

Abstract

Paramagnetic metal complexes gained a lot of attention due to their participation in a number of important chemical reactions. In most cases, these complexes are dominated by 17-e metalloradicals that are associatively activated with highly reactive paramagnetic 19-e species. Molybdenum paramagnetic complexes are among the most investigated ones. While some examples of persistent 17-e Mo-centered radicals have been reported, in contrast, 19-e Mo-centered radicals are illusive species and as such could rarely be detected. In this work, the photodissociation of the [Cp(CO)3Mo]2 dimer (1) in the presence of phosphines was revisited. As a result, the first persistent, formally 19-e Mo radical with significant electron density on the Mo center (22%), Cp(CO)3Mo•PPh2(o-C2B10H11) (5b), was generated and characterized by EPR spectroscopy and MS as well as studied by DFT calculations. The stabilization of 5b was likely achieved due to a unique electron-withdrawing effect of the o-carboranyl substituent at the phosphorus center.
Introduction
Organometallic chemistry is mostly dominated by diamagnetic complexes, which obey the 16- and 18-electron rule.1,2 This rule is very useful for predicting the stability and reactivity of diamagnetic metal complexes. Since the 1980s, however, paramagnetic metal complexes began to gain significant attention due to their important role in a variety of chemical reactions.3−7 For instance, paramagnetic metal intermediates of the second and third rows are involved in redox reactions, chain mechanisms, homolytic cleavage, and catalysis of C–C bond formation3,8 as well as in mediating redox reactions in energy-conversion processes and in biomimetic C–H bond activation and epoxidation of hydrocarbons.9 Their intermediacy in industrial processes such as the Wacker reaction is also well-known.10 As a result, the range of organometallic chemistry has expanded to include numerous paramagnetic 17-e complexes, which exist as both stable complexes and short-lived intermediates.3,4,8,11,12 The reactions of paramagnetic 17-e metal complexes are associatively activated with 19-e intermediates or transition states.3,4,11 However, unlike 17-e metal complexes, 19-e metal complexes in which the unpaired electron is primarily metal localized in a M–L antibonding orbital are rare and usually unstable and as such were proposed mostly as illusive intermediates.4,12,13 The 19-e Mo-centered radicals, to the best of our knowledge, have never been observed in chemical reactions, with the exception of femtosecond IR spectroscopy.14,15 Noteworthy, persistent 19-e Mo-centered radicals that are perhaps better described as 18-e complexes with reduced ligands (so-called “18 + δ” complexes) were synthesized previously; however, the spin density on the metal center in these complexes was negligible (<1%).16−19
One of the earliest reactions postulated to involve a 19-e intermediate was the photochemical disproportionation of the [Cp(CO)3Mo]2 dimer (1) in the presence of R3P into the Cp(CO)3Mo– (2) and Cp(CO)3(R3P)Mo+ (3) ion pair (Scheme 1).13−15 The accepted mechanism for this reaction, proposed by Tyler and co-workers,13 proceeds through photoexcitation of 1 leading to Mo–Mo bond cleavage and the formation of two 17-e Cp(CO)3Mo• radicals (4). In the presence of R3P the formation of a highly reducing, transient 19-e intermediate Cp(CO)3(R3P)Mo• (5) is proposed. Electron transfer from 5 to 4 leads to formation of Cp(CO)3(R3P)Mo+ (3) and Cp(CO)3Mo– (2) (Scheme 1). Importantly, it was also shown that this reaction is reversible, and in the dark, this salt over time is converted back to 1 and R3P, either via a single electron transfer (SET) path (Scheme 1a) or directly by substitution of R3P (Scheme 1b) with no clear indication as to which mechanism prevails in this transformation.13,20
Scheme 1. Mechanism of [Cp(CO)3Mo]2 Dimer (1) Dissociation in the Presence of R3P; Formation of Cp(CO)3(R3P)Mo+ (3) and Cp(CO)3Mo– (2).

Noteworthy, when 1 was irradiated in the presence of a bidentate diphosphine-based ligand (2,3-bis(diphenylphosphino)maleic anhydride)), a stable isolable 18 + δ complex was formed with most of the spin density located at the diphosphine ligand (i.e., δ was close to zero).16−18 On the other hand, when R3P in this reaction (Scheme 1) was replaced by an N-heterocyclic carbene (NHC), a persistent 17-e Cp(CO)2(NHC)Mo• radical was formed via substitution of one of the COs by the carbene.21 Noteworthy, neither 17-e 4 nor 19-e 5 was observed or characterized by electron paramagnetic resonance (EPR) spectroscopy.
We, therefore, decided to revisit this reaction (Scheme 1) and see whether radicals of type 5 could be stabilized in this process and studied by EPR spectroscopy. Herein, we report the generation and characterization of the first persistent formally 19-e Mo-based radical with a significant spin density of 22% on the Mo center, Cp(CO)3Mo•PPh2(o-C2B10H11) (5b).
Results and Discussion
We first studied the photochemical reaction of 1 in the presence of Ph3P. Thus, when a toluene solution22 containing 1 and Ph3P (1:10) in the EPR cavity was UV-irradiated (λ > 300 nm) at low temperature (200 K), a strongly low-field shifted singlet with a g-value of 2.082 was measured (Figure 1a), which immediately disappeared when irradiation was stopped. We assumed that this signal corresponded to the transient 17-e Cp(CO)3Mo• radical (4) (Scheme 2a). To support our suggestion, 4 was optimized by using DFT (density functional theory), and its EPR parameters were calculated.23 The calculated g-value of 4 (g = 2.069) is in good agreement with the experimentally observed g-value (Figure 1b). To the best of our knowledge, this is the first time that the “parent” 17-e Cp(CO)3Mo• radical (4) was experimentally observed by EPR spectroscopy.
