

Abstract
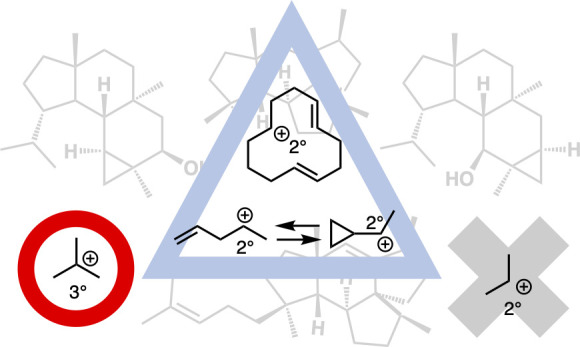
Some experimental observations indicate that a sequential formation of secondary (2°) carbocations might be involved in some biosynthetic pathways, including those of verrucosane-type diterpenoids and mangicol-type sesterterpenoids, but it remains controversial whether or not such 2° cations are viable intermediates. Here, we performed comprehensive density functional theory calculations of these biosynthetic pathways. The results do not support previously proposed pathways/mechanisms: in particular, we find that none of the putative 2° carbocation intermediates is involved in either of the biosynthetic pathways. In verrucosane biosynthesis, the proposed 2° carbocations (II and IV) in the early stage are bypassed by the formation of the adjacent 3° carbocations and by unusual skeletal rearrangement reactions, and in the later stage, the putative 2° carbocation intermediates (VI, VII, and VIII) are not present as the proposed forms but as nonclassical structures between homoallyl and cyclopropylcarbinyl cations. In the mangicol biosynthesis, one of the two proposed 2° carbocations (X) is bypassed by a C–C bond-breaking reaction to generate a 3° carbocation with a C=C bond, while the other (XI) is bypassed by a strong hyperconjugative interaction leading to a nonclassical carbocation. We propose new biosynthetic pathways/mechanisms for the verrucosane-type diterpenoids and mangicol-type sesterterpenoids. These pathways are in good agreement with the findings of previous biosynthetic studies, including isotope-labeling experiments and byproducts analysis, and moreover can account for the biosynthesis of related terpenes.
Keywords: terpene, biosynthesis, carbocation cascade, DFT study, nonclassical structure
Introduction
Terpenes/terpenoids are fascinating compounds due to their complicated and diverse structures and wide range of bioactivities. They are biosynthesized from simple unsaturated hydrocarbons via successive carbocation-mediated reactions triggered by terpene cyclases.1 A detailed knowledge of their biosynthetic mechanisms would be very helpful for understanding the evolution of particular biosynthetic pathways as well as for designing new biosynthetic routes for complex functional molecules. However, the mechanistic issues are extremely difficult to resolve fully by means of experimental studies alone, since terpene cyclization is a domino-type reaction occurring inside a single enzyme (“black box”). We recently established a powerful combination of quantum-chemical calculations with the artificial force induced reaction (AFIR) method2,3 to unveil complicated biosynthetic pathways/mechanisms, such as those leading to trichobrasilenol,4 quiannulatene,5 and cyclooctatin.6,7 In general, experimental and theoretical studies indicate that a terpene biosynthesis involves a sophisticated exothermic cascade of reactions, in which carbocation intermediates are converted into more stable intermediates such as allyl cations, tertiary (3°) carbocations, and cycloalkylcarbinyl cations. On the basis of experimental observations, it has been proposed that secondary (2°) carbocation intermediates play a role in some cases, such as in the biosynthesis of verrucosane-type diterpenoids and mangicol-type sesterterpenoids, but it remains controversial whether such 2° cations are viable intermediates. The stability of carbocations is one of the most important concepts in organic chemistry, and their stability relationships are fundamental to understanding many aspects of the reactivity of organic molecules/intermediates. For example, experimental studies show that sec-Bu+ is 14.5 kcal mol–1 more unstable than tert-Bu+.8−11 Therefore, it is of considerable interest to verify the putative involvement of 2° carbocations in the biosynthesis of natural products.
Verrucosane-type Diterpenoids
Verrucosan-2β-ol (ver) and neoverrucosan-5β-ol (neo), featuring a unique 3,6,6,5-tetracyclic system, are diterpenoids isolated from Chloroflexus aurantiacus, a filamentous, gliding, thermophilic phototrophic bacterium.12 Rieder et al. extensively investigated the biosynthetic pathway of verrucosan-2β-ol by means of an in vivo incorporation of singly or doubly 13C-labeled acetate, and they proposed a biosynthetic pathway involving the formation of many secondary carbocations (II, IV, VI, VII, and VIII) (Figure 1).13,14 Another important issue is to identify the bifurcation mechanism leading to the ver and neo skeletons.15 At the bifurcation point, a unique C–C bond rearrangement via a cyclopropylcarbinyl cation was proposed. A similar skeletal rearrangement is seen in the cyclooctatin biosynthesis.6,7 However, the key cyclopropylcarbinyl/homoallyl cation intermediates in cyclooctatin biosynthesis are categorized as 3° carbocations, whereas those in the proposed verrucosane-type biosynthesis are all 2° cations (VI, VII, and VIII).
Figure 1.

Proposed biosynthetic pathways of verrucosan-2β-ol and neoverrucosan-5β-ol.
Mangicol-type Sesterterpenoids
Mangicols are a family of marine fungal sesterterpenoids with a unique 6,5,5-spirotricyclic skeleton (Figure 2A),16,17 isolated from Fusarium heterosporum, and they show weak cytotoxicity toward various cancer cell lines. On the basis of experimental studies, the biosynthetic pathway shown in Figure 2B was proposed,16,17 involving the successive formation of two 2° carbocations (X and XI).
Figure 2.

