

Abstract

Uranyl-photocatalyzed hydrolysis of diaryl ethers has been established to achieve two types of phenols at room temperature under normal pressure. The single electron transfer process was disclosed by a radical quenching experiment and Stern–Volmer analysis between diphenyl ether and uranyl cation catalyst, followed by oxygen atom transfer process between radical cation of diphenyl ether and uranyl peroxide species. The 18O-labeling experiment precisely demonstrates that the oxygen source is water. Further application in template substrates of 4-O-5 linkages from lignin and 30-fold efficiency of flow operation display the potential application for phenol recovery via an ecofriendly and low-energy consumption protocol.
Keywords: lignin, phenol recovery, photocatalysis, oxygen atom transfer, flow operation
Lignin, one of the largest renewable resource of arenes in nature, is a kind of macromolecule polymerized from aromatic monomers through C–C bonds (e.g., β-5, β-β, β-1, and 5-5 linkages) and C–O bonds (e.g., α-O-4, β-O-4, and 4-O-5 linkages) (Scheme 1a).1 The recovery of aromatic chemicals from lignin would help solve the problems caused by accelerating consumption of fossil resources and corresponding environmental crisis, in which activation of C–O linkages is one of the pivot points.2 The 4-O-5 linkage is the most challenging part owing to its higher bond dissociation energy (BDE = 77.74 kcal/mol) in comparison to α-O-4 (BDE < 57.28 kcal/mol) and β-O-4 (BDE < 69.35 kcal/mol) (Scheme 1b).3,4
Scheme 1. C–O Bond Cleavage of Diphenyl Ether.

Typical studies mainly focused on hydrogenolysis and hydrolysis for C–O bond cleavage in diphenyl ether (DPE: template substrate for 4-O-5 linkage) (Scheme 1c). A series of elegant strategies by Hartwig, Grubbs, Mauriello, and others have been developed for selective hydrogenolysis with or without the assistance of transition-metal catalysts.5−13 Hydrolysis of DPE was reported by Katritzky et al.14 and Lercher and co-workers15−17 for cleaving the 4-O-5 linkage with great significance in lignin-first biorefinery,18 which is thermodynamically challenging with the reaction energy of −7.1 kcal/mol comparing to −14.2 kcal/mol in hydrogenolysis.19 Recently, the degradation of DPE was realized by Li et al. through esterification under light with an acridinium photocatalyst with two molecules of phenols obtained via the following hydrolysis.20 Meanwhile, the cleavage of C–C21 and β-O-422,23 linkages of lignin were achieved with the development of photocatalysis.24−27
Based on our previous study on uranyl species,28 the ligand-to-metal charge transfer (LMCT) mode and superior oxidative ability [Eox = +2.60 V vs SCE]29−32 supply great potential for DPE activation [Eox = +1.88 V vs SCE].33 Herein, the photocatalyzed hydrolysis of diaryl ethers was established to afford two kinds of phenols via uranyl catalysis with visible light stimulation at room temperature and normal pressure (Scheme 1d).
We commenced our study with DPE and uranyl nitrate hexahydrate as the photocatalyst stimulated with 460 nm light at room temperature, in which water was added as the oxygen source and diverse acids as coactivators (Table 1, entries 1–3). Encouragingly, the desired product phenol was afforded in 55% yield with trichloroacetic acid. Considering the solvent effect, acetone turned out to be the optimal one, while tetrahydrofuran and dichloromethane were not due to the tendency of free radical generation (Table 1, entries 4–6).34,35 Increasing the amount of water disturbed the transformation, while decreasing the water helped the reaction (Table 1, entries 7 and 8). It is worth noting that the transformation was greatly promoted even without additional water being supplied (Table 1, entry 9), in which water was supplied by the dimerization and dehydration of acetone. Two equivalents of trichloroacetic acid afforded the optimal results (Table 1, entries 10–12). Control experiments further demonstrated that UO2(NO3)2·6H2O and blue light were essential (Table 1, entries 13 and 14). Compared to the uranyl cation, Ir(ppy)2(dtbbpy)·PF4 [E1/2 = +0.66 V vs SCE],26 Ru(bpy)3Cl2·6H2O [E1/2 = +0.77 V vs SCE],26 or Eosin Y [E1/2 = +0.78 V vs SCE]36 was inefficient for the transformation (Table 1, entry 15).
