

Abstract

The first direct and selective synthesis of substituted itaconimdes by palladium-catalyzed aminocarbonylation of alkynols is reported. Key to the success of this transformation is the use of a novel catalyst system involving ligand L11 and appropriate reaction conditions. In the protocol here presented, easily available propargylic alcohols react with N-nucleophiles including aryl- and alkylamines as well as aryl hydrazines to provide a broad variety of interesting heterocycles with high catalyst activity and excellent selectivity. The synthetic utility of the protocol is demonstrated in the synthesis of natural product 11 with aminocarbonylation as the key step. Mechanistic studies and control experiments reveal the crucial role of the hydroxyl group in the substrate for the control of selectivity.
Keywords: itaconimide, palladium, P ligand, aminocarbonylation, alkynol
Introduction
Succinimides constitute an important class of organic molecules, which widely exist in a large number of five-membered natural products with diverse biological and pharmaceutical activities.1−5 Among these compounds, itaconimide derivatives characterized by exocyclic C=C bonds are found in many natural products, and this structural motif is also present in current drug candidates (Scheme 1).6−11 For example, longimide B I is known for its antitumor activity,2,6 while compounds II and III showed potent GPR119 agonistic7 and antagonist activity,8 respectively. In addition, 3-arylmethylidene pyrrolidine-2,5-diones IVa and IVb were tested as type 2 5α-reductase (T2-5α-reductase) inhibitors.9,10 As a final example, compound V is mentioned here displaying potent anti-Xa activity, whereby both the cyclic structure and the geometry provided by the double bond contribute to the pharmaceutical potential of V.11 Interestingly, related fulgimides, e.g., VI, are also known to be molecular switches with many potential applications in optical materials.12−14
Scheme 1. Importance of Itaconimides: Selected Examples of Natural Compounds, Biologically Active Molecules, and Interesting Postfunctionalizations.

Moreover, itaconimides are recognized as useful synthons VII as they contain condensed functionalities for further scaffold diversification.15−30 For example, they can be readily transformed into relevant pyrrolidines by reduction15 and provide an easy access to the corresponding succinic acid derivatives by ring-opening reactions.16−18 Furthermore, the fairly acidic methylene group shows nucleophilic ability,19−25 which has been used in diazo-transfer reactions,19 allylic additions,20 and Michael addition reactions.21−23 Finally, the activated exocyclic olefin can be further functionalized26−32 through cycloadditions26−29 and asymmetric hydrogenation reactions30 and forms highly stable thio-Michael adducts that resist thiol-exchange-mediated breakdown under physiological conditions.31,32
Although the synthesis of itaconimides attracted significant interest in the area of drug development, relatively few synthetic methodologies to access this class of compounds in a general manner exist. Primarily, activated alkenes33−44 such as α,β-unsaturated anhydrides,33−35 the parent compound,10 and maleimides20,31,36−44 were used for their preparation (Scheme 2a–c). Thus, until today the most commonly used method involves conjugate-addition of triphenylphosphine to endocyclic olefinic maleimides followed by a Wittig reaction with aldehydes, resulting in the inevitable formation of (over)stoichiometric amounts of phosphine oxides.19,31,40−44 In addition, more specific three-component reactions of allenoates, isocyanides, and carboxylic acids and a cycloisomerization/rearrangement cascade of Ugi adducts have been also reported.45,46
Scheme 2. Summary of the Main Synthesis Methods of Itaconimides.

Preferably, any new methodology in this area should be environmentally benign, atom-efficient, and straightforward. On the basis of our general expertise and recent works on carbonylations,47−50 we had the idea that specifically such transformations can be also used to access itaconimides (Scheme 2d). Indeed, as one of the most important homogeneous catalytic processes, transition-metal-catalyzed carbonylation reactions allow for direct conversion of easily available feedstocks into a variety of carbonylated compounds.51−54 In this respect, the aminocarbonylation of propargyl alcohols which are commercially available or easily accessed is an ideal method. However, such methodologies are intrinsically challenging due to the problems associated with the acidity of the active metal hydride catalysts and the basicity of the amine reagent. Moreover, aminocarbonylations are prone to produce many side products such as esters (lactones), acids, and branched or linear α,β-unsaturated amides. Thus, it is not surprising that, to the best of our knowledge, no such process has been reported, and herein, we present the first example of a general and highly selective synthesis of itaconimide derivatives through Pd-catalyzed aminocarbonylation.
Results and Discussion
Model Reaction and Catalyst Optimization
At the beginning of our studies, carbonylation of 1-ethynyl-1-cyclohexanol 1a with 4-fluoroaniline 2a was chosen as a benchmark reaction. As shown in Figure 1, via selective dicarbonylation of 1a, it should be possible to obtain itaconimide 3aa directly. However, a variety of other carbonylated products can be formed easily in this reaction. For example, the branched α,β-unsaturated amide 4, linear α,β,γ,δ-unsaturated amide 5, and linear α,β-unsaturated amide with an allylic hydroxyl group 6.
Figure 1.