Figure 1.

(a) EPR spectrum of 4. (b) DFT calculated Mulliken atomic spin densities and g-value of 4.23
Scheme 2. Photochemical Reaction of Dimer 1 in the Presence of Excess of Ph3P and Formation of an Unstable Cp(CO)3Mo• (4) and a Persistent 6, with Proposed Pathway Leading to 6.

After irradiation (λ > 300 nm) of the same toluene solution (1 and Ph3P (1:10)) at room temperature for 30 min, a high-intensity doublet was measured (14.2 G) with a g-value of 2.044 and a hyperfine coupling a(95,97Mo) = 12.4 G from magnetically active Mo isotopes (Figure 2a). This radical species was persistent with τ1/2 ≈ 180 min and thus allowed us to study its molecular composition using mass spectrometry (MS). Using atmospheric pressure chemical ionization (APCI) MS in positive mode, we were able to detect a mass that corresponds to a 17-e Mo-centered radical (647.9559 (M + H)+) (Figure 2b), Cp(CO)2Mo• ← P(Ph)2–Mo(CO)3Cp (6) (Scheme 2b).
Figure 2.

(a) EPR spectrum of 6 (blue) and its simulation (red). (b) MS of 6 (647.9559 (M + H)+) (red) and its simulation (blue). (c) DFT calculated Mulliken atomic spin densities and EPR parameters in 6.23
DFT calculation of 6 and its EPR parameters23 gave a g-value of 2.053 with hyperfine coupling constants (hfcc) of a(31P) = 30.54 G, a(95,97 Mo) = 20.96, and spin density located mostly on the Mo atom (85%) (Figure 2c). The computed EPR parameters are in good agreement with the experimental values considering the low spin density at the phosphorus center and rather complicated electronic structure of the Mo atom.24,25
We assumed that 6 was formed via unstable 19-e Mo-based intermediate 5a under irradiation (Scheme 2c). The unpaired electron in 5a can migrate to the σ* orbital at the phosphorus center under irradiation, giving the excited species 5a* (Scheme 2d).23,265a* resembles in its electronic structure phosphoranyl radicals (R4P•), which tend to decay via α- or β-scission reactions,27−29 and thus 5a* decays in a similar manner via α-scission of the Ph–P bond, giving 18-e Cp(CO)3MoPPh2 (7) (Scheme 2e).307 may then substitute one of the CO groups at 4 to give 6 (Scheme 2f). A similar type of photoinduced CO substitution was previously reported.13,21
To overcome the problem of instability of 5a, especially under irradiation (see Scheme 2d,e), we decided to replace Ph3P by Ph2P(o-C2B10H11) (8) (Scheme 3).31,32 We envisioned that this substitution will solve a few of the problems that we encountered when using Ph3P (Scheme 2). First, Ph2P(o-C2B10H11) (8) is a weaker donor due to the strong electron-withdrawing effect of the o-carboranyl group33−39 and thus will lead to a less electron-rich Mo center, which would make the desired 19-e complex less reducing and as a result more stable.40 Second, the o-carboranyl substituent at the phosphorus center could help overcome the instability of 5a under irradiation. In contrast to 5a, which under irradiation is excited to a phosphoranyl-type radical 5a*, which decays via α-scission reaction (see Scheme 2d,e), in 5b the photoinduced electron migration would most probably lead to the migration of the spin density into the o-carboranyl cage, an effect that was previously shown by our and other groups.41−45 This may prevent the decay of 5b radical by α-scission (Scheme 2e).
Scheme 3. Reaction of Dimer 1 with Ph2P(o-C2B10H11) (8) to Generate the Persistent Radical 5b; Decay of 5b by Cl Atom Abstraction Giving 9 (b) and Oxidation of 5b by [Ph3C][B(C6F5)4] Giving 3b (a).

The reaction between 1 and 8 (1:10) in toluene under UV irradiation (λ > 300 nm, 30 min) did not produce the desired radicals. However, when the solvent was changed to CH2Cl2,22 and the solution of 1 and 8 (1:2) was irradiated with visible light from a 34 W blue LED lamp (λ = 420–540 nm) for 1 h,46 the desired radical 5b was generated (Scheme 3) and was stable enough to study by EPR spectroscopy and MS methods (Figure 3).
Figure 3.

(a) EPR spectrum of 5b (blue) and its simulation (red). (b) MS of 5b (573.1174 (M–H)−) (red) and its simulation (blue). (c) DFT calculated Mulliken atomic spin densities and EPR parameters in 5b.23
The EPR spectrum of 5b (g = 1.980) is characterized by the hfcc with the 31P nucleus a(31P) = 21.3 G and magnetically active Mo isotopes a(95,97Mo) = 36.0 G (Figure 3a). The geometry of 5b was DFT optimized, and its EPR parameters were calculated.23 The calculated g-value (1.990) is in good agreement with the experimental g-value (1.980), with an hfcc of a(31P) = 10.8 G and a(95,97Mo) = 23.4 G. The spin density is distributed between Mo (22%) and carbon atoms of the CO and Cp substituents (15–18%, for each carbon) (Figure 3c).23 Noteworthy, the spin density on the phosphine ligand (8) is negligible (2.4%).23 The mass corresponding to radical 5b (573.1174 (M–H)−) was found in the MS of the reaction mixture by using APCI MS in negative mode (Figure 3b). Noteworthy in 5b, the lower spin density on the Mo center (22%), as well as the negatively shifted g-value compared to a free electron (Δg = −0.0223), clearly contrasts with the higher spin density and positively shifted Δg of the 17-e Mo-centered radicals 2 (80%, Δg = 0.0797) and 6 (85%, Δg = 0.0417) (Schemes 1 and 2).