Table 1. Condition Optimizationa.

| entry | H2O (equiv) | acid (equiv) | solvent | 2a (%) |
|---|---|---|---|---|
| 1 | 20 | CF3COOH | MeCN | 26 |
| 2 | 20 | CH3COOH | MeCN | 33 |
| 3 | 20 | CCl3COOH | MeCN | 55 |
| 4 | 20 | CCl3COOH | DCM | NR |
| 5 | 20 | CCl3COOH | THF | NR |
| 6 | 20 | CCl3COOH | acetone | 79 |
| 7 | 40 | CCl3COOH | acetone | 68 |
| 8 | 10 | CCl3COOH | acetone | 84 |
| 9 | CCl3COOH | acetone | 96(80)b | |
| 10c | CCl3COOH | acetone | 83 | |
| 11d | CCl3COOH | acetone | 76 | |
| 12 | acetone | 48 | ||
| 13e | CCl3COOH | acetone | NR | |
| 14f | CCl3COOH | acetone | NR | |
| 15g | CCl3COOH | acetone | NR |
General conditions: DPE (0.4 mmol), UO2(NO3)2·6H2O (4 mol %), acid (2 equiv), solvent (2 mL), N2, RT, 48 h, blue light (460 nm), 1H NMR yields with CH2Br2 as the internal standard.
Isolated yields.
Acid (1 equiv).
Acid (4 equiv).
Without light.
Without UO2(NO3)2·6H2O.
Ru(bpy)3Cl2·6H2O, Eosin Y, or Ir(ppy)2(dtbbpy)·BF4 take place of UO2(NO3)2·6H2O. NR = No reaction.
With the optimized conditions in hand, symmetrical diaryl ethers were first investigated as shown in Scheme 2. A broad range of diaryl ethers with electron-neutral, -deficient, and -rich groups at the para-position were well tolerated in this reaction, delivering the desired products with good to excellent yields. The transformation went smoothly for phenol 2b, though it was latent to be oxidized at the benzylic position. C–O bonds were successfully disconnected to obtain the corresponding phenols 2d and 2e with halogen substituents, which were never found in previous reports due to the C–X bond dissociation tendency.9,11 Remarkably, easily hydrolyzed carboxylic ester (2g) and cyano (2h) were preserved during the transformation. Diaryl ethers with substitutions at the meta-position afforded the corresponding phenols 2i–2l as well.
Scheme 2. Hydrolysis of Diaryl Ethers to Phenols.
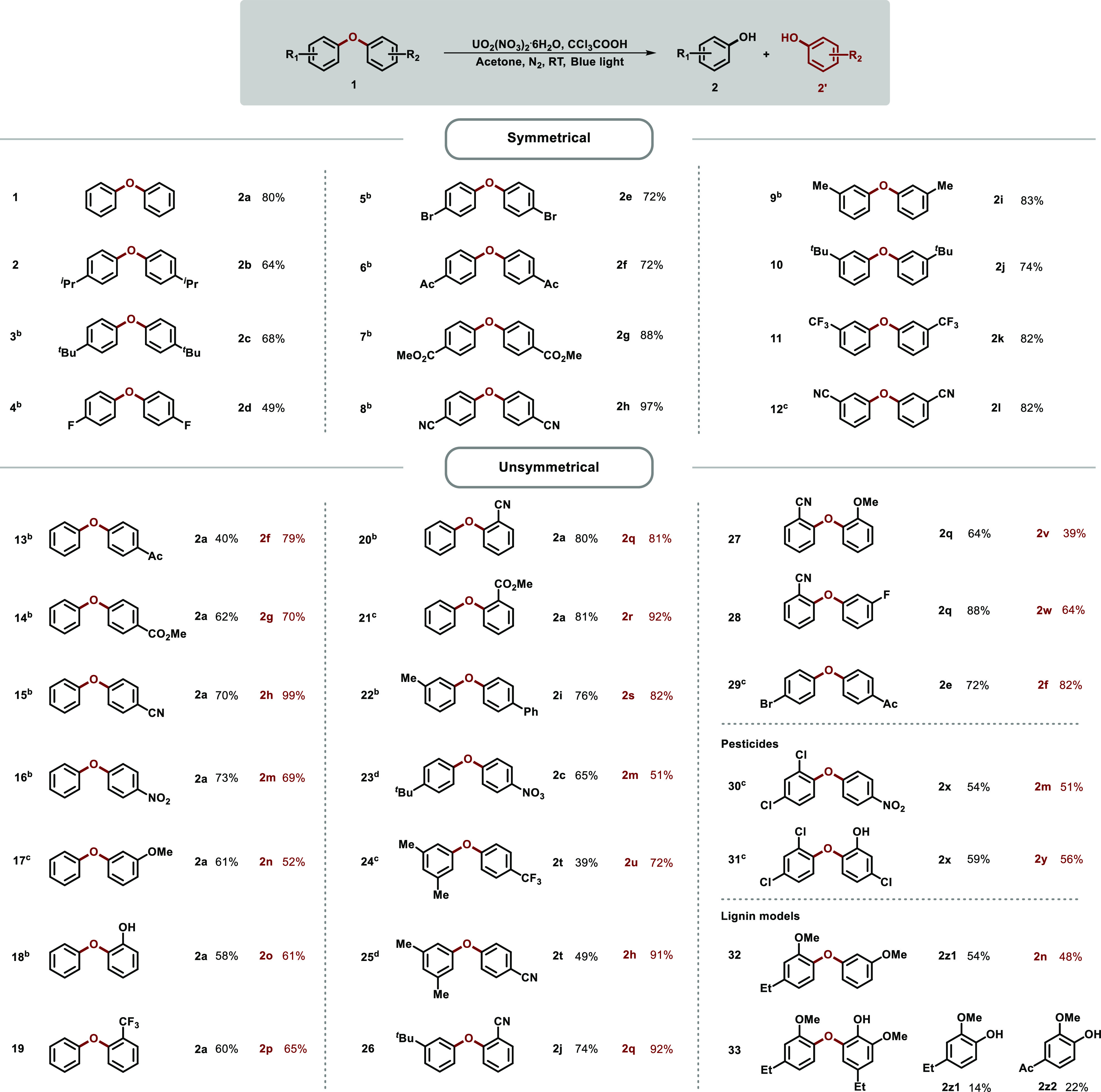
Standard conditions: Diaryl ethers (0.4 mmol), UO2(NO3)2·6H2O (4 mol %), CCl3COOH (0.8 mmol), acetone (2.0 mL), N2, RT, blue light (460 nm), isolated yields.
Diaryl ethers (0.2 mmol).
Blue light (430 nm).
UO2(NO3)2·6H2O (6 mol %), blue light (430 nm).