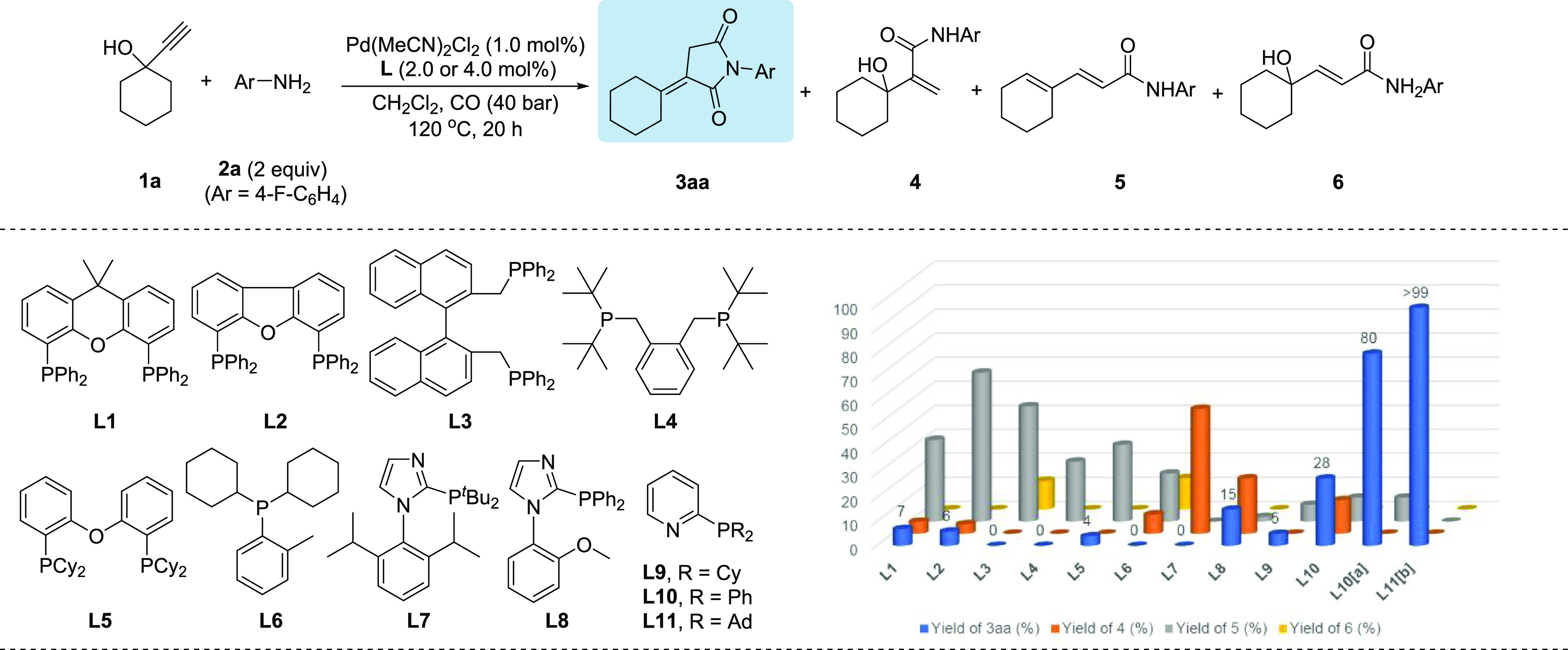
Pd-catalyzed aminocarbonylation of 1-ethynyl-1-cyclohexanol 1a: influence of phosphine ligands. Reaction conditions: 1a (0.5 mmol), 2a (1.0 mmol), Pd(MeCN)2Cl2 (1.0 mol %), bisphosphine ligand (2.0 mol %) or monophosphine ligand (4.0 mol %), CO (40 atm), CH2Cl2 (2.0 mL), 20 h at 120 °C. The yields of products were determined by GC analysis with mesitylene as an internal standard. [a] indicates the following conditions: Pd(cod)Cl2 (1.0 mol %), 2a (0.75 mmol), pentane (2.0 mL). [b] indicates the following conditions: Pd(cod)Cl2 (1.0 mol %), 2a (0.75 mmol), PTSA·H2O (10 mol %), pentane (2.0 mL), 100 °C.
In general, for palladium-catalyzed coupling processes, the choice of ligand is crucial and allows for controlling the selectivity and activity.55−59 Thus, to achieve the desired target product 3aa, we investigated the effect of different bidentate and monodentate phosphines (2 or 4 mol %, respectively) for the carbonylation of 1a in the presence of 1 mol % Pd(MeCN)2Cl2, in dichloromethane at 40 bar CO, 120 °C. Using the classic ligand Xantphos L1, itaconimide 3aa was obtained in only 7% yield; nevertheless, this demonstrated the feasibility of a double carbonylation process. Other bisphosphine ligands with different backbones and chelating units such as DBFphos L2, Naphos L3, 1,2-bis((di-tert-butylphosphan-yl)methyl)benzene L4, and bis(2-dicyclohexyl-phosphinophenyl) ether L5 favored the formation of linear α,β,γ,δ-unsaturated amide 5. No improvement of the chemoselectivity was achieved when monodentate phosphine ligand L6 was tested.
Notably, the imidazole-based ligand L7 showed high chemoselectivity for monoaminocarbonylation of 1a, leading to amide 4 with 52% yield. Interestingly, when ligand L8 was applied, the yield of unsaturated succinimide 3aa was improved to 15%. Using ligands L9 and L10 which bear pyridyl substituents on the phosphorus atoms, ligand L10 gave the best result (28% yield) in our preliminary ligand screening. To optimize the benchmark reaction further, we evaluated the influence of critical reaction parameters in the presence of L10. By variation of catalyst precursors, solvents, and other factors, the yield of 3aa reached 80% with a selectivity of 8/1 (3aa/5) (see Supporting Information Tables S1–S4 for details). Gratifyingly, with ligand L11 in which the phenyl groups are replaced with bulkier adamantyl substituents, the linear side product 5 was completely suppressed, giving 3aa in 87% yield (see Supporting Information Table S4 for details). Lowering the reaction temperature (100 °C) resulted in an improved yield of 3aa (94%), whereas a lower CO pressure (20 bar) had a negative effect on the desired product yield (82%) (see Supporting Information Table S5 for details), and finally, in the presence of 10 mol % PTSA, the yield of 3aa reached >99% with excellent chemoselectivity.
Mechanistic Investigations and Catalytic Cycle
To gain insights into the reaction mechanism, several control experiments were conducted (Scheme 3). First, 1-ethynylcyclohexene 9 and cyclohexylacetylene 10 were examined under the standard conditions, showing the crucial role of the hydroxyl group in 1a to accommodate the conversion of the propargylic alcohol. While 9 led to a complex mixture, cyclohexylacetylene 10 afforded monoaminocarbonylated products (see Supporting Information, Scheme S2).
