

Summary
Rheumatoid arthritis (RA) is a chronic, progressive autoimmune disease characterized by synovitis and symmetrical joint destruction. RA has become one of the key diseases endangering human health, but its etiology is not clear. Therefore, identifying the immunopathogenic mechanisms of RA and developing therapeutic drugs to treat autoimmune diseases have always been difficult. This article mainly reviews the immunopathogenic mechanism of RA and advances in the study of anti-inflammatory drugs in order to provide a reference for the treatment of RA and drug development in the future.
Keywords: rheumatoid arthritis, immunopathogenesis, cytokines, inflammatory drugs
1. Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory, systemic autoimmune disease with an incidence of 5-10 cases per 1,000 people (1,2). Nonsuppurative joint and joint tissue inflammation is a main feature of RA, which mainly manifests as joint synovitis, resulting in damage to the cartilage, ligaments, tendons, and other joint tissues as well as multiple organ damage. The basic pathological changes in RA are synovitis, acute synovial swelling and exudation, chronic granulocyte infiltration, synovial hyperplasia and hypertrophy, and vasculitis. The latter is the pathological basis of joint injury, deformity, and obstruction and causes the disease to progress to the irreversible stage. The initial symptoms of RA are swelling and pain in the joints of the hands and feet, and especially the palms, toes, and proximal interphalangeal joints. Large joints, including the elbows, shoulders, ankles, and knees, can also be involved (1). In addition to joint symptoms, patients with RA often experience other symptoms such as fever, anemia, scleritis, pericarditis, vasculitis, and enlarged lymph nodes, and a variety of autoantibodies can be found in their serum. Without proper treatment, RA mainly affects the small joints of the limbs, such as the hands, feet, and wrists; symptoms are usually symmetrical and can be temporarily relieved. Without systematic treatment, however, RA can occur repeatedly for many years, eventually leading to joint deformities and loss of function.
Treatment of RA has two objectives: symptom relief and maintenance of function, and slowing the process of tissue injury. Currently, drugs used to treat RA are mainly divided into non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs) and glucocorticoids (GCs). NSAIDs play an anti-inflammatory, antipyretic and analgesic role by inhibiting the activity of cyclooxygenase (COX), reducing the generation of prostaglandin (PG), and inhibiting the secretion of various cytokines. DMARDs can interfere with RA symptoms and signs, improve body function, and inhibit the progression of joint injury (3). Currently, IL-6R antibodies and JAK inhibitors are the most effective biological DMARDs (4). Although these drugs have a certain therapeutic effect, there are still some patients who fail to respond to the therapeutic drugs or who do not continue to respond (5). Therefore, there is an urgent need to develop drugs with new targets or new mechanisms to meet the clinical needs of these patients.
This review focuses on the current understanding of the immunopathogenic mechanisms of bone and cartilage damage caused by inflammatory disorders and progress in the use of anti-inflammatory drugs to treat patients with RA.
2. Immune mechanisms of RA
Cascade responses of innate and adaptive immunity are important mechanisms of the RA inflammatory process (6). Many inflammatory cytokines and autoantibodies drive RA-associated inflammation and are maintained by epigenetic changes in fibroblast-like synovial cells, facilitating further inflammation (7,8). During this process, many immune cells (neutrophils, granulocytes, macrophages, and B and T cells) invade the synovium and the synovial fluid. This invasion results in the release of many cytokines, chemokines, autoantibodies, and reactive oxidative species (ROS) in the synovial and joint spaces, leading to joint injury. The serological markers of the disease are the presence of high titers of rheumatoid factor (RF) and anti-citrullinated peptide antigens and antibodies (ACPAs) (9,10). This complex pathogenic mechanism will be discussed in more detail below.
2.1. Immune cells
Synovial inflammation reflects subsequent immune activation, which is characterized by leukocyte invasion by innate immune cells (e.g., monocytes, macrophages, dendritic cells, and neutrophils) and adaptive immune cells (e.g., Th1, Th2, Th17 cells, B cells, and plasma cells) (11,12).
2.1.1. T cells
T cells play an important role in the RA immune-mediated inflammatory response. In experimental models of collagen-induced RA, activated T cells aggregate in the inflamed joints as the disease progresses (13,14). Naive CD4+T helper cells (Th) can differentiate into different cell lines (Th1, Th2, and Th17), characterized by the specific expression of transcription factors and proinflammatory cytokines in the system under antigen stimulation (15,16).
In the past, the pathogenesis of RA was generally believed to involve the abnormal differentiation of CD4+T lymphocytes, which mainly manifested as a Th1/Th2 imbalance. As the pathogenesis of RA has been better understood and key transcription factors in the differentiation and development of different T cell subsets have been examined, Th17 and regulatory T cells (Tregs) have been found to play an important role in mediating the inflammatory response, articular cartilage and bone destruction, and bone erosion in RA (17,18).
Th17 cells can secrete interleukin-17 (IL-17) as well as cytokines such as IL-21 and IL-22. IL-17 can aggravate the inflammatory response and it participates in many autoimmune diseases. IL-17 expression increased significantly in the serum and joint fluid of patients with RA, which promoted synovial cells to secrete a variety of inflammatory cells to make chondrocytes to synthesize matrix, enhance osteoclast activity, and cause bone erosion (19). Tregs are a subgroup of CD4+ T cells with immunosuppressive activity. Treg cells can inhibit T cells and antigen-presenting cells by releasing the cytokines IL-10 and TGF-β and by reducing the production of inflammatory cytokines and antibody secretion, thereby exhibiting an immunosuppressive effect. Th17 and Treg cells can transform each other under specific cytokine microenvironment conditions. CD4+T cells can differentiate into Treg cells when induced with TGF-β alone. When IL-6 is also present, it can induce RORγt expression, inhibit Treg cell production, and promote the differentiation of initial CD4+T cells into Thl7 cells (20). Therefore, the body's immune status can be regulated and the pathogenesis and progression of RA can be managed by controlling differing factors in the Th17 cell environment, inhibiting Th17 cell differentiation and proinflammatory cytokine expression, enhancing Treg activity, and regulating the balance of Th17 cells/Tregs in the body. This may provide a new therapeutic direction for prevention and control of RA.
2.1.2. B cells
In patients with RA, citrulline antigen-oriented B cells and B cells that react with citrulline antigens have significant effects in vitro (21). This citrullinated antigen-directed B cell response contributes to the initiation and persistence of inflammatory processes. Thus, the ACPA response is the major humoral immune response associated with RA (22). An abnormal dynamic between immune cells leads to abnormal aggregation of activated T cells, B cells, mast cells, neutrophils, macrophages, and cells entering APCs, which contribute to the cellular immune response in the course of RA (23).
