

Histone citrullination by PADI4 is required for hypoxia-induced gene expression.
Abstract
Hypoxia-inducible factors (HIFs) activate transcription of target genes by recruiting coactivators and chromatin-modifying enzymes. Peptidylarginine deiminase 4 (PADI4) catalyzes the deimination of histone arginine residues to citrulline. Here, we demonstrate that PADI4 expression is induced by hypoxia in a HIF-dependent manner in breast cancer and hepatocellular carcinoma cells. PADI4, in turn, is recruited by HIFs to hypoxia response elements (HREs) and is required for HIF target gene transcription. Hypoxia induces histone citrullination at HREs that is PADI4 and HIF dependent. RNA sequencing revealed that almost all HIF target genes in breast cancer cells are PADI4 dependent. PADI4 is required for breast and liver tumor growth and angiogenesis in mice. PADI4 expression is correlated with HIF-1α expression and vascularization in human breast cancer biopsies. Thus, HIF-dependent recruitment of PADI4 to target genes and local histone citrullination are required for transcriptional responses to hypoxia.
INTRODUCTION
Reduced O2 availability (hypoxia) is a characteristic feature of the tumor microenvironment. Measurements of the partial pressure of oxygen in locally advanced breast cancers revealed a median PO2 of 10 mmHg (~1.4% O2), with one-quarter of all tumor regions having a PO2 of <2.5 mmHg, as compared to normal breast tissue, which had a median PO2 of 65 mmHg (~9.3% O2) and no regions with a PO2 of <15 mmHg (1). Liver cancers are also highly hypoxic, with three-quarters of tumors assayed having a PO2 of 10 mmHg or less (1). Intratumoral hypoxia is a driving force for breast and liver cancer progression (2–4) that is mediated by hypoxia-inducible factors (HIFs), which are heterodimeric proteins consisting of an O2-regulated HIF-1α, HIF-2α, or HIF-3α subunit and a constitutively expressed HIF-1β subunit (5). Increased HIF-1α expression in primary tumor biopsies is associated with decreased disease-free, progression-free, and overall survival of breast cancer patients (6–10) and is associated with lymph node metastasis and decreased overall survival of patients with hepatocellular carcinoma (HCC) (11, 12).
In response to hypoxia, HIFs activate transcription of hundreds of genes that play key roles in angiogenesis, metabolic reprogramming, extracellular matrix remodeling, invasion and metastasis, cancer stem cell specification, and immune evasion (2, 13–15). The molecular mechanism by which HIFs activate transcription is an area of active investigation. HIFs bind to hypoxia response elements (HREs) containing the core HIF binding sequence 5′-RCGTG-3′ (R = A or G) in target genes (16) and recruit mediator subunits (17), histone-modifying enzymes (18–22), and chromatin-remodeling complexes (23). It is through recruitment of these proteins that HIFs and other activators increase the rate of transcription initiation and elongation (24). HIFs have also been shown to regulate the transcription of genes that encode coactivator proteins, which are then recruited by HIFs to target genes (21, 25).
It is well known that acetylation of lysine residues in histone tails serves to reduce their net positive charge and thereby decrease interaction with the negatively charged DNA, enabling RNA polymerase II to access the DNA and transcribe it into mRNA. In addition, bromodomain proteins specifically bind to histones with acetylated lysine residues (26). A less well-known posttranslational modification of histones that decreases their net positive charge is the hydrolysis of arginine (Arg) residues to citrulline (Cit), which is catalyzed by the peptidylarginine deiminases PADI2 (27) and PADI4 (28). PADI4 hydrolyzes Arg-3 of histone H4 to Cit as well as catalyzing deimination (citrullination) of Arg residues 2, 8, 17, and 26 of H3; Arg-3 of H2A; and Arg-54 of H1 (29–32). In addition to affecting histone-DNA interactions, citrullination may affect histone-histone interactions and histone interactions with other chromatin proteins (33).
Increased PADI4 expression was demonstrated in multiple cancer types, including breast cancer and HCC (34). Treatment with Cl-amidine (N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide), a pan-PADI inhibitor, impaired the growth of breast tumor xenografts (35). PADI expression was induced in human glioma cells exposed to hypoxia or the HIF inducer dimethyloxalylglycine (36). Forced PADI4 overexpression in an esophageal carcinoma cell line stimulated tumor xenograft growth and increased the expression of CA9 (37), which is a well-known HIF target gene encoding carbonic anhydrase 9 (38). On the basis of these findings, we hypothesized that PADI4 is a HIF target gene product that HIFs recruit to HREs to modify histones and stimulate transcriptional activation under hypoxic conditions.
RESULTS
HIFs activate transcription of the PADI4 gene in hypoxic cancer cells
Triple-negative breast cancers (TNBCs) lack expression of the estrogen receptor, progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) and are aggressive cancers for which effective therapy is lacking. SUM159 human TNBC cells were stably transfected with lentiviral vectors encoding a nontargeting control (NTC) short hairpin RNA (shRNA) or shRNA targeting either HIF-1α (sh1α) or HIF-2α (sh2α) or both [double knockdown (DKD)], and the knockdown efficiency was confirmed by immunoblot assays (Fig. 1A). The subclones were exposed to 20 or 1% O2 for 24 hours, and RNA was isolated for analysis by reverse transcription (RT) and quantitative real-time polymerase chain reaction (qPCR). Hypoxia increased PADI4 mRNA levels by threefold in NTC cells, and this induction was completely abrogated by knockdown of HIF-1α and/or HIF-2α (Fig. 1B). Immunoblot assays of cells exposed to hypoxia for 48 hours revealed decreased PADI4 protein expression in the sh1α and sh2α subclones and complete loss of expression in the DKD subclone (Fig. 1A). A kinetic analysis revealed peak PADI4 protein levels in parental SUM159 cells exposed to 1% O2 for 24 to 48 hours (Fig. 1C). The functional consequence of HIF knockdown on PADI4 gene expression phenocopied the effect on VEGFA (fig. S1A), which encodes vascular endothelial growth factor A, a prototypical HIF target gene (39).
Fig. 1. Hypoxia-induced PADI4 expression is HIF-dependent and is required for HIF target gene transcription.
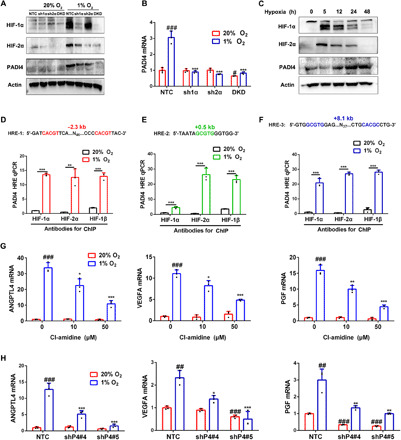
(A) SUM159 subclones expressing a nontargeting control (NTC) shRNA, an shRNA targeting HIF-1α (sh1α) or HIF-2α (sh2α), or shRNAs targeting both HIF-1α and HIF-2α [double knockdown (DKD)] were exposed to 20 or 1% O2 for 6 hours (HIF-1α and HIF-2α) or 48 hours (PADI4 and actin), and immunoblot (IB) assays of whole-cell lysates (WCLs) were performed. (B) SUM159 subclones were exposed to 20 or 1% O2 for 24 hours followed by RT-qPCR (mean ± SD; n = 3). #P < 0.05, ###P < 0.001 versus NTC at 20% O2; ***P < 0.001 versus NTC at 1% O2 [two-way analysis of variance (ANOVA)]. (C) SUM159 cells were exposed to 1% O2, and IB assays were performed. (D to F) SUM159 cells were exposed to 20 or 1% O2 for 16 hours, and ChIP assays were performed. Primers encompassing HIF binding sites located 2.3 kb 5′ (D), 0.5 kb 3′ (E), and 8.1 kb 3′ (F) to the PADI4 TSS were used for qPCR. Results were normalized to the first lane (mean ± SD; n = 3). **P < 0.01, ***P < 0.001 versus 20% O2 (Student’s t test). (G) SUM159 cells were exposed to 20 or 1% O2 for 24 hours followed by RT-qPCR. Results were normalized to lane 1 (mean ± SD; n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001 versus vehicle at 20% O2. **P < 0.01, ***P < 0.001 versus vehicle at 1% O2 (two-way ANOVA). (H) SUM159 subclones were exposed to 20 or 1% O2 for 24 hours followed by RT-qPCR (mean ± SD; n = 3). ##P < 0.01, ###P < 0.001 versus NTC at 20% O2. *P < 0.05, **P < 0.01, ***P < 0.001 versus NTC at 1% O2 (two-way ANOVA).