(A) Chemical structures of mangicol A–G. (B) Proposed mechanism of mangicdiene biosynthesis.
We began our study by evaluating the proposed pathways of verrucosane and mangicol biosynthesis with density functional theory (DFT) calculations. Since the results did not support the involvement of the proposed 2° carbocations, we then comprehensively explored the biosynthetic pathways by means of DFT calculations combined with the AFIR method. On the basis of these findings, we propose new routes that are in good agreement with previous experimental biosynthetic studies and also provide plausible pathways for the formation of other related terpenes/terpenenoids.
Computational Methods
All calculations were performed with GRRM112,18−21 based on the Gaussian 16 program.22 Structure optimizations were performed at the M06-2X level in the gas phase using the 6-31+G(d,p) basis set.23 The vibrational frequencies were computed at the same level to check whether each optimized structure is an energy minimum (no imaginary frequency) or a transition state (single imaginary frequency). Intrinsic reaction coordinates (IRC) calculations24−27 were performed to track minimum energy paths from transition structures to the corresponding local minima. Single-point energies were calculated at the mPW1PW91/6-31+G(d,p) level28,29 based on the structures optimized by the M06-2X method. The Gibbs free energy used for discussion in this study was calculated by adding the gas-phase Gibbs free energy correction.
Results and Discussion
Verrucosane-type Biosynthesis
We first discuss the biosynthetic pathway of verrucosane-type diterpenoids. A systematic search of reaction pathways showed that the reaction IM0 → IM7 depicted in Figure 3A is the most favorable pathway. The dissociation of pyrophosphate followed by an E/Z isomerization30,31 initiates the exothermic carbocation cascade of verrucosane biosynthesis and yields an allylic carbocation (IM0). Then, the C3–C4 single bond rotates with a small activation barrier of 4.9 kcal mol–1 so that the C1 carbon can interact directly with the C6–C7 π bond to afford the 3° carbocation intermediate (IM1). As can be seen from the elongated C1–C6 bond distance (1.63 Å) as well as the natural population analysis (NPA) charges on C3 (+0.04) and C11 (+0.04), the 3° carbocation (+0.48) at the C7 position is partially stabilized by the C1–C6 σ bond and the distal C2–C3/C10–C11 π bonds. Thus, the monocyclic six-membered 3° cation IM1 undergoes a smooth skeletal rearrangement to afford 7,5-bicyclic 3° cation (IM2) in a single step with a large stabilization energy. A close examination of the IRC calculation results revealed that this step is actually concerted but involves two asynchronous events, namely, (1) a reverse (3° → 2° cation) Wagner-Meerwein rearrangement (ring expansion to give the 7-membered ring) and (2) an annulation to yield the 7,5-bicyclic system. Notably, the experimentally supported monocyclic seven-membered secondary carbocation structure (Figure 1, II) is not a minimum on the potential energy surface (PES). A conformational change of IM2 with an activation barrier of only 1.0 kcal mol–1 yields IM3, in which the 3° cation center is further stabilized by a cation-σ bond interaction assisted by the distal C14–C15 π bond, leading to the second skeletal rearrangement to afford 7,6,5-tricyclic 3° carbocation intermediate (IM4). Again, the experimentally supported 7,6-bicyclic 2° carbocation structure (Figure 1, IV) is not located. Thus, the proposed intermediary 2° carbocations in the early stage of the biosynthesis are bypassed by the formation of the adjacent 3° carbocations together with skeletal rearrangement via a kind of nonclassical carbocation interaction with a through-space participation of the C–C σ bond and the distal π bond. Note that, in these skeletal rearrangement processes (IM1 → IM2/IM3 → IM4), a direct attack of the corresponding (C10–C11/C14–C15) π electrons on the 3° carbocation centers (at C7/C11) does not occur due to the destabilization associated with a four-membered ring formation. From IM4, a 1,5-H shift takes place with a reasonable activation energy of 6.7 kcal mol–1 to migrate the isopropyl 3° cation to a homoallyl cation in a seven-membered ring (IM5) with a slight endothermicity. As judged from the NPA charge distributions and the bond lengths, we consider IM5 to be an intermediate structure between the homoallyl cation and the corresponding cyclopropylcarbinyl cation. The cyclopropylcarbinyl cation has highly distorted cyclopropane C–C σ bonds that can effectively stabilize the adjacent carbocation. Indeed, it has been reported that homoallyl/cyclopropylcarbinyl 3° cations play pivotal roles as stable intermediates in various stages of brasilane-type sesquiterpene3 and cyclooctatin-type diterpene biosynthesis.6,7 It is noteworthy that these were all 3° cations, whereas IM5 is a more unstable 2° cation, presumably reflecting the nonclassical intermediate (equilibrium) structure. Finally, an unusual C–C bond rearrangement takes place via a highly distorted bicyclobutonium 3° cation32,33 (IM6) with very low activation energy to give another homoallyl-cyclopropylcarbinyl intermediate structure (IM7). IM7 is slightly more stable than IM5 by 3.7 kcal mol–1, probably owing to hyperconjugation from neighboring moieties. The energy diagram (Figure 3B) immediately suggests that this is a thermodynamically and kinetically favorable biosynthetic reaction cascade: (1) the activation barriers are all low enough for the reactions to proceed smoothly at ambient temperature, (2) the entire energy profile descends as the reactions proceed, and (3) the overall exothermicity is very large (ca. 40 kcal mol–1). Any of the three skeletal rearrangements (IM1 → TS_1–2/IM3 → TS_3–4/IM5 → TS_5–6) could be the rate-determining step.
Figure 3.