To the best of our knowledge, this is the first time that a persistent formally 19-e Mo-based radical complex with significant electron density on Mo (22%) was generated and studied spectroscopically. In contrast, doing the reaction between 1 and Ph3P (1:2) under the same reaction conditions did not yield the 19-e Mo-based radical 5a, but radical 6 was observed by EPR spectroscopy (Scheme 2), meaning that the o-carboranyl substituent at the P center indeed plays a crucial role in stabilizing this type of radical.
Expectedly, in the dark, 5b was not stable over long periods of time (τ1/2 ≈ 48 h), and after a few days only the starting materials, 1 and 8, were detectable by NMR spectroscopy. Notably, when the same reaction mixture was irradiated again (λ = 420–540 nm), 5b was regenerated. Similar to the described reaction in Scheme 1, we assume that 5b is a persistent 19-e Mo radical intermediate of the dissociation reaction of 1 in the presence of 8.13,20 Interestingly, 5b extracted by pentane is more stable than in CH2Cl2 solution with τ1/2 ≈ 100 h.
Oxidation of 5b was achieved by its reaction with [Ph3C][B(C6F5)4], giving the corresponding cation [Cp(CO)3(Ph2(o-C2B10H11)P)Mo]+ (3b) (Scheme 3a), which was also independently synthesized, isolated, and fully characterized (X-ray molecular structure shown in Figure 4a).47 Noteworthy, 3b was also observed by 31P NMR (see Figure S20) in the photodissociation process as consequent reaction of radicals 5b and 4, similarly to the reaction shown in Scheme 1.
Figure 4.

POV-ray depiction of cation 3b (a) and of 9 (b); thermal ellipsoids at the 50% probability level. Hydrogens and the [B(C6F5)4]− anion are omitted for clarity.
Reaction of 5b with Ph3CCl in pentane produced Cp(Cl)(CO)2MoPPh2(o-C2B10H11) (9) (Scheme 3b), the product of Cl atom abstraction and decarbonylation (for the EPR spectrum of this reaction see Figure S22).459 was isolated by crystallization, and its molecular structure was determined by X-ray crystallography (Figure 4b).
The redox chemistry
of 3b was studied by both cyclic
voltammetry (CV) and chemical reduction experiments. The CV of 3b in CH2Cl2 (5.99 mM) using [nBu4N][B(C6F5)4] (0.1 M) as a
supporting electrolyte was collected and revealed irreversible reduction
events centered at  = −0.736 V and
= −0.736 V and 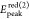 = −1.377 V with a corresponding anodic
event at
= −1.377 V with a corresponding anodic
event at 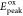 = −0.341 V vs the Ag/Ag+ redox
couple at a scan rate of 100 mV/s (Figure 5).45 The large
peak-to-peak separation (395 mV) between
= −0.341 V vs the Ag/Ag+ redox
couple at a scan rate of 100 mV/s (Figure 5).45 The large
peak-to-peak separation (395 mV) between  and
and 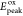 suggests significant
structural reorganization
upon reduction of 3b.
suggests significant
structural reorganization
upon reduction of 3b.
Figure 5.
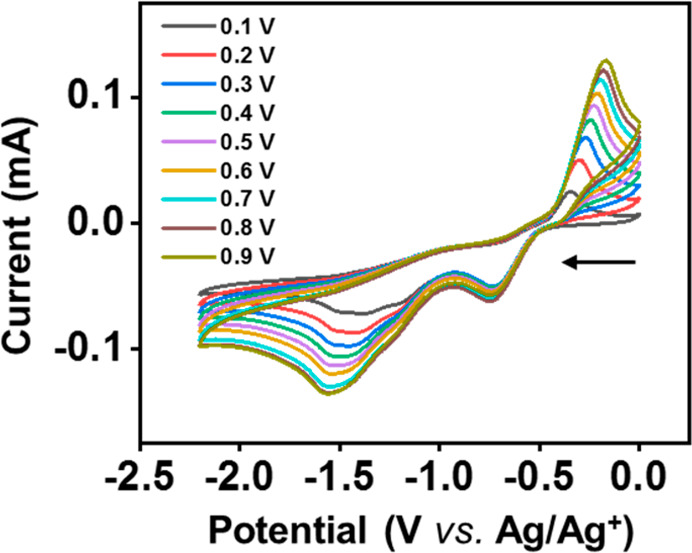
CV of 3b (5.99 mM) in dry 0.1 M [nBu4N][B(C6F5)4]/CH2Cl2 solution obtained at various scan rates with glassy carbon electrodes, Pt wire, and Ag/Ag+ as the working, counter, and reference electrodes, respectively.
On the basis of the CV results, we attempted to isolate the product
of the first reduction event by using FeCp*2 as a reductant
(−1.13 V vs Ag/Ag+ in CH2Cl2). Thus, when 3b was reacted with 1 equiv of FeCp*2 in CH2Cl2, 0.5 equiv of 3b was consumed and 0.5 equiv of free phosphine 8 and
the anion [Cp(CO)3Mo]− (2) were produced. Over time, 3b was consumed totally,
leading to 1 and free 8, likely the result
of the reaction of the remaining 0.5 equiv of 3b with
intermediate 2 (Scheme 4).45 Noteworthy, we did
not observe the formation of 5b in this process by EPR
spectroscopy, suggesting that a rapid 2e reduction process dominates
this transformation. This also suggests that the  peak in the CV is a 2e event. Importantly,
similar 2e reduction processes were reported previously.4,48,49 Because no paramagnetic species
were observed at all stages of this experiment (Scheme 4), we assume that the reaction of 3b and 2 most likely proceeds via a closed-shell pathway
and not through radicals 5b and 4 (see Scheme 1b).
peak in the CV is a 2e event. Importantly,
similar 2e reduction processes were reported previously.4,48,49 Because no paramagnetic species
were observed at all stages of this experiment (Scheme 4), we assume that the reaction of 3b and 2 most likely proceeds via a closed-shell pathway
and not through radicals 5b and 4 (see Scheme 1b).
