

Abstract
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessive disease caused by mutations in the CYP27A1 gene, encoding the sterol 27-hydroxylase. Disruption of the bile acid biosynthesis pathway and accumulation of toxic precursors such as cholestanol cause chronic diarrhea, bilateral juvenile cataracts, tissue deposition of cholestanol and cholesterol (xanthomas), and progressive motor/neuropsychiatric alterations. We have evaluated the therapeutic potential of adeno-associated virus (AAV) vectors expressing CYP27A1 in a CTX mouse model. We found that a vector equipped with a strong liver-specific promoter (albumin enhancer fused with the α1 anti-trypsin promoter) is well tolerated and shows therapeutic effect at relatively low doses (1.5 × 1012 viral genomes [vg]/kg), when less than 20% of hepatocytes overexpress the transgene. This vector restored bile acid metabolism and normalized the concentration of most bile acids in plasma. By contrast, standard treatment (oral chenodeoxycholic acid [CDCA]), while reducing cholestanol, did not normalize bile acid composition in plasma and resulted in supra-physiological levels of CDCA and its derivatives. At the transcriptional level, only the vector was able to avoid the induction of xenobiotic-induced pathways in mouse liver. In conclusion, the overexpression of CYP27A1 in a fraction of hepatocytes using AAV vectors is well tolerated and provides full metabolic restoration in Cyp27a1−/− mice. These features make gene therapy a feasible option for the etiological treatment of CTX patients.
Keywords: cerebrotendinous xanthomatosis, CTX, adeno-associated virus, gene therapy, chenodeoxycholic acid, cholestanol
Graphical abstract
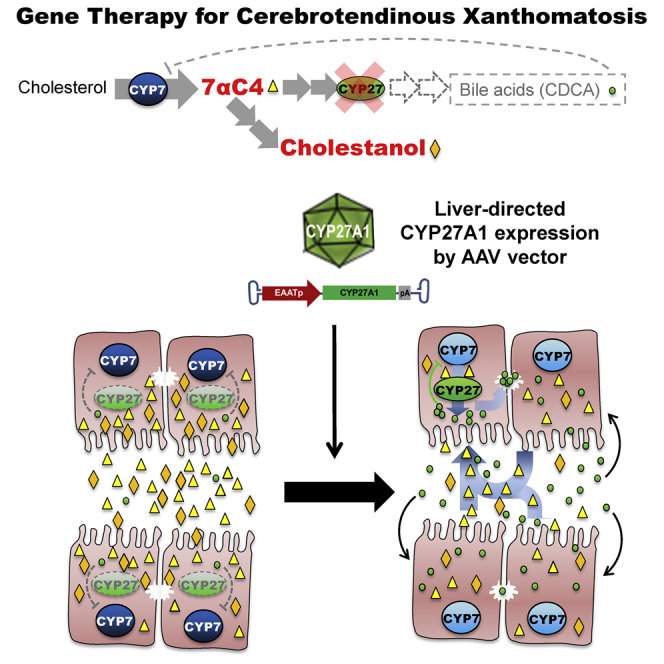
Cerebrotendinous xanthomatosis (CTX) is caused by mutations in the CYP27A1 gene, which is involved in the biosynthesis of bile acids. We found that a relatively low intravenous dose of an AAV vector expressing this gene under the control of a strong liver-specific promoter restores bile acid metabolism in knockout mice.
Introduction
Cerebrotendinous xanthomatosis (CTX, OMIM 213700) is an autosomal recessive disease caused by mutations in the CYP27A1 gene,1,2 which encodes a mitochondrial sterol 27-hydroxylase (CYP27A1, EC 1.14.13.15).3 This enzyme is involved in cholesterol metabolism and particularly in different steps of bile acid (BA) biosynthetic pathways.4 CTX patients present complex and heterogeneous clinical manifestations, including chronic diarrhea with infantile onset, juvenile cataracts, tendon xanthomas, osteoporosis, premature atherosclerosis, mild respiratory insufficiency, and progressive neuropsychiatric alterations (ataxia, peripheral neuropathy, epilepsy, depression, and cognitive deterioration).5 Some reports describe cases of severe infantile cholestasis,6, 7, 8 although liver damage is not a common characteristic of this disease. The onset and severity of symptoms differ among CTX patients, which contributes to the fact that their diagnosis is usually delayed more than 15 years.9,10 Recent analyses based on pathogenic allele frequencies predict an approximate incidence of 1:150,000 in Europeans; 1:250,000 in Africans; 1:70,000 in Americans; 1:65,000 in East Asians; and 1:35,000 in South Asians.11 These figures suggest that CTX is underdiagnosed and markedly under-reported, since only a few hundred cases have been described in the literature.4 The physiopathology of CTX is complex and not fully understood.12,13 Although CYP27A1 is expressed in most cells, its absence in hepatocytes explains the deficiency of chenodeoxycholic acid (CDCA) observed in patients. Through the classical (neutral) pathway, cholesterol is first converted into 7α-hydroxycholesterol by the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1) and then CYP27A1 is involved in further steps of side-chain hydroxylation. In the alternative (acidic) pathway, CYP27A1 directly oxidizes cholesterol into 27-hydroxycholesterol, which is subsequently hydroxylated by other enzymes.4 Decreased CDCA levels result in CYP7A1 upregulation, which together with the deficiency of 27-hydroxylase activity causes accumulation of precursor metabolites including 7α-hydroxy-4-cholesten-3-one (7αC4), cholestanol, and bile alcohols.14 The xanthomas in CTX consist of tissue depositions of cholesterol and cholestanol crystals surrounded by foamy macrophages and fibroblasts. Apart from tendons, these lesions can be found in the white matter, especially in the cerebellum, which justifies the inclusion of CTX in the group of leukodystrophic diseases.15 The mouse model available at the moment is based on a homozygous truncation of the Cyp27a1 gene.16 Although it recapitulates key biochemical traits of the human disease and has been instrumental for physiopathological studies,13 the elevation of cholestanol is milder than in most CTX patients.17 This is in part due to the presence of alternative bile acid biosynthetic routes and stronger detoxification systems in mice compared with humans.18 As a consequence, no xanthomas and no consistent neurological manifestations have been described in this model. The standard treatment for CTX is oral CDCA supplementation, which efficiently inhibits CYP7A1 expression and avoids accumulation of cholestanol.19 Although this pharmacological approach is able to control most clinical manifestations, some biochemical parameters remain altered.14 In the present study, we have developed an alternative treatment based on gene supplementation of CYP27A1 in the liver. Extensive preclinical and clinical studies have demonstrated that adeno-associated virus (AAV) vectors are safe and efficient for in vivo liver transduction.20 Here, we describe an AAV8 vector expressing the human CYP27A1 coding sequence under the control of a strong liver-specific promoter. Using the Cyp27a1 knockout mouse model, we have compared the biological effect of the AAV8-EAAT-CYP27A1 vector and the standard CDCA treatment. Whereas both approaches showed therapeutic effect, we observed different mechanisms of action. CDCA administration caused deep inhibition of Cyp7a1 expression, strong reduction of cholestanol, and sup ra-physiological levels of CDCA in plasma. However, persistent activation of the hepatic enzymes involved in xenobiotic-induced responses suggests that some toxic metabolites are still present. By contrast, a single administration of AAV8-EAAT-CYP27A1 at a clinically feasible dose achieved restoration of bile acid metabolism together with normalization of bile acid pool composition, as suggested by serum bile acid profile.