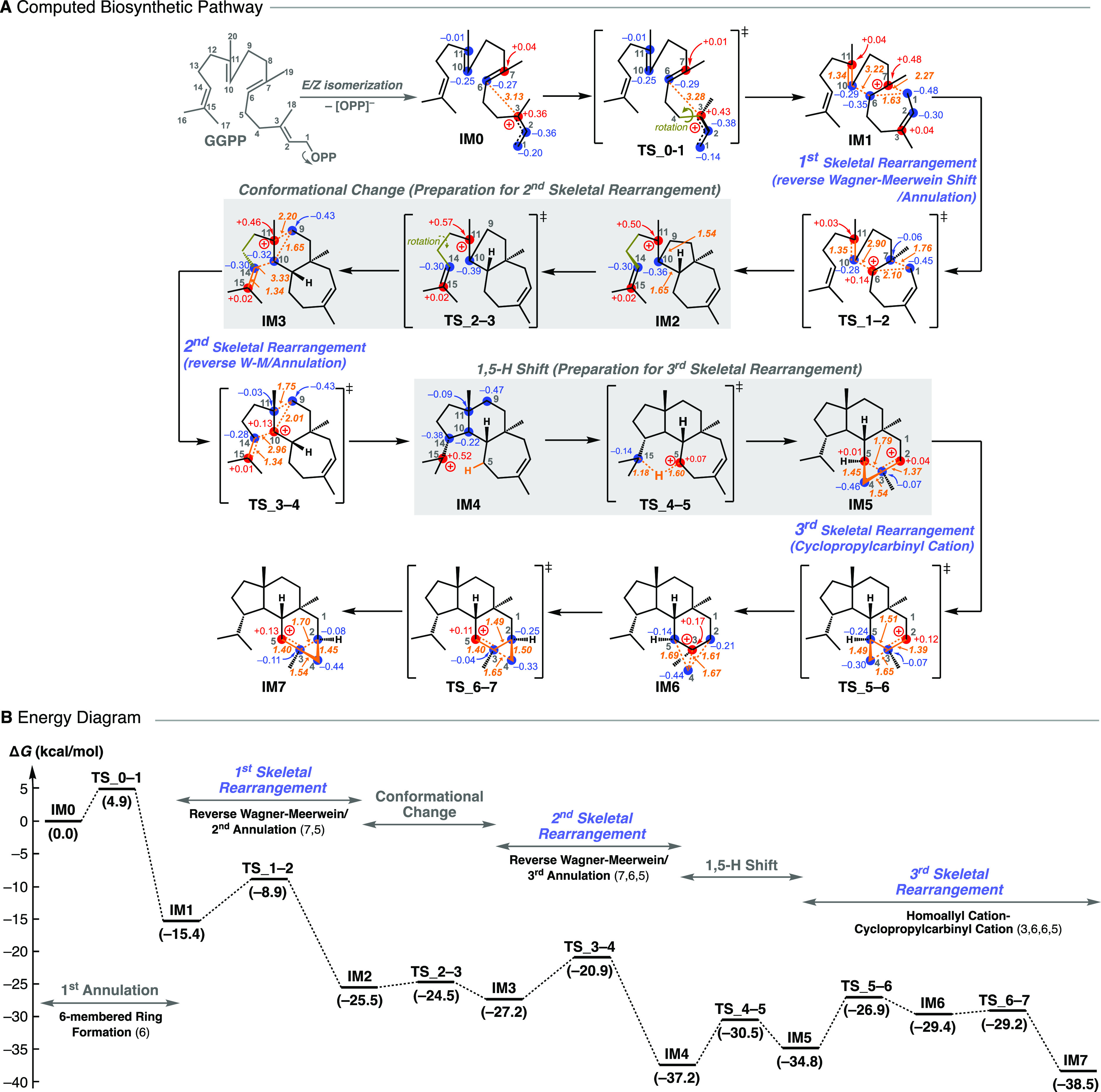
Results of a DFT evaluation of (A) the whole biosynthetic pathway and (B) the energy diagram of verrucosane. IM stands for intermediate. TS stands for transition state. Potential energies (kcal mol–1, Gibbs free energies calculated at the mPW1PW91/6-31+G(d,p) level based on M06-2X/6-31+G(d,p) geometries) relative to IM0 are shown in parentheses.