Scheme 4. Reduction Reaction of Cation 3b by FeCp*2.

Conclusion
To conclude, in this work the photodissociation of 1 in the presence of Ph3P and 8 was performed, and thorough EPR studies were done. The photochemical reaction of 1 with Ph3P in toluene led to the formation of the persistent 17-e Mo-centered radical complex 6 via a transient “parent” 17-e complex Cp(CO)3Mo• (4), which was detected by EPR for the first time. 6 is presumably formed under irradiation which induces α-scission reaction of a P–Ph bond, followed by adduct formation with 4. To overcome this problem, 8 was used instead of Ph3P, which in reaction with 1 in CH2Cl2 under irradiation at λ = 420–540 nm gave the persistent formally 19-e Mo-based radical 5b. Accessing what previously had only been a hypothesized intermediate in Mo chemistry allowed us to carry out some preliminary reactivity studies. Oxidation of 5b by [Ph3C][B(C6F5)4] gave the corresponding cation 3b. The reaction of 5b with alkyl chlorides gave 9 via Cl atom abstraction and decarbonylation. The electrochemical reduction of 3b proceeds via two irreversible reduction events. To study this reduction process, 3b was reacted with FeCp*2 which via the 2e reduction process gave intermediate anion 2, which further led to dimer 1 and free 8; no paramagnetic species were observed in this process. We continue to study the chemistry of 5b and still search for its isolable analogues.
Experimental Section
General Considerations
All preparations were performed under an anhydrous N2 atmosphere by using standard Schlenk and glovebox techniques (Vac.-Atmospheres Nexus II equipped with a −35 °C freezer). Toluene, dichloromethane, and hexane were dried by using a Vac. Atm. Solvent purification system. o-Difluorobenzene and CDCl3 were dried over CaH2 for several days prior to distillation. All solvents were degassed by freeze–pump–thaw and stored on activated 4 Å molecular sieves prior to use. All glassware was oven-dried and cooled under vacuum before use. Commercial reagents were purchased from Sigma-Aldrich, Strem, or Apollo Scientific and used without further purification unless indicated otherwise.
Spectroscopic Analyses
NMR spectra were recorded at room temperature by using a Bruker AvanceIII-400 MHz spectrometer and referenced to residual solvent, or externally (11B: BF3·Et2O; 19F: CFCl3; 31P: 85% H3PO4) in some of the cases the tubes were equipped with DMSO-d6 capillary as external standard. Data for 1H NMR are reported as follows: chemical shift (δ ppm), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sep = septet, m = multiplet), coupling constant (Hz). The EPR spectra were recorded on a Bruker EMX-10/12 X-band (ν = 9.3 GHz) digital EPR spectrometer equipped with a Bruker N2 temperature controller. The spectra were recorded at a microwave power of 100–200 mW and a 100 kHz magnetic field modulation of 0.1–3.0 G amplitude (unless otherwise specified). The digital field resolution was 2048 points per spectrum. This allowed all hyperfine splittings to be measured directly with accuracy better than 0.1 G. Spectra processing and simulation were performed with Bruker WIN-EPR and SimFonia software. When the reactions were performed under UV irradiation, a high-pressure mercury lamp (1000 W) (ARC lamp power supply model 69920) was used, with the output being focused onto the sample with a quartz lens. When the reactions were performed under visible light irradiation (λ = 420–540 nm), a blue LED lamp (34 W) (Kessil, Model No. H150-BLUE) was used.
Electrochemical Measurements
The cyclic voltammetry (CV) measurements were performed by using a CHI760E electrochemical workstation. A 3 mm glassy carbon was used as the working electrode, Ag wire was used as the reference electrode, and a Pt wire was used as the counter electrode. [nBu4N][B(C6F5)4] in CH2Cl2 (0.1 M) was used as a supporting electrolyte. All electrochemical measurements were performed under an inert atmosphere in a glovebox. All electrodes were rinsed with the electrolyte solution prior to use. For all CVs measurements, the first scan cycle was discarded.
X-ray Crystallography
Data were collected on a Bruker KAPPA APEX II diffractometer equipped with an APEX II CCD detector using a TRIUMPH monochromator with a Mo Kα X-ray source (α = 0.71073 Å). The crystals were mounted on a cryoloop with Paratone oil, and all data were collected at 100(2) K. Crystal structures were solved by direct methods and refined by full matrix least-squares. All hydrogen atom positions were idealized and rode on the atom of attachment. Structure solution, refinement, graphics, and creation of publication materials were performed by using a SHELXT-2014 and a SHELXL-2014.
Synthesis of Cp(CO)3Mo•PPh2(o-C2B10H11) (5b)
Inside the glovebox a J Young NMR tube was charged with [Cp(CO)3Mo]2 (1) (0.05 g, 0.10 mmol) and Ph2P(o-C2B10H11) (8) (0.07 g, 0.2 mmol), and 1 mL of CH2Cl2 was added. This solution was then placed under irradiation at λ = 420–540 nm in a water bath, and the progress of the reaction was monitored by EPR and NMR spectroscopy. After 1 h, 5b was measured by EPR spectroscopy, and [Cp(CO)3(Ph2(o-C2B10H11)P)Mo]+ (3b) and free 8 were measured by 31P NMR. CH2Cl2 was then removed under vacuum, and 5b was extracted by pentane. Yield: 4%. The EPR spectrum of 5b was recorded in pentane (Figure 3a). HRMS (APCI): m/z calcd for C22H25B10P1O3Mo1: 573.1596 (M–H)−; found: 573.1174 (Figure 3b).