Results
An AAV vector equipped with a liver-specific promoter achieves efficient expression of CYP27A1 in CTX mice
Our therapeutic approach is based on expression of CYP27A1 in the liver. For the design of the expression cassette, we compared the performance of two different regulatory sequences, depicted in Figure 1A: (1) a well-established hybrid liver-specific promoter (EAAT)21 and (2) the endogenous CYP27A1 regulatory sequence comprising 2,024 base pairs (bp) upstream of the translation start site22,23 (referred hereinafter as C27P). The purpose was to determine whether the C27P promoter was suitable for regulation of transgene expression. Both sequences were first incorporated in luciferase (Luc) reporter plasmids and transfected into different liver-derived cell lines from human (HuH-7, HepG2, and Hep3B) and mouse origin (Hepa1-6 and AML12). As a reference, we used the reporter plasmid containing the cytomegalovirus (CMV) promoter. The promoter-less plasmid pGL3-Basic was used to determine background luciferase activity. We obtained different results depending on the cell line. Whereas the EAAT promoter was more potent than C27P in HepG2 and HuH-7 cells, no difference was observed in AML12, and both promoters showed relatively low activity in Hep3B and Hepa1-6 cells (Figure 1B). In order to obtain more relevant information, we performed in vivo transfection of plasmids in C57BL/6 mice by means of hydrodynamics injection. Quantification of light emission by bioluminescence imaging (BLI) 48 h after injection revealed a strong transcriptional activity of the EAAT promoter and a relatively low strength of the C27P sequence (Figure 1C), as expected based on the abundance of endogenous albumin and Cyp27a1 transcripts (https://gtexportal.org/home/). Both promoters were used to control the transcription of the CYP27A1 coding sequence, in the context of an AAV vector genome, giving rise to the AAV8-EAAT-CYP27A1 and AAV8-C27P-CYP27A1 vectors (Figure 2A). Seven-week-old CTX mice were treated with intravenous injections of the vectors at doses ranging from 5 × 1011 to 5 × 1013 viral genomes [vg]/kg. Animals were sacrificed two weeks later, and transgene expression (CYP27A1) was analyzed by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) in liver extracts. As a reference for physiological expression, the endogenous mouse Cyp27a1 messenger RNA (mRNA) was quantified using primers targeted to exon 8 (Table 1). As expected, the full-length mouse Cyp27a1 mRNA was only detected in wild-type (WT) mice when these primers were used (Figure 2B). Mice treated with the AAV8-C27P-CYP27A1 vector showed CYP27A1 mRNA levels above background only when the dose reached 5 × 1012 vg/kg. Even at the highest dose tested (5 × 1013 vg/kg), the mRNA content was below the physiological level detected in WT mice. Although direct comparison of mouse and human mRNAs is not straightforward using qRT-PCR, these data indicate that the C27P regulatory sequence is weaker than the endogenous CYP27A1 promoter in its genomic context. By contrast, treatment with the AAV8-EAAT-CYP27A1 vector obtained a robust expression of CYP27A1 even at the lowest dose tested (5 × 1011 vg/kg, equivalent to 1 × 1010 vg per mouse). A fraction of liver samples was processed for protein extraction, and western blot was performed in order to determine CYP27A1 content. Of note, we used an antibody capable of detecting the mouse and human enzymes, but not the truncated protein expressed by the CTX mice. The result partially confirms the strong expression in mice treated with the AAV8-EAAT-CYP27A1 vector. However, a global increase of CYP27A1 above WT levels was only observed when the dose of vector was 1.5 × 1012 vg/kg or higher (Figure 2C). In agreement with the qRT-PCR results, the AAV8-C27P-CYP27A1 vector only achieved detectable CYP27A1 protein at the highest dose tested. In order to determine the percentage of transduced hepatocytes corresponding to these vector doses, liver samples were processed for immunohistochemistry. In mice treated with AAV8-EAAT-CYP27A1, hepatocytes overexpressing CYP27A1 could be readily detected, preferentially in the centrilobular zone (Figure 2D). We observed that a global increase of CYP27A1 protein could be obtained when less than 20% of hepatocytes overexpress the transgene (1.5 × 1012 vg/kg vector dose). However, limitations in the sensitivity of the antibody precluded detection of hepatocytes expressing low levels, which resulted in a misleadingly low percentage of positive hepatocytes in mice treated with the AAV8-C27P-CYP27A1 vector. We could not obtain consistent detection of mouse Cyp27a1 protein in WT mice using the same antibody (data not shown). To elucidate the suspected under-estimation of hepatocyte transduction, a new set of mice were treated with intravenous injections of the AAV8-EAAT-GFP vector, which expresses the reporter gene GFP under the control of the EAAT promoter. The high specificity and sensitivity of GFP immunohistochemical detection allowed confirmation that a dose of 1.5 × 1012 vg/kg vector transduces approximately 10% of mouse hepatocytes, whereas the 1.5 × 1013 vg/kg dose reaches close to 80% (Figure S1A).
Figure 1.

The EAAT promoter shows stronger activity than the CYP27A1 5′ UTR in some cell lines and in vivo
(A) Schematic representation of luciferase reporter plasmids containing the CMV promoter, the hybrid liver-specific promoter EAAT (albumin enhancer linked to the α1 anti-trypsin promoter), and the 5′ UTR from the human CYP27A1 gene (C27P). The promoter-less plasmid pGL3-Basic was used as a negative control. (B) The plasmids were transfected in the indicated liver-derived cell lines from human (HuH-7, HepG2, and Hep3B) and mouse (Hepa1-6 and AML12) origin. Luciferase activity was measured in cell extracts obtained 48 h after transfection. The result is expressed as percentage of activity, considering the CMV promoter as a reference. (C) The plasmids were injected in C57BL/6 mice (n = 6) by hydrodynamic injection, and light emission was quantified by BLI 48 h later. Bars represent means ± SEM for each group. ∗p < 0.05, Kruskal-Wallis.
Figure 2.

