

Abstract
Dried blood spot (DBS) metabolite analysis is a central tool for the clinic, e.g., newborn screening. Instead of applying multiple analytical methods, a single liquid chromatography-mass spectrometry (LC–MS) method was developed for metabolites spanning from highly polar glucose to hydrophobic long-chain acylcarnitines. For liquid chromatography, a diphenyl column and a multi-linear solvent gradient operated at elevated flow rates allowed for an even-spread resolution of diverse metabolites. Injecting moderate volumes of DBS organic extracts directly, in contrast to evaporation and reconstitution, provided substantial increases in analyte recovery. Q Exactive MS settings were also tailored for sensitivity increases, and the method allowed for analyte retention time and peak area repeatabilities of 0.1–0.4 and 2–10%, respectively, for a wide polarity range of metabolites (log P −4.4 to 8.8). The method’s performance was suited for both untargeted analysis and targeted approaches evaluated in clinically relevant experiments.
Keywords: metabolomics, dried blood spots, LC−MS, inborn errors of metabolism
1. Introduction
Reliable and accurate analyses of biological samples are essential for clinical purposes. Untargeted analyses of small biomolecules, i.e., metabolomics, provide the advantage of enabling the detection of large numbers of compounds at the same time. The approach has been described as having no discrimination in terms of which compounds can be detected within certain instrumental limitations, e.g., mass range.1,2 In human clinical samples, these metabolites include endogenous molecules, all the exogenous molecules that we are voluntarily and involuntarily exposed to, and a myriad of metabolites generated by our microbiomes.
Inborn errors of metabolism (IEMs) represent a large and diverse group of diseases.3−5 An IEM is typically caused by genetic mutations in a single gene, leading to changes in function or quantity of a vital enzyme. This in turn leads to deviations in the patient’s metabolism. IEMs can have serious consequences such as severe brain damage or death and are included in newborn screening programs in most countries. For IEM diagnostics, targeted analyses are mostly used, i.e., monitoring a limited number of predefined analytes. Untargeted analyses can also be of great importance for these diseases, as symptoms are often diverse and unspecific, making diagnostics based on targeted analyses difficult and potentially very time-consuming.5
Dried blood spots (DBS) are increasingly used in diagnostics due to ease of sampling, storage, and transportation, and higher stability of most analytes compared with, e.g., plasma or whole blood samples.6−8 Palmer et al. reported detecting more metabolites in DBS than in plasma samples, likely due to red and white blood cell metabolites.9 DBS are also widely used in newborn screening,10,11 allowing newborn screening even in remote areas, as samples can be gathered anywhere in the world and sent to laboratories elsewhere for analysis.
Mass spectrometry (MS) is a key tool in metabolomics.12−14 MS enables reliable and accurate identification of compounds and generally provides increased sensitivity compared to, e.g., NMR spectroscopy techniques.15 Regarding clinical metabolic screening, methods may be built upon direct infusion mass spectrometry due to, e.g., speed and simplicity.10,16 On the other hand, applying liquid chromatography (LC) upstream to MS has the advantage of providing additional separation of compounds, thereby increasing the number of identifiable compounds and reduction of matrix effects such as ion suppression and ion enhancement.14,17 Applying LC–MS also increases the chance of identification of isomers and provides the additional parameter of retention time for identification purposes.14,18
A key challenge with analyses of biological samples is the wide polarity range of clinically relevant metabolites, ranging from, e.g., hydrophobic fatty acids to polar amino acids, often addressed by using several analytical methods/platforms, e.g., employing gas chromatography in addition to hydrophilic interaction liquid chromatography (HILIC) and reversed phase LC, to cover a broad part of the metabolome.19−23 Although comprehensive analysis is provided, multi-separation approaches can be time-consuming and laborious, sometimes requiring extensive sample preparation procedures. Several single metabolomics methods have been developed for, e.g., plasma and urine analysis, with excellent results (see, e.g., refs (24) and (25)). Broad range single methods are even beginning to merge different omics; He et al. have recently combined lipidomics and proteomics with a single-shot technology.26 We set out to develop and optimize a single method suited for covering a broad range of metabolites focusing on dried blood spots while still ensuring simplicity for practical clinical use, e.g., IEM diagnostics. We were interested if a single LC–MS method would have satisfactory analytical performance (for example, robust retention times and peak area measurements) for metabolites ranging from sugars to lipids.
In this work, we present optimizations and demonstrations of a metabolomics LC-Orbitrap MS method for DBS analysis intended to “bridge” the hydrophobic and polar metabolome in a single run. We focus on factors including retention time/peak area stability, selectivity/peak capacity, DBS extraction, and MS parameter settings. We demonstrate the method for both untargeted analysis and a targeted approach.
2. Materials and Methods
2.1. Equipment
Filter paper cards used were Whatman 903 Protein Saver cards (GE Healthcare Life Sciences, Chicago, IL, USA). A manual puncher from McGill (Advantus Corp., Jacksonville, FL, USA) was used to punch the DBS. Microtubes were obtained from Sarstedt (Nümbrecht, Germany). For extraction, a Thermomixer Comfort (Eppendorf, Hamburg, Germany) was used. Glass tubes for evaporation to dryness were obtained from VWR (Radnor, PA, USA), and the evaporator used was a TurboVap LV (Caliper Life Sciences, Waltham, MA, USA). HPLC vials, caps, and inserts were from La-Pha-pack (Thermo Scientific, Waltham, MA, USA).