2.1.3. Macrophages
Macrophages are full-time antigen-presenting cells that activate T cells through their costimulatory molecules such as CD80/86 and CD40. Macrophages play an important role in many inflammatory responses, and their number is strongly associated with symptoms of RA and joint damage (24). Macrophages abound in the synovium and cartilage pannus of inflamed joints. The increased number of macrophages in RA may be due to the lack of apoptosis. Macrophages in synovial fluid of patients with RA overexpress the FADD-like IL-1 invertase inhibitor protein (FLIP), which prevents tumor necrosis factor receptor FAS-mediated macrophage apoptosis. Moreover, macrophage activation, such as through overexpression of MHCⅡ molecules, produces proinflammatory cytokines, chemokines, macrophage inflammatory protein-1 (MIP-1), monocyte chemoattractant protein-1 (MCP-1), matrix metalloproteinases (MMPs), and neopterin, which can exacerbate inflammatory responses (25).
2.2. Factors related to RA
A variety of cytokines play important roles in the development and progression of RA. Cytokines such as interleukin-1 (IL-1), IL-6, IL-17, and tumor necrosis factor α (TNF-α) promote osteoclast production, whereas cytokines such as interferon-α (IFN-α), IFN-β, and IFN-γ antagonize this cell production, thereby regulating the bone balance and participating in bone and cartilage destruction and repair. In addition, cytokines can directly or indirectly regulate immune active cells or regulatory T cells and participate in regulation of the inflammatory response. A variety of cytokine-targeting biological agents has been developed to achieve disease relief in patients with RA.
2.2.1. Interleukins (IL)
IL-6 is produced by a variety of cells such as endothelial cells, fibroblasts, keratinocytes, chondrocytes, some tumor cells, and immune cells including monocytes, macrophages, T cells, and B cells. High levels of IL-6 are detected in the blood and synovial fluid of most patients with RA. IL-6 promotes the secretion of ROS and protease by neutrophils, increases inflammation, and causes joint injury (26). In addition, IL-6 stimulates osteoclast differentiation by activating RANKL-dependent or independent mechanisms (27). Hence, IL-6 may be related to osteochondral destruction and osteoporosis in patients with RA. A current RA therapy blocks IL-6 and IL-6R (28,29). Humanized anti-IL-6R antibodies can block the binding of IL-6 and IL-6R and affect the role of IL-6. Therefore, interfering with IL-6 activity is a treatment approach for RA (30).
IL-37 levels in plasma or peripheral blood mononuclear cells (PBMCs) in patients with RA are significantly higher than those in healthy controls and increase with increased disease activity (31,32). IL- 37 levels in plasma of patients with RA are positively correlated with levels of TNF-α, IL-6, IL-17A, and C-reactive protein as well as the Disease Activity Score in 28 joints (DAS28) but are significantly reduced after DMARD treatment. Wang et al. (33) found higher levels of IL-37+CD4+ T cells, total IL-37+ lymphocytes, IL-18Rα+CD4+ cells, IL-18Rα+ CD4- cells, and total IL-18Rα + lymphocytes in the PBMCs of patients with RA than in those in the healthy control group. Patients with RA have higher IL-37 levels than healthy individuals, but in vitro and in vivo experiments indicated that IL-37 has anti-inflammatory action. When patients receive DMARD, their IL- 37 level decreases, indicating that the increase in IL- 37 expression in RA is a feedback increase, that is, a response that limits disease severity.
The cytokine IL-34 has recently been found to have multiple effects on the immune system. Although research is still in the preliminary stage, the IL-34 produced by epithelial cells is indispensable for the development of tissue macrophage-like cells (34). Interestingly, recent studies indicated that IL-34 is also expressed in synovial fibroblasts and the sublining and intimal lining of the synovium in patients with RA. IL-34 expression is also significantly correlated with synovitis severity (35). IL-34 levels in fibroblast-like synoviocytes (FLS), serum, and synovial fluid are significantly increased in patients with RA compared to healthy individuals and patients with osteoarthritis (OA) (36-39), and IL-34 levels are associated with total leukocytes in synovial fluid (35). In addition, serum IL-34 levels in patients with RA are positively correlated with rheumatoid factor and anti-cyclic citrullinated peptide antibody titers (40). Therefore, an abnormal level of IL-34 may be an effective marker of RA activity, and real-time fluorescence quantitative PCR may reveal a high level of IL-34 expression in osteoblasts. These studies have clearly indicated that IL-34 plays a role in the pathogenesis of RA.
2.2.2. TNF-α
Animal experiments (41) have demonstrated that TNF-α overexpression can cause severe arthritis in mice and that TNF-α suppression can prevent its development. Drugs that block TNF-α activity can alleviate the clinical symptoms of RA. In the affected joints of patients with RA, TNF-α promotes IL-6 production in synovial cells and co-induces vascular endothelial growth factor. TNF-α is encoded in the major histocompatibility complex (MHC). The presentation of peptides by the MHC is dictated by the TNF-α gene, which may be related to the therapeutic effect of blocking TNF-α.
2.2.3. Chemokines
Chemokines are inducible pro-inflammatory cytokines and are divided into four subgroups: CXC, CC, C, and CX3C (42). Chemokines and chemokine receptors play a key role in leukocyte migration into inflammatory tissues. Chemokines CCL5 and CCL15 belong to the CC subgroup. The increased specificity of CCL5 and CCL15 in RA may be related to the infiltration and aggregation of Th1 cells in inflamed joints. CXCL16 of the chemokine CXC subfamily increases in the synovial membrane and plays an important role in T cell aggregation and synovial inflammation. Therefore, CXCL16 may become a new target for RA therapy. IL-8 normal T cells in in the serum of patients with rheumatoid synovitis have significantly higher levels of regulatory activation chemokines (RANTES) and McP-1 than those in patients with other types of synovitis, and serum levels of IL-8 and RANTES are associated with rheumatic synovitis in different tissue types (43).
2.2.4. Interferon (IFN)
IFN-γ has a wide range of immunomodulatory actions that can activate NK cells, improve their killing ability, and induce the expression of macrophages, T cells, B cells and other cells, thus improving their ability to present antigens. The level of serum IFN-γ in patients with RA is reported to be significantly higher than that in healthy controls (44).
2.3. JAK/STAT signaling pathway
Four members (JAK1, JAK2, JAK3, and TYK2) of the JAK family and seven members (STAT 1-4, STAT 5A/B, and STAT 6) of the STAT family are found in mammals. They share a structurally and functionally common region, called the JAK homologous (JH) region (Figure 1). JAK/STAT proteins are ubiquitous, and different combinations of them respond to specific cytokine or growth factor signals, guaranteeing a high level of specificity with different roles in vivo (45-47). IL-6/JAK/STAT mechanisms of signaling cascades allow direct communication between transmembrane receptors and nuclei, which can be summarized in the following steps (Figure 2): IL-6 ligands bind IL-6r-Gp130 receptor complexes and activate JAK tyrosine kinases recruited to their receptor intracellular regions. Once a JAK protein is activated, it undergoes dimerization, it phosphorylates tyrosines, and it activates its main substrate, the STAT protein. Tyrosine-phosphorylated STAT proteins homo- or hetero- dimerize and shift to the nucleus, where they interact with coactivators and bind to specific regulatory elements in the promoter regions of thousands of different target protein-coding genes, as well as micro-RNAs and long noncoding RNAs. STAT activity is regulated by phosphorylation, acetylation, and methylation, promoting STAT dimer stabilization, DNA binding, interaction with transcription costimulatory factors, and target cell expression (48-50). Negative regulators of JAK/STAT signaling provide further levels of control, guaranteeing cell feedback inhibition that can induce specific cytokine receptor signaling (45,51,52). Indeed, a soluble IL-6 receptor (SIL-6R), including its extracellular portion, can bind IL-6 and IL- 6-SIL-6R complexes and activate gp130 homodimers in cells lacking membrane-bound IL-6R (53,54). Hence, JAK/STAT signaling cascades provide a significant direct and tuned translation of extracellular signals into transcriptional responses in many cells.
