

Abstract
Pyruvate kinase deficiency (PKD), an autosomal-recessive disorder, is the main cause of chronic non-spherocytic hemolytic anemia. PKD is caused by mutations in the pyruvate kinase, liver and red blood cell (PKLR) gene, which encodes for the erythroid pyruvate kinase protein (RPK). RPK is implicated in the last step of anaerobic glycolysis in red blood cells (RBCs), responsible for the maintenance of normal erythrocyte ATP levels. The only curative treatment for PKD is allogeneic hematopoietic stem and progenitor cell (HSPC) transplant, associated with a significant morbidity and mortality, especially relevant in PKD patients. Here, we address the correction of PKD through precise gene editing at the PKLR endogenous locus to keep the tight regulation of RPK enzyme during erythropoiesis. We combined CRISPR-Cas9 system and donor recombinant adeno-associated vector (rAAV) delivery to build an efficient, safe, and clinically applicable system to knock in therapeutic sequences at the translation start site of the RPK isoform in human hematopoietic progenitors. Edited human hematopoietic progenitors efficiently reconstituted human hematopoiesis in primary and secondary immunodeficient mice. Erythroid cells derived from edited PKD-HSPCs recovered normal ATP levels, demonstrating the restoration of RPK function in PKD erythropoiesis after gene editing. Our gene-editing strategy may represent a lifelong therapy to correct RPK functionality in RBCs for PKD patients.
Keywords: hematopoietic stem cell, gene editing, hemolytic anemia, pyruvate kinase deficiency, gene therapy, CRISPR-Cas9, AAV6
Graphical abstract
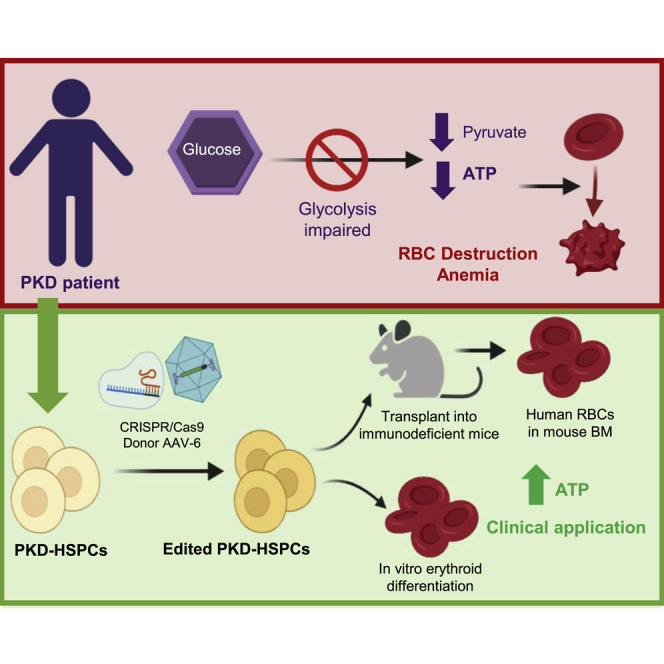
Combination of CRISPR-Cas9 ribonucleoprotein electroporation and donor AGAVE transduction in human hematopoietic stem and progenitor cells from pyruvate kinase deficiency patients allows efficient gene editing with restoration of erythrocyte energetic balance. Maintenance of stem cell properties without detectable off-targets indicates clinical applicability of gene editing for this inherited hemolytic anemia.