We also generated NTC, sh1α, sh2α, and DKD subclones of Hepa1-6 mouse HCC cells (fig. S1, B to E) and demonstrated a fourfold induction of PADI4 mRNA in hypoxic NTC cells that was again completely abrogated by knockdown of HIF-1α and/or HIF-2α (fig. S1F). In addition, previously established knockdown subclones of MDA-MB-231 human TNBC cells (40) were analyzed, revealing hypoxic induction of PADI4 mRNA in NTC but not in sh1α, sh2α, or DKD subclones (fig. S1G). Immunoblot assays revealed similarly increased levels of PADI4 protein in Hepa1-6 and SUM159 cells exposed to 1% O2 for 48 hours (fig. S1H).
To determine whether HIFs directly bind to the PADI4 gene to activate its transcription, SUM159 cells were exposed to 20 or 1% O2 for 16 hours and chromatin immunoprecipitation (ChIP) assays were performed. Using antibodies against HIF-1α, HIF-2α, or HIF-1β, we demonstrated hypoxia-induced binding of HIF-1 (HIF-1α + HIF-1β) and HIF-2 (HIF-2α + HIF-1β) to consensus HIF binding sites located 2.3 kb 5′ to the transcription start site (TSS) and 0.5 and 8.1 kb 3′ to the TSS (Fig. 1, D to F). The −2.3-kb site contained two consensus HIF binding site sequences as direct repeats separated by 52 base pairs (bp) (Fig. 1D, in red), whereas the +8.1-kb site contained an inverted repeat of the consensus sequence separated by 43 bp (Fig. 1F, in blue). In contrast, there was no hypoxia-induced binding of HIF subunits to the RPL13A gene (fig. S1I), which is not regulated by hypoxia or HIFs. Similar ChIP results were obtained using Hep3B human HCC cells (fig. S2). Together, these results demonstrate that hypoxia induces PADI4 expression that is directly mediated by HIF-1 and HIF-2.
Hypoxia-induced PADI4 is required for HIF target gene transcription
To investigate whether PADI4 is, in turn, required for HIF target gene transcription, SUM159 and Hepa1-6 cells were exposed concomitantly to hypoxia and different concentrations of Cl-amidine, an inhibitor of PADI4 catalytic activity (41, 42), for 24 hours followed by RT-qPCR analysis of HIF-regulated mRNAs encoding the angiogenic factors angiopoietin-like 4 [ANGPTL4; (40)], VEGFA (39), and placental growth factor [PGF; (43)]. The hypoxia-induced expression of all three genes was significantly decreased by Cl-amidine treatment of SUM159 (Fig. 1G) and Hepa1-6 (fig. S3A) cells. In contrast, RPL13A mRNA expression was not induced by hypoxia or inhibited by Cl-amidine in SUM159 (fig. S1J) or Hepa1-6 (fig. S3A) cells. Hypoxia-induced VEGFA mRNA expression was also inhibited by Cl-amidine in MDA-MB-231 cells (fig. S3B) and Hep3B human HCC cells (fig. S3C).
To complement the results of pharmacological inhibition of PADI4, we knocked down PADI4 expression by stably transfecting SUM159, Hepa1-6, MDA-MB-231, and Hep3B cells with lentiviral vectors encoding five different shRNAs that targeted PADI4 (fig. S3, D to H). Analysis of NTC and two different shRNA PADI4-knockdown (shP4) subclones of SUM159 and Hepa1-6 revealed significantly decreased expression of ANGPTL4, VEGFA, and PGF mRNA under hypoxic conditions (Fig. 1H and fig. S3I). Hypoxia-induced ANGPTL4 mRNA expression was also significantly inhibited by PADI4 knockdown in MDA-MB-231 (fig. S3J) and Hep3B (fig. S3K) cells. A time course analysis of HIF target gene expression in NTC and PADI4-knockdown subclones of SUM159 revealed induction of ANGPTL4, PGF, and VEGFA mRNA (but not RPL13A mRNA), which was maximal after 5 hours at 1% O2 and was maintained through 48 hours of hypoxic exposure in the NTC subclone (Fig. 2). The data from the NTC subclone indicate that there is sufficient PADI4 to maximally induce HIF target gene expression even at the earliest hypoxic time point (5 hours), which is before the hypoxia-induced augmentation of PADI4 expression (Fig. 1C). Data from the knockdown subclones demonstrate that PADI4 is essential for hypoxia-induced gene transcription, even at the earliest time point (Fig. 2). Together, Figs. 1 and 2 and figs. S1 to S3 demonstrate that PADI4 expression is induced by hypoxia in a HIF-dependent manner in breast and liver cancer cell lines and that, in turn, PADI4 is required for activation of HIF target gene transcription.
Fig. 2. Kinetics of HIF target gene expression and dependence on PADI4 in SUM159 cells.
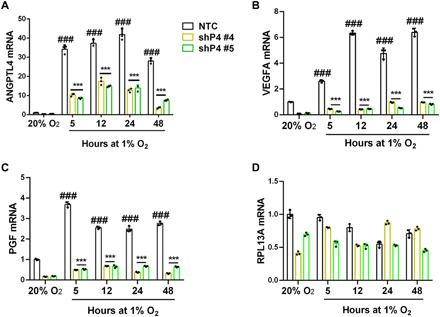
(A to D) NTC and PADI4-knockdown subclones (shPADI4 #4 and #5) were exposed to 20 or 1% O2 for 5 to 48 hours and analyzed by RT-qPCR for ANGPTL4 (A), VEGFA (B), PGF (C) or RPL13A (D) mRNA expression. Data are shown as mean ± SEM (n = 3). ###P < 0.001 versus NTC at 20% O2; ***P < 0.001 versus NTC at 1% O2 (two-way ANOVA).
PADI4 is recruited to HREs by HIFs and stabilizes HIF occupancy of HREs
To test whether PADI4 physically interacts with HIF-1α or HIF-2α to mediate target gene transcriptional activation, V5 epitope–tagged PADI4 was constitutively expressed in SUM159 cells, which were exposed to 20 or 1% O2 for 8 hours, and whole-cell lysates (WCLs) were prepared. Using anti–HIF-1α antibody, coimmunoprecipitation of V5-PADI4 selectively from WCLs of hypoxic cells was demonstrated (Fig. 3A). Conversely, using anti-V5 antibody, coimmunoprecipitation of HIF-2α selectively from WCLs of hypoxic cells was demonstrated (Fig. 3B). Thus, PADI4 interacts with both HIF-1α and HIF-2α in hypoxic breast cancer cells. Next, we tested whether endogenous PADI4 is recruited to HREs of HIF target genes under hypoxic conditions. ChIP assays using antibody against PADI4 revealed hypoxia-induced occupancy of HREs at the ANGPTL4, VEGFA, and PGF genes (but not at the RPL13A gene) in parental SUM159 (fig. S4A) and Hep3B (fig. S4B) cells. Hypoxia-induced PADI4 recruitment to HREs was also observed in the NTC (but not in the DKD) subclone of SUM159 cells (Fig. 3C). Thus, HIFs recruit PADI4 to HREs of HIF target genes under hypoxic conditions.
Fig. 3. PADI4 is recruited to HREs by HIFs and stabilizes HIF occupancy at HREs.
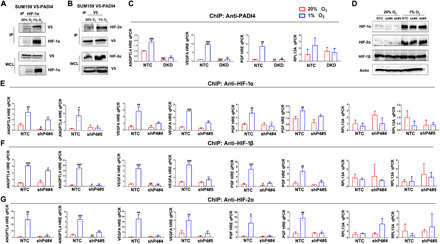
(A and B) V5 epitope–tagged PADI4 was expressed in SUM159 cells, which were exposed to 20 or 1% O2 for 8 hours, and WCLs were prepared for immunoprecipitation using antibody (Ab) against HIF-1α and IB assays with Ab against V5 or HIF-1α (A) or Ab against V5 and IB assays with Ab against V5 or HIF-2α (B). (C) SUM159 subclones were exposed to 20 or 1% O2 for 16 hours, and ChIP assays were performed using anti-PADI4 Ab. Primers encompassing HRE sites in the ANGPTL4, VEGFA, or PGF genes were used for qPCR, and non-HIF target gene RPL13A was analyzed as a negative control. Results were normalized to NTC at 20% O2 (mean ± SD; n = 3). ##P < 0.01, ###P < 0.001 versus NTC at 20% O2; **P < 0.01, ***P < 0.001 versus NTC at 1% O2 (two-way ANOVA). (D) SUM159 subclones were exposed to 20 or 1% O2 for 6 hours, and IB assays were performed using Ab against HIF-1α, HIF-2α, HIF-1β, or actin. (E to G) SUM159 subclones were exposed to 20 or 1% O2 for 16 hours, and ChIP assays were performed using Ab against HIF-1α (E), HIF-1β (F), or HIF-2α (G) and qPCR of ANGPTL4, VEGFA, and PGF HREs and RPL13A. Results were normalized to NTC at 20% O2 (mean ± SD; n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001 versus NTC at 20% O2; *P < 0.05, **P < 0.01, ***P < 0.001 versus NTC at 1% O2 (two-way ANOVA).