Subsequently, the cleavage of C–O bond with unsymmetrical diaryl ethers was tested, starting with diaryl ethers possessing unilateral groups. Phenol derivatives bearing electron-withdrawing substitutions (2f–2h and 2m) at the para-position along with phenol were readily obtained with good efficiency as expected. Phenol 2n with the methoxy group substituted at the meta-position was also compatible. Remarkably, substrates with ortho-position substituted groups could be degraded to corresponding products (2o–2r). Furthermore, diaryl ethers bearing different substitutions varied on both sides of the aromatics were successfully transformed. Good yield was obtained when both parts of the diaryl ether possessed electron-donating groups (Scheme 2, 22). The substituted diaryl ethers with electron-deficient and -rich groups on both sides could be converted to corresponding phenols in middle to good yields (Scheme 2, 23–27). Good to excellent yields could be obtained when both parts of the diaryl ether possessed electron-withdrawing groups (Scheme 2, 28, 29). It is noteworthy that nitrofen (herbicide) and triclosan (antibiotic) with multiple substituents were transformed smoothly (Scheme 2, 30, 31), showing the potential for the degradation of pesticide waste. For our lignin degradation target, models of 4-O-5 linkage in lignin were evaluated. For trisubstituted substrates, 2z1 and 2n were obtained in 48% and 54% yield, respectively. The cleavage of the C–O bond of the pentasubstituted one, whose presence was confirmed in lignin by 2D NMR spectroscopy,37,38 was achieved at room temperature and normal pressure for the first time. 2z1, widely used for food additives and spices, was obtained in 14% yield. And 2z2, the midbody for Tinib anticancer drugs, was obtained in 22% yield.
To further demonstrate the application potential of the strategy, a flow device was designed for the transformation of lignin templates (Scheme 3). Compared to tube operation, the desired products were obtained in up to 30 times efficiency with a flow device, in which the reaction mixture was pumped into light permeable PTFE tube wrapping around a high reflective aluminum bar, surrounded by 60 tandem LED lamps (430 nm), despite the residue volume of the flow pipeline being about 4.7 mL.
