or IgG as control and were immunoblotted with mouse anti-HA and rabbit anti–lamin B1 antibodies. Bottom: Proteins from cellular lysates were subjected to immunoprecipitation with mouse antibodies against HA or IgG as control and were immunoblotted with mouse anti-HA and rabbit anti–lamin B1 antibodies. Same input (same extract used for coimmunoprecipitation) was loaded for top and bottom panels. (C) Representative PLA between HA-IRIF 53BP1 and lamin B1, with negative controls using either one of the two antibodies. Scale bar, 10 μm. (D) Scheme of HA-tagged 53BP1 constructs (left) and PLA between endogenous lamin B1 and 53BP1 constructs (right). Cells transfected with vectors expressing different HA-53BP1 domains were fixed after 48 hours. PLA was then performed using antibodies directed against lamin B1 and HA. Unpaired t test value ***P < 0.0001. Ab, antibody.
The interaction of 53BP1 with lamin B1 controls its recruitment to damaged DNA
To assess whether the defect of 53BP1 recruitment upon lamin B1 overexpression is due to 53BP1 sequestration, we identified the region of lamin B1 involved in the interaction with 53BP1.
In vitro GST pull-down, using various purified truncated lamin B1 constructions (Fig. 7A), shows that the N-terminal part of lamin B1, and more precisely the Head Coil 1 domain (amino acids 1 to 243), interacts directly with the IRIF domain of 53BP1 (Fig. 7B). In contrast, the Coil 2 domain (amino acids 244 to 424) and the C-terminal part (amino acids 398 to 586) of lamin B1 do not reproducibly interact with the 53BP1 IRIF domain.
Fig. 7. Identification of the lamin B1–interacting domain with 53BP1.
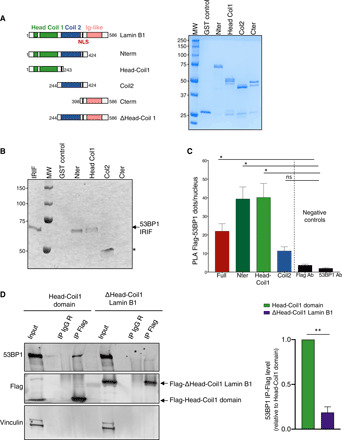
(A) Scheme of lamin B1 constructions used for GST pull-down experiment (left) and Coomassie-stained samples of purified recombinant proteins used for interaction studies (right). (B) Interaction between Head Coil 1 domain of Lamin B1 with IRIF domain of 53BP1 detected by GST pull-down experiment. IRIF 53BP1 was incubated with GST or GST–lamin B1 constructions in the presence of Benzonase and the protein complexes that formed were separated by 10% SDS-PAGE and analyzed by Western blot using anti-53BP1 antibody. The left lane corresponds to 100 ng of purified IRIF 53BP1 used in these pull-down experiments. Asterisk corresponds to an unspecific trapping of antibody to GST-Coil2. (C) PLA between endogenous 53BP1 and Flag–lamin B1 constructs: PLA spots were counted in Flag-expressing cells using Cell Profiler and KNIME software in five independent experiments. One-way analysis of variance (ANOVA) test: *P < 0.05 (D) Coimmunoprecipitation of Flag- Head Coil 1 domain of Lamin B1 and endogenous 53BP1. Proteins from cellular lysates of SV40-fibroblasts transfected with Flag–Lamin B1 constructions were subjected to immunoprecipitation with rabbit antibodies against Flag tag or IgG as control and were immunoblotted with mouse anti-53BP1 and rabbit anti-Flag tag antibodies (left). Quantification of from three independent experiments (right). Unpaired t test value **P < 0.005. Error bars, SEM.
We aimed to confirm in cellulo and by another approach which lamin B1 region interacts with 53BP1. For this purpose, vectors expressing different lamin B1 fragments were first expressed in human fibroblasts (Fig. 7A and fig. S8B), and their in situ interaction with endogenous 53BP1 was tested by PLA. A higher number of PLA dots per nucleus was observed in cells transfected with the N-terminal region of lamin B1 (amino acids 1 to 424) and with the Head Coil1 domain (amino acids 1 to 243) compared to cells transfected with the Coil 2 domain of lamin B1 (amino acids 244 to 424) and to PLA-negative controls (Fig. 7C). We next expressed Flag-Head-Coil 1 domain or Flag–lamin B1 devoid of this interaction domain. Endogenous 53BP1 is revealed by Western blot in Immunoprecipitation of Flag-Head-Coil1. In contrast, 53BP1 is almost undetectable in Delta-Head-coil1 lamin B1 immunoprecipitation (Fig. 7D). PLA experiments confirmed this result. PLA dot numbers corresponding to 53BP1-delta Head-Coil1 are very low compared to 53BP1-HeadCoil1 domain dots (fig. S8C). These results confirm the data obtained by GST pull-down and show that the region encompassing the Head and Coil 1 domains of lamin B1 is sufficient for the interaction with 53BP1 in cellulo.
Then, to further confirm that lamin B1–53BP1 interaction is key in the decrease of 53BP1 foci observed upon lamin B1 overexpression, we analyzed both 53BP1 foci formation upon expression of the minimal interaction domain of lamin B1 (Head-Coil1) and upon expression of lamin B1 lacking its interaction domain (Δ-Head-Coil1). We observed that the expression of the interaction domain of lamin B1 (Head-Coil1) mimics the effect of overexpressed full-length lamin B1 protein and leads to a decrease in 53BP1 foci (Fig. 8A and fig. S9). In contrast, the expression of lamin B1 protein lacking the interaction domain (Δ-Head-Coil1) has no impact on 53BP1 foci formation. We verified that the relative expression of full protein, Head-Coil1 domain, and lamin B1 lacking this region are equivalent by Western blot (fig. S8B), and the number of 53BP1 foci was quantified in cells with comparable expression (measured by immunofluorescence) of the different lamin B1 constructions (fig. S9). Together, our data demonstrate that lamin B1 directly interacts with the 53BP1 IRIF domain, involved in the recognition of damaged chromatin, and suggest that lamin B1 overexpression affects the recruitment of 53BP1 to damage through the trapping of 53BP1. To investigate, the impact of 53BP1 sequestration by lamin B1 on DNA damage persistence, we analyzed the γH2AX foci persistence after IR and the number of breaks on metaphases upon expression of either the interaction domain of lamin B1 or the lamin B1 lacking its interaction domain with 53BP1 (Fig. 8, B and C). The expression of the interaction domain mimics the impact of the expression of full lamin B1: persistence of γH2AX foci and elevated number of DNA breaks on metaphase. In contrast, the lamin B1 lacking the interaction domain does not trigger a significant DNA damage persistence observed either by measurement of γH2AX foci (Fig. 8B) or number of breaks per metaphase (Fig. 8C). These data show that the sequestration of 53BP1 by lamin B1 is the key in DNA damage persistence upon lamin B1 dysregulation.
Fig. 8. Lamin B1–53BP1 interaction is key for the 53BP1 recruitment defect observed upon lamin B1 overexpression.