The following LC columns were evaluated: Polaris C18-Ether and Pursuit XRs Diphenyl (both from Agilent Technologies (Santa Clara, CA, USA)), C18-Pentafluorophenyl (PFP) from ACE Technologies (Aberdeen, Scotland), Aeris Peptide XB-C18 (Phenomenex (Torrance, CA, USA)), and Raptor Biphenyl (Restek (Bellefonte, PA, USA)). For column specifications, see Table 1.
Table 1. Specifications of Evaluated Columns.
| column | length, mm | diameter, mm | particle size, μm | pore size, Å | surface area, m2/g | carbon load, % |
|---|---|---|---|---|---|---|
| Polaris C18-Ether | 250 | 2.0 | 3.0 | 180 | 200 | 12 |
| Pursuit XRs Diphenyl | 250 | 2.0 | 3.0 | 100 | 440 | 15 |
| ACE C18-PFP | 250 | 2.1 | 3.0 | 100 | 300 | 14 |
| Aeris Peptide XB-C18 | 250 | 2.1 | 3.6 | 100 | 200 | 10 |
| Raptor Biphenyl | 150 | 2.1 | 2.7 | 90 | 150 | 7 |
2.2. Chemicals and Solvents
All water used was of type 1 (>18 MΩ cm), obtained from a MilliQ ultrapure water purification system (Merck Millipore, Darmstadt, Germany). Methanol was obtained from Rathburn Chemicals (Walkerburn, Scotland). Formic acid (98%) was obtained from Merck.
Tobramycin, acylcarnitines C2, C12, and C16, D2 glycolic acid, D6 glucose, and acylcarnitines D3 C2, D3 C12, and D3 C16 were obtained from Larodan (Solna, Sweden). D4 succinic acid, 13C creatine, and uric acid were purchased from Sigma (Darmstadt, Germany). Vancomycin (1000 mg powder) was obtained from MIP Pharma GmbH (Blieskastel, Germany). 13C2 guanidinoacetate was obtained from Dr. H Ten Brink (VU University Medical Center, Amsterdam, The Netherlands). Creatinine was obtained from Merck. Creatine was obtained from Nutritional Biochemical Corporation (Cleveland, OH, USA).
2.3. Method Development
The following parameters were evaluated and optimized: MS parameters, with regard to signal intensity: electrospray voltage (evaluated values: 1, 2, 3.5, 4, 5, 6, and 7 kV), electrospray needle position (evaluated positions: A–D, A being the closest to the inlet), resolution (evaluated values: 17500, 35,000, 70,000, and 140,000 FWHM (at m/z 200)), and automatic gain control (AGC) target value (evaluated values: 2E4, 5E4, 1E5, 2E5, 5E5, 1E6, 3E6, and 5E6 ion counts). The evaluation of using either a broad (and split) scan range of m/z 50–750 and 750–1700 (performed with one analysis) or only one range of m/z 50–750 was done with regard to peak area.
LC parameters, with regard to peak distribution, are as follows: LC column (see Table 1 for names and specifications) and gradient elution profile (see Figure 1 for evaluated profiles; more details are described below). LC parameters, with regard to peak capacity, are as follows: injection volume (evaluated volumes: 2, 10, and 20 μL) and mobile phase flow rate (evaluated rates: 150 and 300 μL/min).
Figure 1.

Gradient profiles tested. Mobile phase A: water with 0.1% formic acid. Mobile phase B: methanol with 0.1% formic acid.
Figure 1 shows the evaluated gradient elution profiles (evaluated using a Pursuit XRs Diphenyl column). The original gradient was the starting point of the gradient profile optimization, while gradients 1–6 were defined based on when compounds in a spiked DBS eluted. Total analysis time was later reduced to 32.5 min.
Sample preparation optimization is as follows: evaluation of evaporating samples to dryness and re-solving in 2, 40, and 80% methanol, respectively, versus injection of an 80% methanol extract without evaporation to dryness, with regard to peak area. DBS punch location (evaluated locations: A–D, A being in the center of the spot and D in the perimeter (see Figure 6 in the Results and Discussion section)), with regard to standard deviation of the measured peak area (10 spots were used, providing 10 punches from each location) was evaluated.
Figure 6.

Top: punch locations tested. Ten spots were used, providing 10 punches from each location. Bottom: peak areas with RSD % of selected compounds in DBS samples prepared with punches taken from locations A, B, C, and D.
2.4. Sample Preparation
For optimization experiments, whole blood from a healthy volunteer was used. The whole blood sample was either spotted onto a filter paper card directly or mixed with aqueous standards (50:50 v/v) and spotted onto a filter paper card. The same blood sample was used in all experiments for optimization of a chosen parameter. The whole blood sample was either mixed with the standards directly before spotting or stored in a freezer at −80 °C and thawed before mixing and spotting.
Dried blood spots were either made immediately prior to each experiment, or prepared samples were stored at −80 °C before use. The following steps constitute the final sample preparation: 3.2 mm punches were punched from DBS (∼3 μL of whole blood or ∼1.5 μL of whole blood for the samples consisting of whole blood mixed with aqueous standards) and extracted in a microtube with 100 μL of 80% aqueous methanol with 0.1% formic acid using a thermomixer for 45 min (at 45 °C, 700 rpm). Samples were transferred to an HPLC vial for analysis directly after extraction.
2.5. Instrumentation
LC instrumentation used was a Dionex Ultimate 3000 UHPLC quaternary system pump, column department, and autosampler, from Thermo Scientific. The MS used was a Q Exactive Orbitrap (Thermo Scientific). The ionization source was an electrospray, and samples were analyzed in both positive and negative modes (in separate injections).