Figure 1.
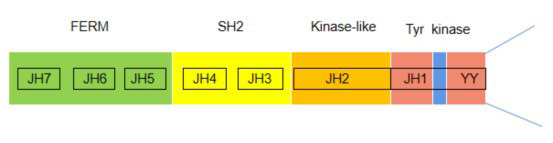
JH domains and JAK3 phosphorylation sites found in JAK/STAT proteins. FERM, four-point.1-ezrin-radaxin-moesin domain; JAK, Janus kinase; JH, JAK homology; kinase-like, pseudokinase domain; SH2, Src homology domain; Tyr kinase, tyrosine kinase domain.
Figure 2.
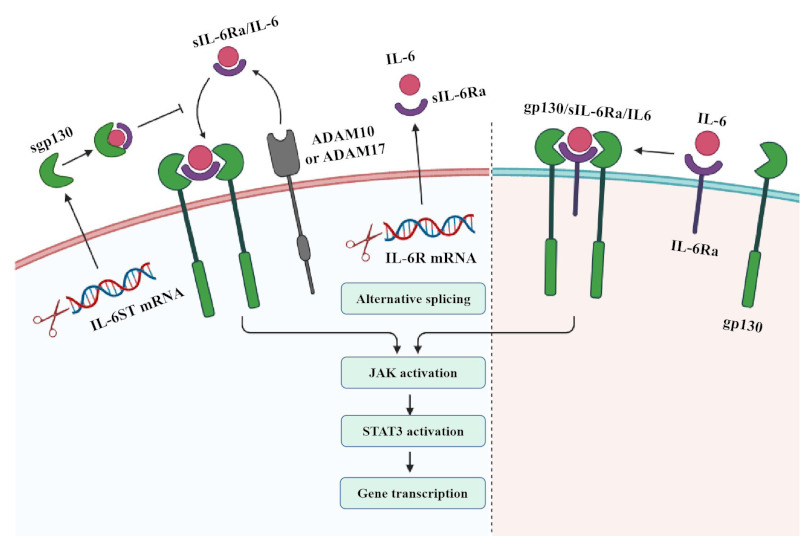
IL-6 signaling pathways (58).
Levels of cytokines IL-6, IL-15, IFN, and granulocyte-macrophage colony stimulating factor (GM-CSF), which are involved in pathogenesis of synovial inflammation and joint destruction, increase significantly in patients with RA (55). These factors can activate JAK/ STAT1 signaling pathways, IL-6, IL-15, IL-10, and IFN binding to JAK1, as well as platelet-derived factor (PDGF), EGF, GM-CSF, and IL-6 binding to JAK3. Using immunohistochemistry, Kasperkovitz et al. (56) verified that the total STAT1 protein level in the synovial tissue of patients with RA was significantly higher than that in patients with OA and was mainly expressed in T cells and B cells at the site of inflammatory infiltration as well as in FLS in the intimal lining of the synovium. Activation of the STAT signaling pathway in the synovial membrane may be achieved by inducing STAT1 expression to promote synovial inflammation. However, Krause et al. (57) found that a STAT3 deficiency induces accelerated apoptosis of RA-FLS, suggesting an important role of STAT3 in RA-FLS. Thus, STAT may have dual regulatory effects on exacerbating symptoms and protecting joints in synovial inflammation associated with RA.
IL-6 binds to membrane-bound IL-6 receptors (IL- 6R), inducing the formation of a heterodimer complex consisting of two molecules each of IL-6, IL-6R, and IL-6 receptor subunits (gp130). Formation of this complex leads to activation of the JAK/STAT3 signaling pathway, resulting in target gene transcription. Soluble IL-6R (SIL-6R) binds to IL-6 in the signaling pathway. SIL-6R can be produced by alternative splicing of IL6R mRNA or cleavage of disintegrin and metalloprotein-containing domain protein 170 (ADAM10) or cleavage of IL-6R by ADAM17. When IL-6 binds to SIL-6R, the complex is able to bind to gp130 and induce its dimerization, thereby activating downstream signaling pathways. While IL-6R is expressed in limited cell types, gp130 is widely expressed. IL-6 acts on cells with limited or missing IL-6R expression through SIL-6R transfer. IL-6 transduction signals can be negatively regulated by soluble gp130 (sgp130), which is produced by alternative splicing. Gp130 competes with the membrane binding of IL-6-SIL-6R complexes, thereby inhibiting IL-6 signal transduction but not classical IL-6 signaling pathways.
3. Immunotherapy and therapeutic drugs for RA
3.1. NSAIDs
NSAIDs are commonly used in autoimmune diseases such as RA and ankylosing spondylitis (AS) and can effectively reduce the clinical symptoms and signs of disease and eliminate local joint inflammation. However, such drugs can only treat the symptoms rather than the causes of disease and cannot control the activity or progression of the disease. Common adverse reactions to NSAIDs include central nervous system symptoms (pain, dizziness, tinnitus, etc.), cardiovascular damage (high blood pressure, edema, myocardial infarction, heart failure, etc.), gastrointestinal symptoms (abdominal pain, poor appetite, vomiting, ulcers, bleeding, etc.), changes in the hematopoietic system (thrombocytopenia), liver and kidney dysfunction, asthma, and skin eruptions. Following aspirin, many NSAIDs have been developed for clinical use (59).
3.2. Conventional DMARDs
Conventional DMARDs commonly used in clinical practice include methotrexate (MTX), leflunomide (LEF), cyclophosphamide (CTX), azathioprine (AZA), cyclosporin A (CsA), mycophenolate mofetil (MMF), tacrolimus (FK506), and salazosulfapyridine (60). These drugs are widely used in autoimmune diseases, chronic kidney disease, transplant rejection, and tumors. Although the chemical structures and pharmacological mechanisms of the various conventional DMARDs differ, they work in a similar slow-acting manner, inhibiting the progression of RA after a few weeks or months and allowing the symptoms and signs of the disease to remain relatively stable for a long time. LEF mainly inhibits the activity of dihydroorotate dehydrogenase, it affects the synthesis of lymphocytic pyrimidine, and it alleviates the clinical symptoms and improves the laboratory markers of RA.