(A) Impact of the lamin B1 interaction domain on radiation-induced 53BP1 foci, 30 min after irradiation (2 Gy). 53BP1 foci were counted in Flag-expressing cells using ImageJ software and foci per nuclei plotted in three classes according to the Flag-expression level of the nuclei (right) in four independent experiments. One-way ANOVA test: *P < 0.05. (B) Analysis of persistent γH2AX foci kinetics after expression of the lamin B1 interaction domain. γH2AX foci were counted in Flag-expressing cells using ImageJ software and plotted for cells with similar Flag-expression level. Histograms show the percentage of radio-induced foci persisting 3 hours after irradiation (0.5 Gy) from three to four independent experiments. (C) Chromosomal aberrations were scored in metaphase spreads prepared from Flag–Lamin B1 constructions expressing cells or control cells, under basal conditions (NI) or irradiation (2 Gy) 24 hours before cell harvest. Histogram shows the mean number of chromatid breaks scored per metaphase (n = 199 to 425). P value above bar compare to matching NIR or IR “empty vector” datasets. (D) Model of 53BP1 recruitment control by lamin B1. In unchallenged cells, 53BP1 interacts with lamin B1 via its minimal region of foci formation including the Tudor domains and the UDR domain. Our data suggest an ATM activity–dependent dissociation between these proteins occurs upon DNA damage, thus enabling 53BP1 to be recruited to damaged chromatin. However, in the case of lamin B1 overexpression, 53BP1 is sequestered and its recruitment to damaged chromatin is precluded. For (B) and (C), unpaired t test P values *P < 0.05; **P < 0.005; ***P < 0.0001. Error bars, SEM.
DISCUSSION
53BP1 recruitment at DSB sites is essential for its function in the DDR and must be finely controlled. 53BP1 plays a critical role in the DSB repair pathway choice; however, uncontrolled 53BP1 binding or recruitment to damage sites leads to genetic instability (18, 19). Several sophisticated mechanisms have been described for the binding of 53BP1 to damaged chromatin (20). The minimal region of 53BP1 required for this interaction includes its Tudor domains and UDR motif. Once the chromatin is damaged, 53BP1 binds to H4K20me2 through its Tudor domains and to H2AK15Ub through its UDR motif (46, 55, 56). After DSB induction, the RNF8 and RNF168 proteins play essential roles in 53BP1 recruitment to damaged chromatin by unmasking H4K20me2 through the ubiquitination and subsequent proteasomal degradation of JMJD2A and L3MBTL1 proteins (57–59). In addition, various posttranslational modifications are involved in the regulation of 53BP1 recruitment to DNA damages. UDR motif phosphorylation has been shown to impede the binding of 53BP1 to H2AUb in the mitotic chromatin, thus avoiding genetic instability during chromosome segregation (18, 19). However, there are emerging data suggesting that mechanisms are also in place to control 53BP1 at distance from damaged chromatin.
Our results unveil that the lamin B1 protein directly interacts with the 53BP1 region encompassing the Tudor and UDR domains (IRIF domain), which are required for 53BP1 to interact with damaged chromatin and for the control of its recruitment (46, 55, 56). These data suggest that, in addition to sequestering 53BP1, lamin B1 could also mask the region of 53BP1 required for its recruitment to damaged chromatin. However, we were not able to detect any recruitment of lamin B1 at DNA damage, and lamin B1–53BP1 PLA dots seem not be colocalized with 53BP1 foci, suggesting a control of 53BP1 recruitment by lamin B1 at distance from damage. Recent reports suggested that the TIRR protein binds to the IRIF domain of 53BP1 and controls the 53BP1 recruitment to DNA damage (also at distance of DNA damage) (60, 61). These data, along with our results, highlight again the key role of the IRIF domain for 53BP1 regulation after induction of DNA damage. This domain, by being masked by proteins, such as lamins or TIRR proteins (and likely by other proteins remaining to be discovered), controls the arrival of 53BP1 to the damage. The IRIF domain is also responsible for the direct binding to damaged chromatin. In addition, 53BP1 binding to DNA damage at G2/M is also regulated by posttranslational modifications of the IRIF domain (18, 19). Recently, it has been shown that the nucleoskeleton NuMA protein, which is proposed to be structurally related to nuclear lamins, also controls 53BP1 recruitment at distance of DNA damage, through its interaction with 53BP1 protein (62). Interactions between 53BP1 and TIRR, NuMA, or lamin B1 are severed after IR induced DNA damages (18, 19). TIRR protein has been recently reported to be an important inhibitor of the 53BP1-P53 complex and therefore controls p53-mediated cell fate program (63). Recent studies indicated that 53BP1 nuclear compartments show features of liquid-liquid phase separation (64, 65). Modulation of phase separation is also a way to modulate 53BP1 function. Recent reports showed that AHNAK, a scaffold protein, which interact with 53BP1 in G1, can also modulate the p53-dependent cell fate, by limiting the accumulation of 53BP1 on chromatin and by restraining 53BP1 oligomerization and phase separation (66). In the presence of DNA damage, in contrast with lamin B1, the interaction between AHNAK and 53BP1 on chromatin is enhanced and probably limiting p53-P21 response. The potential relationship, competition, or cross-talk between lamins and TIRR, Numa, and AHNAK is not known yet and should be very interesting to further explore.
The dissociation of TIRR proteins from 53BP1 is controlled by an ATM-dependent DDR and requires 53BP1 phosphorylation. NuMA phosphorylation by ATM has been also proposed for the release of its 53BP1 interaction after DNA damage, since 53BP1 recruitment on laser track is abolished after expression of a nonphosphorylable NuMA mutant (62). Our data also suggest that ATM could also regulate the association/dissociation of 53BP1 from lamin B1, likely through phosphorylation of 53BP1. This regulation by ATM strongly reinforces the physiological importance of these interactions.
Although our data suggest that the 53BP1 phosphorylation could control the dissociation between lamin B1 and 53BP1 upon IR, we do not exclude that other mechanisms could be participate in the regulation of the interaction between these proteins. Therefore, it would be interesting to investigate whether some posttranslational modifications of lamin B1 could regulate the association-dissociation with 53BP1. Many different posttranslational modifications (such as phosphorylation, ubiquitination, acetylation…) of lamin B1 have been described. In the course of the revision of this manuscript, it has been published and proposed that acetylation of lamin B1 may participate to localization of 53BP1 at NE, and the authors suggested that it could play a role on the balance between DSB repair (DSBR) mechanisms (67). The impact of the acetyltransferase or deacetylase potentially responsible for lamin B1 acetylation could be further investigate on 53BP1 interaction and on DNA damage persistence or on genome (in)stability.
While lamin A defect has been reported to affect 53BP1 stability (30), we did not observe any change in the 53BP1 level after lamin B1 overexpression. Moreover, we showed that the impaired 53BP1 recruitment to DNA damage induced by lamin B1 is not associated with a change of lamin A and lamin C proteins. In our study, we identified a direct interaction between lamin B1 and 53BP1 proteins and we identified the interaction domains. The expression of lamin B1 lacking its interacting domain does not affect either 53BP1 recruitment or persistent γH2AX foci after IR or number of breaks observed on metaphases. Together, these observations suggest that the defect of 53BP1 recruitment and subsequent DNA damage persistence after lamin B1 overexpression is mainly due to the direct sequestration of 53BP1 by lamin B1.
A previous study revealed that the lamin A and 53BP1 proteins could coimmunoprecipitate (31). Here, we confirmed a close vicinity between both proteins by PLA. It has not yet been reported whether the lamin A could interact directly with 53BP1. The interaction between lamin A and 53BP1 seems to require the presence of lamin B1 (or a properly organized lamin B1 meshwork), as we found that lamin B1 depletion decreases the number of dots when the PLA was performed to analyze the in situ interaction between endogenous lamin A and 53BP1 proteins (fig. S5B).