2.6. Settings and Details
The following settings constitute the final method. The LC column used was a Pursuit XRs Diphenyl (see Table 1 for details). The injection volume was 2 μL. The mobile phase (A: water with 0.1% formic acid and B: methanol with 0.1% formic acid) flow rate was 300 μL/min. The gradient elution profile was profile 6 in Figure 1. Column temperature was 30 °C, and total analysis time was 32.5 min. Re-equilibration time was 10 min.
The scan type was full MS (scan range m/z 50–750). The resolution was 70,000 FWHM (at m/z 200). The AGC target value was 1,000,000 ion counts. Maximum injection time was 250 ms. Electrospray settings are the following: sheath gas (N2) flow rate: 40 (a.u.), auxiliary gas (N2) flow rate: 10 (a.u.), sweep gas (N2) flow rate: 2 (a.u.), capillary temperature: 250 °C, S-lens RF level: 50, auxiliary gas heater temperature: 300 °C, electrospray voltage: 3.5 kV, and electrospray needle position: C.
Equation 1 was used for calculation of peak capacity (Pc)
| 1 |
where tG is the gradient elution time, n is the number of peaks, and w is the peak width at the baseline (13.4% peak height) for each peak.
2.7. Computer Software
Software used was Xcalibur (Version 4.2.47), Tune (version 2.11), and SII for Xcalibur 1.5, all from Thermo Scientific. Compound Discoverer 2.1 (Thermo Scientific) was used for data processing.
2.8. Approval by the Regional Committee for Medical and Health Research Ethics
The use of whole blood from healthy volunteers was approved by the Regional Committee for Medical and Health Research Ethics (case no.: 173346).
3. Results and Discussion
An LC-Orbitrap MS method for metabolomics analyses of DBS was optimized regarding sample preparation, chromatographic properties, and MS conditions for coverage of a broad range of metabolites and a high degree of sensitivity. A selection of endogenous and isotopically labeled metabolites (spiked in controlled amounts) was used for the optimization experiments. Included in this list were hydrophobic acylcarnitines (key biomarkers in newborn screening) as well as more polar amino acids including valine and tyrosine (biomarkers of maple syrup urine disease and tyrosinemia, respectively). Using the same methods, proof-of-concept demonstrations were performed for both targeted and untargeted DBS applications. Below is a more detailed presentation of the optimizations, method evaluation, and proof-of-concept experiments.
3.1. Method Development
3.1.1. MS Optimization
Q Exactive mass spectrometry parameters were optimized for metabolites from DBS with regard to signal intensity. For optimization of MS parameters, we used a standard mix of metabolites (about 5 μmol/L each) with a range of molecular weights, structures, and polarities (see Figure 2). In these experiments, all monitored standards were isotopically labeled to ensure that observed changes in signal intensities were caused only by parameter settings without interference from endogenous contributions (except for the drug vancomycin). Vancomycin was added to include a compound with a relatively large mass that is also analyzed in our routine laboratory, making method comparison possible. Optimization experiments were primarily performed using an aqueous mix of standards mixed with whole blood (50:50 v/v) and spotted onto a filter paper card. For these samples, the following MS settings were considered to be the best choices: electrospray voltage of 3.5 kV, electrospray needle position C (options were A–D, A being the closest to the inlet, with a difference between the positions of approximately 3.5 mm), resolution of 70,000 FWHM (at m/z 200), and automatic gain control (AGC) target value of 1,000,000 ion counts. See Figure 2 for comparison to other settings. AGC target values and electrospray voltage were of highest significance regarding sensitivity. Importantly, sensitivity increased significantly (92–109% increase in the peak area of investigated compounds) when changing from a split scan range (m/z 50–750 and 750–1700) to one scan range (m/z 50–750).