Scheme 3. Flow Reaction.
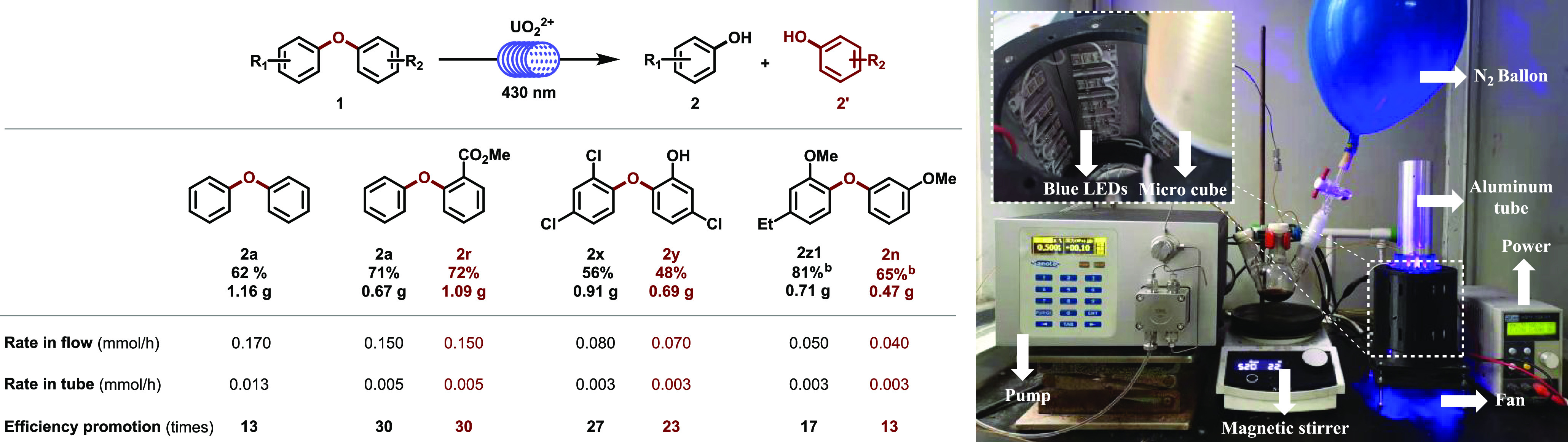
Reaction conditions: Diaryl ethers (10 mmol), UO2(NO3)2·6H2O (4 mol %), CCl3COOH (2 equiv), acetone (50 mL), N2, RT, blue light (430 nm), isolated yields.
Based on starting material recovery.
Mechanistic studies were carried out to understand the process. First, radical quenching experiments with 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO) and butylated hydroxytoluene (BHT) suggested the radical property of the system (Scheme 4a). The uranyl cation was approved to serve as the photosensor via UV–vis absorption at 424 nm (Scheme 4b), and it was also confirmed to interact with DPE directly via Stern–Volmer analysis (Scheme 4c). Based on the above results, this transformation was initiated with excited uranyl cations through a single electron transfer (SET) process between DPE under blue light irradiation.
Scheme 4. Mechanistic Studies.