We show here that pre–lamin A overexpression leads to defect of 53BP1 recruitment and persistence of DNA damage revealed by γH2AX and an increase of breaks on metaphase. However, the mechanism(s) involved appear to be different from the mechanism that we have described here after overexpression of lamin B1. The overexpression of pre–lamin A has no impact on localization of its interaction with 53BP1 (no relocalization to NE) and does not prevent either its dissociation from 53BP1 after DNA damages. These data suggest that lamin A overexpression, in contrast to lamin B1 overexpression, does not impede 53BP1 recruitment by a sequestration mechanism, but likely due to defect in 53BP1 transport to nucleus as it has been reported (32). Different links between lamin A and DNA repair have been established (68), thus many other consequences of lamin A dysregulation could also account for the persistence of DNA damage (68). It has been also proposed that lamin A affects foci stability and could interact with γH2AX (69), the overexpression of lamin A may lead to a prolonged γH2AX signal. This may also explain the apparent γH2AX persistence in the case of overexpression of lamin A.
Upon lamin B1 depletion, dysregulated expression of different DNA repair genes has been reported (33). These transcriptional alterations also seem to lead to a defect in DNA damage handling. In contrast, upon lamin B1 overexpression, we found no difference in the expression of proteins involved in NHEJ. Our data show that lamin B1 overexpression affects DSB repair not by altering key DNA/NHEJ repair gene expression but directly by sequestering the 53BP1 protein. This association is physiologically and tightly regulated since genotoxic treatment leads to a dissociation of endogenous 53BP1 from endogenous lamin B1. Together, these data reinforce the idea that the control of lamin B1 levels is of particular importance for genome stability, not only in the course of tumorigenesis but also during senescence or in pathology such as HGPS.
Persistence of DNA damage has been proposed to participate in the deleterious effects of progerin expression in HGPS. It would be interesting to investigate whether progerin could also interact with 53BP1 to characterize this potential interaction. Our data also raise the question of the potential role of the decrease in lamin B1 observed in progeria (70) on DNA damage persistence. We report here that the interaction between lamin A and 53BP1 is affected by the depletion of lamin B1. Decreased level of lamin B1 may potentially have an importance for the persistence of the DNA damage observed in the HGPS pathology. Therefore, the role of lamin B1 decrease and of potential 53BP1 defect in progeria should be more investigated in future.
Altered nuclear shape and increased lamin B1 levels are observed in many tumors and seem to be associated with advanced grades of tumorigenesis and poor outcome (40–44). We show here that lamin B1 up-regulation leads to DSB accumulation that may ultimately lead to chromosomal rearrangements and genetic instability, both of which being hallmarks of tumorigenesis. It is thus tempting to speculate that the lamin B1 dysregulation could contribute to tumor progression through 53BP1 sequestration and/or DNA damage accumulation. A decrease of lamin A has also been observed in various tumors. A-type lamin depletion leads to 53BP1 destabilization due to increased activity of the cathepsin L proteasome pathway (71). It has been reported that pre–lamin A overexpression leads to 53BP1 cytoplasmic accumulation and subsequent recruitment defect to DNA damage because of NUP153 mislocalization (32). Hence, the dysregulation of either A-type or B-type lamin affects the DNA damage repair through an alteration of 53BP1, which is, however, mediated through distinct mechanisms. On the one hand, lamin A dysregulation affects the level of 53BP1 through degradation or mislocalization, and on the other hand, lamin B1, when overexpressed, sequesters 53BP1 away from repair sites. Then, in both situations, the dysregulation of lamins may participate in the genetic instability, which is associated with tumorigenesis. Exciting future studies should be performed to directly address this hypothesis.
In summary, the present data can be unified in the following model (Fig. 8D): In normal cells and in the absence of DNA damage, lamin B1 interacts with 53BP1 and can serve as “a reservoir” for 53BP1, away from repair sites but quickly available in response to genotoxic stresses. Upon DNA damage, 53BP1 dissociates from lamin B1, thus allowing its rapid recruitment to damaged chromatin. In the case of overexpression (or pathological situations associated with the lamin B1 up-regulation), lamin B1 sequesters 53BP1, impeding its recruitment to DNA damage sites. This impedance could have an impact on the frequencies of chromosomal rearrangements and thus may contribute to tumor evolution. However, because both 53BP1 deficiency and lamin B1 overexpression lead to radiosensitivity, dysregulation of the lamin B1 level may also represent an Achilles’ heel of tumors.
MATERIALS AND METHODS
Cell lines
SV40-transformed human fibroblasts (GM0639 cell line) and U2OS and derivative cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal calf serum, 2 mM glutamine, and penicillin (200 IU/ml) at 37°C with 5% CO2. U2OS 53BP1 knockout (KO) were provided by D. Durocher (6).
Survival assay
Forty-eight hours after the transfection, SV40-transformed fibroblast cells were platted at 100, 500, 1000, or 2000 cells per well. After adhesion, cells were treated with different doses of gamma ray (iBL637 irradiator; 0.5, 1, or 2 Gy at 1.77 Gy/min) and incubated for 8 to 11 days at 37°C before staining with gentian violet and colony counting.
Constructions of vectors expressing the different 53BP1 and Lamin B1 fragments
Two sets of constructs have been made depending on the experiments to be performed. For in vitro direct interaction tests, the different domains of either human 53BP1 (53BP1) or human lamin B1 (Lamin B1) were cloned by site-directed recombination using the Gateway technology (Thermo Fisher Scientific, Waltham, MA). For in vivo experiments, the domains of lamin B1 and 53BP1 were cloned by either by sequence- and ligation-independent cloning (SLIC) or by restriction-ligation. Open reading frames (ORFs) to be cloned were amplified by polymerase chain reaction (PCR) using specific primers (table S1). Briefly, two sequential PCR reactions were performed for the first set of constructs. The first PCR was performed with a template-specific forward primer that brought a tobacco etch virus (TEV) protease recognition site-encoding sequence for further cleavage. The second PCR was performed with a forward generic primer including the attB1 recombination site sequence. Both PCRs of the first set were performed with a template-specific reverse primer including the attB2 recombination site. The resulting PCR products were recombined by BP reaction to generate entry clones used for further LR reactions (first set of constructs) or to release the inserted domain by restriction-ligation (second set of constructs). For the second set of constructs, the ORF to be cloned by SLIC was amplified with sequence-specific primers extended at the 5′ and 3′ ends with a homology sequence for a modified pcDNA3 vector, and SLIC cloning was performed. All PCRs were performed using Phusion DNA polymerase following the manufacturer’s instructions. After amplification, PCR products were purified using a GeneJET PCR purification kit (Thermo Fisher Scientific). The Gateway cloning was performed following the manufacturer’s instructions. The BP reaction was performed using the pDONOR207 donor vector (Life Technologies), and LR reactions were performed using either pGGWA or pHGWA (72) for 53BP1 fragments or full-length lamin B1, respectively. All primers were designed to have a Tm ≈ 60°C. Restriction enzymes were obtained from New England Biolabs (Ipswich, MA) and antibiotics from Sigma-Aldrich (St Louis, MO). The sequences of all constructs were verified using universal T7 primers from GATC (T7, pcDNA3.1-RP/1, pcDNA3.1-RF, pDONOR-FP, and pDONOR-RP, depending on the construct) (table S2).
Ds-Red lamin B1–expressing vector is described in (73). Untagged lamin B1 full length was purchased from Origene (#SC 116661). Flag–lamin B1 was constructed by cloning a PCR product (primers: lower: GGGGTACCCCTTACATAATTGCACAGCTTCTATTGGATGCTCTTGGGGTTCCCTGCTGGTGGAAAA; upper: CCGCTCGAGCTAGCCACCATGGACTACAAGGACGACGATGACAAGGCGGCCGCGGCGACT-GCGACCCCCGTGCCGCGGARGGGCAGCCGCGCTGGCGGCCCCACCACG) amplified from pCMV6-XL4-LMNB1 (#SC 116661, Origene) into pcDNA3-puromycin vector Xho I and Kpn I sites.