Mangicol-type Biosynthesis
Next, we discuss in detail the biosynthesis of mangicol-type sesterterpenoids. Only a few C20/C25 terpenoids based on 5,11-bicyclic systems, such as ophiobolin,34,35 cotylenin A,36 and fusicoccadiene,37 have been reported,38 in contrast to those derived from 5,15-systems, such as quiannulatene,5,39 sesterfisherol,40,41 (+)-astellatene,42,43 arathanadiene derivatives, sestermobaraenes,44 and aspergildienes.45 In the present study, IM8 was adopted as the simplest chemical model (Figure 4).46−49 The reaction pathway for the conversion of IM8 into IM11 is shown in Figure 4A, together with the relative energies with respect to IM8. The generation of allyl cation IM8, upon the loss of pyrophosphate of SM, triggers the carbocation cyclization cascade and affords a 5,11-bicyclic intermediate IM9a, which is a 3° carbocation intermediate, as generally found in terpene biosynthesis. After the conformational change of IM9a to IM9b with an activation barrier of only 0.2 kcal mol–1, an unusual C–C bond cleavage to afford the monocyclic 15-membered 3° carbocation IM10a proceeds in a single step from the 5/11 bicyclic structure via TS_9b–10a with an activation energy of 11.3 kcal mol–1. Notably, the experimentally supported 6,11-bicyclic 2° carbocation structure (X) is not a minimum on the PES.16,17 We anticipated that the present skeletal rearrangement would involve two asynchronous events, namely, (1) the 1,2-alkyl shift (ring expansion to give 6,11-bicyclic skeleton) and (2) the unusual C–C bond cleavage. Indeed, we found the corresponding peaks (shoulder_9b–10a-1 and shoulder_9b–10a-2, respectively) located before and after the main peak (TS_9b–10a) upon a close examination of the IRC analysis results (Figure 4B). As judged from the elongated C10–C11 (1.65 Å) and C13–C15 (1.65 Å) bond distances as well as the near linearity of the C10–C14–C15 moiety (dihedral angles of empty orbital of C14 with C10–C11 and C13–C15: 165.8° and 176.5°, respectively), we consider that shoulder_9b–10a-1 is close to the putative 2° carbocation structure in which the carbocation at C14 is partially stabilized by hyperconjugative interactions with the C10–C11 and C13–C15 σ bonds. If the C13–C15 bond were broken, a primary (1°) carbocation would be generated at C13, which would be very unstable.50,51 Hence, the C10–C11 bond is elongated (while the C13–C15 bond is shortened) to provide TS_9b–10a, leading to the formation of a stable 3° carbocation at C11 (IM10). Note that the distance between the C10–C14 double bond and 3° cation center C11 in IM10a is more than 3 Å, and there are NPA charges at C10 (−0.18) and C14 (−0.25), suggesting little or no interaction between these locations. IM10a has a homoallylic 3° carbocation moiety. Thus, IM10a is converted via TS_10a–10b to a more stable cyclopropylcarbinyl 3° cation IM10b, of a type that is often observed in various stages of brasilane-type sesquiterpene,4 cyclooctatin-type diterpene,6,7 and isoafricanol biosynthesis.52 Thus, IM10 has three equilibrium structures within ca. 4 kcal mol–1, and interconversion (conformational change) between homoallyl carbocations IM10a and IM10c proceeds via viacyclopropylcarbinyl cation IM10b. Then, a cation-mediated transannular cyclization takes place from the reactive conformer IM10c to form the 5,12-bicyclic system IM11 with a very low activation energy (2.6 kcal mol–1). Intermediate IM11 has an elongated C14–C15 single bond (1.66 Å) and C10–C14 double bond (1.41 Å), reflecting the partial breakdown of the C–C σ bond (1.54 Å) and π bond (1.34 Å), respectively. This suggests a nonclassical structure8,53 between the monocyclic 3° cation and bicyclic humulyl 2° cations, leading to the following multiple annulations (vide infra). A similar reaction is also found in variediene biosynthesis.54,55
Figure 4.

(A) Reaction pathways and potential energy changes from IM8 to IM11 (early stage of mangicol biosynthesis). The numberings are derived from those of GFPP. Potential energies (kcal mol–1, Gibbs free energies calculated at the mPW1PW91/6-31+G(d,p) level based on M06-2X/6-31+G(d,p) geometries) relative to IM8 are shown in parentheses. (B) A representative example of the evolution of key bond lengths in the conversion of IM9b to IM11.
We next located the later stage of mangicol biosynthesis, that is, the formation of the spirotricyclic structure (Figure 5). Route A is very similar to the previously proposed pathway, in which the annulation of IM11 takes place to give 5,9,5-tricyclic intermediate IM12 having B- and D-rings with a slight endothermicity, followed by 1,2-H shift via TS_12–13 with a large stabilization energy to give the bridgehead 3° carbocation IM13. From IM13, a smooth annulation involving conformational change of the nine-membered ring and 1,2-H shift proceeds to construct the 6,5,5-spirotricyclic skeleton IM14a in a single step with a large exothermicity. Then, IM14a is subjected to deprotonation to give the mangicol core skeleton (PD). The energy profile is consistent with the previously proposed pathway (Figure 2B), with reasonable activation barriers (all low enough for the reactions to proceed smoothly at ambient temperature) and with an overall large exothermicity (over 35 kcal mol–1). However, another unprecedented route, Route B (via 5,6,5,5-tetracyclic intermediate IM17 having A- and D-rings), which was located by application of the AFIR method for comprehensive searching of reaction paths, turned out to be the most favorable (Figure 5B). As supported by the NPA charge distribution of IM11, where C10 (+0.26) and C3 (+0.04) are more positive than the C6 and C7 carbons, cation-stitching multiple annulation initiates this pathway with a very low activation energy (3.8 kcal mol–1) to form the 5,5,7,4-tetracyclic intermediate IM15a with a large exothermicity (22.2 kcal mol–1), in which the highly distorted cyclobutane C3–C6 bond effectively stabilizes the adjacent C7 carbocation.56 Thus, after the interconversion from IM15a to IM15c, a 1,2-H shift takes place smoothly to give a 5,5,9-tricyclic 3° cation (IM16) stabilized by a cation-π interaction3,57 with the newly generated C6–C7 double bond. This is followed by another smooth cation-mediated annulation to give the 5,5,6,5-tetracyclic skeleton IM17, which undergoes a 1,2-H shift via TS_17–18a to give IM18a. Then a C–C bond rearrangement takes place to yield the 5,6,5,5-tetraspirocyclic mangicol core skeleton (IM14b), which is subjected to a deprotonation to form PD. Notably, this new route is not only kinetically and thermodynamically the most favorable pathway but also appears to provide a versatile biosynthetic pathway leading to the formation of related terpenes/terpenoids, including tsukubadiene,58 variediene,54,55 deoxyconidiogenol/conidiogenone,59,60 and phomopsene/methyl phomopsenonate61,62 (Figure 5B,C). A careful structural comparison of the terpene cyclases responsible for the formation of mangicol, tsukubadiene, variediene, deoxyconidiogenol, and phomopsene could be helpful to clarify how the structural diversification is controlled.
Figure 5.