Figure 2.
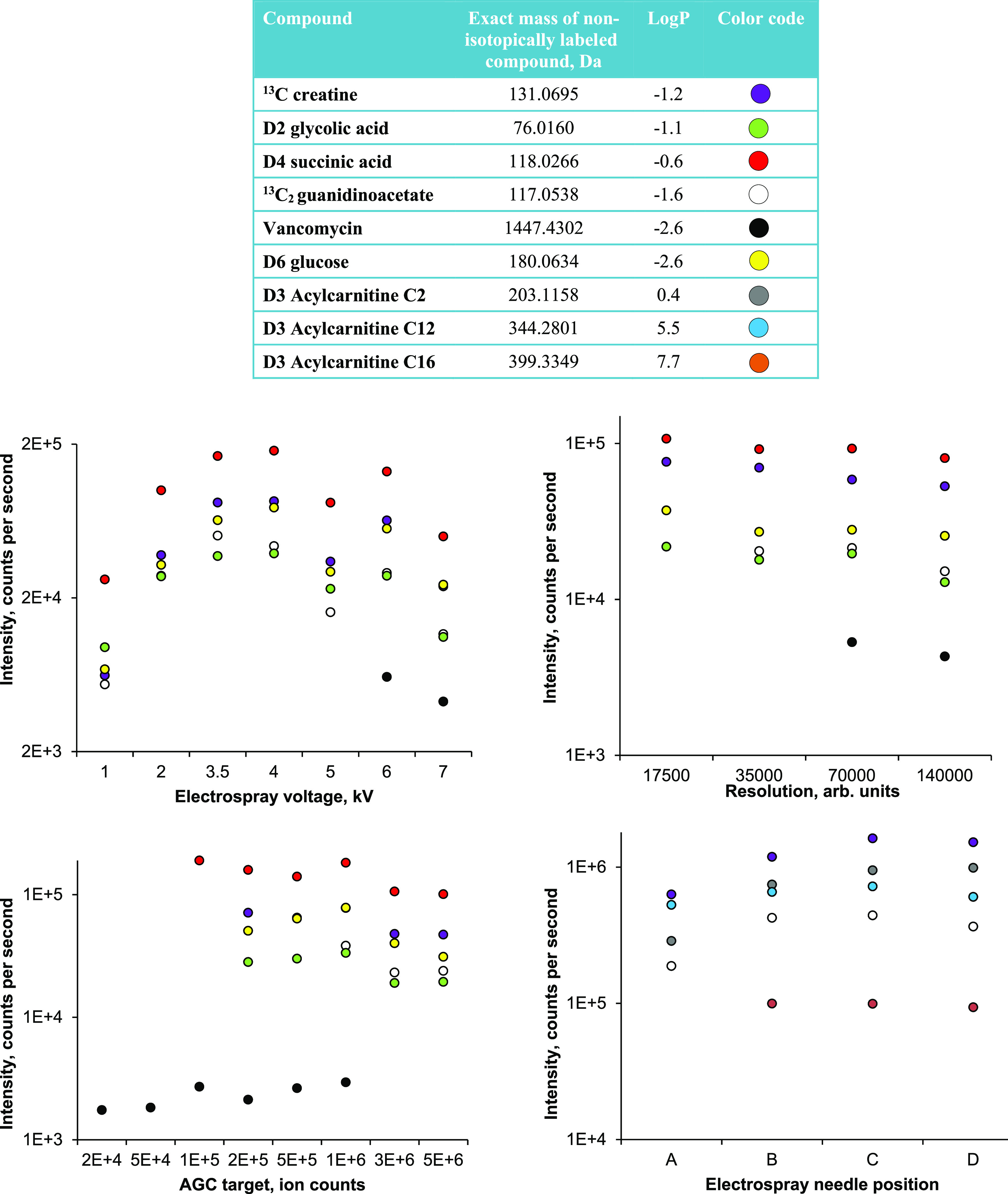
Top: exact mass and log P values of compounds used for MS optimization experiments. Exact mass and log P values were found at Pubchem.27 Colored dots show the color of each compound in the plots. Bottom: peak intensity with tested settings.
3.1.2. LC Optimization
Chromatographic parameters were optimized for metabolites from DBS first with regard to selectivity (here focusing on the ability to separate neighboring polar compounds while separating neighboring nonpolar compounds from each other) and subsequently peak capacity. In these experiments, the monitored standards were non-isotopically labeled metabolites but related to those in the MS experiments (see Figure 3). Optimization experiments were primarily performed using an aqueous mix of standards mixed with whole blood (50:50 v/v) and spotted onto a filter paper card. The following LC parameter settings were considered to be the best choices (see Figure 3 for comparison to other settings): analytical column: Pursuit XRs Diphenyl (hydrophobic + π–π interactions provided a modest increased selectivity of the model polar analytes), multi-linear gradient elution profile 6 in Figure 1 (interpreted as the best in maintaining the diphenyl column’s selectivity within a shortened analysis time), injection volume of 2 μL (associated with the highest peak capacity when using gradient profile 6, e.g., due to not overloading the column), and mobile phase flow rate of 300 μL/min (no large difference in peak capacity between tested flow rates, allowing a further reduction of the analysis time).
Figure 3.

Top: exact mass and log P values of compounds used for LC optimization experiments. Exact mass and log P values were found at Pubchem.27 Colored dots show the color of each compound in the plots. Bottom: average retention time (n = 3) of compounds for tested columns (A). All columns tested provided sharp chromatographic peaks (see the Supporting Information, Figures S1–S4), but the diphenyl SP variant was interpreted as having a modestly best selectivity for polar compounds (0–10 min area). (B) Retention times for gradient elution profiles using the diphenyl SP, with gradient 6 arguably preserving selectivity at a shorter analysis time. Average peak capacity (n = 3) for tested injection volumes (C) and tested mobile phase flow rates (D).
3.1.3. Sample Preparation Optimization
Sample preparation parameters were optimized for metabolites from DBS with regard to recovery/detection capability. The following steps and settings constitute the final sample preparation protocol: extraction of one punch from a DBS with 100 μL of 80% aqueous methanol with 0.1% formic acid and thermo-mixing for 45 min at 45 °C (700 rpm). The solution was directly transferred to an HPLC vial, as this gave improved recovery and detection (28–40% increase) for all compounds tested compared to evaporating to dryness and re-solving in the starting mobile phase (Figure 4). Although a centrifugation step was not included, we did not experience blockages of the LC column. The simplified procedure of injecting higher organic contents did not lead to substantial changes in retention time for any of the compounds investigated.
Figure 4.

Improved sensitivity with an increased amount of methanol. Peak area in DBS samples prepared with 2, 40, and 80% aqueous methanol in the reconstitution solution and with the exclusion of the evaporation and reconstitution steps. Retention times of the compounds for each organic solvent concentration are shown.