Transfections
For the various experiments, 1.8 × 105 cells were plated 1 day before transfection. SV40 fibroblasts and 53BP1 KO U2OS cells were transfected using the jetPEI transfection reagent under the conditions specified by the manufacturer (polyplus transfection), and U2OS cells were transfected using Lipofectamine (Lipofectamine 2000, Life Technologies). For the repair foci analysis, the proximity ligation, and the survival assays, 3 × 10−13 mol of p-CMV-LMNB1 plasmid or the same amount of the different 53BP1 or LMNB1 constructs, WT 53BP1 plasmid or 28A mutant 53BP1 plasmids (Addgene), or the empty plasmid was used to transfect the cells 48 hours before treatment and/or fixation. To perform NHEJ quantification assay, meganuclease I-SceI expression was achieved by transient transfection of the cells with 7.5 × 10−13 mol of the pCMV–Hemagglutinin (HA)–I-SceI plasmid, together with either 2.5 × 10−13 mol of the p-CMV-LMNB1 plasmid (lamin B1 overexpression) or the empty plasmid (control conditions). The U2OS cells used for the microirradiation experiments were transfected with 1.5 μg of GFP-53BP1 plasmid and 1.5 μg of either DsRed-LMNB1 plasmid or empty plasmid. In the PLA assays, cells were transfected with plasmids as described above or with 20 μM specific siRNA to lamin B1, lamin A, or 53BP1 (ON-TARGET plus SMARTpool, Dharmacon) or with control siRNA (NEG05, Eurogentec) using INTERFERin (Polyplus Transfection) under conditions specified by the manufacturer.
Nonhomologous end joining assay
SV40-transformed human fibroblasts cell line (GC92) containing the pCOH-CD4 intrachromosomal NHEJ substrate described in Fig. 2 was used to determine NHEJ frequency as described previously (48). Briefly, at 72 hours after transfection, cells were harvested in phosphate-buffered saline (PBS) with 20 mM EDTA and then washed in PBS (without Ca2+) and fixed in PBS with 2% (w/v) paraformaldehyde for 20 min at room temperature. After the cells were saturated with PBS and 2% (w/v) bovine serum albumin (BSA) for 15 min, they were stained for 45 min with 0.5 μg of anti-CD4–fluorescein isothiocyanate (FITC) or anti-CD4–Alexa Fluor 647 antibodies (BD Pharmingen). The cells were washed twice in PBS before fluorescence-activated cell sorting analyses.
Cellular fractionation
The cytosolic proteins from SV40-transformed fibroblasts were extracted by incubation in a buffer containing 1 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol (DTT), 0.1% Triton X-100, and protease and phosphatase inhibitors (Roche) at 4°C for 10 min, followed by a centrifugation at 5000g for 2 min at 4°C. The nuclear fraction was obtained by extraction of the remaining pellet in a tris buffer [10 mM tris (pH 7.5), 1% SDS, and protease and phosphatase inhibitors].
Western blotting analysis
SV40-transformed fibroblasts were lysed in a buffer containing 10 mM tris (pH 7.5), 1% SDS, and protease and phosphatase inhibitors. We resolved the denatured protein extracts (25 to 50 μg) using 10% SDS–polyacrylamide gel electrophoresis (PAGE) or 3 to 8% NuPAGE tris-acetate gels (for the large proteins, such as 53BP1), transferred the proteins onto a nitrocellulose membrane, and probed the membrane with the following primary antibodies: anti–lamin B1 (rabbit, ab16048, Abcam), anti-53BP1 (rabbit, #4937S, Cell Signaling), anti-HA (mouse, MMS-101P, Covance), anti-γH2AX (mouse, #05636, Upstate), anti-actin (rabbit, 057 K4803, Sigma-Aldrich), and anti-vinculin (mouse, ab18058, Abcam). Immunoreactivity was visualized using an enhanced chemiluminescence detection kit (EZ-ECL, Biological Industries), and signals were quantified using ImageJ software. For the GST pull-down experiments, for the control of siRNA depletion and fragments or GFP-tagged constructs expression, we used secondary fluorescent antibody [anti-rabbit IR800 (R-05060), anti-rabbit IR700 (R-05054), anti-mouse IR800 (R-05061), and anti-mouse IR700 (R-05055), DIAGOMICS]. Fluorescent signals were acquired with Odyssey imager (LI-COR Biosciences) and quantified with Image Studio software (LI-COR Biosciences).
Protein purification
His–Lamin B1 and His-IRIF 53BP1
The gene encoding His-protein gene was placed under the T7 promoter in the vector pHGWAHsLaminB, which was introduced into Escherichia coli strain BL21 (DE3). His-protein expression was induced by 0.5 mM isopropyl-β-d-thiogalactoside (IPTG). All of the subsequent protein purification steps were carried out at 4°C. Cells were harvested, resuspended in lysis buffer [50 mM tris-HCl (pH 8), 500 mM NaCl, 1 mM DTT, 10% glycerol, 1× Triton X-100, lysozyme (1 mg/ml), 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 10 mM benzamidine, and 2 μM pepstatin] and disrupted by sonication. The extract was cleared by centrifugation at 186,000g for 1 hour at 4°C and then complemented with imidazole (20 mM final) before being loaded onto a 2-ml nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen) equilibrated with 50 mM tris-HCl (pH 8 at 4°C) and 500 mM NaCl. The column was washed with 30 ml of buffer A [50 mM tris-HCl (pH 8 at 4°C), 500 mM NaCl, 1 mM DTT, and 20 mM imidazole] followed by a second wash with 30 ml of buffer B [50 mM tris-HCl (pH 8 at 4°C), 100 mM NaCl, 1 mM DTT, and 20 mM imidazole]. Protein was eluted with buffer B containing 500 mM imidazole. Fractions containing His-protein were pooled and applied to a 1-ml Resource Q column (GE Healthcare) equilibrated with buffer B without imidazole. Protein was eluted with a 12-ml linear gradient of 0.05 to 0.5 M NaCl. Purified His-protein was stored at −80°C.
GST-53BP1 fragment purification and GST–lamin B1 fragment purification
The plasmids pGGWAHs53BP1 fragment (containing the GST-53BP1 fragment fusion gene) and plasmid pGGWAHs lamin B1 fragment (containing the GST–lamin B1 fragment fusion gene) under the control of IPTG-inducible T7 promoter were introduced into E.coli strain BL21 (DE3). Overnight culture was diluted 100-fold in LB and incubated at 37°C until OD600 (optical density at 600 nm) reached 1. At that time, IPTG was added to 0.5 mM, and incubation was pursued for 3 hours at 37°C. Cells were harvested; resuspended in 50 mM tris-HCl (pH 8 at 4°C), 500 mM NaCl, 1 mM DTT, 10% glycerol, 1× Triton X-100, lysozyme (1 mg/ml), 1 mM 4-(2-aminoethyl) benzenesulphonyl fluoride, 10 mM benzaminide, and 2 μM pepstatin; and disrupted by sonication. Extracts were cleared by centrifugation at 186,000g for 1 hour at 4°C and then loaded onto a 1-ml Glutathione Sepharose 4B (GE Healthcare) column equilibrated with buffer A [50 mM tris-HCl (pH 8 at 4°C), 500 mM NaCl, 1 mM DTT, 0.5 mM EDTA, and 10% glycerol]. After the columns were washed extensively [40 ml of buffer A followed by 80 ml of buffer B (buffer A containing 100 mM NaCl)], GST proteins were eluted with elution buffer [50 mM tris-HCl (pH 8 at 4°C), 100 mM NaCl, 1 mM DTT, 0.5 mM EDTA, 10% glycerol, and 30 mM glutathione]. Fractions containing GST proteins were concentrated using Vivaspin concentrator and then loaded onto a Superdex 75 10/300 GL (GE Healthcare) equilibrated with buffer B. Purified proteins were stored at −80°C.