A potential mechanism by which PADI4 binding might promote HIF-1α transcriptional activity is by inhibiting HIF-1α degradation, as has been reported for many HIF-1α–interacting proteins (44). However, immunoblot assays showed no change in the hypoxia-induced expression of HIF-1α, HIF-2α, or HIF-1β in PADI4-knockdown as compared to NTC cells (Fig. 3D). We next investigated whether the occupancy of HREs by HIFs was affected by PADI4 expression. ChIP assays revealed a significant decrease in the hypoxia-induced occupancy of the ANGPTL4, VEGFA, and PGF HREs (but not RPL13A) by HIF-1α (Fig. 3E), HIF-1β (Fig. 3F), and HIF-2α (Fig. 3G) in PADI4-knockdown SUM159 cells, using two independent subclones (shP4#4 and shP4#5). Thus, PADI4 facilitates or stabilizes the binding of HIF-1 and HIF-2 to HREs in chromatin of hypoxic cancer cells.
Hypoxia induces histone citrullination at HREs that is PADI4 and HIF dependent
Given that inhibition of PADI4 catalytic activity impaired HIF target gene expression, we next investigated the role of PADI4 in modifying histones. Using an antibody against citrullinated histone H3 (Cit-H3), we analyzed bulk histone protein by immunoblot assays of WCLs. Hypoxia markedly increased the levels of Cit-H3 in SUM159 cells without changing total H3 levels, and treatment of the cells with 50 μM Cl-amidine blocked the hypoxia-induced increase in Cit-H3 levels (Fig. 4A). Similarly, hypoxia induced increased Cit-H3 levels in bulk chromatin in NTC but not in PADI4-knockdown (shP4#4 and shP4#5; Fig. 4, B and C) or HIF-DKD (Fig. 4D) SUM159 cells. Hypoxia also induced increased Cit-H4 levels, which was abrogated by Cl-amidine treatment (Fig. 4E) or by knockdown of PADI4 (Fig. 4, F and G) or HIFs (Fig. 4H).
Fig. 4. Hypoxia induces histone citrullination at HREs that is PADI4 and HIF dependent.
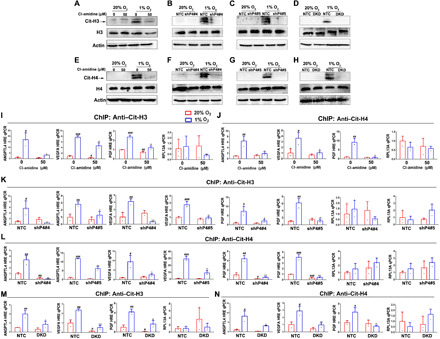
(A to D) SUM159 cells (A) or subclones (B to D) were exposed to 20 or 1% O2 for 48 hours, and IB assays were performed using Ab against citrullinated histone H3 (Cit-H3) or total H3. (E to H) SUM159 cells (E) or subclones (F to H) were exposed to 20 or 1% O2 for 48 hours, and IB assays were performed using Ab against citrullinated Arg-3 of histone H4 (Cit-H4) or total histone H4 (H4). (I to N) SUM159 cells were exposed to 20 or 1% O2 for 16 hours, and ChIP assays were performed using Ab against Cit-H3 (I, K, and M) or Cit-H4 (J, L, and N). Results were normalized to the first lane (mean ± SD; n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001 versus first lane; *P < 0.05, **P < 0.01, ***P < 0.001 versus second lane (two-way ANOVA).
Next, we performed ChIP assays to analyze Cit-H3 and Cit-H4 levels specifically at the ANGPTL4, VEGFA, and PGF HREs. Hypoxia induced increased Cit-H3 (Fig. 4I) and Cit-H4 (Fig. 4J) marks at the HREs (but not at RPL13A) in parental SUM159 cells, and the hypoxic induction was attenuated by treatment with Cl-amidine. The hypoxic induction of Cit-H3 marks at the ANGPTL4, VEGFA, and PGF HREs (but not RPL13A) was observed in NTC cells and was abrogated in PADI4-knockdown cells (Fig. 4K). Similar results were observed for Cit-H4 (Fig. 4L). In addition, the hypoxic induction of Cit-H3 (Fig. 4M) and Cit-H4 (Fig. 4N) marks at the ANGPTL4, VEGFA, and PGF HREs (but not at RPL13A) was observed in NTC cells and was abrogated in HIF-DKD cells. Total H3 and H4 levels at HREs were not significantly increased by hypoxia or significantly decreased by PADI4 or HIF knockdown (fig. S4, C to H). Together, the data indicate that hypoxia-induced and HIF-mediated induction of PADI4 expression and recruitment of PADI4 to HREs leads to the hypoxia-induced citrullination of histones H3 and H4 at HREs and increased HIF occupancy of HREs.
PADI4 is required for hypoxia-induced histone methylation and acetylation at HREs
We next investigated whether impaired citrullination in PADI4-knockdown cells has an impact on other histone modifications at HREs. We focused on trimethylation of lysines 4 and 36 of H3, because H3K4me3 and H3K36me3 are marks of actively transcribed chromatin (45, 46). H3K4me3 (Fig. 5A) and H3K36me3 (Fig. 5B) marks were increased at the ANGPTL4, VEGFA, and PGF HREs (but not at RPL13A) in response to hypoxia in NTC but not in two independent PADI4-knockdown subclones of SUM159. Similarly, H3K4me3 (Fig. 5C) and H3K36me3 (Fig. 5D) marks were increased at the ANGPTL4, VEGFA, and PGF HREs (but not at RPL13A) in response to hypoxia in NTC but not in HIF-DKD cells. We also explored the effects of PADI4 knockdown on acetylation of histones H3 at lysine-4 (H3K4ac) and H4 at lysine-5 (H4K5ac). ChIP assays revealed that H3K4ac (Fig. 5E) and H4K5ac (Fig. 5F) marks were increased at the ANGPTL4, VEGFA, and PGF HREs (but not at RPL13A) in response to hypoxia in NTC but not in two independent PADI4-knockdown subclones of SUM159. These results provide further evidence of the critical role of PADI4 expression and recruitment to HREs for hypoxia-induced transcriptional activation.
Fig. 5. PADI4 is required for histone methylation and acetylation at HREs.
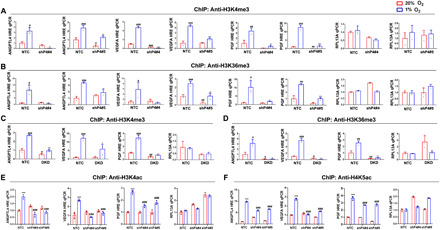
(A to D) NTC and PADI4-knockdown (shP4#4 and shP4#5) subclones (A and B) or NTC and HIF-DKD subclones (C and D) were exposed to 20 or 1% O2 for 16 hours, and ChIP assays were performed using Ab against H3K4me3 (A and C) or H3K36me3 (B and D). Results were normalized to NTC at 20% O2 (mean ± SD; n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001 versus NTC at 20% O2; *P < 0.05, **P < 0.01, ***P < 0.01 versus NTC at 1% O2 (two-way ANOVA). (E and F) SUM159 subclones were exposed to 20 or 1% O2, and ChIP assays were performed using Ab against H3K4ac (E) or H4K5ac (F). Results were normalized to NTC at 20% O2 (mean ± SD; n = 3). ***P < 0.001 versus NTC at 20% O2; ###P < 0.01 versus NTC at 1% O2 (two-way ANOVA).
PADI4 is required for hypoxia-induced expression of most HIF target genes
To obtain a more global view of the role of PADI4 in the transcriptional response to hypoxia, NTC, PADI4-knockdown, and HIF-DKD subclones of SUM159 were exposed to 20 or 1% O2 for 24 hours and RNA was isolated for high-throughput RNA sequencing (RNA-seq). In response to hypoxia, 1307 mRNAs were significantly induced by hypoxia in NTC but not in HIF-DKD cells (Fig. 6A), and of these mRNAs, the hypoxic induction of 1135 mRNAs (87%) was also lost in PADI4-knockdown cells. Furthermore, 817 mRNAs were significantly repressed by hypoxia in NTC but not in HIF-DKD cells (Fig. 6B), and of these mRNAs, the hypoxic repression of 723 mRNAs (88%) was also lost in PADI4-knockdown cells. Thus, the vast majority of HIF target genes require both HIFs and PADI4 for efficient transcriptional regulation in response to hypoxia in SUM159 breast cancer cells.