3.3. Glucocorticoids (GCs)
GCs are widely used in RA and can have potent anti-inflammatory and immunomodulatory actions, reduce the number of mononuclear macrophages in the circulatory system, reduce inflammatory factor and prostaglandin synthesis, and reduce Fc receptor expression (61). At the same time, GCs can prevent inflammatory cell exudation, reduce osteoclast formation, and reduce articular cartilage destruction. GCs have potent therapeutic action on immune cells, humoral factors, osteoblasts, and chondrocytes. GCs are divided into endogenous GCs and exogenous GCs. Endogenous GCs are a class of steroid hormones secreted by the adrenal cortex in the physiological state and include cortisone and hydrocortisone. Exogenous GCs such as dexamethasone and methylprednisolone are often used to treat RA.
3.4. Biological agents
Biological agents act as therapeutic agents by blocking key inflammatory cytokines or cell surface molecules, such as monoclonal antibodies targeting IL-1, IL-6, TNF-α, and IL-17, anti-CD20 monoclonal antibodies, B lymphocyte-stimulating factor (BAFF) inhibitors, T cell inhibitors, integrin monoclonal antibodies, and selective adhesion molecule inhibitors (4).
3.4.1. T cell inhibitors
Abatacept, a fusion protein consisting of the Fc region of IgG1 and the extracellular domain of CTLA4, is a selective T-cell co-stimulation inhibitor. Abatacept inhibits T cell activation by binding to CD80 and CD86 on antigen-presenting cells, thereby inhibiting the production of inflammatory factors such as TNF-α, IFN-γ, and IL-2. It can be used clinically to treat patients with moderate to severe active RA who have not sufficiently responded to one or more conventional DMARDs, as well as patients with juvenile idiopathic arthritis (JIA). Abatacept can reduce serum LL-6, RF, C-reactive protein, MMP-3, and TNF- levels, delay the process of structural destruction of tissue, and reduce the symptoms and signs of RA.
3.4.2. Targeted B-cell therapy
In 2004, the first randomized, double-blind, placebo-controlled trial of rituximab in patients with long-term active RA noted significant results when rituximab was combined with MTX or CTX (62). In addition, a clinical study by the current authors examined the efficacy and safety of different doses of rituximab combined with MTX (with or without glucocorticoids) in patients with active RA who did not respond to conventional DMARDs; both low and high doses of rituximab were effective and well-tolerated (63,64).
Rituximab combined with MTX in one course of treatment can significantly slow the clinical progression of disease activity and alleviate radiation injury in patients with RA not sufficiently responding to anti- TNF-α therapy (65). An open-label prospective study further confirmed that rituximab is a therapeutic option for patients, and especially for seropositive patients (CCP-or RF-positive patients), with no response to single-dose TNF-α inhibitors (66).
3.4.3. IL-6 inhibitors
Tozumab is an anti-IL-6 receptor monoclonal antibody that can inhibit IL-6-mediated signaling by binding to IL-6 transmembrane receptors and inhibiting the production of autoantibodies such as rheumatoid factor (RF) and ACPA. It is mainly used to treat moderate and severe RA as well as JIA. With the success of TOCili- Zumab, multiple biological agents targeting the IL-6 signaling pathway are being developed for treatment of RA. The main adverse reactions to IL-6 inhibitors include an infusion reaction, infection, tumor risk, gastrointestinal ulcer, dyslipidemia, elevated liver transaminase, and neutropenia (67).
Tofacitinib is a novel oral Janus kinase (JAK) inhibitor mediated by JAK1, JAK3, STAT1, and STAT3 via the IL-6/GP130/STAT3 signaling pathway. Tofacitinib is effective in relieving arthritis symptoms in patients with RA, and both the Food and Drug Administration (FDA) and European Medicines Agency (EMA) have approved oral administration of tofacitinib for the treatment of RA (27,68). In addition, tofacitinib can down-regulate the production of pro-inflammatory cytokines IL-17 and IFN-r and the proliferation of CD4+T cells in patients with RA (69,70).
Global data have indicated that patients with RA with an inadequate or poorly tolerated response to anti- TNF-α inhibitors can usually be effectively managed by switching to drugs with new mechanisms of action, such as IL-6R inhibitors (71). IL-6 blockade of signaling pathways (via tocilizumab, which is a monoclonal antibody that binds to IL-6 receptors) can enhance Tregs and inhibit monocyte IL-6 mRNA expression, thereby inducing monocyte apoptosis (72-74). Samalizumab, a monoclonal antibody against IL-6R in humans, was effective and safe in patients with RA with a limited response to MTX in randomized clinical trials (75,76). Other IL-6 inhibitors are shown in Table 1.
Table 1. Biological agents targeting cytokines.
| Cytokine | Drug | Mechanism of action | Phase |
|---|---|---|---|
| IL-6 | Tocilizumab | Inhibit IL-6-mediated signaling involving ubiquitous signal-transducing | Appeared on the market in 2010 |
| Sarilumab | gp130 and STAT3 | Phase III | |
| Clazakizumab | Phase II B | ||
| ALX-0061 | Phase II | ||
| IL-1 | Anakinra | Blocks IL-1 binding to IL-1RI, resulting in intracellular signaling | Appeared on the market in 2001 |
| IL-12/23 | Ustekinumab | Bind to the cytokines IL-12 and IL-23 and down-modulate lymphocyte | Appeared on the market in 2005 |
| Canakinumab | function | Appeared on the market in 2009 | |
| TNF-α | Infliximab | Induce antibody-dependent cytotoxicity (ADCC); the complement | Appeared on the market in 1998 |
| Adalimumab | pathway triggers cell-dependent cytotoxicity (CDC) and targets immune | Appeared on the market in 2002 | |
| Etanercept | cell apoptosis | Appeared on the market in 1998 | |
| Golimumab | Appeared on the market in 2009 | ||
| Certolizumab | Appeared on the market in 2008 |
3.4.4. Anti-IL-12/23 monoclonal antibody
TGF-β, IL-23, and pro-inflammatory cytokines play a role in driving and regulating the human Th17 response in RA (77,78). In addition, an increased Th17 cell count and poor clinical outcomes in patients with RA are associated with IL4R gene variation (79). Therefore, IL-12 and IL-23 participate in the pathogenesis of RA and may be considered potential molecules for immune targeting of RA. Currently, the most widely used anti- IL-12/23 antibody is ustekinumab, which was approved by the US FDA for the treatment of psoriasis in 2009 and which has clinical efficacy significantly superior to that of other biological agents (80). Other anti-IL-12/23 monoclonal antibodies are shown in Table 1.
3.4.5. TNF-α inhibitors
The strategy of blocking TNF-α was introduced into clinical practice at the end of the last century and revolutionized the treatment of RA and many other inflammatory conditions. Steeland et al. recently conducted an impressive review of the successful use of tumor necrosis factor inhibitors including etanercept, infliximab, adamab, cetuximab, and golimumab in RA therapy (81). Infliximab, adalimumab, and golimumab are full-length monoclonal antibodies. In addition to blocking the growth of tumor cells, they act as Fc effectors. They induce antibody-dependent cellular cytotoxicity (ADCC), trigger complement pathways that lead to cell-dependent cytotoxicity (CDC), and target immune cell apoptosis. Etanercept is a soluble TNF receptor that contains truncated Fc domains and that does not contain IgG1 CH1 domains; therefore, etanercept induces less potent ADCC and CDC than monoclonal antibodies such as infliximab (82).