Synthesis of [Cp(CO)3(Ph2(o-C2B10H11)P)Mo][B(C6F5)4] (3b)
Oxidation of 5b by [Ph3C][B(C6F5)4]
Inside the glovebox a J Young NMR tube was charged with 5b in CH2Cl2 solution, and a pinch of [Ph3C][B(C6F5)4] was added. The EPR spectrum was recorded after 10 min, showing complete disappearance of 5b and generation of Ph3C•. 31P NMR was recorded after 30 min, showing the formation of 3b with a typical chemical shift at δ 73.21 ppm.
Independent Synthesis
3b was synthesized from CpMo(CO)3H,50 which was prepared by the following procedure: Mo(CO)6 (1.00 g, 3.79 mmol) was dissolved in 30 mL of CH3CN, and the mixture was refluxed for 12 h. All volatiles were then evaporated under high vacuum, giving the yellow solid Mo(CO)3(CH3CN)3. Mo(CO)3(CH3CN)3 was dissolved in THF (30 mL), and freshly distilled cyclopentadiene (5 mL) was added to this solution and heated for 1 h at 50 °C. After that time, all volatiles were removed, and the remaining solid was sublimed at 60 °C under a high vacuum, giving a yellow crystalline product. [Cp(CO)3MoH]2 dimer is also formed in this reaction (ca. 10%), as reported in the literature.50 The estimated yield for this reaction is ca. 60%. 1H NMR (400 MHz; CDCl3): δ −5.55 (1H, s, Mo–H), 5.42 (5H, s, C5H5). 13C NMR (100 MHz; CDCl3): δ 90.05 (C5H5), 191.12 and 226.87 (CO).
A freshly prepared CpMo(CO)3H (0.25 g, 1 mmol) dissolved in 10 mL of CH2Cl2 was treated with [Ph3C][B(C6F5)4] (0.92 g, 1.00 mmol) at −30 °C. The reaction mixture was allowed to warm to room temperature and stirred for another hour, forming a dark violet solution. To this dark violet solution, 8 (0.33 g, 1.00 mmol) dissolved in 5 mL of CH2Cl2 was added dropwise. The solution was allowed to stir for another hour, turning from violet to red. All the volatiles were evaporated under vacuum, and the residue was washed with (3 × 10) mL of toluene, affording a red solid upon drying. The target compound was crystallized from a CH2Cl2/benzene (1:10) mixture in 70% yield. 1H NMR (400 MHz; o-difluorobenzene, DMSO-d6 capillary): δ 0.99–2.71 (10H, br, B–H), 3.31 (1H, s, C–H), 4.78 (5H, s, C5H5), 6.88–7.08 (10H, m). 13C NMR (100 MHz; CH2Cl2, DMSO-d6 capillary): δ 63.19 (cage C–H), 69.35 (d, JP,C = 18.4 Hz, cage C–P), 95.36 (C5H5), 129.53 (d, JP,C = 10.9 Hz, Ph), 133.84 (b, Ph), 134.37 (b, C6F5), 136.30 (t, JF,C = 13.5 Hz, C6F5), 136.81(b, C6F5), 138.74 (t, JF,C = 13.5 Hz, C6F5), 146.25 (b, Ph), 148.64 (b, Ph), 222.76 and 224.04 (CO). 31P NMR (162 MHz; CH2Cl2, DMSO-d6 capillary): δ 73.21 (s). 19F NMR (376.5 MHz, CH2Cl2, DMSO-d6 capillary): δ −133.96 (b, 8F), −164.49 (t, 4F, J = 20.1 Hz), −168.36 (b, 8F). 11B NMR (128 MHz; CH2Cl2, DMSO-d6 capillary): δ −0.17, −1.50, −2.62, −7.91, −12.68, −17.37. HRMS (ESI+): m/z calcd for C22H26B10P1O3Mo1: 574.1713 (M+); found: 574.1708.
Synthesis of Ph2P(o-C2B10H11) (8)31,32
o-Carborane (1.00 g, 6.93 mmol) dissolved in 50 mL of dimethoxyethane (DME) was reacted with n-BuLi in hexane (2.91 mL, 7.28 mmol) at −15 °C and stirred at this temperature for 1 h. After that time the reaction mixture was allowed to warm to room temperature and stirred for another hour. A 10 mL dimethoxyethane solution of chlorodiphenylphosphine (1.28 mL, 6.93 mmol) was added to the stirring solution at −15 °C. The solution was allowed to warm to room temperature and stirred for 1 h followed by 1 h reflux. All the volatiles were evaporated, and the residue was extracted with Et2O which afforded a white solid upon drying. The target compound was purified by column chromatography on silica gel (60–200 mesh) eluted with CH2Cl2–hexane (1:5). Yield: 80%. 1H NMR (400 MHz; CDCl3), δ 1.75–2.86 (10H, br, B–H), 3.53 (1H, s, C–H), 7.49–7.54 (6H, m), 7.81 (4H, m). 13C NMR (100 MHz; CDCl3): δ 63.6 (d, JP,C = 15.4 Hz, cage C–H), 72.78 (d, JP,C = 75.85 Hz, cage C–P), 128.85 (d, JP,C = 9.6 Hz, Ph), 131.23 (s, Ph), 131.98 (d, JP,C = 15.92 Hz, Ph), 134.99 (d, JP,C = 26.54 Hz, Ph). 31P NMR (162 MHz; CDCl3): δ 25.02 (s). 11B NMR (128 MHz; CDCl3), δ −1.26, −2.38, −6.92, −8.10, −9.83, −11.69, −12.96, −14.15, −15.39.