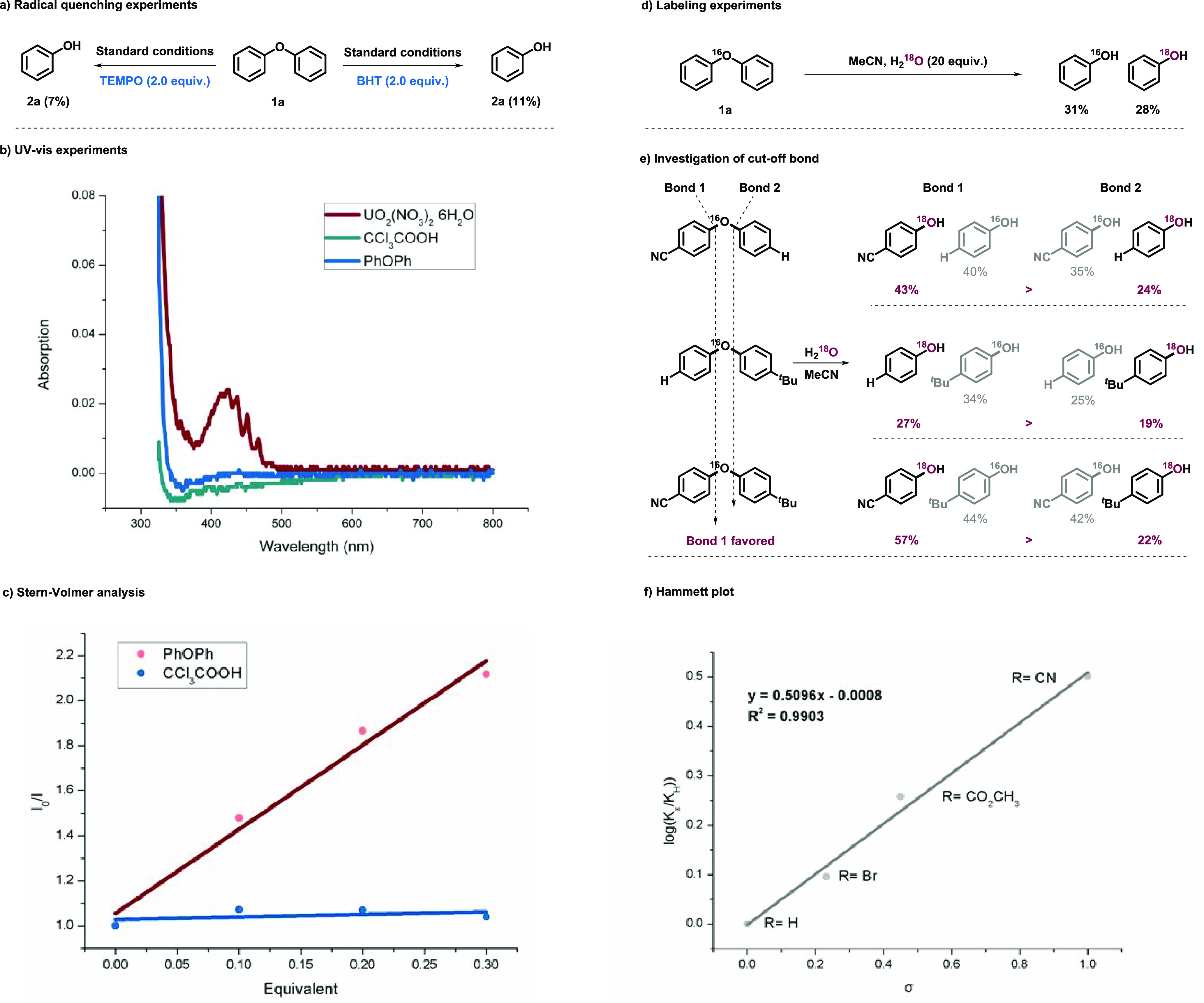
(a) Radical quenching experiments. (b) UV–vis experiments. (c) Stern–Volmer analysis. (d) Labeling experiments. (e) Investigation of cut-off bond. (f) Hammett plot.
18O labeling experiments unambiguously demonstrated the oxygen source from water (Scheme 4d). According to the previous study,39,40 uranyl peroxide complexes were obtained from uranyl photolysis of water, which is responsible for the oxygen atom transfer (OAT) process. Then, the bias of C–O bond breaking was investigated (Scheme 4e). Whether diaryl ethers possessed a tert-butyl or cyano group at the para-position with the other side unsubstituted or a tert-butyl group on one side and a cyano group on the other, both phenols were labeled with 18O, indicating that the cleavage of the C–O bond took place on both bond 1 and 2 as shown in Scheme 4e. Moreover, the ratio of the different phenols depicted the cleavage trend on the bias of the low electron cloud density side. A Hammett plot was carried out to illustrate the relationship between the reaction rate and substituent effect (Scheme 4f). The ρ-value of +0.5096 for reactions showed that the electron-withdrawing groups promoted the transformation and the decisive step was the process of negative charge accumulation.41
Therefore, a possible reaction pathway is depicted in Scheme 5. The uranyl photoredox catalysis was stimulated by blue light through the LMCT process, generating *UO22+. The SET process between *UO22+ and diaryl ether 1 produced UO2+ along with radical cation A, which was attacked by the uranyl peroxide dimer afforded via water-splitting,39,40 yielding the radical cation of phenol B and phenyl oxygen anion C, accompanied by cleavage of the original C–O bond and formation of the new one. The protonation of C, together with the SET process between B and UO2+ [Eox(B•+/2) = + 1.56 V vs SCE > Eox(UO2+/UO22+) = + 0.32 V vs SCE]34,42 afforded desired product 2 and regenerated the catalyst.