Scheme 3. (a) Control Experiments, (b) Kinetic Monitoring of Intermediates over Time, and (c) Proposed Catalytic Cycle.

Standard conditions: Pd(cod)Cl2 (1.0 mol %), L11 (4.0 mol %), PTSA·H2O (10 mol %), pentane (2.0 mL), CO (40 atm), 100 °C, 20 h, 1a or 4–10 (0.5 mmol), 4-fluoroaniline (0.75 mmol). The yields of products were determined by crude 1H NMR analysis using dibromomethane as the internal standard.
Additionally, no conversion of 8 could be achieved when it was tested with amine 2a under standard conditions, excluding the possibility of condensation of α,β-unsaturated anhydrides with the corresponding amines.
To determine the key intermediate which itaconimide 3aa was generated from, amides 4, 5, 6, and 7 were isolated and reacted with CO under standard conditions, separately. No conversion was noted using linear α,β,γ,δ-unsaturated amide 5 as the starting material. Furthermore, 6 and 7 could not be transformed to 3aa either. However, testing the branched amide 4 with an allylic hydroxyl group in the carbonylation reaction provided 3aa in 32% yield. The observed lower yield of 3aa may result from different concentrations of 4 in the control experiment compared to the catalytic process (Scheme 3a, see Supporting Information Schemes S2 and S3 for details).
Next, the kinetic progress of the reaction between alkynol 1a and 4-fluoroaniline 2a was examined under the optimal conditions. As shown in Scheme 3b, 1a is initially converted into the monoaminocarbonylation product 4. Then, 3aa is generated along with the consumption of the reaction intermediate 4. Starting material 1a is fully converted, and the formation of the branched amide 4 achieves a maximum yield of 48% after 40 min. In the following 60 min, the yield of desired product 3aa increases quickly and reaches >99% yield after 100 min. Over the course of the reaction, formation of 7 is observed only in small amounts (maximum 10% yield), which could be explained by the equilibrium with 4.
On the basis of these findings and previous mechanistic studies of Pd-catalyzed aminocarbonylations,55−59 we propose the following main reaction pathway (Scheme 3c). Initially, the stable PdII salt is in situ reduced to give Pd0 phosphine complexes A in the presence of an excess amount of ligand.60−63 After protonation, active complex B is afforded. Then, the carbon–carbon triple bond of alkynol 1a coordinates to Pd to form complex C, and subsequent triple bond insertion affords regioselectively the branched alkenyl-Pd intermediate D. Pd complex D directly undergoes a facile CO insertion process to give the corresponding acyl Pd species E. Next, aminolysis of intermediate E leads to the mono-aminocarbonylation product 4 and regenerates the palladium hydride species B. Compound 4 is also prone to dehydration to form 7, which can be a reversible process. After cycle I, complex B reacts with 4, forming the π-allyl-palladium intermediate F. This intermediate undergoes a fast equilibrium with the corresponding σ-palladium complexes G. Finally, CO insertion and aminolysis lead to the desired product 3aa.
Substrate Scope
With optimized reaction conditions established, we examined the scope of this three-component process with respect to alkynols. As shown in Table 1a, diverse propargylic alcohols can be applied in the carbonylation transformation, demonstrating a practical strategy utilizing abundant alkynol feedstocks directly as robust surrogates for the construction of itaconimides. Different substituents (tert-butyl, phenyl) at the 4-position of cyclohexyl group were compatible with the reaction conditions, giving rise to 3ba and 3ca in high yield (77% and 88%, respectively). This transformation also displayed good functional group tolerance. Hence, the carbonylations of substrates 1d and 1e containing heteroatoms (sulfur, nitrogen) were successful, providing 3da and 3ea in good yields (67% and 87%). Moreover, a wide array of carbocyclic ring systems (4-, 5-, and 12-membered) were amenable to this approach, delivering the corresponding unsaturated succinimides 3fa–3ha in 76–98% yields. Notably, sterically crowded alkynol 1i also reacted smoothly, giving 3ia in 84% yield. Noncyclic alkynols 1j–1n bearing different alkyl and benzyl groups furnished the corresponding desired products 3ja–3na with 35–98% yields. When α-monoalkyl-substituted propargyl alcohols 1o–1q and the α-monoaryl-substituted alkynol 1r were subjected to the optimized conditions, this catalytic system exhibited good activities (53–76% yields) and >20/1 E/Z selectivities. In addition, the diaryl-substituted propargyl alcohol 1s was well-tolerated by the catalyst to afford 3sa in 79% yield. The effectiveness of this methodology is showcased by the late-stage modification of biorelevant derivatives, which provides easy access to diverse itaconimide derivatives, highlighting the substrate scope of this protocol and its utility in organic synthesis. More specifically, tropinone-derived propargylic alcohol 1t could be applied to afford the desired product 3ta in decent yield. Pentoxifyllin, a drug with anti-inflammatory properties, can be transformed to the corresponding product 3ua in 73% yield (E/Z = 1/1).
Table 1. Pd-Catalyzed Aminocarbonylation of Alkynols 1a–v with Amines 2a–pa.


Standard reaction conditions: 1 (0.5 mmol), Pd(cod)Cl2 (1.0 mol %), L11 (4.0 mol %), PTSA·H2O (10.0 mol %), 2 (0.75 mmol), CO (40 atm), pentane (2.0 mL), 20 h at 100 °C. Isolated yields were given within the parentheses. The NMR yields (values before the parentheses) were determined by crude 1H NMR analysis using dibromomethane as the internal standard.
1n (0.1 mmol), 2a (0.15 mmol), Pd(cod)Cl2 (5.0 mol %), L11 (20.0 mol %), PTSA·H2O (0.1 mmol).
1a (0.5 mmol), BnNH22p (1 mmol), PdBr2 (1.0 mol %), L10 (4.0 mol %), TFA (10.0 mol %), CO (40 atm), CH2Cl2 (2.0 mL), 20 h at 120 °C.