GST pull-down assays
GST-53BP1 fragment (10 μg) [GST-Nterm, GST-IRIF, GST-Cterm or GST-Nterm lamin B1, GST-Head-Coil1 lamin B1, GST-Coil2, GST-Cter lamin B1 or GST (10 μg)] was immobilized on 20 μl of Glutathione Sepharose 4B in 100 μl of buffer A [50 mM tris-HCl (pH 8 at 4°C), 150 mM NaCl, 1 mM DTT, 0.5 mM EDTA, 10% glycerol, 2 mM MgCl2, and 25 U Benzonase] for 120 min at 4°C. The beads were collected by centrifugation and washed three times with 300 μl of buffer B (buffer A + 0.05% NP-40). His–lamin B1 (10 μg in 100 μl of buffer B) or His-IRIF 53BP1 (10 μg) was then added, and incubation was pursued for 120 min at 4°C with gentle agitation. The supernatant was removed and the beads were washed two times with 300 μl of buffer B. Proteins bound to the beads were then eluted by the addition of 30 μl of 50 mM tris-HCl (pH 8 at 4°C), 150 mM NaCl, 1 mM DTT, and 30 mM glutathione, and the presence of His–lamin B1 or His-IRIF 53BP1 in the eluate was determined by Western blot using respectively anti–lamin B1 antibody (ab16048, Abcam) or anti-53BP1 antibody (#4937S, Cell Signaling).
Immunofluorescence
Forty-eight hours after transfection, the SV40-transformed fibroblasts grown on glass coverslips were fixed in 4% (w/v) paraformaldehyde for 15 min and washed three times in PBS before a permeabilization step with 0.5% Triton X-100 in PBS for 15 min. After permeabilization, saturation was performed with PBS-BSA 2% containing 0.05% Tween 20 for 1 hour at room temperature. For 53P1 foci analysis, coverslips were fixed for 5 min in ice-cold methanol and washed three times in PBS before saturation performed as described above. Cells were then immunolabeled using the following antibodies diluted with PBS-1%BSA containing 0.05% Tween 20 for 90 min at room temperature: anti–lamin B1 (rabbit, ab16048, Abcam), anti–lamin B1 (mouse, 56144 8D1, Santa-Cruz), anti-Flag (rabbit, F7425, Sigma-Aldrich), anti-53BP1 (mouse, 612522, BD Biosciences), anti-γH2AX (mouse, #05636, Upstate), and anti-MDC1 (rabbit, ab11169, Abcam). Cells were then probed with a secondary anti-mouse or anti-rabbit antibody coupled to Alexa Fluor 488 or 594, respectively (Life Technologies). Counterstaining was performed with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml) in PBS, and slides were mounted with fluorescent mounting medium (Fluoromount-G, Southern Biotech). Images were obtained on a Leica DM5500 equipped with a 63× objective and a CoolSNAP HQ2 camera and were further processed with MetaMorph software (Molecular Devices). Digital image processing for nuclei and foci segmentation and fluorescence measurement was performed using ImageJ software or an in-house script developed in the MetaMorph software. Lamin B1–transfected fibroblasts were classified into two categories according to their lamin B1 expression level: cells expressing moderate (up to 5-fold) or higher (between 5- and 10-fold) levels of lamin B1, as compared to its basal expression level in nontransfected cells. For 53BP1 foci formation after Flag–lamin B1 fragment overexpression, nuclei were classified according to their Flag expression intensity as a fold compared to the maximal Flag antibody background intensity of nonoverexpressing nuclei measured on empty vector–transfected cells. Two classes with similar Flag expression intensity for each lamin B1 construct were defined: two- to fourfold or four- to sixfold intensity over the maximum background intensity. An average of between 25 and 80 cells per conditions and per replicate was analyzed.
Live cell microirradiation experiments
U2OS cells were plated on 35-mm glass bottom petri dishes. At 48 hours before microirradiation, cells were cotransfected with the 53BP1-GFP plasmid and either the lamin B1 plasmid or the empty plasmid. The nuclei were stained with Hoescht (1 μg/ml; Hoescht 33342, 4082, Cell Signaling). Live cell imaging was performed using a PLAN APO 60× (numerical aperture: 1.4) objective on a Nikon A1 confocal laser scanning microscope system attached to an inverted ECLIPSE Ti (Nikon Corp., Tokyo, Japan) maintained at 37°C under 5% of CO2 and 20% O2 atmosphere. The microirradiation was performed for selected regions for 2 s with a 405 nm-laser diode set to 2% of the maximum power. Time-lapse images were acquired every 30 s during 75 min. The analysis of protein recruitment kinetics was performed using the NIS-Elements software (Nikon Corp.) by measuring the change in the fluorescence intensities in the irradiated region over time. Intensity values were normalized by the background level in the same region before microirradiation.
Cytogenetic analysis
SV40-transformed fibroblasts were transfected with lamin B1, Head-Coil1 domain, lamin B1 deleted of the Head-Coil1 domain, with pre–lamin A or control vectors and cultured for 48 hours, then irradiated (IBL637 irradiator, 2 Gy, 1.77 Gy/min) or not, and replated for 24 hours. During the last 2 hours, colchicine (0.1 μg/ml; Sigma-Aldrich) was added to the cells. The trypsinized cell pellet was swollen in hypotonic KCl solution and incubated at 37°C for 20 min. Cells were fixed in ethanol-acetic acid (3:1) and spread on precooled glass slides. After the slides were air-dried, staining with 4% Giemsa was performed for 10 min. After the slides were mounted, metaphase spreads were captured in a blind fashion using a bright-field microscopy (Leica DM5500) with a 63× objective lens.
Coimmunoprecipitation
The cellular proteins from SV40-transformed fibroblasts were extracted 48 hours after division for endogenous conditions or 48 hours after transfection using 50 mM tris-HCl (pH 7), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and a protease inhibitor cocktail (Roche). For the interaction analysis upon ATM inhibitor, cells treated were pretreated for 2 hours with either 10 μM ATM inhibitor (ATMi; KU60019, S1570, Selleckchem) or dimethyl sulfoxide. One hour after irradiation (IBL637 irradiator, 2 Gy, 1.77 Gy/min) or not, cells were collected and proteins were extracted using the same buffer as above with 5% NP-40. The protein cell extract was incubated with the RQ1 nuclease (Promega) or Benzonase nuclease (0.5 U/μl; E1014, Sigma-Aldrich) supplemented with 10 mM MgCl2 for 1 hour at room temperature. The Dynabeads protein G kit was used for the coimmunoprecipitation according to the manufacturer’s recommendations (Life Technologies). In total, 2.5 μg of antibodies was coupled with Dynabeads at room temperature for 1 hour. The beads were subsequently washed three times with 0.05% Tween 20 in PBS before the incubation step with 1 mg of protein cell extract (1 hour and 30 min at room temperature). Laemmli buffer (2×) with 4% β-mercaptoethanol was used to dissociate and denature the beads-antibodies-proteins complexes. Western blot analysis was performed to reveal the proteins as described above. As coimmunoprecipitation controls, a specific mouse and rabbit immunoglobulins (IgGs) were used [mouse IgG (sc2025 Santa Cruz) or rabbit IgG (sc2027 Santa Cruz or 12-370 Merck Millipore)].