Fig. 6. Analysis of HIF- and PADI4-dependent gene expression in SUM159 cells by RNA-seq.

(A and B) Heat maps were prepared showing relative mRNA levels in NTC and HIF-DKD cells exposed to 20 or 1% O2 for 24 hours for 1307 HIF-dependent hypoxia-induced genes (A) and 817 HIF-dependent hypoxia-repressed genes (B). (C to H) HIF-dependent hypoxia-induced genes (C and D), PADI4-dependent hypoxia-induced genes (E and F), and HIF- and PADI4-dependent hypoxia-induced genes (G and H) were subjected to Gene Ontology (GO) biological process analysis (C, E, and G) and gene pathway enrichment analysis (D, F, and H). The top 10 categories for each analysis are shown.
Gene Ontology (GO) biological process enrichment analysis of HIF-dependent, hypoxia-induced genes revealed “axon guidance” as the most significantly enriched category, with “axonogenesis” and “postsynaptic density assembly” also among the top 10 categories (Fig. 6C). Pathway enrichment analysis of the HIF target genes identified in SUM159 cells revealed, as expected, a significant enrichment of genes involved in “HIF-1 transcriptional activity” as the top category (Fig. 6D). PADI4-dependent, hypoxia-induced genes were also enriched for axon guidance (Fig. 6E) and HIF-1 transcriptional activity (Fig. 6F). Similar results were obtained for hypoxia-induced genes that were both HIF and PADI4 dependent (Fig. 6, G and H). PADI4 was required for the hypoxic induction of 35 of 37 genes (95%) present in one or more of these three categories (Table 1). To determine whether these genes are HIF-regulated in human breast cancers, we compared the expression of each of these genes to a 10-gene HIF signature (47) using Pearson’s test and RNA expression data from more than 1200 breast cancers in The Cancer Genome Atlas (TCGA). Expression of the majority of genes (57%) was significantly correlated with the HIF signature (Table 1), suggesting that they are part of the HIF transcriptome in human breast cancers. Inspection of the 1307 HIF target genes revealed a remarkable number of other neuron-related genes (Table 1).
Table 1. Hypoxia-induced and HIF-dependent genes involved in axon guidance (GO: 7411), axonogenesis (GO: 7409), and postsynaptic density assembly (GO: 0097107).
| Gene name | FC* | PADI† | TCGA‡ |
| ARTN | 4.01 | Yes | Yes |
| BRSK2 | 1.78 | Yes | Yes |
| DOCK7 | 1.56 | Yes | Yes |
| DOK4 | 1.77 | Yes | Yes |
| DOK5 | 2.3 | Yes | Yes |
| EFNA1 | 2.1 | Yes | Yes |
| EFNA2 | 4.08 | No | Yes |
| EFNA3 | 4.99 | Yes | Yes |
| EFNB3 | 3.71 | Yes | No |
| EVL | 1.75 | Yes | No |
| GRB10 | 3.92 | Yes | Yes |
| L1CAM | 9.47 | Yes | Yes |
| LINGO3 | 83.7 | Yes | No |
| MAPK3 | 1.82 | Yes | No |
| MAPK7 | 2.34 | Yes | Yes |
| MAPK11 | 1.54 | Yes | Yes |
| NAV1 | 1.77 | Yes | No |
| NCAM1 | 1.74 | Yes | Yes |
| NLGN2 | 1.79 | Yes | Yes |
| NRXN2 | 2.62 | Yes | No |
| NTN1 | 1.57 | Yes | No |
| NTN3 | 10.2 | Yes | No |
| NTNG2 | 2.06 | No | Yes |
| PAK3 | 2.48 | Yes | Yes |
| PLXNA3 | 3.11 | Yes | No |
| PLXNB3 | 9.23 | Yes | Yes |
| RAB3A | 1.9 | Yes | No |
| RAP1GAP | 2.21 | Yes | No |
| RNF165 | 3.59 | Yes | No |
| SEMA3F | 1.82 | Yes | No |
| SHANK1 | 3.02 | Yes | No |
| SHANK3 | 1.61 | Yes | No |
| SHC3 | 1.69 | Yes | Yes |
| SPTBN5 | 2.53 | Yes | No |
| TNFRSF8 | 3.17 | Yes | Yes |
| VEGFA | 3.46 | Yes | Yes |
| VLDLR | 10.1 | Yes | Yes |
*Fold change in expression in SUM159 cells exposed to 1% O2 compared to 20% O2.
†Hypoxic induction is also PADI4 dependent.
‡Gene expression is significantly correlated with expression of HIF signature in TCGA breast cancer database (P < 0.01, Pearson’s test).
Other GO biological processes that were enriched among HIF target genes in SUM159 cells comprised genes involved in “cytokine-mediated signaling pathway,” “cellular response to type I interferon,” and “type I interferon signaling pathway” (Fig. 6C and Table 2). Again, the vast majority (85%) of the 67 genes in these categories were PADI4 dependent, and expression of the majority (66%) of these genes was significantly correlated with expression of the HIF signature in human breast cancers.
Table 2. Hypoxia-induced and HIF-dependent genes involved in cytokine-mediated signaling (GO: 19221) and type I interferon signaling (GO: 60337 and GO: 71357).
| Gene name | FC* | PADI† | TCGA‡ |
| AIM2 | 1.59 | Yes | Yes |
| ANXA2 | 1.57 | Yes | Yes |
| BCL6 | 1.93 | Yes | No |
| CARD14 | 4.22 | Yes | Yes |
| CCL5 | 7.96 | No | Yes |
| CCL24 | 2.55 | Yes | Yes |
| CEACAM1 | 12.7 | No | No |
| CRLF1 | 2.65 | Yes | Yes |
| CRLF2 | 3.71 | Yes | Yes |
| CSF1R | 2.44 | No | Yes |
| DUOX2 | 141 | Yes | Yes |
| EBI3 | 6.68 | Yes | Yes |
| EGR1 | 2 | Yes | No |
| EPO | 4.28 | Yes | Yes |
| FLRT1 | 2.67 | Yes | No |
| FOS | 5.45 | Yes | No |
| HCK | 6.14 | Yes | Yes |
| ICAM1 | 1.72 | Yes | Yes |
| IFIT1 | 5.66 | Yes | No |
| IFIT2 | 11.5 | Yes | Yes |
| IFIT3 | 4.4 | No | Yes |
| IFITM1 | 2.03 | Yes | Yes |
| IFITM2 | 1.91 | Yes | No |
| IFNE | 2.85 | Yes | Yes |
| IFNL1 | 25.5 | No | Yes |
| IL2RG | 19.5 | Yes | Yes |
| IL16 | 3.35 | No | No |
| IL17RE | 2.92 | Yes | Yes |
| IRF9 | 2.39 | Yes | No |
| ISG15 | 1.71 | No | Yes |
| ISG20 | 4.33 | Yes | Yes |
| ITGAX | 5.14 | Yes | Yes |
| ITGB2 | 1.83 | Yes | Yes |
| LAMA5 | 1.61 | Yes | No |
| LBP | 3.38 | Yes | Yes |
| LRRC3 | 4.25 | Yes | Yes |
| MAP3K8 | 2.1 | Yes | Yes |
| MAPK3 | 1.82 | Yes | No |
| MUC1 | 10.3 | Yes | No |
| NCAM1 | 1.74 | No | Yes |
| NOD2 | 4.48 | Yes | Yes |
| NUMBL | 1.79 | Yes | Yes |
| OASL | 13.1 | Yes | Yes |
| PDCD4 | 1.97 | Yes | No |
| PDGFB | 4.87 | Yes | No |
| PODNL1 | 2.62 | Yes | Yes |
| PRTN3 | 30.8 | Yes | No |
| RHOU | 2.15 | Yes | No |
| RORA | 114 | Yes | No |
| RORC | 36 | Yes | No |
| RSAD2 | 27.2 | Yes | Yes |
| RTN4RL2 | 2.16 | Yes | No |
| SOCS1 | 1.5 | Yes | Yes |
| STAT4 | 2.52 | Yes | Yes |
| STAT6 | 1.52 | Yes | No |
| TGFB1 | 2.07 | Yes | Yes |
| TNFRSF8 | 3.17 | Yes | Yes |
| TNFRSF14 | 3 | Yes | No |
| TNFRSF25 | 2.91 | Yes | Yes |
| TNFSF11 | 7.61 | Yes | Yes |
| TNFSF12 | 1.54 | Yes | No |
| TNFSF13 | 2 | No | No |
| TNFSF14 | 2.02 | Yes | Yes |
| TRADD | 2.56 | Yes | Yes |
| VEGFA | 3.46 | Yes | Yes |
| VIM | 1.71 | Yes | Yes |
| XAF1 | 2.14 | No | Yes |
*Fold change in expression in SUM159 cells exposed to 1% O2 compared to 20% O2.