The total number of B cells in the blood of patients with RA is lower than that in healthy controls but it is significantly higher (normal) in patients receiving antitumor necrosis factor therapy. Cardiovascular disease, including heart failure and infection, is the leading cause of disability and death in patients with RA (83). Patients treated with anti-TNF or MTX alone appear to have a further risk of severe infection, such as tuberculosis (84,85). Therefore, anti-TNF-α inhibitory therapy is contraindicated in all patients with heart failure, which represents a considerable proportion of patients with RA (86). Despite the risks associated with anti-TNF-α therapy, it is the treatment of choice for patients with RA when MTX does not provide relief. Other TNF-α inhibitors are shown in Table 1.
3.5. Small molecule inhibitors targeting JAK
3.5.1. Decernotinib
Decernotinib is a next-generation jakinib, and kinase assays revealed its 5-fold selectivity for JAK3 compared to JAK1, JAK2, and TYK2 (87). Decernotinib yielded satisfactory results in animal models of autoimmune diseases (88) and thus entered clinical trials for treatment of RA.
Decernotinib appears to offer promise in the treatment of RA. Phase II trials indicated that a 50-150 mg dose of decernotinib BID improved the American College of Rheumatology (ACR) response criteria and DAS28 joint count for RA with CRP (DAS28-CRP) compared to a placebo. Adverse events reported were similar to those caused by first-generation jakinibs, such as infection, rhinitis, and hyperlipidemia (89-91). Anemia was not observed, which is consistent with decernotinib's selectivity for JAK3 over JAK2. Surprisingly, many patients developed neutropenia, which indicates that the drug may have some off-target effects (87). Recent phase IIb studies have indicated that decernotinib with conventional DMARDs can alleviate synovitis and osteitis in patients with RA (92).
3.5.2. Filgotinib (GLPG0634)
Filgotinib inhibits JAK1 and JAK2 in CBC and kinase assays, but is 30-fold more selective for JAK1 (89). In vitro studies also demonstrated its dose-dependent inhibition of Th1, Th2 and, to a lesser extent, Th17 cell differentiation.
Filgotinib is currently being studied as a potential treatment for RA (93). A phase IIa study indicated that filgotinib was more effective than the placebo at a daily dose of 30 mg or higher (89,94). This was followed by two phase IIb trials: Darwin 1 and Darwin 2. Darwin 1 was a study of 595 patients with RA receiving MTX who were also given filgotinib in a dose ranging from 50 to 100 mg per day. The Darwin 2 study evaluated filgotinib monotherapy in 280 patients with RA at doses ranging from 50 to 200 mg per day (89). In both studies, filgotinib outperformed the placebo in controlling disease activity according to the ACR 20/50 criteria, DAS28-CRP, the Simplified Disease Activity Index (SDAI), and the Clinical Disease Activity Index (95,96). Other small molecule inhibitors targeting JAK are shown in Table 2.
Table 2. Small molecule inhibitors targeting JAK.
| JAK inhibitor | Molecular target | Mechanism of action | Phase |
|---|---|---|---|
| Tofacitinib | JAK1, JAK3 | Interferes with the binding of IL-6 to the IL-6Rα/gp130 complex, STAT proteins | Appeared on the market in 2017 |
| Baricitinib | JAK1, JAK2 | Blocks intracellular signaling, facilitates the turnover of active (phosphorylated) STAT1 and STAT3 | Appeared on the market in 2018 |
| Filgotinib | JAK1 | Blocks intracellular signaling, facilitates the turnover of active (phosphorylated) STAT1 | Phase III |
| Peficitinib | JAK1, JAK3 | Interferes with the binding of IL-6 to the IL-6Rα/gp130 complex, STAT proteins | Phase III |
| SHR0302 | JAK1 | Blocks intracellular signal transduction, facilitates the turnover of active (phosphorylated) STAT1 | Phase II |
4. Summary and prospects for the future
NSAIDs, GCs, conventional DMARDs, biological agents, and other drugs for treatment of RA have definite efficacy but are associated with adverse reactions such as immunosuppression, infection, and the development of new tumors. Therefore, development of anti-inflammatory immunomodulatory drugs for soft regulation of inflammatory immune responses (SRIIR) is important. SRIIR drugs selectively control physiological tissue and cell function and promote recovery from pathological gene and protein changes. Their mechanism may involve one or more key signaling molecules regulating abnormal signaling pathway activity, thus appropriately restoring the static balance of the human body. When SRIIR drugs are used clinically, they can reduce adverse reactions without diminishing physiological function. Paeoniflorin - 6-oxybenzenesulfonic acid ester (code name CP-25) comes from the structural modification of paeoniflorin, an active ingredient of an herbal medicine (97). Cp- 25 can suppress inflammation associated with adjuvant arthritis in rats and collagen-induced arthritis in mice by down-regulating inflammatory mediator production and the immune response, reducing bone damage (98,99). In vitro, CP-25 can inhibit TNF-α or PGE2 stimulation of mature dendritic cells by regulating the expression of CD40, CD80, CD83, CD86, and MHC- Ⅱ. Cp-25 can down-regulate BAFF-stimulated proliferation of B cells, including CD19+ B cells, CD19+ CD20+ B cells, CD19+ CD27+ B cells, and CD19+CD20+CD27+ B cells, and inhibit the expression of BAFFR, TRAF2, and P52. Compared to etanercept and rituximab, CP-25 moderately down-regulates the abnormal rise in B-cell proliferation.
In conclusion, further understanding of the pathological mechanism of autoimmune diseases and the discovery of new drug targets has led to the rapid development of new biological agents targeting cytokines and cell surface molecules in addition to NSAIDs, SAIDs and conventional DMARDs. Biological agents such as monoclonal antibodies targeting IL-1, IL-6, TNF-α, IL-17, and CD20, BAFF inhibitors, T cell inhibitors, integrin monoclonal antibodies, and selective adhesion molecular inhibitors exhibit therapeutic action by blocking inflammatory cytokines or cell surface molecules.
Several small molecule drugs targeting the JAK/ STAT signaling pathway such as tofacitinib, baricitinib, upadacitinib, and filgotinib (see Table 2) have also been developed and used in clinical practice in recent years. Although these drugs are effective, they also cause adverse reactions such as gastrointestinal symptoms, immunosuppression, myelosuppression, and infection. The focus now is on developing an SRIIR with anti-inflammatory immunomodulatory action. Cp-25 may be a new SRIIR with the potential to treat autoimmune diseases. SRIIRs, which control excessive activation of inflammatory immune response-related cells without harming their physiological function, are a new therapeutic strategy and a major direction for development of drugs to treat autoimmune diseases.