Displacement ellipsoids correspond to 30% probability. See Supporting Information Section 2.5 for details.
Next, we evaluated the scope of this aminocarbonylation process with respect to amines. As shown in Table 1b, a variety of aromatic amines with electron-neutral, electron-deficient, and electron-rich substituents led to the corresponding carbonylative products in good yields (51–98%). The substituents on the arylamines had no real impact on the catalysis. Specifically, the reactions of arylamines bearing phenyl, methoxy, and chlorine substituents proceeded smoothly, providing the corresponding products 3ab–3ae in 91–98% isolated yields. Notably, bromine-substituted arylamines, which are known to be sensitive to palladium catalysis, also worked well and afforded 3vf in 51% yield. Interestingly, the 4-aminoacetophenone 2g and 4-aminophenol (2h) which contain functional groups reacted well to give 3ag and 3ah in 74% and 50% yield, respectively. The position of substituents on the phenyl ring has no influence on the reaction outcome. Hence, arylamines 2i–2k afforded 3ai–3ak in 65–82% yields. Moreover, arylamines 2l–2n containing multiple substituents proved to be efficient coupling partners and gave the corresponding products in 80–93% yields. To be noted, heteroaromatic amine 2o with an easily functionalized ester group led to 3ao in quantitative conversion with excellent chemoselectivity.
The carbonylation in the presence of aliphatic amines continues to be a highly challenging goal due to their stronger basicity compared with arylamines.64 Nevertheless, using benzyl amine 2p as a representative example gave the desired 3ap as the major product (44% yield; see Supporting Information Tables S6–S8 for details).
Finally, we became interested in the use of other N-nucleophiles instead of amines. In this respect, specifically aryl hydrazines attracted our interest; these constitute important synthons in the synthesis of many biologically active and industrially important organic compounds.65,66 Indeed, when phenylhydrazine 2q was reacted with 1a under the previously optimized conditions, the corresponding unsaturated succinimide 3aq was isolated as the sole product in excellent yield (90%). Apart from phenylhydrazine, 4-hydrazinylbenzonitrile 2r, o-tolylhydrazine 2s, and 3-chlorophenylhydrazine 2t were employed as efficient N-nucleophiles (Table 2). In all cases, >99% NMR yields were obtained, and the chemoselectivities for double carbonylative products 3ar–3as were excellent. Notably, these novel hydrazine derivatives offer interesting possibilities for the straightforward synthesis of other heterocycles.67−71
Table 2. Pd-Catalyzed Aminocarbonylation of 1a with Aryl Hydrazines 2q–ta.


Standard reaction conditions: 1a (0.5 mmol), Pd(cod)Cl2 (1.0 mol %), L11 (4.0 mol %), PTSA·H2O (10.0 mol %), 2 (0.75 mmol), CO (40 atm), pentane (2.0 mL), 20 h at 100 °C. Isolated yields were given within the parentheses. The NMR yields (values before the parentheses) were determined by crude 1H NMR analysis using dibromomethane as the internal standard.
Displacement ellipsoids correspond to 30% probability. See Supporting Information Section 2.5 for details.
The synthetic utility of our protocol is further demonstrated in the straightforward synthesis of natural product 11, which was isolated from an endophytic fungus.72 Itaconimide 3jq was obtained without additional optimization by aminocarbonylation of 2-methyl-3-butyn-2-ol 1j and benzylic amine 2q. Subsequent removal of the N-p-methoxybenzyl protecting group with CAN gave natural product 11 (Scheme 4).
Scheme 4. Synthesis of Natural Product 11 with Aminocarbonylation as Key Step.

Conclusion
In summary, we report a new catalytic domino transformation which allows an efficient synthesis of itaconimides from easily available propargylic alcohols. Key to this effective methodology is a straightforward aminocarbonylation reaction in the presence of a novel palladium catalystinvolving ligand L11. This protocol complements the currently known methods for carbonylation reactions in organic synthesis, allowing access to this so far overlooked class of compounds. In fact, 40 out of 41 molecules prepared here have not been described before to the best of our knowledge.
The general applicability of this highly selective protocol is demonstrated by reacting more than 20 versatile alkynols including structurally complex and biologically active molecules. Furthermore, diverse N-nucleophiles including arylamines, alkylamines, and aryl hydrazines can be easily employed. Control experiments reveal the presence of the hydroxyl group to be essential for the selective synthesis of the desired products. Mechanistic studies further suggest a novel sequential double carbonylation process with the branched amide 4 as a key intermediate.
Acknowledgments
This work is supported by the state of Mecklenburg-Western Pommerania and the BMBF (Bundesministerium für Bildung und Forschung) in Germany. We thank the analytical team of LIKAT. F.Y. thanks the National Natural Science Foundation of China (21801056) for financial support. Dedicated to Professor Dr. Pierre H. Dixneuf on the occasion of his birthday and excellent work in organometallic catalysis.
Glossary
Abbreviations
- PTSA·H2O
p-toluenesulfonic acid monohydrate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00221.