Expression of CYP27A1 from AAV vectors
(A) Schematic representation of vector genomes. ITR, inverted terminal repeat; pA, polyadenylation signal. (B) The AAV8-EAAT-CYP27A1 and AAV8-C27P-CYP27A1 vectors (abbreviated as EAAT and C27P, respectively) were administered intravenously to 7-week-old CTX mice at the indicated doses; 2 weeks later, mice were sacrificed for quantification of human and mouse CYP27A1 mRNA by qRT-PCR. WT littermates were included as a reference. Data are represented as relative mRNA content, using the housekeeping gene 36b4 as a reference and multiplied by a factor of 1,000 for easier visualization (WT and CTX controls, n = 10; treated CTX, n = 5). Data include pooled male and female mice because no difference was observed between the sexes. (C) One portion of liver samples was used to detect the CYP27A1 protein by western blot. Image shows a representative blot together with quantification of all samples (WT and CTX controls, n = 10; treated CTX, n = 5). Note that the antibody is able to detect both mouse and human protein using this technique. (D) Immunohistochemistry for detection of CYP27A1 (representative images) and quantification of positive hepatocytes. Bars represent means ± SEM for each group. ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Kruskal-Wallis with Dunn’s post test.
Table 1.
List of primers
| Gene | Primer | Sequence |
|---|---|---|
| Human CYP27A1 | forward | 5′-TGTGCTTAAGGAGACTCTGCG-3′ |
| reverse | 5′-ATAGTGGCAGAACACAAACTGG-3′ | |
| Mouse Cyp27a1 exons 1/2 | forward | 5′-ACAAGGCTATGTGCTGCACTTG-3′ |
| reverse | 5′-TGATCCATGTGGTCTCTTATTG-3′ | |
| Mouse Cyp27a1 exons 8/9 | forward | 5′-ATGGCTGAGGAAGAAAGAGG-3′ |
| reverse | 5′-ACACAGTCTTTACTTCTCCCATC-3′ | |
| Mouse Cyp7a1 | forward | 5′-GAAGCAATGAAAGCAGCCTC-3′ |
| reverse | 5′-GTAAATGGCATTCCCTCCAG-3′ | |
| Mouse Cyp3a11 | forward | 5′-TGAATATGAAACTTGCTCTCACTAAAA-3′ |
| Reverse | 5′-CCTTGTCTGCTTAATTTCAGAGGT-3′ | |
| Mouse 36b4 | Forward | 5′-AACAATCTCCCCCTTCTCCTT-3′ |
| Reverse | 5′-GAAGGCCTTGACCTTTTCAG-3′ |
The AAV8-EAAT-CYP27A1 vector normalizes cholestanol and 7αC4 levels in CTX mice
In order to assess the biological effect of the CYP27A1-expressing vectors, blood was collected from CTX mice two weeks after a single intravenous administration. Analysis of cholestanol and 7αC4 in plasma showed that AAV8-EAAT-CYP27A1 was able to normalize the metabolite levels at doses equal to or higher than 1.5 × 1012 vg/kg in female and male mice (Figure 3). The lowest dose of this vector (5 × 1011 vg/kg) achieved only a partial reduction, which was more evident in the case of 7αC4. The same trend was observed when the AAV8-C27P-CYP27A1 vector was used at the highest dose (5 × 1013 vg/kg), in line with the relatively low expression of the transgene. The effects of gene therapy were compared with the standard treatment. To this aim, CTX mice were fed with diets enriched in CDCA at different percentages (0.1, 0.5, or 1% of chow weight) and followed for one month. We observed a dose-dependent reduction of cholestanol and 7αC4 in plasma (Figure 3). The decrease in cholestanol levels was especially intense, reaching values below those of WT mice at 0.5% CDCA or higher. In fact, we found that the therapeutic dose in the CTX model was 0.5%, since this dose was required to achieve a significant reduction of 7αC4 in mice (both male and female). Increasing the dose to 1% CDCA caused weight loss (Figure S2) and was not further evaluated.
Figure 3.

AAV8-EAAT-CYP27A1 and CDCA reduce cholestanol and 7αC4 in CTX mice
(A–D) The AAV8-EAAT-CYP27A1 or AAV8-C27P-CYP27A1 vectors were administered intravenously to 7-week-old CTX mice at the indicated doses. CDCA was mixed in mouse chow at 0.1, 0.5, or 1%. Blood was collected 2 weeks after vector administration or 1 month after initiation of CDCA diet. The concentrations of cholestanol in (A) females and (B) males and 7αC4 in (C) females and (D) males were determined in mouse plasma. Untreated CTX mice and WT littermates were included as a reference (WT, n = 10; CTX, n = 15; treated CTX, n = 5 each sex). Bars represent means ± SEM for each group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 versus untreated CTX mice. #p < 0.05; ###p < 0.001 versus WT mice. Kruskal-Wallis with Dunn’s post test.
In order to determine the stability of transgene expression and therapeutic effect, additional groups of CTX mice treated with AAV8-EAAT-CYP27A1 at 1.5 × 1012 or 1.5 × 1013 vg/kg were sacrificed 2 weeks and 5 months after treatment. The analysis of liver and blood samples confirmed sustained transgene expression and correction of metabolites (Figures 4A and 4B, respectively). The AAV8-EAAT-CYP27A1 vector was well tolerated in CTX mice, with no elevation of serum transaminases (Figure 4C) and absence of histopathological abnormalities in the liver (Figure 4D). Untreated CTX mice showed higher alanine aminotransferase (ALT) levels compared with WT littermates, but this mild elevation could be related to the presence of hepatomegaly, as discussed below.
Figure 4.

AAV8-EAAT-CYP27A1 is well tolerated and achieves sustained therapeutic effect after a single administration
(A–D) The AAV8-EAAT-CYP27A1 vector (EAAT) was administered intravenously to 7-week-old CTX mice at the indicated doses (×1012 vg/kg). (A) Some mice from each group (n = 4) were sacrificed 15 days after vector administration, and the rest (n = 7) were maintained for 5 months. CYP27A1 mRNA was measured by qRT-PCR in liver samples. Data are represented as relative mRNA content, using the housekeeping gene 36b4 as a reference and multiplied by a factor of 1,000 for easier visualization. Blood samples were obtained at the indicated times for measurement of cholestanol and 7αC4 (B) and the transaminase ALT (C). Untreated CTX mice and WT littermates were included as a reference. Data correspond to equilibrated groups of female and male mice. (D) Representative images of liver histology (hematoxylin and eosin staining) of mice 5 months after initiation of treatment. Symbols represent the mean ± SEM for each group.∗p < 0.05; ∗∗∗p < 0.001 versus untreated CTX mice. Kruskal-Wallis with Dunn’s post test.