†Hypoxic induction is also PADI4 dependent.
‡Gene expression is significantly correlated with expression of HIF signature in TCGA breast cancer database (P < 0.01, Pearson’s test).
PADI4 promotes breast cancer and HCC tumor growth
To directly investigate the role of PADI4 in breast cancer, we injected 2 × 106 MDA-MB-231 subclone cells into the mammary fat pad of adult female severe combined immunodeficiency (Scid) mice. Compared to tumors derived from NTC cells, tumors derived from cells expressing either of two different shRNAs targeting PADI4 (shP4#4 or shP4#5) showed significantly decreased tumor growth (Fig. 7A), and the final tumor weights were significantly decreased compared to NTC tumors (Fig. 7B). Expression of ANGPTL4, VEGFA, and PGF mRNA was significantly decreased in PADI4-knockdown compared to NTC tumors (Fig. 7C). Immunohistochemistry using antibodies against CD31, which is expressed by vascular endothelial cells, and α-smooth muscle actin (SMA), which is expressed by vascular smooth muscle cells and pericytes, revealed that PADI4 knockdown markedly reduced intratumoral blood vessel density (Fig. 7D). Immunohistochemistry using antibodies against PADI4 and CA9, a canonical HIF target gene product (38), revealed high expression of both proteins in NTC tumors and markedly decreased expression in PADI4-knockdown tumors (Fig. 7D).
Fig. 7. PADI4 promotes breast cancer and HCC tumor growth and vascularization.
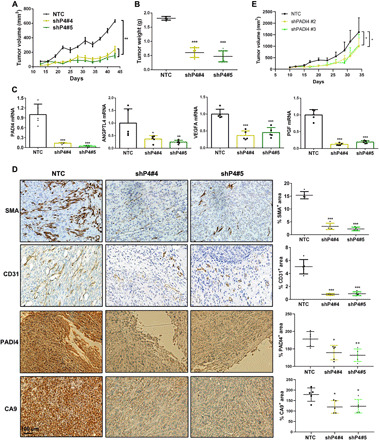
(A) MDA-MB-231 subclone cells were implanted in the mammary fat pad, and tumor volume was measured every 3 days (mean ± SEM; n = 5). **P < 0.01 versus NTC (two-way ANOVA with Sidak’s t test). (B) Tumors were harvested on day 44 and weighed (mean ± SD; n = 5). ***P < 0.001 versus NTC (two-way ANOVA). (C) Tumor RNA was isolated, and RT-qPCR analysis of PADI4, ANGPTL4, VEGFA, and PGF mRNA was performed. *P < 0.05, **P < 0.01, ***P < 0.01 versus NTC (two-way ANOVA). (D) Tumor sections were analyzed by immunohistochemistry (left) for α-smooth muscle actin (SMA), CD31, PADI4, and CA9. The stained area in 10 fields was quantified by ImageJ software, and the percentage of total area that was positive for staining is shown (right) (mean ± SD; n = 5 mice per group). *P < 0.05, ***P < 0.01 versus NTC (two-way ANOVA). (E) Hepa1-6 NTC and PADI4-knockdown subclones were subcutaneously injected into the flank of C57L/J mice. Tumor volume was measured every 2 to 3 days (mean ± SEM; n = 5). *P < 0.05 versus NTC (two-way ANOVA with Tukey’s t test).
To complement data on human cancer cells implanted in immunodeficient mice, we analyzed mouse Hepa1-6 HCC cells injected into syngeneic and immunocompetent C57L mice. Again, PADI4 knockdown significantly decreased tumor growth (Fig. 7E). Thus, PADI4 is required for optimal growth of MDA-MB-231 human breast cancer cells injected into the mammary fat pad of immunodeficient mice and Hepa1-6 mouse hepatoma cells injected subcutaneously into immunocompetent mice, which is consistent with its role in regulating HIF-dependent expression of angiogenic factors in both cell types.
PADI4 is associated with HIF-1α expression and vessel density in human breast cancers
We next used immunohistochemistry to analyze HIF-1α and PADI4 expression in primary human breast cancer biopsies. Both HIF-1α and PADI4 protein were localized to cell nuclei, and expression of these proteins in tumor tissues was much higher than in adjacent normal tissue (Fig. 8A). HIF-1α and PADI4 immunohistochemistry signals were quantified by image analysis and were significantly correlated by Pearson’s test (Fig. 8B). In addition, HIF-1α expression was significantly associated with PR status, HER2 status, and tumor size (table S1). PADI4 expression was significantly associated with histologic grade and tumor size (table S2). Immunohistochemistry was performed with anti-SMA and anti-CD31 antibodies and quantified by image analysis, which revealed that SMA and CD31 staining were significantly decreased in human breast cancers with low PADI4 expression (Fig. 8C). PADI4 expression was significantly correlated with both SMA and CD31 staining by Pearson’s test (Fig. 8D). Thus, both preclinical and clinical data demonstrate that PADI4 expression promotes breast cancer vascularization.
Fig. 8. PADI4 promotes breast cancer angiogenesis and tumor growth.
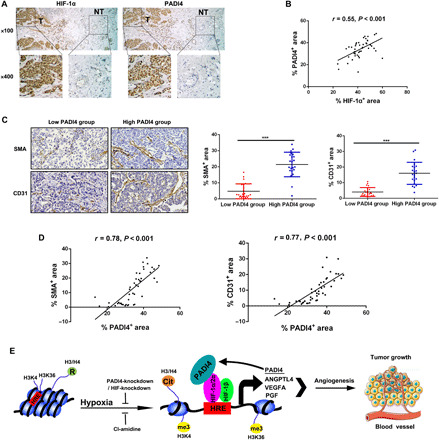
(A) Immunohistochemistry for HIF-1α and PADI4 was performed with consecutive sections of human breast cancer tissue, each of which included both tumor (T) and adjacent nontumor (NT) tissue. Magnification: ×100 for top panels and ×400 for bottom panels. (B) The stained area in five fields from each tumor section was quantified by ImageJ software and represented as the percentage of total area that was positive for HIF-1α or PADI4 staining for each case (n = 45). Data were analyzed using Pearson’s correlation test. (C) SMA and CD31 immunohistochemistry was performed with consecutive sections of human breast cancer tissue. The 45 cases were divided into low (n = 23) and high (n = 22) PADI4 groups based on the median percentage of total area that was positive for PADI4 (left, magnification: ×200). The stained area in five fields from each tumor section was quantified by ImageJ software and represented as the mean percentage of total area positive for SMA or CD31. Staining for SMA (middle) or CD31 (right) was compared in the low and high PADI4 groups (mean ± SD). ***P < 0.001 (Student’s t test). (D) Data were analyzed using Pearson’s correlation test to compare PADI4-positive staining percentage with SMA-positive (left) or CD31-positive (right) staining percentage in tumor sections (n = 45). (E) Summary of results: PADI4 recruitment drives HIF transcriptional activity and tumor vascularization. Hypoxia induces HIF-dependent PADI4 expression. PADI4 protein is recruited by HIFs to HREs and deiminates histone arginine residues to citrulline, leading to chromatin decondensation. PADI4 also stimulates HRE occupancy by HIFs and trimethylation of H3K3 and H3K36. The integrated effect of this chromatin remodeling is to activate transcription of hundreds of HIF target genes.
DISCUSSION
Rapid responses to hypoxia are critical to maintain cellular viability, and the HIF system is poised to respond: Cells constantly synthesize and degrade HIF-1α and HIF-2α so that when O2 levels decline, protein stabilization can rapidly increase levels of the proteins, leading to transcriptional activation. A key feature of the hypoxia regulatory network is that multiple HIF target genes encode coactivator proteins that are recruited by HIFs to HREs at other target genes to further stimulate transcriptional activation. In this study, we have identified a previously unidentified regulatory feature in which HIF-dependent PADI4 gene transcription and HIF-dependent recruitment of PADI4 to target genes leads to increased HIF occupancy of HREs, histone citrullination, additional histone modifications, and transcriptional activation (Fig. 8E). Loss of histone citrullination also resulted in loss of histone methylation marks (H3K4me3 and H3K36me3) and acetylation marks (H3K4ac and H4K5ac). It is not clear whether histone methylation or acetylation is dependent on previous histone citrullination or whether the loss of histone methylation or acetylation at HIF target genes in PADI4-knockdown cells is an indirect effect of impaired gene transcription. Thus, further studies are required to determine whether PADI4-mediated deimination of H3 arginine residues 2, 8, 17, and 26 is directly required for trimethylation of H3 lysine residues 4 and 36 or acetylation of lysine residue 4. HIF/PADI4-dependent methylation of H3K4 and H3K36 complements decreased activity of the O2-dependent histone demethylase KDM5A under hypoxic conditions (48). The deimination of histone lysine residues is an irreversible reaction, and it will be interesting to determine whether reoxygenation leads to replacement of citrulline-containing histones with arginine-containing histones and, if so, the mechanism by which this occurs.