Introduction
Pyruvate kinase deficiency (PKD) is an inherited autosomal-recessive metabolic disorder produced by mutations in the Liver and Erythroid Pyruvate Kinase gene (PKLR). PKLR encodes for liver (LPK) and erythroid (RPK) pyruvate kinase proteins, expressed in liver and in red blood cells (RBCs), respectively. RPK is implicated in the last step of the anaerobic glycolysis pathway, the main source of energy in RBCs. To date, >200 different mutations in the PKLR gene have been linked to PKD.1,2 The disease becomes clinically relevant when the protein activity decreases below 25% of the normal activity in erythrocytes.3 Most frequent clinical signs are mild to very severe anemia, reticulocytosis, splenomegaly, and iron overload, being life-threatening in severely affected patients.4 PKD is considered the most common cause of chronic non-spherocytic hemolytic anemia (CNSHA), with an estimated prevalence of ∼1:20,000, being higher in certain populations, such as the Amish community, as a result of a founder effect.5,6
Treatment for PKD is mostly palliative. The most extended treatment is RBC transfusion2, although it can have related adverse effects, such as alloimmunization against donor blood cells and worsening of iron overload, which can result in liver and heart organ damage.2,7 Spleen removal aims to prevent RBC destruction to increase the number of oxygen-transporting cells. Splenectomy does not arrest hemolysis but can increase hemoglobin (Hb) values up to 1–3 g/dL.2 However, this treatment carries risks of encapsulated bacterial infections and of increased venous thrombosis. Moreover, ∼14% of splenectomized PKD patients remain dependent on blood transfusion after splenectomy. Currently, treatment with AG-348 (Mitapivat), an allosteric activator of RPK that increases RPK enzymatic activity and ATP levels, in PKD patients with a sufficient level of expression of RPK protein is being evaluated in clinical trials.8 However, the only curative treatment for PKD patients is allogeneic hematopoietic stem cell transplantation (HSCT), although it is not considered as routine treatment in PKD patients, because of the limitation of human leukocyte antigen (HLA)-compatible donors and the severe adverse effects, such as graft-versus-host disease (GvHD), which can be particularly severe in PKD patients.9, 10, 11
Autologous HSCT of genetically corrected cells could overcome these limitations. This strategy has been used in several hematological genetic diseases,12,13 including hemoglobinopathies,14, 15, 16, 17 being already approved for clinical application in Europe (Zynteglo; https://t.ly/sBSg). We have recently developed a lentiviral vector to genetically correct PKD.18 This lentiviral-mediated gene therapy approach would offer a durable and curative clinical benefit with a single treatment, as shown by the preliminary results obtained in the first two patients already infused with transduced autologous hematopoietic stem cells (HSCs) (NCT04105166).19
Despite the promising results of conventional gene therapy, the ideal gene therapy approach should lead to the specific correction of the mutated gene, maintaining the endogenous regulation and eliminating the integration of exogenous DNA material elsewhere. Gene editing can be used as a therapeutic approach to conduct precise homology directed repair (HDR). Repair of a variety of genetic defects affecting non-hematopoietic tissues20, 21, 22 and the hematopoietic system, such as X-linked SCID,23 SCD,24,25 X-linked chronic granulomatous disease (X-CGD),26,27 and Fanconi anemia28,29 among others, has been successfully attempted by gene editing. Moreover, gene editing is already showing promising clinical results, as recently shown in hemoglobinopathies.30, 31, 32, 33 The improvement in the condition of two β-thalassemia and SCD patients enrolled in the CTX001 clinical trial34 paves the way to consideration of gene editing as a promising approach for RBC disorders, such as PKD. Previous studies demonstrated that gene editing in the PKLR locus is feasible in induced pluripotent stem cells (iPSCs)35 and in hematopoietic stem and progenitor cell (HSPCs),36 although the efficacy achieved was far from being clinically relevant. In this work, we have implemented CRISPR/Cas9 nuclease system to induce double-stranded breaks (DSBs) at the translation start site of RPK isoform and recombinant adeno-associated vectors (rAAVs) to deliver a therapeutic DNA donor as an alternative gene editing approach to address PKD correction. Using this new strategy, we have been able to correct the phenotype of erythroid cells derived from PKD-HSPCs and to achieve clinically relevant levels of functional correction that envision the treatment of PKD patients by gene editing.