The AAV8-EAAT-CYP27A1 vector restores bile acid metabolism in CTX mice
After confirming the biological effect of gene therapy and CDCA treatment on biochemical markers of CTX, we studied the impact on the expression of key enzymes involved in bile acid metabolism. To this aim, mRNA was extracted from liver samples collected 2 weeks after vector administration or 1 month after initiation of the CDCA treatment. First, we studied the impact of the treatments on the transcriptional control of the endogenous Cyp27a1 gene. Since CTX mice present truncation of the gene at the penultimate exon (number 8),16 we used primers targeting exons 1/2 in order to detect the WT or truncated transcripts. In contrast with results shown in Figure 2B (in which primers were designed in exons 8/9), Cyp27a1 mRNA could be detected in CTX mice. The transcripts were less abundant than in WT littermates, probably because of nonsense-mediated decay. We observed that supplementation of CP27A1 had no influence on the transcription of the endogenous gene, whereas CDCA treatment caused a moderate inhibition (Figure 5A). Quantification of Cyp7a1 expression confirmed upregulation of this rate-limiting enzyme in CTX mice compared with their WT littermates (Figure 5B). The AAV8-EAAT-CYP27A1 vector demonstrated efficient normalization of Cyp7a1 expression even at the lowest dose tested (5 × 1011 vg/kg), in agreement with the reduction of 7αC4 shown in Figure 3. By contrast, a high dose of the AAV8-C27P-CYP27A1 vector was completely inefficient. Treatment with 0.5% CDCA caused a drastic reduction of Cyp7a1 expression, below physiological levels. Second, we analyzed expression of the Cyp3a11 gene, which encodes a key enzyme in the response to xenobiotics in the liver.24 As previously described,18 CTX mice showed overexpression of this gene (Figure 5C), thanks to the activation of the pregnane X receptor (PXR) pathway. Interestingly, 0.5% CDCA achieved only a slight reduction of Cyp3a11 expression, whereas AAV8-EAAT-CYP27A1 completely normalized mRNA content at all doses. These effects were entirely dependent on the efficient expression of CYP27A1 from AAV8-EAAT-CYP27A1, since an equivalent vector expressing GFP showed no changes compared with untreated CTX mice (Figure S1B). In addition, the AAV8-C27P-CYP27A1 vector showed no significant effect.
Figure 5.

AAV8-EAAT-CYP27A1, but not CDCA, normalizes the expression of Cyp7a1 and Cyp3a11 in CTX mice
The AAV8-EAAT-CYP27A1 or AAV8-C27P-CYP27A1 vectors were administered intravenously to 7-week-old CTX mice at the indicated doses. CDCA was mixed in mouse chow at 0.1 or 0.5%. Untreated CTX and WT littermates were included as a reference. Animals were sacrificed 2 weeks after vector administration, or 1 month after initiation of CDCA diet, and liver samples were obtained for quantification of endogenous Cyp27a1 (A), Cyp7a1 (B), and Cyp3a11 (C) expression. Data are represented as relative mRNA content, using the housekeeping gene 36b4 as a reference and multiplied by a factor of 1,000 for easier visualization (WT and CTX control, n = 15; EAAT and C27P, n = 5; CDCA, n = 8). Bars represent means ± SEM for each group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 versus untreated CTX mice. #p < 0.05; ##p < 0.05; ###p < 0.001 versus WT mice. Kruskal-Wallis with Dunn’s post test.
Activation of the xenobiotic response pathways produces hepatomegaly in CTX mice.25 This alteration was reversed by the AAV8-EAAT-CYP27A1 vector, but only partially by CDCA at 0.5% (Figure 6A).
Figure 6.

AAV8-EAAT-CYP27A1 reverses hepatomegaly in CTX mice
The AAV8-EAAT-CYP27A1 vector was administered intravenously to 7-week-old CTX mice at the indicated doses. CDCA was mixed in mouse chow at 0.1 or 0.5%. Untreated CTX and WT littermates were included as a reference. Mice were maintained for 3 months and then they were sacrificed for determination of relative liver weight, represented as percentage of body weight. Bars represent means ± SEM for each group. ∗p < 0.05; ∗∗∗p < 0.001 versus untreated CTX mice. ##p < 0.01; ###p < 0.001 versus WT mice. Kruskal-Wallis with Dunn’s post test.
The AAV8-EAAT-CYP27A1 vector normalizes bile acid composition in blood
In order to determine whether gene therapy is able to achieve sustained metabolic correction in CTX mice, the bile acid profile was analyzed in the blood of animals treated with AAV8-EAAT-CYP27A1 for 5 months at 1.5 or 15 × 1012 vg/kg. The optimal dose of CDCA (0.5% chow weight) was maintained for the same period and used for comparison. We observed an increase of primary and secondary bile acids in mice treated with the vector at both doses (Figure 7; Table S2), including CDCA. In most cases, the levels were equivalent to those found in WT littermates. Only the concentrations of cholic acid (CA), deoxycholic acid (DCA), and their tauroconjugates showed a moderate increase compared with WT mice at 1.5 × 1012 vg/kg. This tendency was more evident in the high-dose group and included other species such as tauromuricholic acids (TMCAs) and taurohyodeoxycholic acid (THDCA). In sharp contrast, treatment with CDCA caused a drastic increase in this bile acid and its derivatives (1,000-fold above normal levels, corresponding to 51% of total bile acids versus 2% in WT mice), whereas CA and derived species were not restored.
Figure 7.

AAV8-EAAT-CYP27A1, but not CDCA, normalizes bile acid composition in blood of CTX mice
The AAV8-EAAT-CYP27A1 vector was administered intravenously to 7-week-old CTX mice at the indicated doses (×1012 vg/kg). CDCA was mixed in mouse chow at 0.5%. Untreated CTX and WT littermates were included as a reference. Blood was collected 5 months after the initiation of treatment, and the main free bile acids and tauroconjugates were analyzed in plasma. Bars represent means ± SEM for each group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Kruskal-Wallis with Dunn’s post test. aMCA, α-muricholic acid; bMCA, β-muricholic acid; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; HDCA, hyodeoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; THDCA, taurohyodeoxycholic acid; TLCA, taurolithocholic acid; TMCA, tauromuricholic acid; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid.