Another consequence of PADI4 activity is that deimination prevents methylation of H3R17, H4R3, and H3R2 by the arginine methyltransferases CARM1 (30), PRMT1 (28), and PRMT6 (49), respectively. In addition to histones, PADI4 deiminates arginine residues in transcriptional regulatory proteins, including p300, which is a HIF coactivator, but the site of deimination in the GRIP1 domain (50) is not involved in p300 interaction with HIF-1α or HIF-2α (51). In contrast to its activating role in hypoxia-inducible transcription, PADI4 interacts with p53 and represses its transcriptional activity by blocking histone arginine methylation (52). The negative regulation of p53 by PADI4 is of interest, because p53 is a negative regulator of HIF transcriptional activity (53). Further studies are required to determine whether coactivators that are recruited to HREs by HIFs are regulated by PADI4-mediated deimination.
Last, loss of PADI4 impaired the binding of HIF-1 and HIF-2 to HREs in chromatin, a characteristic shared with other coactivators that are recruited by HIFs to target genes, such as JMJD2C/KDM4C (21) and PKM2 (25), which reflects the extensive physical interactions between HIFs, coactivators, cyclin-dependent kinases, mediators, and components of the basal transcriptional machinery (17–23, 44). Histone citrullination by PADI4 may also stabilize binding of HIFs to HREs by increasing access to DNA or through altered interactions with histones.
In addition to delineating the molecular mechanisms governing chromatin remodeling at HIF target genes, we investigated the effect of PADI4 knockdown in an orthotopic model of breast cancer. Tumors contain regions of hypoxia, which is far more severe than in any normal tissue, and thus, HIF activity is critical for cancer progression. Here, we focused on the expression of three HIF target genes (ANGPTL4, PGF, and VEGFA) encoding angiogenic factors that play important roles in tumor vascularization (54–56). We show that HIF-PADI4 interaction is critical for angiogenic factor expression and that PADI4 knockdown impairs their expression in breast tumors, thereby impairing vascularization. In human breast cancers, we found that PADI4 expression was significantly correlated with both HIF-1α expression and tumor vascularization, establishing the clinical relevance of our findings.
The high-level expression of PADI4 in breast and liver cancer is in notable contrast to its expression in normal human tissues, which is restricted to bone marrow and spleen (57). Further studies are required to identify the oncogenic switch that is responsible for PADI4 gene expression in breast and liver cancers. Irrespective of the mechanism by which this occurs, it establishes basal expression that can then be modulated by HIF-dependent transcriptional activation. Even more remarkably, these cancer cells are dependent on PADI4 for hypoxia-inducible gene expression. PADI4 inhibition blocks HIF transcriptional activity, and HIF inhibition blocks PADI4 catalytic activity, suggesting that drugs targeting HIF or PADI activity may have similar and beneficial effects in breast and liver cancer. Because both HIF-1α and PADI4 overexpression are associated with patient mortality in other cancers, further studies are warranted to determine whether this pathway plays an important role in other human cancers.
RNA-seq analysis revealed the remarkable number of genes whose expression is modulated in response to hypoxia and the requirement for both HIF and PADI4 activity to mediate these transcriptional responses. GO analysis unexpectedly revealed that a battery of genes induced by hypoxia in SUM159 cells encoded proteins involved in axonogenesis/axon guidance. This finding suggests that these gene products may allow breast cancer cells to use neurons as a source of survival signals and/or as guidance signals for migration and invasion. Alternatively, the expression of these genes may facilitate the establishment of metastatic foci after seeding of the brain. The overall survival of breast cancer patients after diagnosis of brain metastasis is only 9 months (58). Given the need for greater mechanistic insight of breast cancer metastasis to the brain, these provocative findings warrant further investigation.
Last, both HIF-1α and PADI4 expression are correlated with breast and liver cancer vascularization and patient mortality. Effective therapies for patients with TNBC or advanced HCC are not currently available. The current study suggests that pharmacologic inhibition of HIF and/or PADI4 activity may represent novel therapeutic strategies for these patients.
MATERIALS AND METHODS
Cell culture
MDA-MB-231, Hepa1-6, and Hep3B cells were maintained in high-glucose (4.5 mg/ml) Dulbecco’s modified Eagle’s medium (DMEM), and SUM-159 cells were maintained in DMEM/F12 (50:50). All culture media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were maintained at 37°C in a standard 5% CO2, 95% air tissue culture incubator (20% O2). For hypoxic exposure, cells were placed in a modular incubator chamber (Billups-Rothenberg) that was flushed with a 1% O2/5% CO2/94% N2 gas mixture. Cell line authentication and Mycoplasma testing were performed at the Johns Hopkins University Genetic Resources Core Facility by PCR analysis.
Lentivirus transduction
Expression vectors encoding shRNA targeting human HIF-1α or HIF-2α, as well as the NTC shRNA, were described previously (47). The other shRNA sequences are shown in table S3. LKO.1-puro lentiviral vectors encoding shRNA targeting human or mouse PADI4 mRNA were purchased from Sigma-Aldrich. All lentiviral shuttle vectors were transfected into 293T cells for packaging. Viral supernatant was collected after 48 hours and used for transduction of liver and breast cancer cell lines as described previously (47). Puromycin (2 μg/ml for Hepa1-6, 1 μg/ml for SUM159 and Hep3B, and 0.5 μg/ml for MDA-MB-231) was added to the medium of cells transduced with lentivirus for selection of pools of cells expressing the shRNA.
RT-qPCR assays
As previously described (59), RNA was extracted using TRIzol (Invitrogen) and complementary DNA (cDNA) synthesis was performed according to the manufacturer’s instructions (Applied Biosystems). qPCR was performed using SYBR Green qPCR Master Mix (Bio-Rad). The expression of each target mRNA relative to 18S ribosomal RNA was calculated on the basis of the threshold cycle (Ct) as 2−Δ(ΔCt), where ΔCt = Cttarget − Ct18S and Δ(ΔCt) = ΔCttreatment − ΔCtcontrol. Nucleotide sequences of PCR primers are shown in table S4.
Immunoblot and immunoprecipitation assays
WCLs were prepared in modified radioimmunoprecipitation assay buffer. For immunoblot assays, WCLs were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), blotted onto nitrocellulose membranes, and probed with primary antibodies (table S5). The membranes were incubated with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare), and the chemiluminescent signal was detected using ECL Plus (GE Healthcare). For immunoprecipitation, equal amounts of WCLs (500 μg) were incubated with antibody (2 μg) against HIF-1α (#10006421, Cayman Chemical) or V5 (NB600-381, Novus Biologicals) in the presence of protein G–Sepharose beads (Amersham Biosciences) at 4°C overnight, and the immunoprecipitates were subjected to SDS-PAGE and immunoblot assays.
ChIP assays
As previously described (59), SUM159 and Hep3B cells were incubated at 20 or 1% O2 for 16 hours, cross-linked in 3.7% formaldehyde for 15 min, quenched in 0.125 M glycine for 5 min, and lysed with SDS lysis buffer. Chromatin was sheared by sonication, and lysates were precleared with salmon sperm DNA/protein A agarose slurry (Millipore) for 1 hour and incubated with antibody against HIF-1α, HIF-1β, HIF-2α, PADI4, Cit-H3, H3K4me3, H3K36me3, H3K4ac, H3, Cit-H4, H4K5ac, or H4 (table S5) in the presence of protein A–agarose beads overnight. After serial washing of the agarose beads with low-salt, high-salt, and LiCl buffers, DNA was eluted in 1% SDS with 0.1 M NaHCO3, and cross-links were reversed by addition of 0.2 M NaCl. DNA was purified by phenol-chloroform extraction and ethanol precipitation and analyzed by qPCR using primers shown in table S4.
RNA-seq analysis
SUM159 NTC, HIF-DKD, and PADI4-KD subclones were seeded into six-well plates in three biological replicates and exposed to 20 or 1% O2 for 24 hours. Total RNA was isolated from the subclones using TRIzol (Invitrogen) and treated with deoxyribonuclease (Qiagen). Library preparation and sequencing using the NovaSeq 6000 platform (Illumina) were performed by Genetic Resources Core Facility High Throughput Sequencing Center at the Johns Hopkins University School of Medicine (https://grcf.jhmi.edu/). The FASTQ files were subjected to quality check and analyzed by Genialis Inc. (https://www.genialis.com/). Differential expression results with a false discovery rate of <0.05 and mRNA fold change of >1.5 were used as a cutoff for further downstream analysis.