Reaction pathways and potential energy changes from IM11 to PD (later stage of mangicol biosynthesis, Part 2). See Figure 3 for details. (A) Previously proposed route. (B) New route. (C) Comparison of the energy profiles of route A (dashed line) and route B (solid line). Potential energies (kcal mol–1, Gibbs free energies calculated at the mPW1PW91/6-31+G(d,p) level based on M06-2X/6-31+G(d,p) geometries) relative to IM8 are shown in parentheses.
Conclusion
In conclusion, the current computational study has uncovered in detail the biosynthetic pathways of the verrucosane-type diterpenoids and mangicol-type sesterterpenoids as well as provided new insight into the mechanisms of structure diversification in terpene biosynthesis, especially the exquisite skeletal construction processes and conformational changes (the Cartesian coordinates of the three-dimensional structures of all species are given in the Supporting Information). Remarkably, we found that none of the previously proposed 2° carbocation intermediates was obtained as a minimum on the PES. The verrucosane biosynthetic cascade bypasses the formation of unstable 2° carbocations II and IV by the formation of adjacent 3° carbocations (IM1 and IM3) combined with skeletal rearrangement reactions involving reverse (3° → 2° cation) Wagner-Meerwein rearrangements. Other putative 2° cations were located in equilibrium with nonclassical carbocation intermediates (IM5 and IM7) via viaa bicyclobutonium cation (IM6), and this could be the branching point between the verrucosan-2β-ol (ver) and neoverrucosan-5β-ol (neo) biosyntheses. In the mangicol cyclization cascade, the formation of putative 2° carbocation X is bypassed by breaking the adjacent C–C bond to form the more stable 3° carbocation (IM10). Although C–C bond cleavage is an endothermic reaction, the unfavorable energy loss is compensated by the simultaneous generation of a C=C double bond and a more stable 3° carbocation. Another proposed 2° carbocation XI is avoided as a result of a strong hyperconjugative interaction with the adjacent C–C bond, affording a nonclassical structure (IM11) between a 3° carbocation and a humulyl 2° cation. We further found a new, energetically viable pathway for the 6,5,5-spirotricycle formation in the mangicol biosynthesis, and this can also account for the formation of other terpenes/terpenenoids. A future comparative study of the terpene cyclases responsible for mangicol, tsukubadiene, variediene, deoxyconidiogenol, and phomopsene formations could help to establish the molecular basis of the regulation of the branching biosynthetic pathways by these enzymes. Thus, the results presented here are helpful to complete the picture of verrucosane and mangicol biosynthesis and also offer insights into the stability and reactivity of various carbocations and bonds that should be useful not only in terpene biosynthesis but also in fundamental organic chemistry. In particular, this work underlines that great caution is needed in suggesting the involvement of 2° carbocations (even in the cases of humulyl cations and cycloalkylcarbinyl cations) in the biosynthesis of natural products, since they are very unstable compared with 3° cations. We hope this work will be helpful for future mechanistic investigations of terpenes/terpenoids biosynthesis.
Acknowledgments
This work was supported by JSPS KAKENHI (S) (No. 17H06173 (M.U.)), MEXT Leading Initiative for Excellent Young Researchers (No. JPMXS0320200422 (H.S.)), JSPS Grant-in-Aid for Scientific Research on Innovative Areas (No. 19H04643 (M.U.)), and JST CREST (No. JPMJCR19R2 (M.U.)).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00178.
Computational details, three-dimensional representation of the structure, coordinates and energies for all computed structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Christianson D. W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. 10.1021/acs.chemrev.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Ohno K.; Morokuma K. Systematic Exploration of the Mechanism of Chemical Reactions: the Global Reaction Route Mapping (GRRM) Strategy Using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. 10.1039/c3cp44063j. [DOI] [PubMed] [Google Scholar]
- Isegawa M.; Maeda S.; Tantillo D. J.; Morokuma K. Predicting Pathways for Terpene Formation from First Principles-Routes to Known and New Sesquiterpenes. Chem. Sci. 2014, 5, 1555–1560. 10.1039/c3sc53293c. [DOI] [Google Scholar]