Results
Design of the CRISPR-Cas9-AAV6-donor-transfer system to target PKLR gene in hematopoietic stem and progenitor cells
To develop a gene editing-based universal strategy for PKD patients, we followed a knockin approach to insert a therapeutic donor close to the translation start site of RPK gene in the PKLR locus (Figures S1A and S2A). To promote HDR and favor integration of the therapeutic donor, we induced DSBs close to the RPK translation starting site by a specific CRISPR/Cas9 ribonucleoprotein (RNP). Therapeutic donor was delivered into target cells by adeno-associated vectorization as previously described.24,25,37
First, we designed different guide RNAs (gRNAs) to create DSBs around the start codon of the RPK gene according to two main criteria, (1) the highest on-target score possible (implying high specificity and less probable off-target [OT] activity) and (2) the closest distance to start codon of RPK isoform. Up to 8 different gRNAs were designed with the different web tools available (see Table S1). We assessed the activity of a first set of 6 gRNAs, complexed with wild-type (WT) SpCas9 protein (RNPs), by tracking of insertion/deletions (indels) by decomposition (TIDE) analyses in cord blood (CB) CD34+ cells, 4 days post-editing (Figure 1A). SG1 and SG3 produced the highest frequency of indels at the on-target site (62.7% ± 14.2% and 48.7% ± 30.3%, respectively). SG5 and SG6 were discarded because of their low indel generation efficacy. Then, SG1, SG2, SG3, and SG4 were analyzed to assess OTs by GUIDE-seq unbiased-cell method. Two additional guides, SG7 and SG8, which showed reduced in silico OT activity (using in-house IDT gRNA design checker), were also included in the GUIDE-seq analysis. gRNAs were transfected into HEK293 cells stably expressing WT Cas9, which allowed a stringent detection of OT by GUIDE-seq analysis in vivo.38 Identified OTs are shown in Figure S1B and Tables S2 and S3 (GEO: GSE171935). rhAmpSeq libraries were designed based on GUIDE-seq data to analyze gene editing activity of the three most promising gRNAs, SG1 (the highest on-target activity in CB-CD34+ cells) and SG7 and SG8 (the lowest in silico OT activity). CB-CD34+ cells were nucleofected with SG1-, SG7- or SG8-RNPs using either WT Cas9 protein or HiFi Cas9 protein, an improved version of Cas9 showing lower OT.25 On-target and OT events were studied by the developed rhAmpSeq panels. To get additional information, gene editing modification in CB-CD34+ at on-target sites of SG1, SG7, and SG8 was analyzed in the presence or absence of a double stranded oligonucleotide (dsODN) donors (Figure 1B; Figure S1C and Table S4). rhAmpSeq confirmed the highest on-target activity of SG1, which was not compromised when using HiFi Cas9. Moreover, the percentage of perfect HDR mediated by the dsODN was not altered with the use of HiFi Cas9 (Figure 1B). These data were also verified in CB-CD34+ cells nucleofected with RNPs formed with either WT Cas9 or HiFi Cas9 by TIDE assays (Figure 1C). Finally, OT effect of SG1 in CB-CD34+ cells was analyzed through the rhAmpSeq library of the top 49 OT sites (Table S5). Percentage of gene modification in the most important OTs was low and even reduced below the limit of detection when HiFi Cas9 protein was used. Gene editing of the top nine OT sites in CB-CD34+ cells was below 0.1% total gene editing when HiFi Cas9 was used (Figure 1D). Altogether, these analyses demonstrated that HiFi Cas9/SG1-RNP promotes significant levels of HDR in the on-target site, with a minimal effect in the potential OTs, confirming the safety of the use of this RNP in the PKLR locus.
Figure 1.

Selected single guide RNA displays high on-target efficiency and very low OT incidence
(A) TIDE analysis to quantify the generation of indels in CB-CD34+ cells nucleofected with different guides as RNP, selected around the translation starting site of the RPK. Efficacy in the generation of indels was performed 4 days post-editing. (B) rhAmp-Seq analyses of the HDR versus NHEJ in CB-CD34+ cells edited with homologous double stranded oligonucleotide (dsODN) template and SG1, complexed with either WT Cas9 or HiFi Cas9. Blue, % of NHEJ repair; gray, % of perfect HDR; orange, % of imperfect HDR. (C) TIDE analyses comparing generation of indels in CD34+ cells using SG1, nucleofected as RNP using WT or HiFi Cas9 protein. (D) Analyses of total gene editing efficiency by Cas9 HiFi + SG1 ± HDR-SG1 dsODN template in the top nine sites ranked high to low out of 49 OTs identified by GUIDE-seq. Orange and yellow bars show gene editing caused by SG1 complexed with WT Cas9 or HiFi Cas9, respectively, in the absence of the specific HDR template. Blue and gray bars show gene editing caused by SG1 complexed with WT Cas9 or HiFi Cas9, respectively, in the presence of the specific HDR template. Blue square indicates the limit of detection of the assay (<0.1%). Each experiment was performed using pools of 2 or 3 healthy donor CD34+ samples. At least three experiments per group were performed. Data are represented as mean ± SD. Significance was analyzed by non-parametric two-tailed Mann-Whitney test. ∗, p<0.05.