Funding: None.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Wasserman AM. Diagnosis and management of rheumatoid arthritis. Am Fam Physician. 2011; 84:1245-1252. [PubMed] [Google Scholar]
- 2.Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002; 4 Suppl 3:S265-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smolen JS, van der Heijde D, Machold KP, Aletaha D, Landewe R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann Rheum Dis. 2014; 73:3-5. [DOI] [PubMed] [Google Scholar]
- 4.Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017; 76:960-977. [DOI] [PubMed] [Google Scholar]
- 5.Chandrupatla D, Molthoff CFM, Lammertsma AA, van der Laken CJ, Jansen G. The folate receptor beta as a macrophage-mediated imaging and therapeutic target in rheumatoid arthritis. Drug Deliv Transl Res. 2019; 9:366-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmdahl R, Malmstrom V, Burkhardt H. Autoimmune priming, tissue attack and chronic inflammation - The three stages of rheumatoid arthritis. Eur J Immunol. 2014; 44:1593-1599. [DOI] [PubMed] [Google Scholar]
- 7.Harre U, Schett G. Cellular and molecular pathways of structural damage in rheumatoid arthritis. Semin Immunopathol. 2017; 39:355-363. [DOI] [PubMed] [Google Scholar]
- 8.Mateen S, Zafar A, Moin S, Khan A Q, Zubair S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin Chim Acta. 2016; 455:161-171. [DOI] [PubMed] [Google Scholar]
- 9.Cohen E, Nisonoff A, Hermes P, Norcross BM, Lockie LM. Agglutination of sensitized alligator erythrocytes by rheumatoid factor(s). Nature. 1961; 190:552-553. [DOI] [PubMed] [Google Scholar]
- 10.Scherer HU, Huizinga TWJ, Kronke G, Schett G, Toes REM. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol. 2018; 14:157-169. [DOI] [PubMed] [Google Scholar]
- 11.Cuda CM, Pope RM, Perlman H. The inflammatory role of phagocyte apoptotic pathways in rheumatic diseases. Nat Rev Rheumatol. 2016; 12:543-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bazzazi H, Aghaei M, Memarian A, Asgarian-Omran H, Behnampour N, Yazdani Y. Th1-Th17 ratio as a new insight in rheumatoid arthritis disease. Iran J Allergy Asthma Immunol. 2018; 17:68-77. [PubMed] [Google Scholar]
- 13.Jung SM, Lee J, Baek SY, Lee J, Jang SG, Hong SM, Park JS, Cho ML, Park SH, Kwok SK. Fraxinellone attenuates rheumatoid inflammation in mice. Int J Mol Sci. 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers LK, Stuart JM, Kang AH. A CD4 cell is capable of transferring suppression of collagen-induced arthritis. J Immunol. 1989; 143:3976-3980. [PubMed] [Google Scholar]
- 15.Chiocchia G, Boissier M C, Ronziere M C, Herbage D, Fournier C. T cell regulation of collagen-induced arthritis in mice. I. Isolation of Type II collagen-reactive T cell hybridomas with specific cytotoxic function. J Immunol. 1990; 145:519-525. [PubMed] [Google Scholar]
- 16.Sakaguchi S, Benham H, Cope A P, Thomas R. T-cell receptor signaling and the pathogenesis of autoimmune arthritis: Insights from mouse and man. Immunol Cell Biol. 2012; 90:277-287. [DOI] [PubMed] [Google Scholar]
- 17.Mai J, Wang H, Yang X F. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front Biosci (Landmark Ed). 2010; 15:986-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelchtermans H, Geboes L, Mitera T, Huskens D, Leclercq G, Matthys P. Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann Rheum Dis. 2009; 68:744-750. [DOI] [PubMed] [Google Scholar]
- 19.Zizzo G, De Santis M, Bosello S L, Fedele A L, Peluso G, Gremese E, Tolusso B, Ferraccioli G. Synovial fluid-derived T helper 17 cells correlate with inflammatory activity in arthritis, irrespectively of diagnosis. Clin Immunol. 2011; 138:107-116. [DOI] [PubMed] [Google Scholar]
- 20.Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol. 2011; 31:596-605. [DOI] [PubMed] [Google Scholar]
- 21.Kerkman P F, Rombouts Y, van der Voort E I, Trouw L A, Huizinga T W, Toes R E, Scherer H U. Circulating plasmablasts/plasmacells as a source of anticitrullinated protein antibodies in patients with rheumatoid arthritis. Ann Rheum Dis. 2013; 72:1259-1263. [DOI] [PubMed] [Google Scholar]
- 22.Pelzek AJ, Gronwall C, Rosenthal P, Greenberg JD, McGeachy M, Moreland L, Rigby WFC, Silverman GJ. Persistence of disease-associated anti-citrullinated protein antibody-expressing memory B cells in rheumatoid arthritis in clinical remission. Arthritis Rheumatol. 2017; 69:1176-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malemud C J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2018; 10:117-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Araki Y, Mimura T. The mechanisms underlying chronic inflammation in rheumatoid arthritis from the perspective of the epigenetic landscape. J Immunol Res. 2016; 2016:6290682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol. 2017; 46:112-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narazaki M, Tanaka T, Kishimoto T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev Clin Immunol. 2017; 13:535-551. [DOI] [PubMed] [Google Scholar]
- 27.Tournadre A, Pereira B, Dutheil F, Giraud C, Courteix D, Sapin V, Frayssac T, Mathieu S, Malochet-Guinamand S, Soubrier M. Changes in body composition and metabolic profile during interleukin 6 inhibition in rheumatoid arthritis. J Cachexia Sarcopenia Muscle. 2017; 8:639-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kishimoto T, Kang S, Tanaka T. IL-6: A new era for the treatment of autoimmune inflammatory diseases. In: Innovative Medicine: Basic Research and Development. Edited by Nakao K, Minato N, Uemoto S. Tokyo; 2015:131-147. [PubMed] [Google Scholar]
- 29.Venuturupalli S. Immune mechanisms and novel targets in rheumatoid arthritis. Immunol Allergy Clin North Am. 2017; 37:301-313. [DOI] [PubMed] [Google Scholar]
- 30.Rose-John S. Interleukin-6 family cytokines. Cold Spring Harb Perspect Biol. 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu W D, Zhao Y, Liu Y. Insights into IL-37, the role in autoimmune diseases. Autoimmun Rev. 2015; 14:1170-1175. [DOI] [PubMed] [Google Scholar]
- 32.Zhao P W, Jiang W G, Wang L, Jiang Z Y, Shan Y X, Jiang Y F. Plasma levels of IL-37 and correlation with TNF-alpha, IL-17A, and disease activity during DMARD treatment of rheumatoid arthritis. PLoS One. 2014; 9:e95346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Wang Y, Xia L, Shen H, Lu J. Elevated frequency of IL-37- and IL-18Ralpha-positive T cells in the peripheral blood of rheumatoid arthritis patients. Cytokine. 2018; 110:291-297. [DOI] [PubMed] [Google Scholar]