- Sato H.; Hashishin T.; Kanazawa J.; Miyamoto K.; Uchiyama M. DFT Study of a Missing Piece in Brasilane-Type Structure Biosynthesis: An Unusual Skeletal Rearrangement. J. Am. Chem. Soc. 2020, 142, 19830–19834. 10.1021/jacs.0c09616. [DOI] [PubMed] [Google Scholar]
- Sato H.; Mitsuhashi T.; Yamazaki M.; Abe I.; Uchiyama M. Computational Studies on Biosynthetic Carbocation Rearrangements leading to Quiannulatene: Initial Conformation Regulates Biosynthetic Route, Stereochemistry, and Type of Skeleton. Angew. Chem., Int. Ed. 2018, 57, 14752–14757. 10.1002/anie.201807139. [DOI] [PubMed] [Google Scholar]
- Sato H.; Teramoto K.; Masumoto Y.; Tezuka N.; Sakai K.; Ueda S.; Totsuka Y.; Shinada T.; Nishiyama M.; Wang C.; Kuzuyama T.; Uchiyama M. "Cation-Stitching Cascade”: Exquisite Control of Terpene Cyclization in Cyclooctatin Biosynthesis. Sci. Rep. 2016, 5, 18471–18476. 10.1038/srep18471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y. J.; Tantillo D. J. The Energetic Viability of an Unexpected Skeletal Rearrangement in Cyclooctatin Biosynthesis. Org. Biomol. Chem. 2015, 13, 10273–10278. 10.1039/C5OB01785H. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Prakash G. K. S. In Carbocation Chemistry ;Wiley-VCH: Weinheim, Germany, 2004; pp 1–408. [Google Scholar]
- Carey F. A.; Sundberg R. J. In Advanced Organic Chemistry ;Kluwer Academic: New York, 2000; pp 263–350. [Google Scholar]
- Deno N. C.; Jaruzelski J. J.; Schriesheim A. Carbonium Ions. I. An Acidity Function (C0) Derived from Arylcarbonium Ion Equilibria. J. Am. Chem. Soc. 1955, 77, 3044–3051. 10.1021/ja01616a036. [DOI] [Google Scholar]
- Bittner E. W.; Arnett E. M.; Saunders M. Quantitative Preparation and Enthalpy of Rearrangement of the sec-Butyl Cation. J. Am. Chem. Soc. 1976, 98, 3734–3735. 10.1021/ja00428a072. [DOI] [Google Scholar]
- Hefter J.; Richnow H. H.; Fischer U.; Trendel J. M.; Michaelis W. (−)-Verrucosan-2β-ol from the Phototrophic Bacterium Chloroflexus aurantiacus: First Report of a Verrucosane-type Diterpenoid from a Prokaryote. J. Gen. Microbiol. 1993, 139, 2757–2761. 10.1099/00221287-139-11-2757. [DOI] [Google Scholar]
- Rieder C.; Strauss G.; Fuchs G.; Arigoni D.; Bacher A.; Eisenreich W. Biosynthesis of the Diterpene Verrucosan-2β-ol in the Phototrophic Eubacterium Chloroflexus aurantiacus. J. Biol. Chem. 1998, 273, 18099–18108. 10.1074/jbc.273.29.18099. [DOI] [PubMed] [Google Scholar]
- Eisenreich W. C.; Rieder C.; Grammes C.; Hessler G.; Adam K. P.; Becker H.; Arigoni D.; Bacher A. Biosynthesis of a Neo-epi-verrucosane Diterpene in the Liverwort Fossombronia alaskana. J. Biol. Chem. 1999, 274, 36312–36320. 10.1074/jbc.274.51.36312. [DOI] [PubMed] [Google Scholar]
- Yang Y. L.; Zhang S.; Ma K.; Xu Y.; Tao Q.; Chen Y.; Chen J.; Guo S.; Ren J.; Wang W.; Tao Y.; Yin W. B.; Liu H. Discovery and Characterization of a New Family of Diterpene Cyclases in Bacteria and Fungi. Angew. Chem., Int. Ed. 2017, 56, 4749–4752. 10.1002/anie.201700565. [DOI] [PubMed] [Google Scholar]
- Renner M. K.; Jensen P. R.; Fenical W. Mangicols: Structures and Biosynthesis of a New Class of Sesterterpene Polyols from a Marine Fungus of the Genus Fusarium. J. Org. Chem. 2000, 65, 4843–4852. 10.1021/jo000081h. [DOI] [PubMed] [Google Scholar]
- Bian G.; Han Y.; Hou A.; Yuan Y.; Liu X.; Deng Z.; Liu T. Releasing the Potential Power of Terpene Synthases by a Robust Precursor Supply Platform. Metab. Eng. 2017, 42, 1–8. 10.1016/j.ymben.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Maeda S.; Osada Y.; Morokuma K.; Ohno K.. GRRM11 , ver. 11.03; Institute for Quantum Chemical Exploration, 2012.
- Ohno K.; Maeda S. A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy Surface. Chem. Phys. Lett. 2004, 384, 277–282. 10.1016/j.cplett.2003.12.030. [DOI] [Google Scholar]
- Maeda S.; Ohno K. Global Mapping of Equilibrium and Transition Structures on Potential Energy Surfaces by the Scaled Hypersphere Search Method: Applications to ab initio Surfaces of Formaldehyde and Propyne Molecules. J. Phys. Chem. A 2005, 109, 5742–5753. 10.1021/jp0513162. [DOI] [PubMed] [Google Scholar]
- Ohno K.; Maeda S. Global Reaction Route Mapping on Potential Energy Surfaces of Formaldehyde, Formic Acid, and Their Metal-Substituted Analogues. J. Phys. Chem. A 2006, 110, 8933–8941. 10.1021/jp061149l. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 16 , rev. C.01; Gaussian, Inc.: Wallingford, CT, 2016. (full reference is in the; Supporting Information).