Discussion
The progress of AAV vectors is making gene therapy a realistic option for monogenic diseases involving the liver. However, the clinical feasibility of this approach still requires careful preclinical evaluation. Apart from the size of the expression cassette (which should fit into the 4.7 kilobase [kb] capacity of these vectors), one of the most important parameters is the percentage of transduced hepatocytes required to obtain a relevant therapeutic effect. The requirement of low percentage of hepatocyte transduction increases the chances of success using safe doses of the vectors. Typical examples are diseases in which a functional therapeutic protein can be expressed from the liver and secreted into the bloodstream, such as hemophilia.26 In other cases, such as the copper storage disorder Wilson disease, the protein acts intracellularly, but transduced hepatocytes can act as a sink to eliminate the excess copper.27,28 Our results indicate that CTX could fall into the latter category, provided that the transduced hepatocytes express high enough amounts of the CYP27A1 cytochrome. Our preclinical results indicate that complete biochemical restoration can be obtained with less than 20% hepatocytes transduced by the AAV8-EAAT-CYP27A1 vector. This conclusion is based not only on CYP27A1 immunohistochemistry (which cannot detect hepatocytes expressing low levels) but also on indirect comparison with the AAV8-EAAT-GFP vector, which allows highly specific and sensitive GFP immunodetection. We hypothesize that the excess of the highly permeable 7αC4 metabolite generated in untransduced hepatocytes can penetrate and be metabolized in other cells overexpressing CYP27A1. Still, this “sink effect” seems to have a limit, since the lowest dose of the AAV8-EAAT-CYP27A1 vector achieved a global hepatic content of CYP27A1 protein similar to that of the WT mice, but it obtained only a partial reduction in cholestanol levels. This indicates that the minimal threshold of transduced hepatocytes could be close to 10%, at least in the mouse model. By contrast, the effect of the AAV8-C27P-CYP27A1 vector was marginal even at the highest dose tested, probably because the transcriptional activation conferred by the C27P regulatory sequence was lower than that of the CYP27A1 promoter in its genomic context. Taking into account the size constraints imposed by the AAV cloning capacity, increasing the potency of this sequence would require the addition of enhancers, similar to the hybrid EAAT promoter.21 At this moment, we cannot rule out the possibility that the C27P promoter could be more active in human hepatocytes than mouse hepatocytes, although our results in cell lines do not support this hypothesis. In any case, overexpression of CYP27A1 was well tolerated in CTX mice, suggesting that physiological regulation of transgene expression is not an absolute requisite in this disease. This is another advantageous circumstance in terms of clinical feasibility. The need for alternative therapies for CTX is apparently low because the standard treatment based on lifelong oral administration of CDCA is efficient in controlling cholestanol levels and ameliorates many clinical manifestations such as chronic diarrhea and progression of xanthomas.19 However, in this work we provide evidence that the mechanism of action of gene therapy is different. CDCA at the therapeutic dose caused a marked accumulation of this bile acid in blood, as observed in CTX patients.29,30 We found that Cyp7a1 expression was virtually abrogated at this dose. Despite this drastic effect, the xenobiotic response pathway remained activated in CTX mice, suggesting that the generation of other potentially toxic metabolites was not inhibited. This phenomenon could only be detected using a mouse model, since the PXR pathway is not induced in CTX patients.18 Further investigation is needed to determine whether these metabolites could be responsible for the progressive neurological deterioration observed in many patients despite CDCA treatment.14,31,32 Anecdotal experiences with plasmapheresis favor the hypothesis that complete detoxification is not achieved with CDCA treatment alone.33 According to our preclinical results, dose escalation would not be an option because it is not well tolerated. Development of a mouse model with clear neurological manifestations is needed to assess whether gene therapy is able to address this important aspect of the disease. Despite the existence of some differences between mouse and human bile acid metabolism, we found that Cyp27a1−/− mice are a valuable tool to evaluate different treatments at the biochemical level. For instance, 0.5% CDCA was very efficient in reducing cholestanol, but 7αC4 was not completely normalized, in line with some clinical observations.14 By contrast, gene therapy achieved a parallel reduction of both metabolites. In summary, we provide evidence that CYP27A1 supplementation using an AAV vector could be a safe and feasible alternative for the treatment of CTX, offering the possibility of complete and stable metabolic correction after a single vector administration.
Materials and methods
Cell culture
HuH-7 (JCRB0403), HepG2 (ATCC HB-8065), Hep3B (ATCC HB-8064), 293T (ATCC CRL-3216), Hepa1-6 (ATCC CRL-1830), and AML12 (ATCC CRL-2254) cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM)-high glucose (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Thermo Fisher Scientific, Carlsbad, CA), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, and 1% non-essential amino acids (Gibco, Thermo Fisher Scientific, Waltham, MA). The AML12 cell line (ATCC CRL-2254) was maintained in DMEM/F12 medium (Gibco, Thermo Fisher Scientific), supplemented with 10% FBS, 0.01 mg/mL insulin, 0.005 mg/mL transferrin, 5 ng/mL selenium (Gibco, Thermo Fisher Scientific), 40 ng/mL dexamethasone, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained at 37°C in a 5% CO2 atmosphere.
Luciferase reporter plasmids
The pGL3-Basic plasmid (Promega, Madison, WI) is a promoter-less construct used to determine the background luciferase expression. The pCMV-Luc and pEalbPa1AT-Luc plasmids have been already described.21 The EalbPa1AT-Luc promoter (hereinafter referred to as EAAT) is a liver-specific, hybrid regulatory sequence consisting of the mouse albumin enhancer linked to the human α1-antitrypsin promoter. The pC27P-Luc plasmid contains a regulatory sequence comprising 2,024 bp upstream of the human CYP27A1 translation initiation site,22,23 synthetized by GenScript (Piscataway, NJ) and introduced into the MluI-NheI sites of pGL3-Basic.
Transfection and luciferase assays
All cell lines were seeded in 24-well plates at a density of 105 cells per well; 24 h later, they were transfected with Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) using 1 μg of plasmid DNA and 2 μg of Lipofectamine per well. Five hours later, medium was refreshed and cells were maintained for 48 h before addition of the Passive Lysis Buffer 5X (Promega, Madison, WI). Luciferase activity was measured with the Luciferase Reporter Assay System (Promega) in a Luminat KB 9507 luminometer (Berthold Technologies, Bad Wildbad, Germany). Data were normalized by protein content in each sample (in μg), determined by the Bradford assay (Bio-Rad, Hercules, CA). Promoter activity is represented as percentage of luciferase activity, using the CMV promoter as a reference.
AAV vectors
AAV-EAAT-CYP27A1 and AAV-C27P-CYP27A1 are AAV8 vectors containing the CYP27A1 complementary DNA (cDNA) under the control of the EAAT or CYP27A1 promoters, respectively. For the construction of the AAV-EAAT-CYP27A1 genome (pAAV-EAAT-CYP27A1 plasmid), the CYP27A1 coding sequence (NCBI ID: CCDS2423.1) was synthetized by GenScript (Leiden, the Netherlands). This DNA fragment was introduced using NheI and XbaI sites into a plasmid containing the EAAT promoter and a polyadenylation site, flanked by inverted terminal repeats (ITRs) from AAV2. The pAAV-EAAT-Luc plasmid contains the Firefly luciferase under the control of the EAAT promoter. For construction of the pAAV-C27P-CYP27A1 plasmid, the CYP27A1 promoter was excised from the pC27P-Luc plasmid using MluI and NheI sites and introduced into the same sites of pAAV-EAAT-CYP27A1, thus replacing the EAAT promoter. For viral particle (VP) production, the plasmids were transfected together with the pDP8-ape helper plasmid (Plasmid Factory, Bielefeld, Germany) in 293T cells, using polyethyleneimine (Polysciences, Warrington, PA). Three days later, culture media and cells were separated by centrifugation. VPs were extracted from the cell pellet by addition of lysis buffer (50 mM Tris-Cl, 150 mM NaCl, 2 mM MgCl2, and 0.1% Triton X-100) and 3 cycles of freezing and thawing (−80°C). VPs in the culture media were precipitated using polyethylene glycol solution (PEG8000, 8% [v/v] final concentration, Sigma-Aldrich) for 48–72 h at 4°C and further centrifugation at 1,378 × g for 15 min. The pellet was resuspended in lysis buffer and kept at −80°C. VPs obtained from culture medium and cell lysates were purified by ultracentrifugation at 350,000 × g during 2.5 h in a 15%–57% iodixanol gradient. Finally, the purified viruses were concentrated using Amicon Ultra Centrifugal Filters-Ultracel 100K (Millipore, Burlington, MA). Quantification of AAV vectors was performed by qPCR. To this end, VPs were treated with DNase and then viral genomes were extracted using the High Pure Viral Nucleic Acid Kit (Roche, Indianapolis, IN). Primers are listed in Table 1.