In situ PLA
SV40-transformed fibroblasts grown on slides were fixed in methanol for 10 min, washed in PBS, and stored in PBS (4°C). The slides were then saturated and stained as described above for immunofluorescence labeling using the following antibodies couples: anti-53BP1 (mouse, 612522, BD Biosciences) and anti–lamin B1 (rabbit, ab16048, Abcam) or anti-HA.11 (mouse, MMS 101P, Covance) and anti–lamin B1 (rabbit, ab16048, Abcam) or anti-53BP1 (mouse, 612522, BD Biosciences) and anti-Flag (rabbit, F7425, Sigma-Aldrich) or anti-53BP1 (mouse, 612522, BD Biosciences) and anti–lamin A (rabbit, L1293, Sigma-Aldrich) antibodies. PLAs were performed using a Duolink in situ detection kit (Sigma-Aldrich) according to the manufacturer’s protocol. For PLA after Flag–Lamin B1 fragment overexpression, analysis was made among populations with similar Flag expression intensity for each Lamin B1 construct. For PLA assays in the 53BP1 KO U2OS cells after expression of WT or 28A mutant 53BP1, nuclei populations of similar level of 53BP1 expression were analyzed. Digital images were acquired with an SPE Leica DMRxA2 confocal microscope using a 63× objective lens. Images were further processed with the Leica and ImageJ software. To study the localization of the endogenous interactions, PLA dots were acquired on multiple Z-plan on a confocal microscope. Dots were then scored on the focus plane and categorized as being at the membrane (colocalization with lamin staining) or in the nuclear interior, using ImageJ software.
Microscope equipment and acquisition settings
For immunofluorescence assays, the images were acquired using a Leica DM5500B epifluorescence microscope. A Lumen 200-W mercury lamp (Microvision) was used as a light source. The used Leica filters are as follows: A4 excitation filter band-pass (BP), 360/40 nm; long-pass dichroic (LPDC) mirror, 400 nm; emission filter, BP 470/40 nm (#11504162, Leica); L5 excitation filter, BP 480/40 nm; LPDC mirror, 505 nm, emission filter BP 527/30 (#11504166, Leica); TX2 excitation filter BP 560/40 nm, LPDC mirror 595 nm, emission filter BP 645/75 nm (#11504166, Leica). A monochrome CoolSNAP HQ2 14-bit camera with a charge-coupled device sensor pixel size 6.45 × 6.45 μm was used. Images were acquired at a magnification of ×63 oil immersion (numerical aperture: 1.30). The acquired image size was 1344 × 1024 pixels, with a 14-bit depth, and an x-y dimension of 0.1024 μm per pixel.
For PLAs, the images were acquired using a Leica SPE laser scanning confocal microscope, equipped with a photomultiplier tube and 405-, 488-, and 532-nm excitation lasers. Commercial software LAS AF (Leica) was used for image acquisition. Image acquisition was performed with a magnification of ×63 immersion oil (numerical aperture: 1.30). The acquired image size was 512 × 512 pixels, with a 12-bit depth, and an x-y dimension of 0.227 μm per pixel. All included images in the figures have a linear gamma (= 1 curve) and their display ranges have been equally rescaled.
Digital microscopy image analyses
Raw data images acquired with linear and full display range were iteratively deconvolved using the Wiener filter preconditioned Landweber method (parallel iterative 2D deconvolution plugin v1.9 with ImageJ software) for deblurring. All digital images, except for 53BP1 foci formation after lamin B1 fragment overexpression and PLA assays in 53BP1 KO U2OS cells, were processed in batch with the CellProfiler software. The segmentation of each structure of interest (i.e., nuclei, foci, and PLA dots) was based on an adaptive labeling–dependent threshold. The consistency of the automatic segmentation was controlled visually on the original images supplemented with the outlines of identified objects. Measurement data for each object were collected by CellProfiler during the pipeline run and then further analyzed using the KNIME analytics platform for calculation of per nuclei parameters. 53BP1 foci formation after lamin B1 fragment overexpression and PLA assays in 53BP1 KO U2OS cells after expression of WT or 28A mutant 53BP1 were analyzed with ImageJ software. After nuclei segmentation on DAPI counterstaining, foci or PLA dots were counted as local maxima, respectively, on the 53BP1 or PLA field and matched to the intensity of, respectively, lamin B1 fragment expression or 53BP1 expression for the analyzed nuclei. Nuclei were classified according to their lamin B1 fragment or 53BP1 intensity as compared to the maximal background intensity of nonoverexpressing nuclei measured on empty vector–transfected cells. For the 53BP1 foci, two classes with similar expression intensity for each lamin B1 construct were defined: low- or moderate-expressing cells, corresponding respectively to two- to fourfold or four to sixfold expression over the maximum background intensity. For PLA dots, populations with similar expression of LMNB1 fragment or 53BP1 were compared.
Statistical analysis
Statistical analyses [unpaired t test, multiple t test, two-tailed Mann-Whitney test, one-way analysis of variance (ANOVA)] were performed using GraphPad Prism 7.0 (GraphPad Software Inc.).