Additional experimental results and procedures and characterization data (PDF)
Author Contributions
§ Y.G. and F.Y. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Miller C. A.; Long L. M. Anticonvulsants. I. An Investigation of N-R-α-R1-α-Phenylsuccinimides. J. Am. Chem. Soc. 1951, 73, 4895–4898. 10.1021/ja01154a126. [DOI] [Google Scholar]
- Sashidhara K. V.; Singh S. P.; Kant R.; Maulik P. R.; Sarkar J.; Kanojiya S.; Ravi Kumar K. Cytotoxic cycloartane triterpene and rare isomeric bisclerodane diterpenes from the leaves of Polyalthia longifoliavar. pendula. Bioorg. Med. Chem. Lett. 2010, 20, 5767–5771. 10.1016/j.bmcl.2010.07.141. [DOI] [PubMed] [Google Scholar]
- Rybka S.; Obniska J.; Rapacz A.; Filipek B.; Zmudzki P. Synthesis and evaluation of anticonvulsant properties of new N-Mannich bases derived from pyrrolidine-2,5-dione and its 3-methyl-, 3-isopropyl, and 3-benzhydryl analogs. Bioorg. Med. Chem. Lett. 2017, 27, 1412–1415. 10.1016/j.bmcl.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Kaminski K.; Obniska J.; Chlebek I.; Liana P.; Pekala E. Synthesis and biological properties of new N-Mannich bases derived from 3-methyl-3-phenyl- and 3,3-dimethyl-succinimides. Part V. Eur. J. Med. Chem. 2013, 66, 12–21. 10.1016/j.ejmech.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Socała K.; Mogilski S.; Pieróg M.; Nieoczym D.; Abram M.; Szulczyk B.; Lubelska A.; Latacz G.; Doboszewska U.; Wlaź P.; Kamiński K. KA-11, a Novel Pyrrolidine-2,5-dione Derived Broad-Spectrum Anticonvulsant: Its Antiepileptogenic, Antinociceptive Properties and in Vitro Characterization. ACS Chem. Neurosci. 2019, 10, 636–648. 10.1021/acschemneuro.8b00476. [DOI] [PubMed] [Google Scholar]
- Luo K.; Bao Y.; Liu F.; Xiao C.; Li K.; Zhang C.; Huang R.; Lin J.; Zhang J.; Jin Y. Synthesis and biological evaluation of novel benzylidene-succinimide derivatives as noncytotoxic antiangiogenic inhibitors with anticolorectal cancer activity in vivo. Eur. J. Med. Chem. 2019, 179, 805–827. 10.1016/j.ejmech.2019.06.094. [DOI] [PubMed] [Google Scholar]
- Kim H.; Cho S. J.; Yoo M.; Kang S. K.; Kim K. R.; Lee H. H.; Song J. S.; Rhee S. D.; Jung W. H.; Ahn J. H.; Jung J.-K.; Jung K.-Y. Synthesis and biological evaluation of thiazole derivatives as GPR119 agonists. Bioorg. Med. Chem. Lett. 2017, 27, 5213–5220. 10.1016/j.bmcl.2017.10.046. [DOI] [PubMed] [Google Scholar]
- López-Rodríguez M. L.; Morcillo M. J.; Rovat T. K.; Fernández E.; Vicente B.; Sanz A. M.; Hernández M.; Orensanz L. Synthesis and Structure-Activity Relationships of a New Model of Arylpiperazines. 4.1 1-[ω-(4-Arylpiperazin-1-yl)alkyl]-3-(diphenylmethylene)-2,5-pyrrolidinediones and −3-(9H-fluoren-9-ylidene)-2,5pyrrolidinediones: Study of the Steric Requirements of the Terminal Amide Fragment on 5-HT1A Affinity/Selectivity. J. Med. Chem. 1999, 42, 36–49. 10.1021/jm980285e. [DOI] [PubMed] [Google Scholar]
- Nolan C. J.; Evans B. A. J.; Smith H. J.; Ahmadi M.; Nicholls P. J.; Hewlins M. J. E. Inhibitors of Type 2 5α-Reductase from Human Genital Skin Fibroblasts Based on 3-Phenylmethylene Pyrrolidine-2, 5-dione. Pharm. Sci. 1997, 3, 249–257. [Google Scholar]
- Riemer N.; Shipman M.; Wessig P.; Schmidt B. Iterative Arylation of Itaconimides with Diazonium Salts through Electrophilic Palladium Catalysis: Divergent β-H-Elimination Pathways in Repetitive Matsuda-Heck Reactions. J. Org. Chem. 2019, 84, 5732–5746. 10.1021/acs.joc.9b00627. [DOI] [PubMed] [Google Scholar]
- Song Y.; Clizbe L.; Bhakta C.; Teng W.; Wong P.; Huang B.; Tran K.; Sinha U.; Park G.; Reed A.; Scarborough R. M.; Zhu B.-Y. Design and synthesis of factor Xa inhibitors and their prodrugs. Bioorg. Med. Chem. Lett. 2003, 13, 297–300. 10.1016/S0960-894X(02)00921-6. [DOI] [PubMed] [Google Scholar]
- Pal B.; Chang C.-H.; Zeng C.-J.; Lin C.-H. Template-Assisted Benzannulation Route to Pentacene and Tetracene Derivatives and its Application to Construct Amphiphilic Acenes That Self-Assemble into Helical Wires. Chem. - Eur. J. 2017, 23, 17542–17548. 10.1002/chem.201703084. [DOI] [PubMed] [Google Scholar]
- Remón P.; Bälter M.; Li S.; Andréasson J.; Pischel U. An All-Photonic Molecule-Based D Flip-Flop. J. Am. Chem. Soc. 2011, 133, 20742–20745. 10.1021/ja2100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andréasson J.; Straight S. D.; Moore T. A.; Moore A. L.; Gust D. Molecular All-Photonic Encoder-Decoder. J. Am. Chem. Soc. 2008, 130, 11122–11128. 10.1021/ja802845z. [DOI] [PubMed] [Google Scholar]