Once SG1 was selected, we designed AAV donors to promote HDR at this on-target site. Two AAV donors were designed, (1) a reporter donor (AAV-GFP), carrying a turbo-GFP cDNA under the regulation of Ubiquitin C promoter (UBC), and (2) a therapeutic donor (AAV-coRPK), which carried a codon optimized version of the WT cDNA of the RPK gene (coRPK cDNA), without any exogenous promoter, to allow the endogenous PKLR promoter to drive the expression of the therapeutic coRPK cDNA (Figure S2A and additional details in Supplemental methods). Donor sequences were flanked with AAV Inverted terminal repeats (ITRs) and packaged into AAV-6 serotype. The combination of SG1 RNPs and AAV vectors was used for subsequent experiments
Homology directed repair in the PKLR locus with the developed tools is effective in erythroleukemic cells and in primary human hematopoietic progenitors
K562 human erythroleukemia cells were electroporated with the SG1-RNP and transduced with either reporter or therapeutic rAAV donor at a viral concentration of 1 × 104 genome copies per microliter (gc/μL). Cells transduced with the reporter donor were analyzed by flow cytometry, 21 days post-transduction (dpt). Up to 5% GFP+ cells were identified (Figure S2B). DNA from samples edited with either of the donors was analyzed by specific PCRs amplifying the genomic junctions between the endogenous and exogenous DNA at both 5′ and 3′ ends to verify the correct integration of the transgene (Figure S2C). Results confirmed HDR of reporter and therapeutic donors at the on-target site (Figure S2D). Furthermore, we quantified the expression of WT endogenous RPK and exogenous therapeutic coRPK transcripts through qRT-PCR. Transduced and untransduced cells expressed endogenous RPK mRNA. However, coRPK transcripts were exclusively detected in K562 cells transduced with the therapeutic vector (Figures 2E and 2F).
Figure 2.

The combination of SG1/Cas9 RNPs with rAAV donor delivery does not show toxicity and allows efficient gene editing in human hematopoietic progenitors at the RPK translation starting site
(A) CFU assay shows number and types of hematopoietic progenitors after gene editing. GEMM, granulocyte/erythrocyte/monocyte/megakaryocyte progenitors; BFU-E, burst forming unit-erythroid; CFU-GM, colony forming unit-erythroid. Mean ± SD is indicated. (B) Representative agarose gel of PCR analyses of the specific integration of coRPK in individually picked colonies by amplification of the left (5′ PCR, 516 bp) and right (3′ PCR, 852 bp) homology arms of the donor construct. (C) Representative Sanger sequencing of one of the positive amplicons showing correct HDR. (D) Percentage of correct HDR in CFUs assessed by PCR when cells were edited with GFP-AAV or coRPK-AAV. The figure shows the data from 3 independent experiments, in which up to 96 CFUs were analyzed per condition in each experiment. Three independent experiments were performed with pools of 3 or 4 different healthy donors per experiment. Significance was analyzed by non-parametric two-tailed Mann-Whitney or two-way ANOVA tests.
Next, we assessed the targeting efficiency in human HSPCs. CB-CD34+ cells were nucleofected with SG1 RNP, formed with HiFi Cas9 protein, and then transduced with either of the two AAV donors. Forty-eight hours after nucleofection and transduction, clonogenic potential of edited progenitors was assessed. No differences in the number or type of hematopoietic colonies were found when cells were edited with either donor (Figure 2A). GFP+ colonies from CD34+ cells edited with the reporter donor (Figure S3A) reached values up to 38% GFP+ colony forming units (CFUs) (21.1% ± 17.4% average) (Figure S3B). Correct integration of both donors was verified by PCR in individually picked colonies. Specific PCR bands, demonstrating correct integration of the donor sequences, were obtained in CFUs derived from edited CB-CD34+ cells (Figure 2B; Figure S3C). PCR amplicons of individual CFUs were Sanger sequenced, and the results confirmed the correct integration of the reporter and therapeutic donors in human progenitor cells (Figure 2C; Figure S3D). Percentage of HDR positive colonies, which displayed correct integration in both ends of the reporter and the therapeutic donor sequences, was 35.5% ± 4.7% and 38.5% ± 7.4%, respectively (Figure 2D). Altogether, the results evidence the efficient knockin of the donors at the translation starting site of RPK isoform in human hematopoietic progenitor cells.
PKLR locus is efficiently targeted in long-term hematopoietic repopulating stem cells
To study whether the more primitive HSCs were also targeted with the described strategy, transduction efficiency in CD34+ subpopulations using the AAV-GFP donor was assessed by flow cytometry. Forty-eight hours post electroporation and transduction, transduction efficiency was 14.5% ± 4.7% in CD34+ cells. Within the CD34+CD38−CD90+ primitive HSCs, 6.7% ± 5.2% were GFP+ (Figure 3A; Figure S4A).