Synthesis of Cp(Cl)(CO)2MoPPh2(o-C2B10H11) (9)
The pentane solution of 5b was reacted with an excess of Ph3CCl. The EPR spectrum was recorded right after, showing almost complete disappearance of 5b and formation of Ph3C• (see Figure S22). Overnight red crystals of 9 were formed from this solution in 92% yield. Noteworthy, 9 is not stable in CHCl3 or C6H6 solutions for a long period of time. 1H NMR (400 MHz; CDCl3): δ 1.65–3.35 (10H, br, B–H), 4.60 (1H, s, C–H), 5.23 (5H, s, C5H5), 7.45–7.49 (5H, m), 7.57–7.58 (1H, m), 7.69–7.73 (2H, m), 8.06 (2H, t, J = 9.12 Hz). 13C NMR (100 MHz; CDCl3): δ 66.46 (d, JP,C = 8.6 Hz, cage C–H), 95.56 (C5H5), 127.69 (d, JP,C = 9.6 Hz, Ph), 127.75 (d, JP,C = 9.6 Hz, Ph), 130.92, 132.47, 133.06 (d, JP,C = 10.2 Hz, Ph), 136.75 (d, JP,C = 11.5 Hz, Ph). 31P NMR (162 MHz; CDCl3): δ 70.69 (s). 11B NMR (128 MHz; CDCl3): δ 0.74, −0.21, −1.42, −2.91, −4.2, −7.29, −8.42, −11.86, −12.93. HRMS (ESI+): m/z calcd for C21H26B10P1O2Mo1: 547.1727 (M–Cl)+; found: 547.1728.
Acknowledgments
This work was supported by the US–Israel Binational Science Foundation, Grant 2018221, and Israeli Science Foundation, Grant 237/18. We also thank Arunavo Chakraborty for the help with electrochemistry data interpretation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03568.
NMR, EPR, CV, experimental and computational details (PDF)
Accession Codes
CCDC 2057570–2057572 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Tolman C. A. The 16 and 18 Electron Rule in Organometallic Chemistry and Homogeneous Catalysis. Chem. Soc. Rev. 1972, 1, 337–353. 10.1039/cs9720100337. [DOI] [Google Scholar]
- Elschenbroich C.; Salzer A.. Organometallics: A Concise Introduction, 1st ed.; VCH: Weinheim, 1989. [Google Scholar]
- Kochi J. K.Preface. In Organometallic Mechanisms and Catalysis; Kochi J. K., Ed.; Academic Press: 1978; pp xiii–xiv. [Google Scholar]
- Astruc D. Nineteen-Electron Complexes and Their Role in Organometallic Mechanisms. Chem. Rev. 1988, 88, 1189–1216. 10.1021/cr00089a010. [DOI] [Google Scholar]
- Song J. S.; Bullock R. M.; Creutz C. Intrinsic Barriers to Atom Transfer (Abstraction) Processes; Self-Exchange Rates for Cp(CO)3M• Radical/Cp(CO)3M-X Halogen Couples. J. Am. Chem. Soc. 1991, 113, 9862–9864. 10.1021/ja00026a029. [DOI] [Google Scholar]
- Schild D. J.; Drover M. W.; Oyala P. H.; Peters J. C. Generating Potent C–H PCET Donors: Ligand-Induced Fe-to-Ring Proton Migration from a Cp*FeIII–H Complex Demonstrates a Promising Strategy. J. Am. Chem. Soc. 2020, 142, 18963–18970. 10.1021/jacs.0c09363. [DOI] [PubMed] [Google Scholar]
- Rottschäfer D.; Ghadwal R.; Danés S.; Sharma M. K.; Neumann B.; Stammler H.-G.; Andrada D. M.; van Gastel M.; Hinz A. Metalloradical Cations and Dications Based on Divinyldiphosphene and Divinyldiarsene Ligands. Chem. - Eur. J. 2021, 27, 5803–5809. 10.1002/chem.202100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappert M. F.; Lednor P. W.. Free Radicals in Organometallic Chemistry. In Advances in Organometallic Chemistry; Stone F. G. A., West R., Eds.; Academic Press: 1976; Vol. 14, pp 345–399. [Google Scholar]
- Sheldon R. A.; Kochi J. K.. Preface. In Metal-catalyzed Oxidations of Organic Compounds; Sheldon R. A., Kochi J. K., Eds.; Academic Press: 1981; pp xiii–xv. [Google Scholar]
- Smidt J.; Hafner W.; Jira R.; Sedlmeier J.; Sieber R.; Rüttinger R.; Kojer H. Katalytische Umsetzungen von Olefinen an Platinmetall-Verbindungen Das Consortium-Verfahren zur Herstellung von Acetaldehyd. Angew. Chem. 1959, 71, 176. 10.1002/ange.19590710503. [DOI] [Google Scholar]
- Stiegman A. E.; Tyler D. R. Reactivity of Seventeen- and Nineteen-Valence Electron Complexes in Organometallic Chemistry. Comments Inorg. Chem. 1986, 5, 215–245. 10.1080/02603598608079840. [DOI] [Google Scholar]
- Baird M. C. Seventeen-Electron Metal-Centered Radicals. Chem. Rev. 1988, 88, 1217–1227. 10.1021/cr00089a011. [DOI] [Google Scholar]
- Stiegman A. E.; Stieglitz M.; Tyler D. R. Mechanism of the Low-Energy Photochemical Disproportionation Reactions of [(η5-C5H5)2Mo2(CO)6]. J. Am. Chem. Soc. 1983, 105, 6032–6037. 10.1021/ja00357a012. [DOI] [Google Scholar]
- Kling M. F.; Cahoon J. F.; Glascoe E. A.; Shanoski J. E.; Harris C. B. The Role of Odd-Electron Intermediates and In-Cage Electron Transfer in Ultrafast Photochemical Disproportionation Reactions in Lewis Bases. J. Am. Chem. Soc. 2004, 126, 11414–11415. 10.1021/ja046223x. [DOI] [PubMed] [Google Scholar]