- Ohki S.; Ozawa N.; Yabe Y.; Matsuda H. Anticholinergic Agents. Synthesis of 1, 1, 4-Trimethyl- and 1, 1, 5-Trimethyl-3-diphenylmethyl-enepyrrolidinium Halides. Chem. Pharm. Bull. 1976, 24, 1362–1370. 10.1248/cpb.24.1362. [DOI] [PubMed] [Google Scholar]
- Mangaleswaran S.; Argade N. P. An Easy Access to (E)-Alkylidenesuccinic Acids. Synthesis 2003, 2003, 343–345. 10.1055/s-2003-37352. [DOI] [Google Scholar]
- Haval K. P.; Argade N. P. Haval-Argade contrathermodynamic rearrangement of alkylidenesuccinimides to alkylmaleimides via the corresponding isoimides: a general approach to alkyl and dialkyl substituted maleimides. Tetrahedron 2006, 62, 3557–3563. 10.1016/j.tet.2006.01.091. [DOI] [Google Scholar]
- Haval K. P.; Argade N. P. Synthesis of Natural Fimbrolides. Synthesis 2007, 2007, 2198–2202. 10.1055/s-2007-983755. [DOI] [Google Scholar]
- Chupakhin E.; Gecht M.; Ivanov A.; Kantin G.; Dar’in D.; Krasavin M. (E)-3-Arylidene-4-diazopyrrolidine-2,5-diones: Preparation and Use in RhII-Catalyzed X-H Insertion Reactions towards Novel, Medicinally Important Michael Acceptors. Synthesis 2021, 53, 1292–1300. 10.1055/s-0040-1706556. [DOI] [Google Scholar]
- Wang J.; Liu H.; Fan Y.; Yang Y.; Jiang Z.; Tan C.-H. Bicyclic Guanidine-Catalyzed Direct Asymmetric Allylic Addition of N-Aryl Alkylidene-Succinimides. Chem. - Eur. J. 2010, 16, 12534–12537. 10.1002/chem.201002183. [DOI] [PubMed] [Google Scholar]
- Zhao B.-L.; Du D.-M. Organocatalytic cascade Michael/Michael reaction for the asymmetric synthesis of spirooxindoles containing five contiguous stereocenters. Chem. Commun. 2016, 52, 6162–6165. 10.1039/C6CC00705H. [DOI] [PubMed] [Google Scholar]
- Zhao B.-L.; Zhang D.; Liu L.; Du D.-M. Organocatalytic asymmetric Michael addition of α-alkylidene succinimides to nitrostyrenes. Org. Biomol. Chem. 2016, 14, 6337–6345. 10.1039/C6OB00711B. [DOI] [PubMed] [Google Scholar]
- Tehri P.; Peddinti R. K. DBU-catalyzed [3 + 2] cycloaddition and Michael addition reactions of 3-benzylidene succinimides with 3-ylidene oxindoles and chalcones. Org. Biomol. Chem. 2019, 17, 3964–3970. 10.1039/C9OB00385A. [DOI] [PubMed] [Google Scholar]
- Song T.; Li L.; Zhou W.; Zheng Z.-J.; Deng Y.; Xu Z.; Xu L.-W. Enantioselective Copper-Catalyzed Azide-Alkyne Click Cycloaddition to Desymmetrization of Maleimide-Based Bis(alkynes). Chem. - Eur. J. 2015, 21, 554–558. 10.1002/chem.201405420. [DOI] [PubMed] [Google Scholar]
- Chen M.-Y.; Xu Z.; Chen L.; Song T.; Zheng Z.-J.; Cao J.; Cui Y.-M.; Xu L.-W. Catalytic Asymmetric Huisgen Alkyne-Azide Cycloaddition of Bisalkynes by Copper(I) Nanoparticles. ChemCatChem 2018, 10, 280–286. 10.1002/cctc.201701336. [DOI] [Google Scholar]
- Luo W.; Hu H.; Nian S.; Qi L.; Ling F.; Zhong W. Phosphine-catalyzed [3 + 2] annulation reaction: highly regio- and diastereoselective synthesis of 2-azaspiro[4.4]nonene-1,3-diones. Org. Biomol. Chem. 2017, 15, 7523–7526. 10.1039/C7OB01957B. [DOI] [PubMed] [Google Scholar]
- Yang W.-L.; Liu Y.-Z.; Luo S.; Yu X.; Fossey J. S.; Deng W.-P. The copper-catalyzed asymmetric construction of a dispiropyrrolidine skeleton via 1,3-dipolar cycloaddition of azomethine ylides to α-alkylidene succinimides. Chem. Commun. 2015, 51, 9212–9215. 10.1039/C5CC02362A. [DOI] [PubMed] [Google Scholar]
- Kaur A.; Kaur M.; Singh B. One-pot Regioselective Synthesis of Novel 1-N-Methyl-spiro[2,3′]oxindole-spiro[3,3″]-1″-N-arylpyrrolidine-2″,5″-dione-4-arylpyrrolidines through Multicomponent 1,3-Dipolar Cycloaddition Reaction of Azomethine Ylide. J. Heterocyclic Chem. 2015, 52, 827–833. 10.1002/jhet.2199. [DOI] [Google Scholar]
- Haddad S.; Boudriga S.; Porzio F.; Soldera A.; Askri M.; Knorr M.; Rousselin Y.; Kubicki M. M.; Golz C.; Strohmann C. Regio- and Stereoselective Synthesis of Spiropyrrolizidines and Piperazines through Azomethine Ylide Cycloaddition Reaction. J. Org. Chem. 2015, 80, 9064–9075. 10.1021/acs.joc.5b01399. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang W. Iridium-catalyzed asymmetric hydrogenation of α-alkylidene succinimides. Angew. Chem., Int. Ed. 2013, 52, 2203–2206. 10.1002/anie.201209126. [DOI] [PubMed] [Google Scholar]
- Kalia D.; Malekar P. V.; Parthasarathy M. Exocyclic Olefinic Maleimides: Synthesis and Application for Stable and Thiol-Selective Bioconjugation. Angew. Chem., Int. Ed. 2016, 55, 1432–1435. 10.1002/anie.201508118. [DOI] [PubMed] [Google Scholar]
- Kim H. Y.; Wiles J. A.; Wang Q.; Pais G. C. G.; Lucien E.; Hashimoto A.; Nelson D. M.; Thanassi J. A.; Podos S. D.; Deshpande M.; Pucci M. J.; Bradbury B. J. Exploration of the Activity of 7-Pyrrolidino-8-methoxyisothiazoloquinolones against Methicillin-Resistant Staphylococcus aureus (MRSA). J. Med. Chem. 2011, 54, 3268–3282. 10.1021/jm101604v. [DOI] [PubMed] [Google Scholar]