Animals and husbandry
A mouse strain with truncation of Cyp27a1 exon 8 was obtained from The Jackson Laboratory (Bar Harbor, ME) (B6.129-Cyp27a1tm1Elt/J, reference [ref.] 009106).16 Mice homozygous for this mutation (referred hereinafter to as Cyp27a1−/− or CTX mice) were maintained in a C57BL6/J background by crossing heterozygous individuals. The offspring was genotyped after weaning as indicated by the repository.
Animals were group housed, up to 6 animals per cage (male or female), provided with food and water ad libitum, and maintained with a 12 h light-dark cycle. The average age for initiation of studies was 7 weeks. AAV vectors were administered intravenously by retro-orbital injection in a final volume of 150 μL of saline solution. CDCA (Sigma-Aldrich, ref. C9377-25G) was supplemented in standard chow at 0.1, 0.5, and 1 g CDCA/100 g of chow (0.1, 0.5, and 1.0% CDCA diets, respectively). Blood was collected by submandibular venous puncture using 1.3 mL ethylenediaminetetraacetic acid (EDTA) tubes (Sarstedt, Nümbrecht, Germany), except for end-time points/terminal procedures, in which cardiac puncture was performed in anesthetized mice. Once animals were euthanized, liver samples were collected for histological and gene expression analyses.
All procedures were performed and approved by the ethical Committee of the Universidad de Navarra, according to the Spanish Royal Decree 53/2013.
Hydrodynamic injection and BLI
For in vivo liver transfection, 20 μg of reporter plasmids diluted in 1.8 mL of saline was injected as a bolus through the lateral tail vein.21 Luciferase activity was determined 48 h later by BLI. To this end, mice were briefly anesthetized with an injection of a ketamine/xylazine mixture (80:10 mg/kg, intraperitoneally [i.p.]). The substrate D-luciferin (REGIS Technologies, Morton Grove, IL) was administered i.p. (100 μL of a 30 μg/μL solution in PBS). Light emission was detected 5, 20, and 30 min later using a PhotonImager Optima apparatus (BioSpace Lab, Nesles-la-Vallée, France). Data were analyzed using the M3Vision software (BioSpace Lab), representing the maximal value obtained for each animal.
Biochemical analyses in plasma
Blood was centrifuged at 10,000 × g for 5 min at room temperature. Plasma was treated with 20 μM butylhydroxytoluene (Sigma-Aldrich) in a N2 atmosphere to protect from oxidation before storage at −80°C in opaque tubes. Sterol extractions were performed using 100 μL of plasma for quantification of cholestanol and 7αC4 concentrations by high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) as previously described.34 Bile acid profiling in serum was carried out after acetonitrile precipitation/extraction,35 using an adaptation36 of a previously described method for bile acid measurement by HPLC-MS/MS37 on a 6420 Triple Quad liquid chromatography (LC)-MS device (Agilent Technologies, Santa Clara, CA).38 ALT was quantified in 40 μL plasma samples using a Cobas C311 automated chemistry analyzer (Roche Diagnostics, Basel, Switzerland).
qPCR
RNA was extracted from frozen liver samples using the Maxwell 16 LEV simplyRNA Cells Kit (Promega) following the manufacturer’s recommendations. Two micrograms of RNA, treated with DNase I, was retro-transcribed using Moloney Murine Leukemia Virus (M-MLV) retro-transcriptase (Invitrogen) and random primers (Life Technologies, Thermo). cDNA was amplified, and relative gene expression was determined by qPCR using iQTM SYBR Green Supermix reagent (Bio-Rad) in CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Table 1 contains the sequence of primers specific for the transgene (CYP27A1) and the mouse genes Cyp7a1, Cyp3a11, Cyp27a1 (exons 1/2), Cyp27a1 (exons 8/9), and 36b4 (used as a housekeeping gene).
Delta cycle threshold (ΔCt) values using 36b4 mRNA levels as reference gene were corrected with the efficiency of amplification of each pair of primers and multiplied by 1,000 to facilitate graphical representation.
Western blot
Total proteins were isolated from liver samples using Radio-Immunoprecipitation Assay (RIPA) buffer (200 mM NaCl, 100 mM HEPES, 10% glycerol, 200 mM NaF, 2 mM Na4P2O7, 5 mM EDTA, 1 mM EGTA, 2 mM DTT [Invitrogen], 0.5 mM phenylmethanesulfonylfluoride [PMSF], 1 mM Na3VO4, and Complete Protease Inhibitor Cocktail [Roche]). Twenty micrograms of total protein extracts was boiled for 1 min and electrophoresed on a 10% polyacrylamide gel. Transfer to nitrocellulose membranes was performed at 340 mA current intensity for 3 h at 4°C. Next, membranes were incubated for 1 h at room temperature in blocking solution (5% BSA in Tris-buffered saline [TBS]-Tween) followed by overnight incubation at 4°C with primary antibodies diluted in 1% BSA, 0.05% Tween 20, and 0.5% sodium azide in TBS. Primary antibodies are anti-CYP27a1 (Abcam, Cambridge, UK, catalog [cat.] no. EPR7529, cat. no. ab126785; 1:1,000) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, Danvers, MA; 1:5,000). After washing with 0.1% Tween 20 in TBS, membranes were incubated for 1 h with anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP) conjugate secondary antibody (GE Healthcare, Chicago, IL, cat. no. NA934V; 1:10,000). Images were acquired with a ChemiDoc system (Bio-Rad), and Image Lab software (Bio-Rad) was used for quantification. See Table S1 for a list of antibodies used in this study.
Immunohistochemistry
For detection of CYP27A1 in hepatocytes, 3 μm thick sections cut from liver samples fixed in 4% paraformaldehyde and embedded in paraffin were deparaffinized with xylene; hydrated with decreasing concentrations of ethanol; and incubated with 3% hydrogen peroxide to block endogenous peroxidase. Antigen retrieval was performed by heating in 10 mM citrate buffer (pH 6) or 10 mM Tris-EDTA buffer for 20 min before incubation with antibody for CYP27A1 (Abcam, Cambridge, UK, cat. no. ab126785; 1:250) and β-catenin (Cell Signaling Technology, Danvers, MA, cat. no. 8480; 1:250), respectively. HRP-conjugated Envision secondary antibody (K4003, Dako, Glostrup, Denmark) followed by diaminobenzidine (DAB) reagent (K3468, Dako) were applied for the detection procedure. Tissue sections were counterstained with hematoxylin (Sigma-Aldrich) and dehydrated. Negative controls were included omitting primary antibodies. Quantification of hepatocytes overexpressing the protein was performed in 5 fields per mouse (311 × 311 μm) using ImageJ software (NIH, Bethesda, MD).