- Cahoon J. F.; Kling M. F.; Schmatz S.; Harris C. B. 19-Electron Intermediates and Cage-Effects in the Photochemical Disproportionation of [CpW(CO)3]2 with Lewis Bases. J. Am. Chem. Soc. 2005, 127, 12555–12565. 10.1021/ja052221g. [DOI] [PubMed] [Google Scholar]
- Mao F.; Philbin C. E.; Weakley T. J. R.; Tyler D. R. Generation of the 19-Electron (18 + δ) Adducts CpMo(CO)3(L2-P) and CpMo(CO)2(L2-P,P’) (Cp = η5-CH3C5H4, η5-C5Ph4H, η5-C5Ph5; L2 = 2,3-bis(diphenylphosphino)maleic anhydride). Crystal structure of the (η5-C5Ph4H)Mo(CO)2L2 Complex. Organometallics 1990, 9, 1510–1516. 10.1021/om00119a024. [DOI] [Google Scholar]
- Meyer R.; Schut D. M.; Keana K. J.; Tyler D. R. Generation and Spectroscopic Characterization of New 18+δ Electron Complexes. Relationship Between the Stability of 18+δ Electron Organometallic Complexes and Their Ligand Reduction Potentials. Inorg. Chim. Acta 1995, 240, 405–412. 10.1016/0020-1693(95)04561-9. [DOI] [Google Scholar]
- Schut D. M.; Keana K. J.; Tyler D. R.; Rieger P. H. Measurement and Manipulation of the Unpaired Electron Density in 18+.delta. Complexes. Correlation of the Charge Density with Reactivity. J. Am. Chem. Soc. 1995, 117, 8939–8946. 10.1021/ja00140a007. [DOI] [Google Scholar]
- Lomont J. P.; Nguyen S. C.; Harris C. B. Direct Observation of a Bent Carbonyl Ligand in a 19-Electron Transition Metal Complex. J. Phys. Chem. A 2013, 117, 2317–2324. 10.1021/jp311732t. [DOI] [PubMed] [Google Scholar]
- Philbin C. E.; Goldman A. S.; Tyler D. R. Back-Reactions in the Photochemical Disproportionation of Cp2Mo2(CO)6 (Cp = C5H4CH3) and the Wavelength-Dependent Photochemistry of the Cp2Mo2(CO)6 Complex With PPh3. Inorg. Chem. 1986, 25, 4434–4436. 10.1021/ic00244a030. [DOI] [Google Scholar]
- Tumanskii B.; Sheberla D.; Molev G.; Apeloig Y. Dual Character of Arduengo Carbene–Radical Adducts: Addition versus Coordination Product. Angew. Chem., Int. Ed. 2007, 46, 7408–7411. 10.1002/anie.200702297. [DOI] [PubMed] [Google Scholar]
- Although CH2Cl2 was reported to work better for these reactions, it is an undesirable solvent for EPR experiments due to its polarity, which leads to a decrease in the Q-factor of the resonator and a decrease in the sensitivity of the spectrometer. In addition, under UV irradiation CH2Cl2 is not innocent. Therefore, toluene was initially chosen for this reaction.
- All calculated structures were optimized by using Gaussian09. The geometries 4, 5b, and 6 were optimized at the uwB97XD/def2-SVP level of theory; Mo was calculated with the Stuttgart/Dresden core electron pseudopotential (SDD) and the def2-SVP basis set. The g factors, the hfcc, and spin densities were calculated by using the ORCA 4.0 software; Neese, F. Max Planck Institute for Bioinorganic Chemistry, Mulheim an Ruhr, Germany, 2018. See the Supporting Information for full computational details.
- Braden D. A.; Tyler D. R. Density Functional Calculations of 19-Electron Organometallic Molecules. A Comparison of Calculated and Observed Anisotropic Hyperfine Coupling Constants for the CpCo(CO)2- Anion. Implications for Determining Orbital Spin Populations from EPR Data. J. Am. Chem. Soc. 1998, 120, 942–947. 10.1021/ja971800l. [DOI] [Google Scholar]
- Hillenbrand J.; van Gastel M.; Bill E.; Neese F.; Fürstner A. Isolation of a Homoleptic Non-oxo Mo(V) Alkoxide Complex: Synthesis, Structure, and Electronic Properties of Penta-tert-Butoxymolybdenum. J. Am. Chem. Soc. 2020, 142 (38), 16392–16402. 10.1021/jacs.0c07073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for TD-DFT calculation and analysis of 5a.
- Marque S.; Tordo P.. Reactivity of Phosphorus Centered Radicals In New Aspects in Phosphorus Chemistry V. Topics in Current Chemistry; Majoral J. P., Ed.; Springer: Berlin, 2005; Vol. 250. [Google Scholar]
- Leca D.; Fensterbank L.; Lacôte E.; Malacria M. Recent Advances in the Use of Phosphorus-Centered Radicals in Organic Chemistry. Chem. Soc. Rev. 2005, 34, 858–865. 10.1039/b500511f. [DOI] [PubMed] [Google Scholar]
- Livshits-Kritsman Y.; Tumanskii B.; Ménard G.; Dobrovetsky R. Isolable Cyclic (Alkyl)(Amino)Carbene–Phosphonyl Radical Adducts. Chem. Commun. 2020, 56, 1341–1344. 10.1039/C9CC09244G. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for C60 radical trap experiments.