Acknowledgments
We acknowledge K. Dubrana, S. Marcand, E. Soler, F. Boussin, and B. Lopez for helpful discussions and comments on the manuscript. We thank D. Durocher for providing the 53BP1-deficient U2OS cell line. The manuscript was edited by American Journal Experts. Funding: This work was supported by the Ligue Nationale Contre le Cancer (Ile de France committee), Association for Research against Cancer (Fondation ARC) number of grant if required (ARCPJA32020070002430), AT Europe Association, Radiobiology program CEA grant, EDF grant, AFM-Téléton grant number of grant if required : (n 21566), INCA grant, and INSERM house funding (SGCSR unit). L.E. was supported by a Ligue Nationale Contre le Cancer fellowship (Haut-de-Seine committee) and by INSERM. A.M. is recipient of a CEA DSV IRTELIS international fellowship and of a Fondation ARC fellowship. D.G. was a recipient of a CEA DSV IRTELIS international fellowship and of a Fondation ARC fellowship. E.R. was recipient of a Fondation pour la Recherche Medicale fellowship. S.W. is recipient of CEA DRF “Phare” fellowship and of a Fondation ARC fellowship. P.W. is recipient of a Ligue Nationale Contre le Cancer fellowship. J.P. is recipient of a PhD fellowship form University Paris Saclay (ED CBMS). Author contributions: P.B. and E.R. wrote the paper. L.E., D.G., A.M., E.R., G.P., and P.B. conceived the experiments. L.E., D.G., A.M., E.R., and G.P. performed most of the experiments. S.W., C.C.-O., P.W., and J.P. performed some experiments. B.T. developed several in-house scripts for imaging analysis and performed some experiments. A.B. performed initial DSB repair experiment. J.D. and X.V. performed GST pull-down assay. E.D. and D.B. constructed the vectors expressing different 53BP1 and lamin B1 domains. L.I., T.K., and A.C. helped with live laser microirradiation experiments. C.L.C. assisted for the development of several in-house scripts in Metamorph software. S.Z.-J. for helpful discussions and especially for design of truncated lamin B1 constructions/proteins. R.S. provided its expertise and biological reagents. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabb3799/DC1
REFERENCES AND NOTES
- 1.Lieber M., The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guirouilh-Barbat J., Huck S., Bertrand P., Pirzio L., Desmaze C., Sabatier L., Lopez B. S., Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 14, 611–623 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Bunting S. F., Nussenzweig A., End-joining, translocations and cancer. Nat. Rev. Cancer 13, 443–454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betermier M., Bertrand P., Lopez B., Is non-homologous end-joining really an inherently error-prone process? PLOS Genet. 10, e1004086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holland A. J., Cleveland D. W., Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 18, 1630–1638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noordermeer S. M., Adam S., Setiaputra D., Barazas M., Pettitt S. J., Ling A. K., Olivieri M., Álvarez-Quilón A., Moatti N., Zimmermann M., Annunziato S., Krastev D. B., Song F., Brandsma I., Frankum J., Brough R., Sherker A., Landry S., Szilard R. K., Munro M. M., McEwan A., Goullet de Rugy T., Lin Z.-Y., Hart T., Moffat J., Gingras A.-C., Martin A., van Attikum H., Jonkers J., Lord C. J., Rottenberg S., Durocher D., The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dev H., Chiang T.-W. W., Lescale C., de Krijger I., Martin A. G., Pilger D., Coates J., Sczaniecka-Clift M., Wei W., Ostermaier M., Herzog M., Lam J., Shea A., Demir M., Wu Q., Yang F., Fu B., Lai Z., Balmus G., Belotserkovskaya R., Serra V., O’Connor M. J., Bruna A., Beli P., Pellegrini L., Caldas C., Deriano L., Jacobs J. J. L., Galanty Y., Jackson S. P., Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 20, 954–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Findlay S., Heath J., Luo V. M., Malina A., Morin T., Coulombe Y., Djerir B., Li Z., Samiei A., Simo-Cheyou E., Karam M., Bagci H., Rahat D., Grapton D., Lavoie E. G., Dove C., Khaled H., Kuasne H., Mann K. K., Klein K. O., Greenwood C. M., Tabach Y., Park M., Côté J.-F., Masson J.-Y., Maréchal A., Orthwein A., SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 37, e100158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mirman Z., Lottersberger F., Takai H., Kibe T., Gong Y., Takai K., Bianchi A., Zimmermann M., Durocher D., de Lange T., 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560, 112–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghezraoui H., Oliveira C., Becker J. R., Bilham K., Moralli D., Anzilotti C., Fischer R., Deobagkar-Lele M., Sanchiz-Calvo M., Fueyo-Marcos E., Bonham S., Kessler B. M., Rottenberg S., Cornall R. J., Green C. M., Chapman J. R., 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bothmer A., Robbiani D., Feldhahn N., Gazumyan A., Nussenzweig A., Nussenzweig M., 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 207, 855–865 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouwman P., Aly A., Escandell J., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q., Haffty B., Tommiska J., Blomqvist C., Drapkin R., Adams D., Nevanlinna H., Bartek J., Tarsounas M., Ganesan S., Jonkers J., 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunting S., Callen E., Wong N., Chen H., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., Xu X., Deng C., Finkel T., Nussenzweig M., Stark J., Nussenzweig A., 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman J. R., Barral P., Vannier J.-B., Borel V., Steger M., Tomas-Loba A., Sartori A. A., Adams I. R., Batista F. D., Boulton S. J., RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A., Feldhahn N., Resch W., Oliveira T. Y., Chait B. T., Nussenzweig A., Casellas R., Robbiani D. F., Nussenzweig M. C., Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabarz A., Guirouilh-Barbat J., Barascu A., Pennarun G., Genet D., Rass E., Germann S., Bertrand P., Hickson I., Lopez B., A role for BLM in double-strand break repair pathway choice: Prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 5, 21–28 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Ward I. M., Minn K., van Deursen J., Chen J., p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23, 2556–2563 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee D., Acharya S., Kwon M., Drane P., Guan Y., Adelmant G., Kalev P., Shah J., Pellman D., Marto J., Chowdhury D., Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 54, 512–525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orthwein A., Fradet-Turcotte A., Noordermeer S., Canny M., Brun C., Strecker J., Escribano-Diaz C., Durocher D., Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Panier S., Boulton S. J., Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 15, 7–18 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Benada J., Burdova K., Lidak T., von Morgen P., Macurek L., Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 14, 219–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng X.-F., Acharya S. S., Choe K. N., Nikhil K., Adelmant G., Satapathy S. R., Sharma S., Viccaro K., Rana S., Natarajan A., Sicinski P., Marto J. A., Shah K., Chowdhury D., A mitotic CDK5-PP4 phospho-signaling cascade primes 53BP1 for DNA repair in G1. Nat. Commun. 10, 4252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dechat T., Adam S. A., Taimen P., Shimi T., Goldman R. D., Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2, a000547 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C., Munnich A., Le Merrer M., Lévy N., Lamin a truncation in Hutchinson–Gilford progeria. Science 300, 2055 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Eriksson M., Brown W., Gordon L., Glynn M., Singer J., Scott L., Erdos M., Robbins C., Moses T., Berglund P., Dutra A., Pak E., Durkin S., Csoka A., Boehnke M., Glover T., Collins F., Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423, 293–298 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X., Huang J. D., Li K. M., Chau P. Y., Chen D. J., Pei D., Pendas A. M., Cadinanos J., López-Otín C., Tse H. F., Hutchison C., Chen J., Cao Y., Cheah K. S., Tryggvason K., Zhou Z., Genomic instability in laminopathy-based premature aging. Nat. Med. 11, 780–785 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Liu Y., Rusinol A., Sinensky M., Wang Y., Zou Y., DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J. Cell Sci. 119, 4644–4649 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varela I., Cadinanos J., Pendas A. M., Gutiérrez-Fernández A., Folgueras A. R., Sánchez L. M., Zhou Z., Rodríguez F. J., Stewart C. L., Vega J. A., Tryggvason K., Freije J. M., López-Otín C., Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564–568 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Gonzalo S., Kreienkamp R., DNA repair defects and genome instability in Hutchinson-Gilford progeria syndrome. Curr. Opin. Cell Biol. 34, 75–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Suarez I., Redwood A., Perkins S., Vermolen B., Lichtensztejin D., Grotsky D., Morgado-Palacin L., Gapud E., Sleckman B., Sullivan T., Sage J., Stewart C., Mai S., Gonzalo S., Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 28, 2414–2427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs-Seymour I., Markiewicz E., Bekker-Jensen S., Mailand N., Hutchison C. J., Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 14, 162–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobb A. M., Larrieu D., Warren D. T., Liu Y., Srivastava S., Smith A. J. O., Bowater R. P., Jackson S. P., Shanahan C. M., Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell 15, 1039–1050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butin-Israeli V., Adam S. A., Jain N., Otte G. L., Neems D., Wiesmüller L., Berger S. L., Goldman R. D., Role of lamin b1 in chromatin instability. Mol. Cell. Biol. 35, 884–898 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu N.