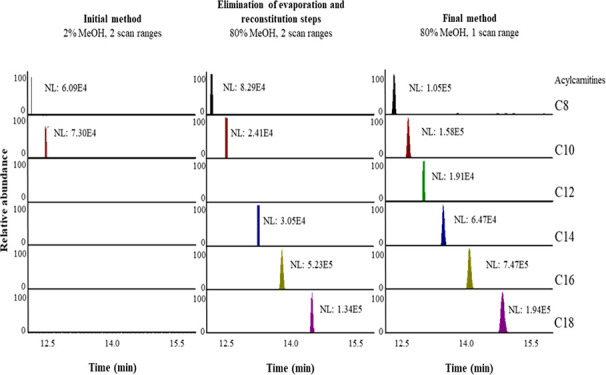
As shown in Figure 5, eliminating the evaporation to dryness step and changing from a broad and split (m/z 50–750 and 750–1700) to a narrow (m/z 50–750) scan range significantly improved detection of acylcarnitines.
Figure 5.
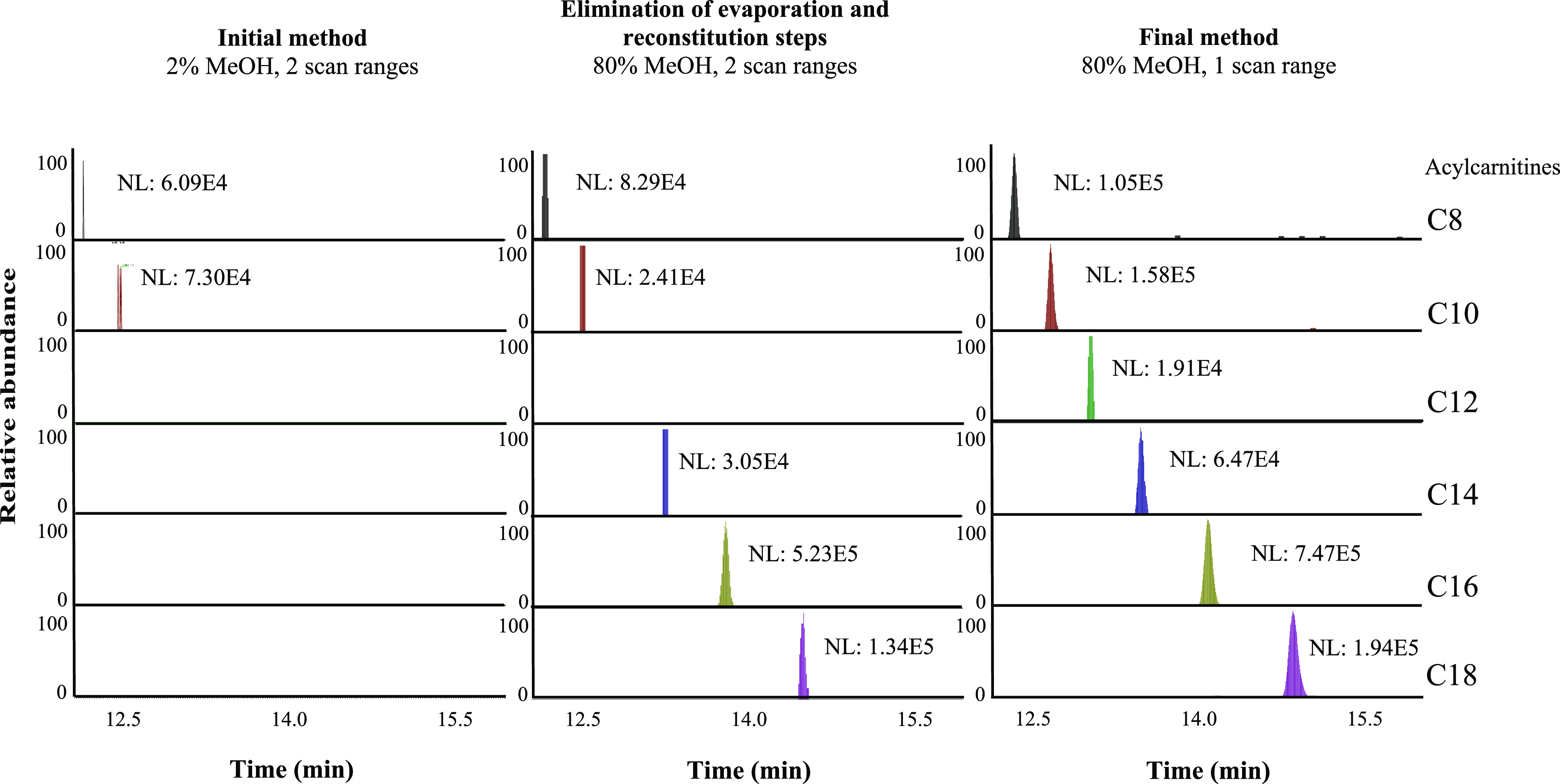
Extracted ion chromatograms of a selection of acylcarnitines showing detection improvement when changing from evaporation to dryness and a broad and split scan range (m/z 50–750 and 750–1700) to direct transfer of the extract to an HPLC vial and a narrow scan range (m/z 50–750).
To evaluate potential differences in punch location within the DBS, four punch locations were investigated (see Figure 6). We observed a larger relative standard deviation (RSD %) in punches taken from the perimeter of the spot compared to the center. Center punches were thus considered to be the best choice.
3.2. Evaluation of Peak Area Linearity
To evaluate the abilities of our DBS MS platform, we investigated the effect on the measured peak area of endogenous metabolites when increasing the number of DBS punches (1, 2, 3, and 4 punches, equivalent to about 3, 6, 9, and 12 μL of whole blood, respectively). Peak area linearity was overall satisfactory for the investigated analytes. As shown in Figure 7, with glycine and alanine as examples, peak area increased linearly with an increasing number of punches (R2 ranging from 0.9358 = ornithine to 0.9994 = alanine). To monitor instrument performance and repeatability, the same DBS sample (healthy volunteer) was injected three times each day during an analysis run of 11 days. Table S1 (Supporting Information) shows a high repeatability regarding average retention time and peak area (0.1–0.4 and 2–10%, respectively).