Figure 3.

Targeted human hematopoietic stem cells efficiently engraft in immunodeficient mice
(A) Percentage of GFP+ cells within the CD34+ compartment and within the HSC subset (CD34+CD38-CD90+), 48 h post-editing with RNP and GFP-AAV. n = 4; 4 healthy donor samples used. (B) Percentage of human cells in the bone marrow (BM) of immunodeficient NSG mice transplanted with AAV-GFP (white dots)- or AAV-coRPK (black dots)-edited cells, 1 and 3 mpt. Triangle dot corresponds to one animal transplanted with irradiated-CD34− cells only. Data are represented as mean ± SD (n = 7 mice in each group from 3 independent experiments). (C) Percentage of the hCD34+ cells within the human population (hCD45+) in mice analyzed in (B). Data are represented as mean ± SD (n = 7 mice in each group from 3 independent experiments). (D) Percentage of the hCD19+ cells within the human population in mice analyzed in (B). (E) Percentage of the hCD33+ cells within the human population in mice analyzed in (B). (F) Percentage of GFP+ cells within the human population (hCD45+) 1 and 3 mpt in mice analyzed in (B). (G) Percentage of GFP+ cells within the human progenitor cells (hCD34+) 1 and 3 mpt in mice analyzed in (B). Data are represented as mean ± SD (n = 7 mice in each group from 3 independent experiments). Significance was analyzed by non-parametric two-tailed Mann-Whitney test. ns, not significant.
To verify the stable gene editing of primitive HSPCs, 8 × 105 to 1 × 106 edited cells were transplanted together with irradiated CD34− cells into sublethally irradiated immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, and hematopoietic engraftment was followed for 1 and 3 months post-transplant (mpt) (Figure 3B). A high and stable engraftment was observed. Mice transplanted with GFP-AAV edited cells reached 87.3% ± 8.4% human chimerism, and mice transplanted with coRPK-AAV edited cells presented similar levels (87.1% ± 5.9%), with no signs of toxicity 3 mpt (Figure 3B; Figure S4B). Multilineage engraftment was also investigated using antibodies against hCD34 for HSPCs, hCD33 for myeloid cells, and hCD19 for lymphoid cells. Edited cells were found within the three human hematopoietic subpopulations (Figures 3C–3E; Figure S4C), confirming the gene editing of HSCs capable of generating myeloid, lymphoid, and progenitor cells. Furthermore, the percentage of GFP+ cells within the human compartment in the bone marrow (BM) of the recipients transplanted with cells transduced with the AAV reporter donor was 2.3% ± 1.9% at 1 mpt and 0.3% ± 0.4% at 3 mpt (Figure 3F; Figure S4B). Additionally, the percentage of GFP+ cells within the hCD34+ compartment analyzed 3 mpt was 2.2% ± 3.6% (Figure 3G; Figure S4C).
To confirm the long-term repopulating capacity of edited cells, BM cells from primary recipients were transplanted into secondary recipients. Flow cytometry analyses of hCD45+ cells in BM of secondary recipients showed that edited cells were able to repopulate efficiently secondary transplanted animals. In recipients infused with engrafted GFP-edited cells from primary animals, 16.9% ± 15.7% of mouse cells were hCD45+ cells. A similar value (17.8% ± 12.6% hCD45+ cells) was observed in the BM of secondary mice infused with cells edited with therapeutic donor (Figure 4A). In all instances, multilineage engraftment was observed (Figures 4B–4D). Human CD45+GFP+ and CD34+GFP+ cells were detected in secondary recipients as well (Figures 4E and 4F), and the percentage of GFP+ cells within the human population was maintained over time (3.3% ± 5.7% total human cells and 0.8% ± 1.4% human CD34+ cells at 3 mpt). Human erythroid compartment, the target compartment of PKD genetic correction, identified as mTer119-hCD235a+hCD71+/−, was also analyzed. GFP+ cells were found within the human erythroid population, and erythroid commitment was not affected when the therapeutic donor was used (Figure S4D). Furthermore, BM cells from secondary recipients infused with cells edited with the therapeutic donor were analyzed by PCR, confirming the specific knockin of the coRPK cDNA in the engrafted human cells (Figure 4G). Taken together, these results demonstrate the efficient knockin gene editing protocol in HSCs, capable of repopulating immunodeficient NSG mice in the long term.
Figure 4.