Scheme 5. Proposed Mechanism.

In summary, Csp2–O bond activation of diaryl ethers affording phenols was achieved at room temperature and normal pressure under blue light stimulation. Both symmetrical and unsymmetrical substrates were achieved with high compatibility. The Csp2–O bond was activated via the SET process between the diaryl ethers and excited uranyl cation. The dimerization of uranyl transferred the oxygen atom from the water to generate easily oxidized phenols with the LMCT process of uranyl excitation. The synergistic mechanism of SET and OAT contributes a straightforward pathway for mild photocatalytic transformation. Further application of the 4-O-5 linkage model with a flow setup indicates the potential for upgrading the degradation of lignin. Further studies on uranyl catalyzed reactions are ongoing in our laboratory.
Acknowledgments
The authors are grateful for financial support provided by NSFC (21971065), STCSM (20XD1421500, 20JC1416800, and 18JC1415600), Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZLCX20180501), and the Fundamental Research Funds for the Central Universities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00168.
Experimental procedures; NMR spectra and analytical data for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Stewart J. J.; Akiyama T.; Chapple C.; Ralph J.; Mansfield S. D. The Effects on Lignin Structure of Overexpression of Ferulate 5-Hydroxylase in Hybrid Poplar. Plant Physiol 2009, 150, 621–635. 10.1104/pp.109.137059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutyser W.; Renders T.; Van den Bosch S.; Koelewijn S. F.; Beckham G. T.; Sels B. F. Chemicals from Lignin: an Interplay of Lignocellulose Fractionation, Depolymerisation, and Upgrading. Chem. Soc. Rev. 2018, 47, 852–908. 10.1039/C7CS00566K. [DOI] [PubMed] [Google Scholar]
- Parthasarathi R.; Romero R. A.; Redondo A.; Gnanakaran S. Theoretical Study of the Remarkably Diverse Linkages in Lignin. J. Phys. Chem. Lett. 2011, 2, 2660–2666. 10.1021/jz201201q. [DOI] [Google Scholar]
- Younker J. M.; Beste A.; Buchanan A. C. III Computational Study of Bond Dissociation Enthalpies for Lignin Model Compounds: β-5 Arylcoumaran. Chem. Phys. Lett. 2012, 545, 100–106. 10.1016/j.cplett.2012.07.017. [DOI] [Google Scholar]
- Sergeev A. G.; Hartwig J. F. Selective, Nickel-Catalyzed Hydrogenolysis of Aryl Ethers. Science 2011, 332, 439–443. 10.1126/science.1200437. [DOI] [PubMed] [Google Scholar]
- Sergeev A. G.; Webb J. D.; Hartwig J. F. A Heterogeneous Nickel Catalyst for the Hydrogenolysis of Aryl Ethers without Arene Hydrogenation. J. Am. Chem. Soc. 2012, 134, 20226–20229. 10.1021/ja3085912. [DOI] [PubMed] [Google Scholar]
- Zaheer M.; Hermannsdoerfer J.; Kretschmer W. P.; Motz G.; Kempe R. Robust Heterogeneous Nickel Catalysts with Tailored Porosity for the Selective Hydrogenolysis of Aryl Ethers. ChemCatChem 2014, 6, 91–95. 10.1002/cctc.201300763. [DOI] [Google Scholar]
- Gao F.; Webb J. D.; Hartwig J. F. Chemo- and Regioselective Hydrogenolysis of Diaryl Ether C-O Bonds by a Robust Heterogeneous Ni/C Catalyst: Applications to the Cleavage of Complex Lignin-Related Fragments. Angew. Chem., Int. Ed. 2016, 55, 1474–1478. 10.1002/anie.201509133. [DOI] [PubMed] [Google Scholar]
- Ren Y.; Yan M.; Wang J.; Zhang Z. C.; Yao K. Selective Reductive Cleavage of Inert Aryl C-O Bonds by an Iron Catalyst. Angew. Chem., Int. Ed. 2013, 52, 12674–12678. 10.1002/anie.201305342. [DOI] [PubMed] [Google Scholar]