Figure 7.

Linear increase in peak area with an increasing number of punches from one spot for glycine and alanine.
3.3. Application/Proof of Concept
3.3.1. Targeted Approach
The method covers a large part of the metabolome. Endogenous metabolites ranging in polarity from log P −4.4 to 8.8 are readily detected; see Table 2 for a list of representative compounds, highlighted here as they are all employed as biomarkers in, e.g., newborn screening and for maple syrup urine disease and tyrosinemia (among other diseases). In addition, Figure 8 shows a total ion chromatogram from a positive ionization dried blood spot analysis with mass spectra and extracted ion chromatograms of eight selected detected endogenous compounds, illustrating the even peak distribution of hydrophilic and hydrophobic compounds along the chromatogram.
Table 2. Examples of Detected Endogenous Compounds with a Wide Range of Polaritiesa.
| amino acid | chemical formula | exact mass [M + H]+, Da | accurate mass [M + H]+, Da | mass error, ppm | log P | acylcarnitine | chemical formula | exact mass, [M + H]+, Da | accurate mass [M + H]+, Da | mass error, ppm | log P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ornithine | C5H12N2O2 | 133.0972 | 133.0970 | –1.50 | –4.4 | C0 | C7H15NO3 | 162.1125 | 162.1122 | –1.85 | –0.2 |
| citrulline | C6H13N3O3 | 176.1030 | 176.1027 | –1.70 | –4.3 | C2 | C9H17NO4 | 204.1230 | 204.1227 | –1.47 | 0.4 |
| arginine | C6H14N4O2 | 175.1190 | 175.1187 | –1.71 | –4.2 | C3 | C10H19NO4 | 218.1387 | 218.1383 | –1.83 | 0.9 |
| glycine | C2H5NO2 | 76.0393 | 76.0393 | 0.00 | –3.2 | C4 | C11H21NO4 | 232.1543 | 232.1539 | –1.72 | 1.2 |
| alanine | C3H7NO2 | 90.0550 | 90.0549 | –1.11 | –3.0 | C5 | C12H23NO4 | 246.1700 | 246.1695 | –2.03 | 1.8 |
| valine | C5H11NO2 | 118.0863 | 118.0861 | –1.69 | –2.3 | C8 | C15H29NO4 | 288.2169 | 288.2162 | –2.43 | 3.4 |
| tyrosine | C9H11NO3 | 182.0812 | 182.0810 | –1.10 | –2.3 | C12 | C19H37NO4 | 344.2795 | 344.2782 | –3.78 | 5.5 |
| methionine | C5H11NO2S | 150.0583 | 150.0581 | –1.33 | –1.9 | C14 | C21H41NO4 | 372.3108 | 372.3103 | –1.34 | 6.6 |
| leucine | C6H13NO2 | 132.1019 | 132.1017 | –1.51 | –1.5 | C16 | C23H45NO4 | 400.3421 | 400.3414 | –1.75 | 7.7 |
| phenylalanine | C9H11NO2 | 166.0863 | 166.0860 | –1.81 | –1.5 | C18 | C25H49NO4 | 428.3734 | 428.3728 | –1.40 | 8.8 |
Log P values were obtained from Pubchem.27
Figure 8.

Mass spectra (A) and extracted ion chromatograms with names, log P values, signal intensities, and structures (B) of a selection of detected endogenous compounds in a dried blood spot (alanine, glycine, phenylalanine, tryptophan, acylcarnitine C16, acylcarnitine C18, LysoPC (C18:1 (9Z)), and 7-ketocholesterol), and total ion chromatogram (C) with asterisks indicating at which points in the chromatogram the spectra in panel (A) are located. Log P values and structures were obtained from Pubchem.27
We also investigated if our method could detect changes in the concentration of one target metabolite out of the thousands of metabolites detected. Six healthy volunteers were taken DBS samples during free intake of coffee and soda for 15 h (during which the participants consumed one to four cups of coffee each) and during no intake of coffee and soda (with the first sample taken after 12 h of no caffeine intake). Caffeine is a suitable compound to monitor as we know that it is exogenous (mostly originating from coffee and soda), and people consume various amounts of the substance. Thus, caffeine was measured in all samples. As shown in Figure 9, the measured amount of caffeine decreased with increasing time since intake.
Figure 9.

Measured amounts of caffeine decreased with time since intake. The spread in the measured peak area is likely caused by different intakes of coffee between the participants.
3.3.2. Detection of Differences in Nutritional States (Untargeted Analysis)
An additional proof of concept study was performed by analyzing DBS samples from six healthy volunteers during free food intake and during fasting. DBS samples were taken during free diet, after 12 h of fasting, and after 36 h of fasting. The volunteers were allowed to drink as much water as they wanted during the fasting period. A principal component analysis plot of samples taken from all volunteers during free diet and after 12 h and 36 h of fasting is shown in Figure 10. The samples from the three nutritional states clearly grouped as three separate clusters, with the apparent exception of the free diet sample from person F, which clustered together with samples taken after 12 h of overnight fasting. However, it turned out that person F was actually omitting breakfast that day, meaning that this point is correctly located together with the overnight fasting samples and should in fact be classified as an overnight fasting sample.
Figure 10.