-A., Sun J., Kono K., Horikoshi Y., Ikura T., Tong X., Haraguchi T., Tashiro S., Regulation of homologous recombinational repair by lamin B1 in radiation-induced DNA damage. FASEB J. 29, 2514–2525 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Parry D. A., Martin C.-A., Greene P., Marsh J. A., Blyth M., Cox H., Donnelly D., Greenhalgh L., Greville-Heygate S., Harrison V., Lachlan K., McKenna C., Quigley A. J., Rea G., Robertson L., Suri M., Jackson A. P., Heterozygous lamin B1 and lamin B2 variants cause primary microcephaly and define a novel laminopathy. Genet. Med. 23, 408–414 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cristofoli F., Moss T., Moore H. W., Devriendt K., Flanagan-Steet H., May M., Jones J., Roelens F., Fons C., Fernandez A., Martorell L., Selicorni A., Maitz S., Vitiello G., der Hoeven G. V., Skinner S. A., Bollen M., Vermeesch J. R., Steet R., Esch H. V., De novo variants in LMNB1 cause pronounced syndromic microcephaly and disruption of nuclear envelope integrity. Am. J. Hum. Genet. 107, 753–762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padiath Q. S., Saigoh K., Schiffmann R., Asahara H., Yamada T., Koeppen A., Hogan K., Ptácek L. J., Fu Y.-H., Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 38, 1114–1123 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Barascu A., Le Chalony C., Pennarun G., Genet D., Imam N., Lopez B., Bertrand P., Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 31, 1080–1094 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donadille B., D’Anella P., Auclair M., Uhrhammer N., Sorel M., Grigorescu R., Ouzounian S., Cambonie G., Boulot P., Laforêt P., Carbonne B., Christin-Maitre S., Bignon Y.-J., Vigouroux C., Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Orphanet J. Rare Dis. 8, 106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butin-Israeli V., Adam S., Goldman A., Goldman R., Nuclear lamin functions and disease. Trends Genet. 28, 464–471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L., Du Y., Kong X., Li Z., Jia Z., Cui J., Gao J., Wang G., Xie K., Lamin B1 is a novel therapeutic target of betulinic acid in pancreatic cancer. Clin. Cancer Res. 19, 4651–4661 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Las Heras J. I., Schirmer E. C., The nuclear envelope and cancer: A diagnostic perspective and historical overview. Adv. Exp. Med. Biol. 773, 5–26 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Radspieler M. M., Schindeldecker M., Stenzel P., Försch S., Tagscherer K. E., Herpel E., Hohenfellner M., Hatiboglu G., Roth W., Macher-Goeppinger S., Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 18, 2654–2660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izdebska M., Gagat M., Grzanka A., Overexpression of lamin B1 induces mitotic catastrophe in colon cancer LoVo cells and is associated with worse clinical outcomes. Int. J. Oncol. 52, 89–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolas N., Chapman J., Nakada S., Ylanko J., Chahwan R., Sweeney F., Panier S., Mendez M., Wildenhain J., Thomson T. M., Pelletier L., Jackson S. P., Durocher D., Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartlerode A. J., Guan Y., Rajendran A., Ura K., Schotta G., Xie A., Shah J. V., Scully R., Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLOS ONE 7, e49211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guirouilh-Barbat J., Rass E., Plo I., Bertrand P., Lopez B. S., Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc. Natl. Acad. Sci. U.S.A. 104, 20902–20907 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B. S., Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 16, 819–824 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Biehs R., Steinlage M., Barton O., Juhász S., Künzel J., Spies J., Shibata A., Jeggo P. A., Löbrich M., DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 65, 671–684.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemaître C., Fischer B., Kalousi A.-S., Hoffbeck A., Guirouilh-Barbat J., Shahar O. D., Genet D., Goldberg M., Betrand P., Lopez B., Brino L., Soutoglou E., The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene 31, 4803–4809 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Malhas A. N., Vaux D. J., Transcription factor sequestration by nuclear envelope components. Cell Cycle 8, 959–964 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Jowsey P., Morrice N. A., Hastie C. J., McLauchlan H., Toth R., Rouse J., Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair 6, 1536–1544 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J., Tkáč J., Cook M., Rosebrock A., Munro M., Canny M., Xu D., Durocher D., A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Botuyan M. V., Lee J., Ward I. M., Kim J.-E., Thompson J., Chen J., Mer G., Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fradet-Turcotte A., Canny M. D., Escribano-Díaz C., Orthwein A., Leung C. Y., Huang H., Landry M.-C., Kitevski-LeBlanc J., Noordermeer S. M., Sicheri F., Durocher D., 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson M. D., Benlekbir S., Fradet-Turcotte A., Sherker A., Julien J.-P., McEwan A., Noordermeer S. M., Sicheri F., Rubinstein J. L., Durocher D., The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Mallette F. A., Mattiroli F., Cui G., Young L. C., Hendzel M. J., Mer G., Sixma T. K., Richard S., RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acs K., Luijsterburg M., Ackermann L., Salomons F., Hoppe T., Dantuma N., The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18, 1345–1350 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Meerang M., Ritz D., Paliwal S., Garajova Z., Bosshard M., Mailand N., Janscak P., Hubscher U., Meyer H., Ramadan K., The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 13, 1376–1382 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Zhang A., Peng B., Huang P., Chen J., Gong Z., The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J. Biol. Chem. 292, 6461–6467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drané P., Brault M.-E., Cui G., Meghani K., Chaubey S., Detappe A., Parnandi N., He Y., Zheng X.-F., Botuyan M. V., Kalousi A., Yewdell W. T., Münch C., Harper J. W., Chaudhuri J., Soutoglou E., Mer G., Chowdhury D., TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 543, 211–216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreno N. S., Liu J., Haas K. M., Parker L. L., Chakraborty C., Kron S. J., Hodges K., Miller L. D., Langefeld C., Robinson P. J., Lelièvre S. A., Vidi P.-A., The nuclear structural protein NuMA is a negative regulator of 53BP1 in DNA double-strand break repair. Nucleic Acids Res. 47, 10475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parnandi N., Rendo V., Cui G., Botuyan M. V., Remisova M., Nguyen H., Drané P., Beroukhim R., Altmeyer M., Mer G., Chowdhury D., TIRR inhibits the 53BP1-p53 complex to alter cell-fate programs. Mol. Cell 81, 2583–2595.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M., Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 38, e101379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pessina F., Giavazzi F., Yin Y., Gioia U., Vitelli V., Galbiati A., Barozzi S., Garre M., Oldani A., Flaus A., Cerbino R., Parazzoli D., Rothenberg E., d’Adda di Fagagna F., Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 21, 1286–1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghodke I., Remisova M., Furst A., Kilic S., Reina-San-Martin B., Poetsch A. R., Altmeyer M., Soutoglou E., AHNAK controls 53BP1-mediated p53 response by restraining 53BP1 oligomerization and phase separation. Mol. Cell 81, 2596–2610.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murray-Nerger L. A., Justice J. L., Rekapalli P., Hutton J. E., Cristea I. M., Lamin B1 acetylation slows the G1 to S cell cycle transition through inhibition of DNA repair. Nucleic Acids Res. 49, 2044–2064 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willaume S., Rass E., Fontanilla-Ramirez P., Moussa A., Wanschoor P., Bertrand P., A link between replicative stress, lamin proteins, and inflammation. Genes 12, 552 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahen R., Hattori H., Lee M., Sharma P., Jeyasekharan A. D., Venkitaraman A. R., A-type lamins maintain the positional stability of DNA damage repair foci in mammalian nuclei. PLOS ONE 8, e61893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scaffidi P., Misteli T., Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11, 440–445 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez-Suarez I., Redwood A. B., Grotsky D. A., Neumann M. A., Cheng E. H., Stewart C. L., Dusso A., Gonzalo S., A new pathway that regulates 53BP1 stability implicates Cathepsin L and vitamin D in DNA repair. EMBO J. 30, 3383–3396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Busso D., Delagoutte-Busso B., Moras D., Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 343, 313–321 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Delbarre E., Tramier M., Coppey-Moisan M., Gaillard C., Courvalin J.-C., Buendia B., The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum. Mol. Genet. 15, 1113–1122 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabb3799/DC1