- Zakharkin L. I.; Zhubekova M. N.; Kazantsev A. V. Synthesis and Reactions of Substituted o-Carboranyldiphenylphosphines. Zh. Obshch. Khim. 1972, 42, 1024–1028. [Google Scholar]
- Godovikov N. N.; Balema V. P.; Rys E. G. Carborane-Containing Organophosphorus Compounds. Synthesis and Properties. Russ. Chem. Rev. 1997, 66 (12), 1017–1032. 10.1070/RC1997v066n12ABEH000311. [DOI] [Google Scholar]
- Hao E.; Fabre B.; Fronczek F. R.; Vicente M. G. H. Syntheses and Electropolymerization of Carboranyl-Functionalized Pyrroles and Thiophenes. Chem. Mater. 2007, 19, 6195–6205. 10.1021/cm701935n. [DOI] [Google Scholar]
- Farha O. K.; Spokoyny A. M.; Mulfort K. L.; Hawthorne M. F.; Mirkin C. A.; Hupp J. T. Synthesis and Hydrogen Sorption Properties of Carborane Based Metal–Organic Framework Materials. J. Am. Chem. Soc. 2007, 129, 12680–12681. 10.1021/ja076167a. [DOI] [PubMed] [Google Scholar]
- Spokoyny A. M. New Ligand Platforms Featuring Boron-Rich Clusters as Organomimetic Substituents. Pure Appl. Chem. 2013, 85, 903–919. 10.1351/PAC-CON-13-01-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu A. R.; Teixidor F.; Viñas C. Metal Promoted Charge and Hapticities of Phosphines: The Uniqueness of Carboranylphosphines. Coord. Chem. Rev. 2014, 269, 54–84. 10.1016/j.ccr.2014.02.016. [DOI] [Google Scholar]
- Grimes R. N.Preface to the Second Edition. In Carboranes, 3rd ed.; Grimes R. N., Ed.; Academic Press: Boston, 2016; p xv. [Google Scholar]
- Liu Y.; Su B.; Dong W.; Li Z. H.; Wang H. Structural Characterization of a Boron(III) η2-σ-Silane-Complex. J. Am. Chem. Soc. 2019, 141, 8358–8363. 10.1021/jacs.9b03213. [DOI] [PubMed] [Google Scholar]
- Jaiswal K.; Volodarsky S.; Kampel V.; Dobrovetsky R. A Self-Catalyzed Reaction of 1,2-Dibenzoyl-o-Carborane with Hydrosilanes – Formation of New Hydrofuranes. Chem. Commun. 2019, 55, 10448–10451. 10.1039/C9CC04780H. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for DFT calculations comparing the stabilities of 5a vs 5b.
- Deng L.; Cheung M.-S.; Chan H.-S.; Xie Z. Reduction of 1,2-(CH2)n-1,2-C2B10H10 by Group 1 Metals. Effects of Bridge Length/Rigidity on the Formation of Carborane Anions. Organometallics 2005, 24, 6244–6249. 10.1021/om050683x. [DOI] [Google Scholar]
- Weber L.; Kahlert J.; Böhling L.; Brockhinke A.; Stammler H.-G.; Neumann B.; Harder R. A.; Low P. J.; Fox M. A. Electrochemical and Spectroelectrochemical Studies of C-Benzodiazaborolyl-ortho-Carboranes. Dalton Trans. 2013, 42, 2266–2281. 10.1039/C2DT32378H. [DOI] [PubMed] [Google Scholar]
- Núñez R.; Tarrés M.; Ferrer-Ugalde A.; de Biani F. F.; Teixidor F. Electrochemistry and Photoluminescence of Icosahedral Carboranes, Boranes, Metallacarboranes, and Their Derivatives. Chem. Rev. 2016, 116, 14307–14378. 10.1021/acs.chemrev.6b00198. [DOI] [PubMed] [Google Scholar]
- Fisher S. P.; Tomich A. W.; Guo J.; Lavallo V. Teaching an Old Dog New Tricks: New Directions in Fundamental and Applied Closo-Carborane Anion Chemistry. Chem. Commun. 2019, 55, 1684–1701. 10.1039/C8CC09663E. [DOI] [PubMed] [Google Scholar]
- Keener M.; Hunt C.; Carroll T. G.; Kampel V.; Dobrovetsky R.; Hayton T. W.; Meńard G. Redox-Switchable Carboranes for Uranium Capture and Release. Nature 2020, 577, 652–655. 10.1038/s41586-019-1926-4. [DOI] [PubMed] [Google Scholar]
- The reported wavelength for the photodissociation reaction of [Cp(CO)3Mo]2 dimer (1) is λ > 525 nm.20
- See the Supporting Information for further CV and chemical reductions with FeCp*2 and CoCp*2 experimental details.
- Bowyer W. J.; Geiger W. E. Electrochemically Induced Changes in Hapticity in Mixed-Sandwich Compounds of Iridium and Rhodium. J. Am. Chem. Soc. 1985, 107, 5657–5663. 10.1021/ja00306a011. [DOI] [Google Scholar]
- Lacoste M.; Varret F.; Toupet L.; Astruc D. Organodiiron “Electron Reservoir” Complexes Containing a Polyaromatic Ligand: Syntheses, Stabilization, Delocalized Mixed Valences, and Intramolecular Coupling. J. Am. Chem. Soc. 1987, 109, 6504–6506. 10.1021/ja00255a042. [DOI] [Google Scholar]
- Keppie S. A.; Lappert M. F. Binuclear Organometallic Compounds. Part V. Insertion Into M–C and H–C Bonds of Coordinatively Unsaturated Transition-Metal Fragments: Synthesis of Group VIA Metal Cyclopentadienyltricarbonyl Metallates (Germanium and Tin) and Hydrides. J. Chem. Soc. A 1971, 0, 3216–3220. 10.1039/J19710003216. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.