Statistical analysis
Prism software (GraphPad) was used for analysis. Datasets following normal distribution (D’Agostino and Pearson normality test) were compared using one-way ANOVA with Sidak’s multiple comparisons tests. Otherwise, groups were compared using Kruskal-Wallis with Dunn’s post test.
Acknowledgments
We thank Enrique and Estefania Aymerich from the AEXCT for their support and inspiration. We are thankful to core services at CIMA, in particular the grant management team, animal facility, and morphology unit. This work has been funded by Gobierno de Navarra (PT038 ref. 0011-1383-2018-000011 and PT013 ref. 0011-1383-2019-000006, XantoGen); Asociacion Española de Xantomatosis Cerebrotendionsa (AEXCT); Fundacion Eugenio Rodriguez Pascual; Fundacion M Torres; Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III,” Spain (PI19/00819, co-funded by European Regional Development Fund/European Social Fund, “Investing in your future”); AECC Scientific Foundation (2017/2020), Spain; “Centro Internacional sobre el Envejecimiento” (OLD-HEPAMARKER, 0348_CIE_6_E), Spain; and Fundació Marato TV3 (ref. 201916-31), Spain. L.M.J. and S.L. are recipients of a Pedro Lopez Berastegui fellowship.
Author contributions
R.H.-A. wrote the original draft and supervised the project. R.H.-A., J.J.G.M., M.A.A., and G.G.-A. contributed to conceptualization, funding acquisition, and review of the manuscript. J.J.G.M., M.A.A., and G.G.-A. provided resources. S.L., A.R., M.J.M., and I.U. performed investigation and reviewed the manuscript. L.B.-R., M.G.-A., M.B., and L.M.-J. performed investigation. I.U., G.G.-A., and A.R. contributed to methodology and resources.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.07.002.
Supplemental information
References
- 1.Björkhem I. Cerebrotendinous xanthomatosis. Curr. Opin. Lipidol. 2013;24:283–287. doi: 10.1097/MOL.0b013e328362df13. [DOI] [PubMed] [Google Scholar]
- 2.Verrips A., Hoefsloot L.H., Steenbergen G.C.H., Theelen J.P., Wevers R.A., Gabreëls F.J.M., van Engelen B.G.M., van den Heuvel L.P.W.J. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain. 2000;123:908–919. doi: 10.1093/brain/123.5.908. [DOI] [PubMed] [Google Scholar]
- 3.Cali J.J., Russell D.W. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J. Biol. Chem. 1991;266:7774–7778. [PubMed] [Google Scholar]
- 4.Salen G., Steiner R.D. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX) J. Inherit. Metab. Dis. 2017;40:771–781. doi: 10.1007/s10545-017-0093-8. [DOI] [PubMed] [Google Scholar]
- 5.Nie S., Chen G., Cao X., Zhang Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2014;9:179. doi: 10.1186/s13023-014-0179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen C.H., Wang Z.X. Liver transplantation due to cerebrotendinous xanthomatosis end-stage liver disease. World J. Pediatr. 2018;14:414–415. doi: 10.1007/s12519-018-0151-9. [DOI] [PubMed] [Google Scholar]
- 7.Gong J.Y., Setchell K.D.R., Zhao J., Zhang W., Wolfe B., Lu Y., Lackner K., Knisely A.S., Wang N.L., Hao C.Z. Severe Neonatal Cholestasis in Cerebrotendinous Xanthomatosis: Genetics, Immunostaining, Mass Spectrometry. J. Pediatr. Gastroenterol. Nutr. 2017;65:561–568. doi: 10.1097/MPG.0000000000001730. [DOI] [PubMed] [Google Scholar]
- 8.Degrassi I., Amoruso C., Giordano G., Del Puppo M., Mignarri A., Dotti M.T., Naturale M., Nebbia G. Case Report: Early Treatment With Chenodeoxycholic Acid in Cerebrotendinous Xanthomatosis Presenting as Neonatal Cholestasis. Front. Pediatr. 2020;8:382. doi: 10.3389/fped.2020.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yahalom G., Tsabari R., Molshatzki N., Ephraty L., Cohen H., Hassin-Baer S. Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin. Neuropharmacol. 2013;36:78–83. doi: 10.1097/WNF.0b013e318288076a. [DOI] [PubMed] [Google Scholar]
- 10.Degos B., Nadjar Y., Amador M. del M., Lamari F., Sedel F., Roze E., Couvert P., Mochel F. Natural history of cerebrotendinous xanthomatosis: a paediatric disease diagnosed in adulthood. Orphanet J. Rare Dis. 2016;11:41. doi: 10.1186/s13023-016-0419-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appadurai V., DeBarber A., Chiang P.W., Patel S.B., Steiner R.D., Tyler C., Bonnen P.E. Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ∼60,000 human exomes. Mol. Genet. Metab. 2015;116:298–304. doi: 10.1016/j.ymgme.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panzenboeck U., Andersson U., Hansson M., Sattler W., Meaney S., Björkhem I. On the mechanism of cerebral accumulation of cholestanol in patients with cerebrotendinous xanthomatosis. J. Lipid Res. 2007;48:1167–1174. doi: 10.1194/jlr.M700027-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Båvner A., Shafaati M., Hansson M., Olin M., Shpitzen S., Meiner V., Leitersdorf E., Björkhem I. On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J. Lipid Res. 2010;51:2722–2730. doi: 10.1194/jlr.M008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mignarri A., Magni A., Del Puppo M., Gallus G.N., Björkhem I., Federico A., Dotti M.T. Evaluation of cholesterol metabolism in cerebrotendinous xanthomatosis. J. Inherit. Metab. Dis. 2016;39:75–83. doi: 10.1007/s10545-015-9873-1. [DOI] [PubMed] [Google Scholar]
- 15.Helman G., Van Haren K., Bonkowsky J.L., Bernard G., Pizzino A., Braverman N., Suhr D., Patterson M.C., Ali Fatemi S., Leonard J. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 2015;114:527–536. doi: 10.1016/j.ymgme.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen H., Reshef A., Maeda N., Lippoldt A., Shpizen S., Triger L., Eggertsen G., Björkhem I., Leitersdorf E. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J. Biol. Chem. 1998;273:14805–14812. doi: 10.1074/jbc.273.24.14805. [DOI] [PubMed] [Google Scholar]