- Abdallah S. M.; Hefny H. A. Microwave synthesis of some new antimicrobial and antiproliferative butenamides and pyrrolidine-2,5-diones. Turk. J. Chem. 2011, 35, 463–474. [Google Scholar]
- Martynov A. V. New Approach to the Synthesis of trans-Aconitic Acid Imides. Russ. J. Org. Chem. 2005, 41, 723–726. 10.1007/s11178-005-0232-9. [DOI] [Google Scholar]
- Abass E. M. Synthesis of Arylmethylene-Butanimides and Butenamides and Evaluation of their Biological Activity. J. Chem. Bio. Phys. Sci. Sec. A 2015, 5 (1), 10–18. [Google Scholar]
- Ballini R.; Bosica G. A direct method for the synthesis of polyfunctionalized unsaturated carbonyl derivatives by Michael addition of nitroalkanes to enediones with the help of DBU. Tetrahedron 1995, 51, 4213–4222. 10.1016/0040-4020(95)00136-V. [DOI] [Google Scholar]
- Morita T.; Akita M.; Satoh T.; Kakiuchi F.; Miura M. Ruthenium-Catalyzed Cross-Coupling of Maleimides with Alkenes. Org. Lett. 2016, 18, 4598–4601. 10.1021/acs.orglett.6b02244. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Han S. H.; Oh Y.; Mishra N. K.; Lee S. H.; Oh J. S.; Kim I. S. Cross-Coupling of Acrylamides and Maleimides under Rhodium Catalysis: Controlled Olefin Migration. Org. Lett. 2016, 18, 2568–2571. 10.1021/acs.orglett.6b00909. [DOI] [PubMed] [Google Scholar]
- Keerthana M. S.; Manoharan R.; Jeganmohan M. Cobalt(III)-Catalyzed Redox-Neutral Coupling of Acrylamides with Activated Alkenes via C-H Bond Activation. Synthesis 2020, 52, 1625–1633. 10.1055/s-0039-1690866. [DOI] [Google Scholar]
- Schirmer M.-L.; Adomeit S.; Spannenberg A.; Werner T. Novel Base-Free Catalytic Wittig Reaction for the Synthesis of Highly Functionalized Alkenes. Chem. - Eur. J. 2016, 22, 2458–2465. 10.1002/chem.201503744. [DOI] [PubMed] [Google Scholar]
- Hedaya E.; Theodoropulos S. The preparation and reactions of stable phosphorus ylides derived from maleic anhydrides, maleimides or isomaleimides. Tetrahedron 1968, 24, 2241–2254. 10.1016/0040-4020(68)88126-8. [DOI] [Google Scholar]
- Haval K. P.; Argade N. P. General Strategy for the Synthesis of Natural and Unnatural Dialkylmaleic Anhydrides. J. Org. Chem. 2008, 73, 6936–6938. 10.1021/jo801284r. [DOI] [PubMed] [Google Scholar]
- Yan L.; Yang W.; Shen Y.; Li L.; Jiang Z. A One-pot Green Synthesis of Alkylidenesuccinimides. Chin. J. Chem. 2011, 29, 1906–1910. 10.1002/cjoc.201180332. [DOI] [Google Scholar]
- Bayat M.; Hosseini S. R.; Asmari E. Simple synthesis of (E) and (Z)-2-(arylmethylidene)-N-phenyl succinimides via Wittig olefination by using PS-TPP resin. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 98–102. 10.1080/10426507.2016.1225739. [DOI] [Google Scholar]
- Huang X.; Sha F. A Novel Three-Component Reaction of Allenoates, Isocyanides, and Carboxylic Acids: Facile Synthesis of Highly Substituted Acryl Imide Derivatives. J. Org. Chem. 2008, 73, 1173–1175. 10.1021/jo702382h. [DOI] [PubMed] [Google Scholar]
- Ramanivas T.; Parameshwar M.; Gayatri G.; Nanubolu J. B.; Srivastava A. K. Asymmetric Synthesis of Functionalized 2,5-Pyrrolidinediones and β-Lactams through Diastereospecific Cycloisomerization/Rearrangement of Chiral Ethanolamine-Derived Ugi Adducts. Eur. J. Org. Chem. 2017, 2017, 2245–2257. 10.1002/ejoc.201700031. [DOI] [Google Scholar]
- Driller K. M.; Klein H.; Jackstell R.; Beller M. Iron-Catalyzed Carbonylation: Selective and Efficient Synthesis of Succinimides. Angew. Chem., Int. Ed. 2009, 48, 6041–6044. 10.1002/anie.200902078. [DOI] [PubMed] [Google Scholar]
- Prateeptongkum S.; Driller K. M.; Jackstell R.; Spannenberg A.; Beller M. Efficient Synthesis of Biologically Interesting 3,4-Diaryl-Substituted Succinimides and Maleimides: Application of Iron-Catalyzed Carbonylations. Chem. - Eur. J. 2010, 16, 9606–9615. 10.1002/chem.201000369. [DOI] [PubMed] [Google Scholar]
- Liu H.; Lau G. P. S.; Dyson P. J. Palladium-Catalyzed Aminocarbonylation of Alkynes to Succinimides. J. Org. Chem. 2015, 80, 386–391. 10.1021/jo502412v. [DOI] [PubMed] [Google Scholar]
- Sha F.; Alper H. Ligand- and Additive-Controlled Pd-Catalyzed Aminocarbonylation of Alkynes with Aminophenols: Highly Chemo- and Regioselective Synthesis of α,β-Unsaturated Amides. ACS Catal. 2017, 7, 2220–2229. 10.1021/acscatal.7b00367. [DOI] [Google Scholar]
- van Leeuwen P. W. N. M.; Claver C.. Rhodium Catalyzed Hydroformylation; Springer: Netherlands, 2002; Vol. 22. [Google Scholar]