Principal component analysis plot of DBS samples from six individuals (A–F) taken after free diet, overnight fasting (12 h), and prolonged fasting (36 h). One point represents metabolites detected in that particular sample. Individual F did not eat before collecting the free diet sample. This point (red arrow) thus clustered correctly together with the overnight fasting samples.
To evaluate the method’s ability to identify discriminating compounds between groups, we used a volcano plot to compare samples taken after overnight fasting (12 h) with samples taken after prolonged fasting (36 h) (Figure 11). Compounds with a significantly lower concentration in prolonged fasting samples were identified: caffeine, theobromine, and paraxanthine, all associated with metabolism of (coffee) drinks.28,29 An upregulated marker of prolonged fasting samples was identified as β-hydroxybutyrate, a ketone body naturally produced during fasting for energy transfer.30 Taken together, the single run platform was well suited for revealing trends of the expected metabolism in this controlled study.
Figure 11.

Volcano plot of DBS samples from six individuals taken after overnight fasting (12 h) and prolonged fasting (36 h). One point represents one compound. Green box: lower concentration measured in samples taken after 36 h than after 12 h of fasting. Red box: higher concentration measured in samples taken after 36 h than after 12 h of fasting.
4. Conclusions
A single LC–MS method has satisfactory analytical performance for a broad range of metabolites with regard to polarity, having evaluated and optimized parameters regarding MS sensitivity, column separation, and sample preparation, demonstrated with proof-of-concept studies regarding both untargeted and targeted metabolite approaches using the same method/settings. We find that a single LC–MS method can indeed be a compromise between multi-method deep profiling and fast “shotgun” approaches. We have here evaluated our platform with regard to key biomarkers of inborn errors of metabolism and are currently exploring the limitations of our platform regarding lipids such as phosphatidylcholines, cholesterol esters, and acylglycerols (employing SPLASH standards), improving the identification abilities of our platform with the use of about 600 metabolite standards (MSMLS by IROA Technologies), and exploring the quantitative abilities of our platform by comparing with standard clinical chemistry.
Acknowledgments
Financial support from UiO:Life Science is gratefully acknowledged. This work was also partially supported by the Research Council of Norway through its Centre of Excellence scheme, project no. 262613.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00326.
(Table S1) Relative standard deviations of retention time and peak area of selected compounds measured in a DBS, number of detected features and annotated/identified metabolites, using Compound Discoverer 2.1, in a dried blood spot positive ionization analysis, and (Figures S1–S4) chromatograms from other evaluated columns (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Patti G. J.; Yanes O.; Siuzdak G. Metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A. D.; Miller M. J.; Beebe K.; Wulff J. E.; Evans A. M.; Miller L. A. D.; Sutton V. R.; Sun Q.; Elsea S. H. Metabolomic Profiling of Human Urine as a Screen for Multiple Inborn Errors of Metabolism. Genetic testing and molecular biomarkers 2016, 20, 485–495. 10.1089/gtmb.2015.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezgu F. Chapter Seven - Inborn Errors of Metabolism. Adv. Clin. Chem. 2016, 73, 195–250. 10.1016/bs.acc.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Saudubray J.-M.; Garcia-Cazorla À. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatr. Clin. North Am. 2018, 65, 179–208. 10.1016/j.pcl.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Ismail I. T.; Showalter M. R.; Fiehn O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, 242. 10.3390/metabo9100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaria R.; Allen K. J.; Koplin J. J.; Roche P.; Greaves R. F. Advantages and Challenges of Dried Blood Spot Analysis by Mass Spectrometry Across the Total Testing Process. EJIFCC 2016, 27, 288–317. [PMC free article] [PubMed] [Google Scholar]
- Prentice P.; Turner C.; Wong M. C. Y.; Dalton R. N. Stability of metabolites in dried blood spots stored at different temperatures over a 2-year period. Bioanalysis 2013, 5, 1507–1514. 10.4155/bio.13.121. [DOI] [PubMed] [Google Scholar]
- Koulman A.; Prentice P.; Wong M. C. Y.; Matthews L.; Bond N. J.; Eiden M.; Griffin J. L.; Dunger D. B. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics : Off. J. Metabolomic Soc. 2014, 10, 1018–1025. 10.1007/s11306-014-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer E. A.; Cooper H. J.; Dunn W. B. Investigation of the 12-Month Stability of Dried Blood and Urine Spots Applying Untargeted UHPLC-MS Metabolomic Assays. Anal. Chem. 2019, 91, 14306–14313. 10.1021/acs.analchem.9b02577. [DOI] [PubMed] [Google Scholar]
- Dénes J.; Szabó E.; Robinette S. L.; Szatmári I.; Szőnyi L.; Kreuder J. G.; Rauterberg E. W.; Takáts Z. Metabonomics of newborn screening dried blood spot samples: a novel approach in the screening and diagnostics of inborn errors of metabolism. Anal. Chem. 2012, 84, 10113–10120. 10.1021/ac302527m. [DOI] [PubMed] [Google Scholar]