- 34.Jin S, Sonobe Y, Kawanokuchi J, Horiuchi H, Cheng Y, Wang Y, Mizuno T, Takeuchi H, Suzumura A. Interleukin-34 restores blood-brain barrier integrity by upregulating tight junction proteins in endothelial cells. PLoS One. 2014; 9:e115981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chemel M, Le Goff B, Brion R, Cozic C, Berreur M, Amiaud J, Bougras G, Touchais S, Blanchard F, Heymann M F, Berthelot J M, Verrecchia F, Heymann D. Interleukin 34 expression is associated with synovitis severity in rheumatoid arthritis patients. Ann Rheum Dis. 2012; 71:150-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia S, Hartkamp L M, Malvar-Fernandez B, van Es I E, Lin H, Wong J, Long L, Zanghi J A, Rankin A L, Masteller E L, Wong B R, Radstake T R, Tak P P, Reedquist K A. Colony-stimulating factor (CSF) 1 receptor blockade reduces inflammation in human and murine models of rheumatoid arthritis. Arthritis Res Ther. 2016; 18:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian Y, Shen H, Xia L, Lu J. Elevated serum and synovial fluid levels of interleukin-34 in rheumatoid arthritis: possible association with disease progression via interleukin-17 production. J Interferon Cytokine Res. 2013; 33:398-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang SJ, Choi B, Kang SS, Chang JH, Kim YG, Chung YH, Sohn DH, So MW, Lee CK, Robinson WH, Chang EJ. Interleukin-34 produced by human fibroblast-like synovial cells in rheumatoid arthritis supports osteoclastogenesis. Arthritis Res Ther. 2012; 14:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang SH, Choi BY, Choi J, Yoo JJ, Ha YJ, Cho HJ, Kang EH, Song YW, Lee YJ. Baseline serum interleukin-34 levels independently predict radiographic progression in patients with rheumatoid arthritis. Rheumatol Int. 2015; 35:71-79. [DOI] [PubMed] [Google Scholar]
- 40.Moon SJ, Hong YS, Ju JH, Kwok SK, Park SH, Min JK. Increased levels of interleukin 34 in serum and synovial fluid are associated with rheumatoid factor and anticyclic citrullinated peptide antibody titers in patients with rheumatoid arthritis. J Rheumatol. 2013; 40:1842-1849. [DOI] [PubMed] [Google Scholar]
- 41.Llanos C, Soto L, Sabugo F, Bastias MJ, Salazar L, Aguillon JC, Cuchacovich M. The influence of -238 and -308 TNF alpha polymorphisms on the pathogenesis and response to treatment in rheumatoid arthritis. Rev Med Chil. 2005; 133:1089-1095. [DOI] [PubMed] [Google Scholar]
- 42.Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheum Dis. 2006; 65:294-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klimiuk PA, Sierakowski S, Latosiewicz R, Skowronski J, Cylwik JP, Cylwik B, Chwiecko J. Histological patterns of synovitis and serum chemokines in patients with rheumatoid arthritis. J Rheumatol. 2005; 32:1666-1672. [PubMed] [Google Scholar]
- 44.Canete JD, Martinez SE, Farres J, Sanmarti R, Blay M, Gomez A, Salvador G, Munoz-Gomez J. Differential Th1/Th2 cytokine patterns in chronic arthritis: Interferon gamma is highly expressed in synovium of rheumatoid arthritis compared with seronegative spondyloarthropathies. Ann Rheum Dis. 2000; 59:263-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002; 296:1653-1655. [DOI] [PubMed] [Google Scholar]
- 46.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002; 285:1-24. [DOI] [PubMed] [Google Scholar]
- 47.Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. 2018; 189:4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal. 2013; 25:1924-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat Rev Cancer. 2014; 14:736-746. [DOI] [PubMed] [Google Scholar]
- 50.Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol. 2016; 54:28-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshimura A, Ito M, Chikuma S, Akanuma T, Nakatsukasa H. Negative regulation of cytokine signaling in immunity. Cold Spring Harb Perspect Biol. 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: The SOCS perspective. Cytokine Growth Factor Rev. 2013; 24:241-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kallen KJ. The role of transsignalling via the agonistic soluble IL-6 receptor in human diseases. Biochim Biophys Acta. 2002; 1592:323-343. [DOI] [PubMed] [Google Scholar]
- 54.Scheller J, Ohnesorge N, Rose-John S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol. 2006; 63:321-329. [DOI] [PubMed] [Google Scholar]
- 55.Walker JG, Smith MD. The Jak-STAT pathway in rheumatoid arthritis. J Rheumatol. 2005; 32:1650-1653. [PubMed] [Google Scholar]
- 56.Kasperkovitz PV, Verbeet NL, Smeets TJ, van Rietschoten JG, Kraan MC, van der Pouw Kraan TC, Tak PP, Verweij CL. Activation of the STAT1 pathway in rheumatoid arthritis. Ann Rheum Dis. 2004; 63:233-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krause A, Scaletta N, Ji JD, Ivashkiv LB. Rheumatoid arthritis synoviocyte survival is dependent on Stat3. J Immunol. 2002; 169:6610-6616. [DOI] [PubMed] [Google Scholar]
- 58.Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/ JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018; 15:234-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bermas B L. Non-steroidal anti inflammatory drugs, glucocorticoids and disease modifying anti-rheumatic drugs for the management of rheumatoid arthritis before and during pregnancy. Curr Opin Rheumatol. 2014; 26:334-340. [DOI] [PubMed] [Google Scholar]
- 60.Combe B, Landewe R, Daien C I, et al. 2016 update of the EULAR recommendations for the management of early arthritis. Ann Rheum Dis. 2017; 76:948-959. [DOI] [PubMed] [Google Scholar]
- 61.Strehl C, Buttgereit F. Optimized glucocorticoid therapy: Teaching old drugs new tricks. Mol Cell Endocrinol. 2013; 380:32-40. [DOI] [PubMed] [Google Scholar]
- 62.Edwards JC, Szczepanski L, Szechinski J, Filipowicz- Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004; 350:2572-2581. [DOI] [PubMed] [Google Scholar]
- 63.Schioppo T, Ingegnoli F. Current perspective on rituximab in rheumatic diseases. Drug Des Devel Ther. 2017; 11:2891-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollenhoven RF, Li NF, Agarwal S, Hessey EW, Shaw TM, Group DS. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: Results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006; 54:1390-1400. [DOI] [PubMed] [Google Scholar]
- 65.Mease PJ, Cohen S, Gaylis NB, Chubick A, Kaell AT, Greenwald M, Agarwal S, Yin M, Kelman A. Efficacy and safety of retreatment in patients with rheumatoid arthritis with previous inadequate response to tumor necrosis factor inhibitors: Results from the SUNRISE trial. J Rheumatol. 2010; 37:917-927. [DOI] [PubMed] [Google Scholar]
- 66.Emery P, Gottenberg J E, Rubbert-Roth A, et al. Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2015; 74:979-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rubbert-Roth A, Furst DE, Nebesky JM, Jin A, Berber E. A review of recent advances using tocilizumab in the treatment of rheumatic diseases. Rheumatol Ther. 2018; 5:21-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Traynor K. FDA approves tofacitinib for rheumatoid arthritis. Am J Health Syst Pharm. 2012; 69:2120. [DOI] [PubMed] [Google Scholar]