Immunohistochemistry
MDA-MB-231 tumor xenografts were fixed in 4% paraformaldehyde and paraffin-embedded, and sections (4 μm thickness) were mounted on glass slides. The sections were deparaffinized and hydrated, and heat-induced antigen retrieval was performed using citrate EDTA buffer followed by incubation at 4°C overnight in primary antibody against SMA (#SC-32251, Santa Cruz Biotechnology), CD31 (#NB100-2284, Novus Biologicals), PADI4 (ab128086, Abcam), or CA9 (NB100-417, Novus Biologicals). The reaction was developed using a biotin-free detection system and visualized using 3,3′-diaminobenzidine. Sections were counterstained with Gill’s II hematoxylin.
Human breast cancer biopsy sections and linked clinical data (tables S1 and S2) were obtained from the Zhejiang Provincial People’s Hospital breast cancer cohort; written informed consent was obtained from all patients at the time of original biopsy sample collection. The present study was approved by the Ethics Committee of Zhejiang Provincial People’s Hospital. Formalin-fixed and paraffin-embedded 4-μm sections were mounted on glass slides. After incubation at 60°C for 1 hour, the slides were deparaffinized with xylene and rehydrated in decreasing concentrations of methanol (100, 95, 85, and 70%). For antigen retrieval, the slides were boiled in 10 mM sodium citrate buffer (pH 6.0) for 10 min. After inhibition of endogenous peroxidase activities by heat-induced recovery with methanol containing 0.3% H2O2 for 10 min, the sections were blocked with Tris-buffered saline with Tween 20 (Sigma-Aldrich) containing 5% normal goat serum for 30 min and incubated overnight at 4°C with primary antibody against HIF-1α (1:40 dilution; #10006421, Cayman Chemical), PADI4 (1:200 dilution; #ab128086, Abcam), SMA (1:1500; #14395-1-AP, ProteinTech), or CD31 (1:800 dilution; #11265-1-AP, ProteinTech) followed by incubation with a biotin-labeled secondary antibody (ZSGB-BIO) for 10 min and horseradish peroxidase–labeled streptavidin buffer (ZSGB-BIO) for 10 min. Color was developed with 3,3′-diaminobenzidine, and instant hematoxylin was used for counterstaining. The stained sections were examined by two pathologists using a multiheaded microscope.
Animal studies
Animal protocols were approved by the Johns Hopkins University Animal Care and Use Committee and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (60). MDA-MB-231 cells (2 × 106) were injected into the mammary fat pad of 6- to 8-week-old female Scid mice (Charles River Laboratories, strain 561). Hepa1-6 cells (1 × 107) were injected subcutaneously into the flank of 6- to 8-week-old C57L/J mice (The Jackson Laboratory). Calipers were used to measure tumor length (L) and width (W), and tumor volume (V) was calculated as V = L × W2 × 0.524. Tumors were harvested, bisected, and processed for immunohistochemistry and RNA analysis.
Statistical analysis
Data are expressed as mean ± SD. For all assays, differences between two groups were analyzed by Student’s t test, whereas differences between multiple groups were analyzed by two-way analysis of variance (ANOVA). Gene pathway enrichment analysis and GO biological process analysis of hypoxia-induced gene expression were generated using the Enrichr web server (http://amp.pharm.mssm.edu/Enrichr). The correlations between HIF-1α or PADI4 expression and clinicopathological characteristics were analyzed by chi-square test. Values of P < 0.05 were considered significant for all analyses.
Acknowledgments
We thank R. Geisler and S. Garcia of Novus Biologicals Inc. for providing antibodies against CA9, H3, H3K4me3, H3K36me3, H3K4ac, H4K5ac, H4, HIF-1α, HIF-1β, HIF-2α, and V5 epitope. We are grateful to W. Luo (University of Texas Southwestern Medical Center) for providing MDA-MB-231 HIF knockout subclones. Funding: This work was supported by grants from the American Cancer Society (122473-RP-13-090-001-COUN), the Armstrong Family Foundation, the Cindy Rosencrans Fund for Triple-Negative Breast Cancer, and the Emerson Collective Cancer Research Fund. G.L.S. is an American Cancer Society Research Professor and the C. Michael Armstrong Professor of Genetic Medicine at the Johns Hopkins University School of Medicine. Y.W. was supported by the China Scholarship Council (grant number 201806280187). Author contributions: G.L.S. and Q.L. designed research; Y.W., Y.L., K.T., Q.X., Y.Y., S.S., N.L., H.L., Y.Z., and R.W. performed research; Y.W., Y.L., Y.Y., C.C., and G.L.S. analyzed data; and Y.W. and G.L.S. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabe3771/DC1
REFERENCES AND NOTES
- 1.Vaupel P., Hockel M., Mayer A., Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 9, 1221–1236 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Semenza G. L., The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta 1863, 382–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiong X. X., Qiu X. Y., Hu D. X., Chen X. Q., Advances in hypoxia-mediated mechanisms in hepatocellular carcinoma. Mol. Pharmacol. 92, 246–255 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Yuen V. W., Wong C. C., Hypoxia-inducible factors and innate immunity in liver cancer. J. Clin. Invest. 130, 5052–5062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza G. L., Pharmacologic targeting of hypoxia-inducible factors. Annu. Rev. Pharmacol. Toxicol. 59, 379–403 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Bos R., van der Groep P., Greijer A. E., Shvarts A., Meijer S., Pinedo H. M., Semenza G. L., van Diest P. J., van der Wall E., Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 97, 1573–1581 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Dales J.-P., Garcia S., Meunier-Carpentier S., Andrac-Meyer L., Haddad O., Levaut M.-N., Allasia C., Bonnier P., Charpin C., Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 116, 734–739 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Giatromanolaki A., Koukourakis M. I., Simopoulos C., Polychronidis A., Gatter K. C., Harris A. L., Sivridis E., c-erbB-2 related aggressiveness in breast cancer is hypoxia-inducible factor 1α dependent. Clin. Cancer Res. 10, 7972–7977 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Schindl M., Schoppmann S. F., Samonigg H., Hausmaninger H., Kwasny W., Gnant M., Jakesz R., Kubista E., Birner P., Oberhuber G.; Austrian Breast and Colorectal Cancer Study Group , Overexpression of hypoxia-inducible factor 1α is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 8, 1831–1837 (2002). [PubMed] [Google Scholar]
- 10.Yamamoto Y., Ibusiki M., Okumura Y., Kawasoe T., Kai K., Iyama K., Iwase H., Hypoxia-inducible factor 1α is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res. Treat. 110, 465–475 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Xiang Z.-L., Zeng Z.-C., Fan J., Tang Z.-Y., He H., Zeng H.-Y., Chang J. Y., The expression of HIF-1α in primary hepatocellular carcinoma and its correlation with radiotherapy response and clinical outcome. Mol. Biol. Rep. 39, 2021–2029 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Xiang Z. L., Zeng Z. C., Fan J., Tang Z. Y., Zeng H. Y., Gao D. M., Gene expression profiling of fixed tissues identified hypoxia-inducible factor-1α, VEGF, and matrix metalloproteinase-2 as biomarkers of lymph node metastasis in hepatocellular carcinoma. Clin. Cancer Res. 17, 5463–5472 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Favaro E., Lord S., Harris A. L., Buffa F. M., Gene expression and hypoxia in breast cancer. Genome Med. 3, 55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semenza G. L., Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 36, 252–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor C. T., Colgan S. P., Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 17, 774–785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza G. L., Jiang B. H., Leung S. W., Passantino R., Concordet J. P., Maire P., Giallongo A., Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 271, 32529–32537 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Galbraith M. D., Allen M. A., Bensard C. L., Wang X., Schwinn M. K., Qin B., Long H. W., Daniels D. L., Hahn W. C., Dowell R. D., Espinosa J. M., HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrero P., Okamoto K., Coumilleau P., O’Brien S., Tanaka H., Poellinger L., Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1α. Mol. Cell. Biol. 20, 402–415 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen R., Xu M., Hogg R. T., Li J., Little B., Gerard R. D., Garcia J. A., The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J. Biol. Chem. 287, 30800–30811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lando D., Peet D. J., Whelan D. A., Gorman J. J., Whitelaw M. L., Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science 295, 858–861 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Luo W., Chang R., Zhong J., Pandey A., Semenza G. L., Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. U.S.A. 109, E3367–E3376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Perri J. I., Dengler V. L., Audetat K. A., Pandey A., Bonner E. A., Urh M., Mendez J., Daniels D. L., Wappner P., Galbraith M. D., Espinosa J. M., The TIP60 complex is a conserved coactivator of HIF1α. Cell Rep. 16, 37–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J. S., Kim Y., Bhin J., Shin H.