- Matern D.; Gavrilov D.; Oglesbee D.; Raymond K.; Rinaldo P.; Tortorelli S. Newborn screening for lysosomal storage disorders. Seminars in Perinatology 2015, 39, 206–216. 10.1053/j.semperi.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Wang S.; Blair I. A.; Mesaros C., Analytical Methods for Mass Spectrometry-Based Metabolomics Studies. In Advancements of Mass Spectrometry in Biomedical Research. Advances in Experimental Medicine and Biology; Woods A. G.; Darie C. C., Eds. Springer International Publishing: Cham, 2019; Vol. 1140, pp. 635–647. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D.; Begley P.; Zelena E.; Francis-McIntyre S.; Anderson N.; Brown M.; Knowles J. D.; Halsall A.; Haselden J. N.; Nicholls A. W.; Wilson I. D.; Kell D. B.; Goodacre R.; Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- Lopes A. S.; Cruz E. C. S.; Sussulini A.; Klassen A., Metabolomic Strategies Involving Mass Spectrometry Combined with Liquid and Gas Chromatography. In Metabolomics: From Fundamentals to Clinical Applications. Advances in Experimental Medicine and Biology; Sussulini A.., Ed. Springer International Publishing: Cham, 2017; Vol. 965, pp. 77–98. [DOI] [PubMed] [Google Scholar]
- Allwood J. W.; Goodacre R. An introduction to liquid chromatography–mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 2010, 21, 33–47. 10.1002/pca.1187. [DOI] [PubMed] [Google Scholar]
- Haijes H. A.; Willemsen M.; Van der Ham M.; Gerrits J.; Pras-Raves M. L.; Prinsen H. C. M. T.; Van Hasselt P. M.; De Sain-van der Velden M. G. M.; Verhoeven-Duif N. M.; Jans J. J. M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. 10.3390/metabo9010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussap M.; Zaffanello M.; Fanos V. Metabolomics: a challenge for detecting and monitoring inborn errors of metabolism. Ann. Transl. Med. 2018, 6, 338–338. 10.21037/atm.2018.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najdekr L.; Blanco G. R.; Dunn W. B., Collection of Untargeted Metabolomic Data for Mammalian Urine Applying HILIC and Reversed Phase Ultra Performance Liquid Chromatography Methods Coupled to a Q Exactive Mass Spectrometer. In Metabolomics: Methods and Protocols. Methods in Molecular Biology; Bhattacharya S. K., Ed. Springer New York: New York, NY, 2019; Vol. 1996, pp. 1–15. [DOI] [PubMed] [Google Scholar]
- Miller M. J.; Kennedy A. D.; Eckhart A. D.; Burrage L. C.; Wulff J. E.; Miller L. A. D.; Milburn M. V.; Ryals J. A.; Beaudet A. L.; Sun Q.; Sutton V. R.; Elsea S. H. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. Journal of inherited metabolic disease 2015, 38, 1029–1039. 10.1007/s10545-015-9843-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrepois K.; Jiang L.; Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)–Mass Spectrometry. Mol. Cell. Proteomics 2015, 14, 1684. 10.1074/mcp.M114.046508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. J.; Rodriguez-Blanco G.; Dunn W. B.; Phillips B. E.; Williams J. P.; Greenhaff P. L.; Smith K.; Gallagher I. J.; Atherton P. J. Untargeted metabolomics for uncovering biological markers of human skeletal muscle ageing. Aging 2020, 12, 12517–12533. 10.18632/aging.103513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen S.; Puurunen J.; Auriola S.; Kullaa A. M.; Kärkkäinen O.; Lohi H.; Hanhineva K. Metabolome of canine and human saliva: a non-targeted metabolomics study. Metabolomics : Official journal of the Metabolomic Society 2020, 16, 90–90. 10.1007/s11306-020-01711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jääskeläinen T.; Kärkkäinen O.; Jokkala J.; Klåvus A.; Heinonen S.; Auriola S.; Lehtonen M.; Group F. C. I.; Hanhineva K.; Laivuori H. A non-targeted LC-MS metabolic profiling of pregnancy: longitudinal evidence from healthy and pre-eclamptic pregnancies. Metabolomics : Off. J. Metabolomic Soc. 2021, 17, 20–20. 10.1007/s11306-020-01752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonte R.; Bongaerts M.; Demirdas S.; Langendonk J. G.; Huidekoper H. H.; Williams M.; Onkenhout W.; Jacobs E. H.; Blom H. J.; Ruijter G. J. G. Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform. Metabolites 2019, 9, 289. 10.3390/metabo9120289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dator R.; Villalta P. W.; Thomson N.; Jensen J.; Hatsukami D. K.; Stepanov I.; Warth B.; Balbo S. Metabolomics Profiles of Smokers from Two Ethnic Groups with Differing Lung Cancer Risk. Chem. Res. Toxicol. 2020, 33, 2087–2098. 10.1021/acs.chemrestox.0c00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Rashan E. H.; Linke V.; Shishkova E.; Hebert A. S.; Jochem A.; Westphall M. S.; Pagliarini D. J.; Overmyer K. A.; Coon J. J. Multi-Omic Single-Shot Technology for Integrated Proteome and Lipidome Analysis. Anal. Chem. 2021, 93, 4217–4222. 10.1021/acs.analchem.0c04764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://pubchem.ncbi.nlm.nih.gov/
- Cornelis M. C.; Erlund I.; Michelotti G. A.; Herder C.; Westerhuis J. A.; Tuomilehto J. Metabolomic response to coffee consumption: application to a three-stage clinical trial. J. Intern. Med. 2018, 283, 544–557. 10.1111/joim.12737. [DOI] [PubMed] [Google Scholar]
- Rothwell J. A.; Fillâtre Y.; Martin J.-F.; Lyan B.; Pujos-Guillot E.; Fezeu L.; Hercberg S.; Comte B.; Galan P.; Touvier M.; Manach C. New Biomarkers of Coffee Consumption Identified by the Non-Targeted Metabolomic Profiling of Cohort Study Subjects. PLoS One 2014, 9, e93474 10.1371/journal.pone.0093474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J. C.; Verdin E. β-hydroxybutyrate: much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. 10.1016/j.diabres.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.