- 69.Gertel S, Mahagna H, Karmon G, Watad A, Amital H. Tofacitinib attenuates arthritis manifestations and reduces the pathogenic CD4 T cells in adjuvant arthritis rats. Clin Immunol. 2017; 184:77-81. [DOI] [PubMed] [Google Scholar]
- 70.Cheung TT, McInnes IB. Future therapeutic targets in rheumatoid arthritis? Semin Immunopathol. 2017; 39:487-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chastek B, Becker LK, Chen CI, Mahajan P, Curtis JR. Outcomes of tumor necrosis factor inhibitor cycling versus switching to a disease-modifying anti-rheumatic drug with a new mechanism of action among patients with rheumatoid arthritis. J Med Econ. 2017; 20:464-473. [DOI] [PubMed] [Google Scholar]
- 72.Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, Ornetti P, Maillefert JF, Miossec P, Bonnotte B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012; 64:2499-2503. [DOI] [PubMed] [Google Scholar]
- 73.Pesce B, Soto L, Sabugo F, Wurmann P, Cuchacovich M, Lopez MN, Sotelo PH, Molina MC, Aguillon JC, Catalan D. Effect of interleukin-6 receptor blockade on the balance between regulatory T cells and T helper type 17 cells in rheumatoid arthritis patients. Clin Exp Immunol. 2013; 171:237-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarantopoulos A, Tselios K, Gkougkourelas I, Pantoura M, Georgiadou AM, Boura P. Tocilizumab treatment leads to a rapid and sustained increase in Treg cell levels in rheumatoid arthritis patients: Comment on the article by Thiolat et al. Arthritis Rheumatol. 2014; 66:2638. [DOI] [PubMed] [Google Scholar]
- 75.Tono T, Aihara S, Hoshiyama T, Arinuma Y, Nagai T, Hirohata S. Effects of anti-IL-6 receptor antibody on human monocytes. Mod Rheumatol. 2015; 25:79-84. [DOI] [PubMed] [Google Scholar]
- 76.Huizinga T W, Fleischmann R M, Jasson M, Radin A R, van Adelsberg J, Fiore S, Huang X, Yancopoulos G D, Stahl N, Genovese M C. Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: Efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis. 2014; 73:1626-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: Results of a phase III study. Arthritis Rheumatol. 2015; 67:1424-1437. [DOI] [PubMed] [Google Scholar]
- 78.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008; 9:650-657. [DOI] [PubMed] [Google Scholar]
- 79.Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. 2014; 141:133-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leipe J, Schramm MA, Prots I, Schulze-Koops H, Skapenko A. Increased Th17 cell frequency and poor clinical outcome in rheumatoid arthritis are associated with a genetic variant in the IL4R gene, rs1805010. Arthritis Rheumatol. 2014; 66:1165-1175. [DOI] [PubMed] [Google Scholar]
- 81.Molinelli E, Campanati A, Brisigotti V, Offidani A. Biologic therapy in psoriasis (part II): Efficacy and safety of new treatment targeting IL23/IL-17 pathways. Curr Pharm Biotechnol. 2017; 18:964-978. [DOI] [PubMed] [Google Scholar]
- 82.Steeland S, Libert C, Vandenbroucke RE. A new venue of TNF targeting. Int J Mol Sci. 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Billmeier U, Dieterich W, Neurath MF, Atreya R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol. 2016; 22:9300-9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levy L, Fautrel B, Barnetche T, Schaeverbeke T. Incidence and risk of fatal myocardial infarction and stroke events in rheumatoid arthritis patients. A systematic review of the literature. Clin Exp Rheumatol. 2008; 26:673-679. [PubMed] [Google Scholar]
- 85.Pala O, Diaz A, Blomberg BB, Frasca D. B lymphocytes in rheumatoid arthritis and the effects of anti-TNF-alpha agents on B lymphocytes: A review of the literature. Clin Ther. 2018; 40:1034-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kroesen S, Widmer A F, Tyndall A, Hasler P. Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-alpha therapy. Rheumatology (Oxford). 2003; 42:617-621. [DOI] [PubMed] [Google Scholar]
- 87.Kotyla P J. Bimodal function of anti-TNF treatment: Shall we be concerned about anti-TNF treatment in patients with rheumatoid arthritis and heart failure? Int J Mol Sci. 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mahajan S, Hogan JK, Shlyakhter D, Oh L, Salituro FG, Farmer L, Hoock TC. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J Pharmacol Exp Ther. 2015; 353:405-414. [DOI] [PubMed] [Google Scholar]
- 89.Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014; 23:1067-1077. [DOI] [PubMed] [Google Scholar]
- 90.Genovese MC, van Vollenhoven RF, Pacheco-Tena C, Zhang Y, Kinnman N. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016; 68:46-55. [DOI] [PubMed] [Google Scholar]
- 91.Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 2015; 67:334-343. [DOI] [PubMed] [Google Scholar]
- 92.Genovese MC, Yang F, Ostergaard M, Kinnman N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: Clinical and MRI findings. Ann Rheum Dis. 2016; 75:1979-1983. [DOI] [PubMed] [Google Scholar]
- 93.Namour F, Diderichsen PM, Cox E, Vayssiere B, Van der Aa A, Tasset C, Van't Klooster G. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015; 54:859-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Namour F, Diderichsen P M, Cox E, Vayssiere B, Van der Aa A, Tasset C, Van't Klooster G. Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clin Pharmacokinet. 2015; 54:859-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko O V, Stanislavchuk M, Greenwald M, Van der Aa A, Vanhoutte F, Tasset C, Harrison P. Filgotinib (GLPG0634/ GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. 2017; 76:1009-1019. [DOI] [PubMed] [Google Scholar]
- 96.Yang XD, Wang C, Zhou P, Yu J, Asenso J, Ma Y, Wei W. Absorption characteristic of paeoniflorin-6'-O-benzene sulfonate (CP-25) in in situ single-pass intestinal perfusion in rats. Xenobiotica. 2016; 46:775-783. [DOI] [PubMed] [Google Scholar]
- 97.Chang Y, Jia X, Wei F, Wang C, Sun X, Xu S, Yang X, Zhao Y, Chen J, Wu H, Zhang L, Wei W. CP-25, a novel compound, protects against autoimmune arthritis by modulating immune mediators of inflammation and bone damage. Sci Rep. 2016; 6:26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen J, Wang Y, Wu H, Yan S, Chang Y, Wei W. A Modified Compound From Paeoniflorin, CP- 25, Suppressed immune responses and synovium inflammation in collagen-induced arthritis mice. Front Pharmacol. 2018; 9:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang F, Shu JL, Li Y, Wu YJ, Zhang XZ, Han L, Tang XY, Wang C, Wang QT, Chen JY, Chang Y, Wu HX, Zhang LL, Wei W. CP-25, a novel anti-inflammatory and immunomodulatory drug, inhibits the functions of activated human B cells through regulating BAFF and TNF-alpha signaling and comparative efficacy with biological agents. Front Pharmacol. 2017; 8:933. [DOI] [PMC free article] [PubMed] [Google Scholar]