- Breit B. In Metal Catalyzed Reductive C@C Bond Formation: A Departure from Preformed Organometallic Reagents; Krische M. J., Ed.; Springer: Berlin, 2007; pp 139–172. [Google Scholar]
- Franke R.; Selent D.; Börner A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Friis S. D.; Lindhardt A. T.; Skrydstrup T. The Development and Application of Two-Chamber Reactors and Carbon Monoxide Precursors for Safe Carbonylation Reactions. Acc. Chem. Res. 2016, 49, 594–605. 10.1021/acs.accounts.5b00471. [DOI] [PubMed] [Google Scholar]
- Liu J.; Han Z.; Wang X.; Wang Z.; Ding K. Highly Regio- and Enantioselective Alkoxycarbonylative Amination of Terminal Allenes Catalyzed by a Spiroketal-Based Diphosphine/Pd(II) Complex. J. Am. Chem. Soc. 2015, 137, 15346–15349. 10.1021/jacs.5b07764. [DOI] [PubMed] [Google Scholar]
- Fang X.; Jackstell R.; Beller M. Selective Palladium-Catalyzed Aminocarbonylation of Olefins with Aromatic Amines and Nitroarenes. Angew. Chem., Int. Ed. 2013, 52, 14089–14093. 10.1002/anie.201308455. [DOI] [PubMed] [Google Scholar]
- Fang X.; Li H.; Jackstell R.; Beller M. Selective Palladium-Catalyzed Aminocarbonylation of 1,3-Dienes: Atom-Efficient Synthesis of β,γ-Unsaturated Amides. J. Am. Chem. Soc. 2014, 136, 16039–16043. 10.1021/ja507530f. [DOI] [PubMed] [Google Scholar]
- Liu J.; Schneider C.; Yang J.; Wei Z.; Jiao H.; Franke R.; Jackstell R.; Beller M. A General and Highly Selective Palladium-Catalyzed Hydroamidation of 1,3-Diynes. Angew. Chem., Int. Ed. 2021, 60, 371–379. 10.1002/anie.202010768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Li H.; Spannenberg A.; Franke R.; Jackstell R.; Beller M. Selective Palladium-Catalyzed Aminocarbonylation of Olefins to Branched Amides. Angew. Chem., Int. Ed. 2016, 55, 13544–13548. 10.1002/anie.201605104. [DOI] [PubMed] [Google Scholar]
- Goldbach V.; Falivene L.; Caporaso L.; Cavallo L.; Mecking S. Single-Step Access to Long-Chain α,ω-Dicarboxylic Acids by Isomerizing Hydroxycarbonylation of Unsaturated Fatty Acids. ACS Catal. 2016, 6, 8229–8238. 10.1021/acscatal.6b02622. [DOI] [Google Scholar]
- Dong K.; Sang R.; Wei Z.; Liu J.; Dühren R.; Spannenberg A.; Jiao H.; Neumann H.; Jackstell R.; Franke R.; Beller M. Cooperative catalytic methoxycarbonylation of alkenes: uncovering the role of palladium complexes with hemilabile ligands. Chem. Sci. 2018, 9, 2510–2516. 10.1039/C7SC02964K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; M’Barki M. A. Evidence of the formation of zerovalent palladium from Pd(OAc)2 and triphenylphosphine. Organometallics 1992, 11, 3009–3013. 10.1021/om00045a012. [DOI] [Google Scholar]
- Amatore C.; Carre E.; Jutand A.; M’Barki M. A. Rates and Mechanism of the Formation of Zerovalent Palladium Complexes from Mixtures of Pd(OAc)2 and Tertiary Phosphines and Their Reactivity in Oxidative Additions. Organometallics 1995, 14, 1818–1826. 10.1021/om00004a039. [DOI] [Google Scholar]
- Hunter E. P. L.; Lias S. G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. 10.1063/1.556018. [DOI] [Google Scholar]
- Balgotra S.; Verma P. K.; Vishwakarma R. A.; Sawant S. D. Catalytic advances in direct functionalizations using arylated hydrazines as the building blocks. Catal. Rev.: Sci. Eng. 2020, 62, 406–479. and references therein 10.1080/01614940.2019.1702191. [DOI] [Google Scholar]
- Huang Y.; Choy P. Y.; Wang J.; Tse M.-K; Sun R. W.-Y.; Chan A. S.-C.; Kwong F. Y. Palladium-Catalyzed Monoarylation of Arylhydrazines with Aryl Tosylates. J. Org. Chem. 2020, 85, 14664–14673. and references therein 10.1021/acs.joc.0c01599. [DOI] [PubMed] [Google Scholar]
- Abdou W. M.; Khidre R. E.; Barghash R. F. Regioselective Condensation of Alkylidenephosphoranes to N-Methoxy- and N-Anilino-1H isoindole-1,3-(2H)-diones. Synth. Commun. 2012, 42, 1967–1978. 10.1080/00397911.2010.551170. [DOI] [Google Scholar]
- King F. D. Synthesis and thermal reactions of 1,2-dihydro-1,2,4-benzotriazines. J. Chem. Soc., Perkin Trans. 1 1988, 3381–3385. 10.1039/p19880003381. [DOI] [Google Scholar]
- Cumming J. G.; Lin X.; Liu H.; Najera I.; Qiu Z.; Sandrin V.; Tang G.; Wu G. Patent WO 2018001948 A1, 2018.
- Sako M. Product class 18: pyridopyridazines. Science of Synthesis 2004, 16, 1109–1153. 10.1055/sos-SD-016-01336. [DOI] [Google Scholar]
- Goldstein D. M.; Rueth M. Patent US 20070219195 A1, 2007.
- Ye K.; Ai H.-L.; Liu J.-K. Identification and bioactivities of secondary metabolites derived from endophytic fungi isolated from ethnomedicinal plants of Tujia in Hubei province: a review. Nat. Prod. Bioprospect. 2021, 11, 185–205. 10.1007/s13659-020-00295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.