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Fukui K. The Path of Chemical Reactions - The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. 10.1021/ar00072a001. [DOI] [Google Scholar]
- Page M.; Doubleday C.; McIver J. W. Jr. Following Steepest Descent Reaction Paths. The Use of Higher Energy Derivatives with ab initio Electronic Structure Methods. J. Chem. Phys. 1990, 93, 5634–5642. 10.1063/1.459634. [DOI] [Google Scholar]
- Ishida K.; Morokuma K.; Komornicki A. The Intrinsic Reaction Coordinate. An ab initio Calculation for HNC→HCN and H– + CH4→CH4 + H–. J. Chem. Phys. 1977, 66, 2153–2156. 10.1063/1.434152. [DOI] [Google Scholar]
- Gonzalez C.; Schlegel H. B. Reaction Path Following in Mass-weighted Internal Coordinates. J. Phys. Chem. 1990, 94, 5523–5527. 10.1021/j100377a021. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange Functionals with Improved Long-range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The mPW and mPW1PW Models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Matsuda S. P. T.; Wilson W. K.; Xiong Q. Mechanistic Insights into Triterpene Synthesis from Quantum Mechanical Calculations. Detection of Systematic Errors in B3LYP Cyclization Energies. Org. Biomol. Chem. 2006, 4, 530–543. 10.1039/b513599k. [DOI] [PubMed] [Google Scholar]
- Croteau R. B.; Shaskus J. J.; Renstrom B.; Felton N. M.; Cane D. E.; Saito A.; Chang C. Mechanism of the Pyrophosphate Migration in the Enzymatic Cyclization of Geranyl and Linalyl Pyrophosphates to (+)- and (−)-Bornyl Pyrophosphates. Biochemistry 1985, 24, 7077–7085. 10.1021/bi00346a009. [DOI] [PubMed] [Google Scholar]
- Hong Y. J.; Tantillo D. J. Quantum Chemical Dissection of the Classic Terpinyl/Pinyl/Bornyl/Camphyl Cation Conundrum - The Role of Pyrophosphate in Manipulating Pathways to Monoterpenes. Org. Biomol. Chem. 2010, 8, 4589–4600. 10.1039/c0ob00167h. [DOI] [PubMed] [Google Scholar]
- Mazur R. H.; White W. N.; Semenow D. A.; Lee C. C.; Silver M. S.; Roberts J. D. Small-ring Compounds. XXIII. The Nature of the Intermediates in Carbonium Ion-type Interconversion Reactions of Cyclopropylcarbinyl, Cyclobutyl and Allylcarbinyl Derivatives. J. Am. Chem. Soc. 1959, 81, 4390–4398. 10.1021/ja01525a073. [DOI] [Google Scholar]
- Hong Y. J.; Giner J.-L.; Tantillo D. J. Bicyclobutonium Ions in Biosynthesis – Interconversion of Cyclopropyl-Containing Sterols from Orchids. J. Am. Chem. Soc. 2015, 137, 2085–2088. 10.1021/ja512901a. [DOI] [PubMed] [Google Scholar]
- Nozoe S.; Morisaki M.; Tsuda K.; Iitaka Y.; Takahashi N.; Tamura S.; Ishibashi K.; Shirasaka M. The Structure of Ophiobolin, a C25 Terpenoid Having a Novel Skeleton. J. Am. Chem. Soc. 1965, 87, 4968–4970. 10.1021/ja00949a061. [DOI] [PubMed] [Google Scholar]
- Chiba R.; Minami A.; Gomi K.; Oikawa H. Identification of Ophiobolin F Synthase by a Genome Mining Approach: A Sesterterpene Synthase from Aspergillus clavatus. Org. Lett. 2013, 15, 594–597. 10.1021/ol303408a. [DOI] [PubMed] [Google Scholar]
- Sassa T.; Ooi T.; Nukita M.; Ikeda M.; Kato N. Structural Confirmation of Cotylenin A, a Novel Fusicoccane-diterpene Glycoside with Potent Plant Growth-regulating Activity from Cladosporium Fungus sp. 501–7W. Biosci. Biotechnol. Biochem. 1998, 62, 1815–1818. 10.1271/bbb.62.1815. [DOI] [PubMed] [Google Scholar]
- Toyomasu T.; Tsukahara M.; Kaneko A.; Niida R.; Mitsuhashi W.; Dairi T.; Kato N.; Sassa T. Fusicoccins are Biosynthesized by an Unusual Chimera Diterpene Synthase in Fungi. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3084–3088. 10.1073/pnas.0608426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami A.; Ozaki T.; Liu C.; Oikawa H. Cyclopentane-forming Di/Sesterterpene Synthases: Widely Distributed Enzymes in Bacteria, Fungi, and Plants. Nat. Prod. Rep. 2018, 35, 1330–1346. 10.1039/C8NP00026C. [DOI] [PubMed] [Google Scholar]
- Okada M.; Matsuda Y.; Mitsuhashi T.; Hoshino S.; Mori T.; Nakagawa K.; Quan Z.; Qin B.; Zhang H.; Hayashi F.; Kawaide H.; Abe I. Genome-Based Discovery of an Unprecedented Cyclization Mode in Fungal Sesterterpenoid Biosynthesis. J. Am. Chem. Soc. 2016, 138, 10011–10018. 10.1021/jacs.6b05799. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Minami A.; Mandi A.; Liu C.; Taniguchi T.; Kuzuyama T.; Monde K.; Gomi K.; Oikawa H. Genome Mining for Sesterterpenes Using Bifunctional Terpene Synthases Reveals a Unified Intermediate of Di/Sesterterpenes. J. Am. Chem. Soc. 2015, 137, 11846–11853. 10.1021/jacs.5b08319. [DOI] [PubMed] [Google Scholar]