- 17.Rizzolo D., Buckley K., Kong B., Zhan L., Shen J., Stofan M., Brinker A., Goedken M., Buckley B., Guo G.L. Bile Acid Homeostasis in a Cholesterol 7α-Hydroxylase and Sterol 27-Hydroxylase Double Knockout Mouse Model. Hepatology. 2019;70:389–402. doi: 10.1002/hep.30612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honda A., Salen G., Matsuzaki Y., Batta A.K., Xu G., Leitersdorf E., Tint G.S., Erickson S.K., Tanaka N., Shefer S. Side chain hydroxylations in bile acid biosynthesis catalyzed by CYP3A are markedly up-regulated in Cyp27-/- mice but not in cerebrotendinous xanthomatosis. J. Biol. Chem. 2001;276:34579–34585. doi: 10.1074/jbc.M103025200. [DOI] [PubMed] [Google Scholar]
- 19.Verrips A., Dotti M.T., Mignarri A., Stelten B.M.L., Verma S., Federico A. The safety and effectiveness of chenodeoxycholic acid treatment in patients with cerebrotendinous xanthomatosis: two retrospective cohort studies. Neurol. Sci. 2020;41:943–949. doi: 10.1007/s10072-019-04169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kramer M.G., Barajas M., Razquin N., Berraondo P., Rodrigo M., Wu C., Qian C., Fortes P., Prieto J. In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol. Ther. 2003;7:375–385. doi: 10.1016/s1525-0016(02)00060-6. [DOI] [PubMed] [Google Scholar]
- 22.Araya Z., Tang W., Wikvall K. Hormonal regulation of the human sterol 27-hydroxylase gene CYP27A1. Biochem. J. 2003;372:529–534. doi: 10.1042/BJ20021651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W., Chiang J.Y.L. Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4α (HNF4α) Gene. 2003;313:71–82. doi: 10.1016/s0378-1119(03)00631-0. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto Y., Yoshida M., Tamura K., Takahashi M., Kodama Y., Inoue K. Dose-dependent difference of nuclear receptors involved in murine liver hypertrophy by piperonyl butoxide. J. Toxicol. Sci. 2015;40:787–796. doi: 10.2131/jts.40.787. [DOI] [PubMed] [Google Scholar]
- 25.Repa J.J., Lund E.G., Horton J.D., Leitersdorf E., Russell D.W., Dietschy J.M., Turley S.D. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J. Biol. Chem. 2000;275:39685–39692. doi: 10.1074/jbc.M007653200. [DOI] [PubMed] [Google Scholar]
- 26.Peyvandi F., Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia. 2019;25:738–746. doi: 10.1111/hae.13816. [DOI] [PubMed] [Google Scholar]
- 27.Murillo O., Luqui D.M., Gazquez C., Martinez-Espartosa D., Navarro-Blasco I., Monreal J.I., Guembe L., Moreno-Cermeño A., Corrales F.J., Prieto J. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016;64:419–426. doi: 10.1016/j.jhep.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Murillo O., Moreno D., Gazquez C., Barberia M., Cenzano I., Navarro I., Uriarte I., Sebastian V., Arruebo M., Ferrer V. Liver Expression of a MiniATP7B Gene Results in Long-Term Restoration of Copper Homeostasis in a Wilson Disease Model in Mice. Hepatology. 2019;70:108–126. doi: 10.1002/hep.30535. [DOI] [PubMed] [Google Scholar]
- 29.Salen G., Batta A.K., Tint G.S., Shefer S. Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism. 1994;43:1018–1022. doi: 10.1016/0026-0495(94)90183-x. [DOI] [PubMed] [Google Scholar]
- 30.Salen G., Tint G.S., Eliav B., Deering N., Mosbach E.H. Increased formation of ursodeoxycholic acid in patients treated with chenodeoxycholic acid. J. Clin. Invest. 1974;53:612–621. doi: 10.1172/JCI107596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pilo-de-la-Fuente B., Jimenez-Escrig A., Lorenzo J.R., Pardo J., Arias M., Ares-Luque A., Duarte J., Muñiz-Pérez S., Sobrido M.J. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur. J. Neurol. 2011;18:1203–1211. doi: 10.1111/j.1468-1331.2011.03439.x. [DOI] [PubMed] [Google Scholar]
- 32.Mignarri A., Dotti M.T., Federico A., De Stefano N., Battaglini M., Grazzini I., Galluzzi P., Monti L. The spectrum of magnetic resonance findings in cerebrotendinous xanthomatosis: redefinition and evidence of new markers of disease progression. J. Neurol. 2017;264:862–874. doi: 10.1007/s00415-017-8440-0. [DOI] [PubMed] [Google Scholar]
- 33.Mimura Y., Kuriyama M., Tokimura Y., Fujiyama J., Osame M., Takesako K., Tanaka N. Treatment of cerebrotendinous xanthomatosis with low-density lipoprotein (LDL)-apheresis. J. Neurol. Sci. 1993;114:227–230. doi: 10.1016/0022-510x(93)90303-g. [DOI] [PubMed] [Google Scholar]
- 34.Baila-Rueda L., Cenarro A., Cofán M., Orera I., Barcelo-Batllori S., Pocoví M., Ros E., Civeira F., Nerín C., Domeño C. Simultaneous determination of oxysterols, phytosterols and cholesterol precursors by high performance liquid chromatography tandem mass spectrometry in human serum. Anal. Methods. 2013;5:2249–2257. [Google Scholar]
- 35.Leníček M., Vecka M., Žížalová K., Vítek L. Comparison of simple extraction procedures in liquid chromatography–mass spectrometry based determination of serum 7α-hydroxy-4-cholesten-3-one, a surrogate marker of bile acid synthesis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016;1033–1034:317–320. doi: 10.1016/j.jchromb.2016.08.046. [DOI] [PubMed] [Google Scholar]
- 36.Nytofte N.S., Serrano M.A., Monte M.J., Gonzalez-Sanchez E., Tumer Z., Ladefoged K., Briz O., Marin J.J.G. A homozygous nonsense mutation (c.214C->A) in the biliverdin reductase alpha gene (BLVRA) results in accumulation of biliverdin during episodes of cholestasis. J. Med. Genet. 2011;48:219–225. doi: 10.1136/jmg.2009.074567. [DOI] [PubMed] [Google Scholar]
- 37.Ye L., Liu S., Wang M., Shao Y., Ding M. High-performance liquid chromatography-tandem mass spectrometry for the analysis of bile acid profiles in serum of women with intrahepatic cholestasis of pregnancy. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007;860:10–17. doi: 10.1016/j.jchromb.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 38.Monte M.J., Martinez-Diez M.C., El-Mir M.Y., Mendoza M.E., Bravo P., Bachs O., Marin J.J.G. Changes in the pool of bile acids in hepatocyte nuclei during rat liver regeneration. J. Hepatol. 2002;36:534–542. doi: 10.1016/s0168-8278(01)00296-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.