- Fedorov A.; Toutov A. A.; Swisher N. A.; Grubbs R. H. Lewis-Base Silane Activation: From Reductive Cleavage of Aryl Ethers to Selective ortho-Silylation. Chem. Sci. 2013, 4, 1640–1645. 10.1039/c3sc22256j. [DOI] [Google Scholar]
- Xu H.; Liu X.; Zhao Y.; Wu C.; Chen Y.; Gao X.; Liu Z. Reductive Cleavage of C-O Bond in Model Compounds of Lignin. Chin. J. Chem. 2017, 35, 938–942. 10.1002/cjoc.201600770. [DOI] [Google Scholar]
- Mauriello F.; Paone E.; Pietropaolo R.; Balu A. M.; Luque R. Catalytic Transfer Hydrogenolysis of Lignin-Derived Aromatic Ethers Promoted by Bimetallic Pd/Ni Systems. ACS Sustainable Chem. Eng. 2018, 6, 9269–9276. 10.1021/acssuschemeng.8b01593. [DOI] [Google Scholar]
- Mauriello F.; Ariga-Miwa H.; Paone E.; Pietropaolo R.; Takakusagi S.; Asakura K. Transfer Hydrogenolysis of Aromatic Ethers Promoted by the Bimetallic Pd/Co Catalyst. Catal. Today 2020, 357, 511–517. 10.1016/j.cattod.2019.06.071. [DOI] [Google Scholar]
- Siskin M.; Katritzky A. R.; Balasubramanian M. Aqueous Organic Chemistry. 4. Cleavage of Diaryl Ethers. Energy Fuels 1991, 5, 770–771. 10.1021/ef00029a028. [DOI] [Google Scholar]
- Roberts V. M.; Knapp R. T.; Li X.; Lercher J. A. Selective Hydrolysis of Diphenyl Ether in Supercritical Water Catalyzed by Alkaline Carbonates. ChemCatChem 2010, 2, 1407–1410. 10.1002/cctc.201000181. [DOI] [Google Scholar]
- He J.; Zhao C.; Lercher J. A. Ni-Catalyzed Cleavage of Aryl Ethers in the Aqueous Phase. J. Am. Chem. Soc. 2012, 134, 20768–20775. 10.1021/ja309915e. [DOI] [PubMed] [Google Scholar]
- Wang M.; Shi H.; Camaioni D. M.; Lercher J. A. Palladium-Catalyzed Hydrolytic Cleavage of Aromatic C-O Bonds. Angew. Chem., Int. Ed. 2017, 56, 2110–2114. 10.1002/anie.201611076. [DOI] [PubMed] [Google Scholar]
- Paone E.; Tabanelli T.; Mauriello F. The Rise of Lignin Biorefinery. Current Opinion in Green and Sustainable Chemistry 2020, 24, 1–6. 10.1016/j.cogsc.2019.11.004. [DOI] [Google Scholar]
- Chatterjee M.; Chatterjee A.; Ishizaka T.; Kawanami H. Rhodium-Mediated Hydrogenolysis/Hydrolysis of the Aryl Ether Bond in Supercritical Carbon Dioxide/Water: An Experimental and Theoretical Approach. Catal. Sci. Technol. 2015, 5, 1532–1539. 10.1039/C4CY01417K. [DOI] [Google Scholar]
- Tan F. F.; He X. Y.; Tian W. F.; Li Y. Visible-Light Photoredox-Catalyzed C-O Bond Cleavage of Diaryl Ethers by Acridinium Photocatalysts at Room Temperature. Nat. Commun. 2020, 11, 6126–6126. 10.1038/s41467-020-19944-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazi S.; Hung Ng W. K.; Ganguly R.; Putra Moeljadi A. M.; Hirao H.; Soo H. S. Selective Photocatalytic C–C Bond Cleavage under Ambient Conditions with Earth Abundant Vanadium Complexes. Chem. Sci. 2015, 6, 7130–7142. 10.1039/C5SC02923F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J. D.; Matsuura B. S.; Stephenson C. R. J. A Photochemical Strategy for Lignin Degradation at Room Temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. 10.1021/ja4113462. [DOI] [PubMed] [Google Scholar]
- Luo J.; Zhang X.; Lu J.; Zhang J. Fine Tuning the Redox Potentials of Carbazolic Porous Organic Frameworks for Visible-Light Photoredox Catalytic Degradation of Lignin β-O-4 Models. ACS Catal. 2017, 7, 5062–5070. 10.1021/acscatal.7b01010. [DOI] [Google Scholar]