-J., Nam H. J., Lee S. H., Yoon J.-B., Binda O., Gozani O., Hwang D., Baek S. H., Hypoxia-induced methylation of a Pontin chromatin remodeling factor. Proc. Natl. Acad. Sci. U.S.A. 108, 13510–13515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roeder R. G., 50+ years of eukaryotic transcription: An expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 26, 783–791 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo W., Hu H., Chang R., Zhong J., Knabel M., O’Meally R., Cole R. N., Pandey A., Semenza G. L., Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen D. J., Omaghi P., Yang J. C., Lowe N., Evans P. R., Ballario P., Neuhaus D., Filetici P., Travers A. A., The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J. 19, 6141–6149 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X., Bolt M., Guertin M. J., Chen W., Zhang S., Cherrington B. D., Slade D. J., Dreyton C. J., Subramanian V., Bicker K. L., Thompson P. R., Mancini M. A., Lis J. T., Coonrod S. A., Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor α target gene activation. Proc. Natl. Acad. Sci. U.S.A. 109, 13331–13336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Wysocka J., Sayegh J., Lee Y.-H., Perlin J. R., Leonelli L., Sonbuchner L. S., McDonald C. H., Cook R. G., Dou Y., Roeder R. G., Clarke S., Stallcup M. R., Allis C. D., Coonrod S. A., Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306, 279–283 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Christophorou M. A., Castelo-Branco G., Halley-Stott R. P., Oliveira C. S., Loos R., Radzisheuskaya A., Mowen K. A., Bertone P., Silva J. C., Zernicka-Goetz M., Nielsen M. L., Gurdon J. B., Kouzarides T., Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuthbert G. L., Daujat S., Snowden A. W., Erdjument-Bromage H., Hagiwara T., Yamada M., Schneider R., Gregory P. D., Tempst P. K., Bannister A. J., Kouzarides T., Histone deimination antagonizes arginine methylation. Cell 118, 545–553 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Hagiwara T., Hidaka Y., Yamada M., Deimination of histone H2A and H4 at arginine 3 in HL-60 granulocytes. Biochemistry 44, 5827–5834 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Kan R., Jin M., Subramanian V., Causey C. P., Thompson P. R., Coonrod S. A., Potential role for PADI-mediated histone citrullination in preimplantation development. BMC Dev. Biol. 12, 19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tessarz P., Kouzarides T., Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Chang X., Han J., Pang L., Zhao Y., Yang Y., Shen Z., Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 9, 40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McElwee J. L., Mohanan S., Griffith O. L., Breuer H. C., Anguish L. J., Cherrington B. D., Palmer A. M., Howe L. R., Subramanian V., Causey C. P., Thompson P. R., Gray J. W., Coonrad S. A., Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 12, 500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sase T., Arito M., Onodera H., Omoteyama K., Kurokawa M. S., Kagami Y., Ishigami A., Tanaka Y., Kato T., Hypoxia-induced production of peptidylarginine deiminases and citrullinated proteins in malignant glioma cells. Biochem. Biophys. Res. Commun. 482, 50–56 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Liu C., Tang J., Li C., Pu G., Yang D., Chang X., PADI4 stimulates esophageal squamous cell carcinoma tumor growth and up-regulates CA9 expression. Mol. Carcinog. 58, 66–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wykoff C. C., Beasley N. J., Watson P. H., Turner K. J., Pastorek J., Sibtain A., Wilson G. D., Turley H., Talks K. L., Maxwell P. H., Pugh C. W., Ratcliffe P. J., Harris A. L., Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 60, 7075–7083 (2000). [PubMed] [Google Scholar]
- 39.Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D., Semenza G. L., Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H., Wong C. C.-L., Wei H., Gilkes D. M., Korangath P., Chaturvedi P., Schito L., Chen J., Krishnamachary B., Winnard P. T. Jr., Raman V., Zhen L., Mitzner W. A., Sukumar S., Semenza G. L., HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Luo Y., Arita K., Bhatia M., Knuckley B., Lee Y. H., Stallcup M. R., Sato M., Thompson P. R., Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 45, 11727–11736 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis V. C., Gizinski A. M., Banda N. K., Causey C. P., Knuckley B., Cordova K. N., Luo Y., Levitt B., Glogowska M., Chandra P., Kulik L., Robinson W. H., Arend W. P., Thompson P. R., Holers V. M., N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 186, 4396–4404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaturvedi P., Gilkes D. M., Wong C. C., Kshitiz, Luo W., Zhang H., Wei H., Takano N., Schito L., Levchenko A. L., Semenza G. L., Hypoxia-inducible factor–dependent breast cancer–mesenchymal stem cell bidirectional signaling promotes metastasis. J. Clin. Invest. 123, 189–205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semenza G. L., A compendium of proteins that interact with HIF-1α. Exp. Cell Res. 356, 128–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buratowski S., Kim T., The role of cotranscriptional histone methylations. Cold Spring Harb. Symp. Quant. Biol. 75, 95–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruthenberg A. J., Allis C. D., Wysocka J., Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 25, 15–30 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Lu H., Samanta D., Xiang L., Zhang H., Hu H., Chen I., Bullen J. W., Semenza G. L., Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. U.S.A. 112, E4600–E4609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batie M., Frost J., Frost M., Wilson J. W., Schofield P., Rocha S., Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Kolodziej S., Kuvardina O. N., Oellerich T., Herglotz J., Backert I., Kohrs N., La Buscató E., Wittmann S. K., Salinas-Riester G., Bonig H., Karas M., Serve H., Proschak E., Lausen J., PADI4 acts as a coactivator of Tal1 by counteracting repressive histone arginine methylation. Nat. Commun. 5, 3995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y. H., Coonrod S. A., Kraus W. L., Jelinek M. A., Stallcup M. R., Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc. Natl. Acad. Sci. U.S.A. 102, 3611–3616 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dames S. A., Martinez-Yamout M., De Guzman R. N., Dyson H. J., Wright P. E., Structural basis for Hif-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. U.S.A. 99, 5271–5276 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao H., Li P., Venters B. J., Zheng S., Thompson P. R., Pugh B. F., Wang Y., Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J. Biol. Chem. 283, 20060–20068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravi R., Mookerjee B., Bhujwalla Z. M., Sutter C. H., Artemov D., Zeng Q., Dillehay L. E., Madan A., Semenza G. L., Bedi A., Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 14, 34–44 (2000). [PMC free article] [PubMed] [Google Scholar]
- 54.Carmeliet P., Moons L., Luttun A., Vincenti V., Compernolle V., De Mol M., Wu Y., Bono F., Devy L., Beck H., Scholz D., Acker T., DiPalma T., Dewerchin M., Noel A., Stalmans I., Barra A., Blacher S., VandenDriessche T., Ponten A., Eriksson U., Plate K. H., Foidart J. M., Schaper W., Charnock-Jones D. S., Hicklin D. J., Herbert J. M., Collen D., Persico M. G., Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 7, 575–583 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Ferrara N., The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res. Treat. 36, 127–137 (1995). [DOI] [PubMed] [Google Scholar]
- 56.Hu K., Babapoor-Farrokhran S., Rodrigues M., Deshpande M., Puchner B., Kashiwabuchi F., Hassan S. J., Asnaghi L., Handa J. T., Merbs S., Eberhart C. G., Semenza G. L., Montaner S., Sodhi A., Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 7, 7816–7828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fagerberg L., Hallström B. M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., Asplund A., Sjöstedt E., Lundberg E., Szigyarto C. A., Skogs M., Takanen J. O., Berling H., Tegel H., Mulder J., Nilsson P., Schwenk J. M., Lindskog C., Danielsson F., Mardinoglu A., Sivertsson A., von Feilitzen K., Forsberg M., Zwahlen M., Olsson I., Navani S., Huss M., Nielsen J., Ponten F., Uhlén M., Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao C., Wang F., Suki D., Strom E., Li J., Sawaya R., Hsu L., Raghavendra A., Tripathy D., Ibrahim N., Effects of systemic therapy and local therapy on outcomes of 873 breast cancer patients with metastatic breast cancer to brain: MD Anderson Cancer Center experience. Int. J. Cancer 148, 961–970 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Lan J., Lu H., Samanta D., Salman S., Lu Y., Semenza G. L., Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc. Natl. Acad. Sci. U.S.A. 115, E9640–E9648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.National Research Council, Guide for the Care and Use of Laboratory Animals (National Academies Press, ed. 8, 2011). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabe3771/DC1