- Sato H.; Narita K.; Minami A.; Yamazaki M.; Wang C.; Suemune H.; Nagano S.; Tomita T.; Oikawa H.; Uchiyama M. Theoretical Study of Sesterfisherol Biosynthesis: Computational Prediction of Key Amino Acid Residue in Terpene Synthase. Sci. Rep. 2018, 8, 2473–2481. 10.1038/s41598-018-20916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A. C.; Kautsar S. A.; Hong Y. J.; Medema M. H.; Bond A. D.; Tantillo D. J.; Osbourn A. Unearthing a Sesterterpene Biosynthetic Repertoire in the Brassicaceae through Genome Mining Reveals Convergent Evolution. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6005–E6014. 10.1073/pnas.1705567114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A. C.; Hong Y. J.; Bond A. D.; Tantillo D. J.; Osbourn A. Diverged Plant Terpene Synthases Reroute the Carbocation Cyclization Path towards the Formation of Unprecedented 6/11/5 and 6/6/7/5 Sesterterpene Scaffolds. Angew. Chem., Int. Ed. 2018, 57, 1291–1295. 10.1002/anie.201711444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou A.; Dickschat J. S. The Biosynthetic Gene Cluster for Sestermobaraenes—Discovery of a Geranylfarnesyl Diphosphate Synthase and a Multiproduct Sesterterpene Synthase from Streptomyces Mobaraensis. Angew. Chem., Int. Ed. 2020, 59, 19961–19965. 10.1002/anie.202010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Cai Y. S.; Cheng F.; Yang C.; Zhang W.; Yu W.; Yan J.; Deng Z.; Hong K. Genome Mining Reveals a Multiproduct Sesterterpenoid Biosynthetic Gene Cluster in Aspergillus Ustus. Org. Lett. 2021, 23, 1525–1529. 10.1021/acs.orglett.0c03996. [DOI] [PubMed] [Google Scholar]
- Hess B. A. Concomitant C-Ring Expansion and D-Ring Formation in Lanosterol Biosynthesis from Squalene without Violation of Markovnikov’s Rule. J. Am. Chem. Soc. 2002, 124, 10286–10287. 10.1021/ja026850r. [DOI] [PubMed] [Google Scholar]
- Hess B. A. Formation of the C Ring in the Lanosterol Biosynthesis from Squalene. Org. Lett. 2003, 5, 165–167. 10.1021/ol027449c. [DOI] [PubMed] [Google Scholar]
- Hess B. A.; Smentek L. Concerted Nature of AB Ring Formation in the Enzymatic Cyclization of Squalene to Hopenes. Org. Lett. 2004, 6, 1717–1720. 10.1021/ol0496125. [DOI] [PubMed] [Google Scholar]
- Hess B. A. Jr.; Smentek L. The Concerted Nature of the Cyclization of Squalene Oxide to the Protosterol Cation. Angew. Chem., Int. Ed. 2013, 52, 11029–11033. 10.1002/anie.201302886. [DOI] [PubMed] [Google Scholar]
- A similar hyperconjugative interaction has been observed in abietadiene biosynthesis, see: Tantillo D. J.The Carbocation Continuum in Terpene Biosynthesis—Where are the Secondary Cations? Chem. Soc. Rev. 2010, 39, 2847–2854. 10.1039/b917107j [DOI] [PubMed] [Google Scholar]
- Hong Y. J.; Tantillo D. J. A Potential Energy Surface Bifurcation in Terpene Biosynthesis. Nat. Chem. 2009, 1, 384–389. 10.1038/nchem.287. [DOI] [PubMed] [Google Scholar]
- Rabe P.; Samborskyy M.; Leadlay P. F.; Dickschat J. S. Isoafricanol Synthase from Streptomyces Malaysiensis. Org. Biomol. Chem. 2017, 15, 2353–2358. 10.1039/C7OB00234C. [DOI] [PubMed] [Google Scholar]
- Scholz F.; Himmel D.; Heinemann F. W.; Schleyer R.; Meyer K.; Krossing I. Crystal Structure Determination of the Nonclassical 2-Norbornyl Cation. Science 2013, 341, 62–64. 10.1126/science.1238849. [DOI] [PubMed] [Google Scholar]
- Rinkel J.; Steiner S. T.; Bian G.; Chen R.; Liu T.; Dickschat J. S. A Family of Related Fungal and Bacterial Di- and Sesterterpenes: Studies on Fusaterpenol and Variediene. ChemBioChem 2020, 21, 486–491. 10.1002/cbic.201900462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y. J.; Tantillo D. J. The Variediene-Forming Carbocation Cyclization/Rearrangement Cascade. Aust. J. Chem. 2017, 70, 362–366. 10.1071/CH16504. [DOI] [Google Scholar]
- Winstein S.; Holness N. J. Neighboring Carbon and Hydrogen. XVIII. Solvolysis of the Nopinyl p-Bromobenzenesulfonates. J. Am. Chem. Soc. 1955, 77, 3054–3061. 10.1021/ja01616a038. [DOI] [Google Scholar]
- Yamato T.; Fujita K.; Shinoda N.; Noda K.; Nagano Y.; Arimura T.; Tashiro M. Through-space Electronic Interactions of [2.2]Metacyclophane Benzyl Cations. Res. Chem. Intermed. 1996, 22, 871–897. 10.1163/156856796X00539. [DOI] [Google Scholar]
- Rabe P.; Rinkel J.; Dolja E.; Schmitz T.; Nubbemeyer B.; Luu T. H.; Dickschat J. S. Mechanistic Investigations of Two Bacterial Diterpene Cyclases: Spiroviolene Synthase and Tsukubadiene Synthase. Angew. Chem., Int. Ed. 2017, 56, 2776–2779. 10.1002/anie.201612439. [DOI] [PubMed] [Google Scholar]
- Roncal T.; Cordobés S.; Sterner O.; Ugalde U. Conidiation in Penicillium Cyclopium is Induced by Conidiogenone, an Endogenous Diterpene. Eukaryotic Cell 2002, 1, 823–829. 10.1128/EC.1.5.823-829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi T.; Kikuchi T.; Hoshino S.; Ozeki M.; Awakawa T.; Shi S. P.; Fujita M.; Abe I. Crystalline Sponge Method Enabled the Investigation of a Prenyltransferase-Terpene Synthase Chimeric Enzyme, Whose Product Exhibits Broadened NMR Signals. Org. Lett. 2018, 20, 5606–5609. 10.1021/acs.orglett.8b02284. [DOI] [PubMed] [Google Scholar]
- Toyomasu T.; Kaneko A.; Tokiwano T.; Kanno Y.; Kanno Y.; Niida R.; Miura S.; Nishioka T.; Ikeda C.; Mitsuhashi W.; Dairi T.; Kawano T.; Oikawa H.; Kato N.; Sassa T. Biosynthetic Gene-Based Secondary Metabolite Screening: a New Diterpene, Methyl Phomopsenonate, from the Fungus Phomopsis amygdali. J. Org. Chem. 2009, 74, 1541–1548. 10.1021/jo802319e. [DOI] [PubMed] [Google Scholar]
- Lauterbach L.; Rinkel J.; Dickschat J. S. Two Bacterial Diterpene Synthases from Allokutzneria albata Produce Bonnadiene, Phomopsene, and Allokutznerene. Angew. Chem., Int. Ed. 2018, 57, 8280–8283. 10.1002/anie.201803800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.