- Ghosh I.; Ghosh T.; Bardagi J. I.; König B. Reduction of Aryl Halides by Consecutive Visible Light-Induced Electron Transfer Processes. Science 2014, 346, 725–728. 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]
- Li X. B.; Tung C. H.; Wu L. Z. Semiconducting Quantum Dots for Artificial Photosynthesis. Nat. Rev. Chem. 2018, 2, 160–173. 10.1038/s41570-018-0024-8. [DOI] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay N. E. S.; Nicewicz D. A. Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 16100–16104. 10.1021/jacs.7b10076. [DOI] [PubMed] [Google Scholar]
- Li Y.; Rizvi S. A.-e.-A.; Hu D.; Sun D.; Gao A.; Zhou Y.; Li J.; Jiang X. Selective Late-Stage Oxygenation of Sulfides with Ground-State Oxygen by Uranyl Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 13499–13506. 10.1002/anie.201906080. [DOI] [PubMed] [Google Scholar]
- Cowie B. E.; Purkis J. M.; Austin J.; Love J. B.; Arnold P. L. Thermal and Photochemical Reduction and Functionalization Chemistry of the Uranyl Dication, [UVIO2]2+. Chem. Rev. 2019, 119, 10595–10637. 10.1021/acs.chemrev.9b00048. [DOI] [PubMed] [Google Scholar]
- Wu L.; Cao X.; Chen X.; Fang W.; Dolg M. Visible-Light Photocatalysis of C(sp3)-H Fluorination by the Uranyl Ion: Mechanistic Insights. Angew. Chem., Int. Ed. 2018, 57, 11812–11816. 10.1002/anie.201806554. [DOI] [PubMed] [Google Scholar]
- Burrows H. D.; Kemp T. J. The photochemistry of the uranyl ion. Chem. Soc. Rev. 1974, 3, 139–165. 10.1039/cs9740300139. [DOI] [Google Scholar]
- West J. G.; Bedell T. A.; Sorensen E. J. The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)-H Fluorination. Angew. Chem., Int. Ed. 2016, 55, 8923–8927. 10.1002/anie.201603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- Capaldo L.; Merli D.; Fagnoni M.; Ravelli D. Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion. ACS Catal. 2019, 9, 3054–3058. 10.1021/acscatal.9b00287. [DOI] [Google Scholar]
- Boehm A.; Bach T. Radical Reactions Induced by Visible Light in Dichloromethane Solutions of Hunig’s Base: Synthetic Applications and Mechanistic Observations. Chem. - Eur. J. 2016, 22, 15921–15928. 10.1002/chem.201603303. [DOI] [PubMed] [Google Scholar]
- Hari D. P.; König B. Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun. 2014, 50, 6688–6699. 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]
- Yue F.; Lu F.; Ralph S.; Ralph J. Identification of 4-O-5-Units in Softwood Lignins via Definitive Lignin Models and NMR. Biomacromolecules 2016, 17, 1909–1920. 10.1021/acs.biomac.6b00256. [DOI] [PubMed] [Google Scholar]
- Li Y.; Akiyama T.; Yokoyama T.; Matsumoto Y. NMR Assignment for Diaryl Ether Structures (4-O-5 Structures) in Pine Wood Lignin. Biomacromolecules 2016, 17, 1921–1929. 10.1021/acs.biomac.6b00257. [DOI] [PubMed] [Google Scholar]
- Petrus E.; Segado M.; Bandeira N. A. G.; Bo C. Unveiling a Photoinduced Hydrogen Evolution Reaction Mechanism via the Concerted Formation of Uranyl Peroxide. Inorg. Chem. 2020, 59, 8353–8360. 10.1021/acs.inorgchem.0c00757. [DOI] [PubMed] [Google Scholar]
- McGrail B. T.; Pianowski L. S.; Burns P. C. Photochemical Water Oxidation and Origin of Nonaqueous Uranyl Peroxide Complexes. J. Am. Chem. Soc. 2014, 136, 4797–4800. 10.1021/ja502425t. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Li C.; Hoffman M. Z. One-Electron Redox Potentials of Phenols in Aqueous Solution. J. Phys. Chem. B 1999, 103, 6653–6656. 10.1021/jp983819w. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.