Gene-edited HSPCs engraft in secondary transplanted immunodeficient mice
(A) Percentage of human CD45+ cells in immunodeficient NSG animals transplanted with total BM of primary transplanted animals with either AAV-GFP (white dots; n = 9)- or AAV-coRPK (black dots; n = 7)-edited cells, analyzed 1 and 3 mpt. (B) Percentage of the hCD34+ cells within the human population (hCD45+) in mice analyzed in (A). (C) Percentage of the hCD19+ cells within the human population in mice analyzed in (A). (D) Percentage of the hCD33+ cells within the human population in mice analyzed in (A). (E) Percentage of GFP+ cells within the human population (hCD45+) in mice analyzed in (A), 1 and 3 mpt. (F) Percentage of GFP+ cells within the human progenitor (CD34+) cells in mice analyzed in (A), 1 and 3 mpt. (G) Representative agarose gel of PCR analysis of the 5′ (516 bp) and the 3′ (852 bp) sequences analyzed in total BM of two different mice, demonstrating correct integration of the donor fragment in these secondary animals. Data are represented as mean ± SD. Kruskal-Wallis multiple comparison test was performed. The significance is represented by p values. ns, not significant.
Optimized gene editing conditions allow clinically relevant efficacies
Unpublished data with lentiviral vectors from our laboratory point out that 25%–30% corrected cells are necessary to observe a clinical improvement in a PKD mouse model (S. Navarro, unpublished data). In order to assess the therapeutic potential of our gene editing system, we intended to maximize the efficacy of the transduction to reach this therapeutic limit. Doses of the reporter AAV ranging from 1 × 101 to 1 × 105 gc/μL were tested for its efficacy, measured by GFP expression, and for its toxicity, assessed in a CFU assay (Figures 5A and 5B; Figure S5). Vector concentrations of 2.5 × 104 and 5 × 104 gc/μL displayed enhanced transduction efficacies of 25.3% ± 0.3% and 33.5% ± 15.1% GFP+ cells, respectively. Remarkably, 14.5% ± 8.9% of the hCD34+hCD38−hCD90+ cells were GFP+ when samples were transduced with 5 × 104 gc/μL AAV (Figure 5A; Figure S5). No significant differences in the number of colonies in any of the viral doses used (Figure 5B) or in the cell expansion post-editing of the cell culture were observed when 5 × 104 gc/μL vector concentration was used (Figure 5C). Gene editing efficiency assessed in the BM of mice transplanted with edited cells was definitely increased (20.4% ± 11.8% with 5 × 104 gc/μL in contrast to 0.3% ± 0.4% achieved with 1 × 104 gc/μL performed in parallel) (Figure 5D). Moreover, the levels of gene editing obtained with 5 × 104 gc/μL were in the range of those required to have clinical benefit to correct PKD.
Figure 5.

Optimized gene editing conditions maintain engraftment potential of HSPCs
(A) Percentage of GFP cells within different subpopulations (%GFP in living cells [white bars], %GFP in hCD34+ cells [gray bars], and %GFP in hCD34+hCD38-hCD90+ [black bars]), 48 h post-transduction, using different MOIs ranging from 1 × 101 to 1 × 105 gc/μL of the reporter donor. One to five different biological replicates were performed using 5 independent HD CB-CD34+ samples. (B) CFU assay of CB-CD34+ cell edited with increased doses of AAVs. Two independent biological replicates from three or four different HD CB-CD34+ cells were used. (C) Cell expansion 48 h post-editing relative to the initial number of edited HSPCs. Four independent biological replicates from different HD CB-CD34+ cells. (D) GFP percentage in hCD45+ cells in the BM of NSG mice transplanted with HSPCs edited with 1 × 104 or 5 × 104 gc/μL GFP-AAV and analyzed 3 mpt. Two independent biological replicates from four different HD CB-CD34+ cells were used. Four to seven mice were analyzed in each conditions. Kruskal-Wallis multiple comparison test was performed. ∗∗, p<0.01. ns, not significant.
ATP deficiency is corrected and long-term engraftment ability is maintained after PKLR gene editing in human stem and progenitor cells from PKD patients
Optimized editing conditions were then tested in BM-CD34+ and peripheral blood CD34+ cells from four PKD patients carrying different mutations in the PKLR gene (Table S6). Twenty-four hours after application of the optimized gene editing procedure, cells were collected and in vitro differentiated toward the erythroid lineage. Erythroid differentiation process was evaluated over time by flow cytometry. As shown in Figure S6A, no differences between healthy and PKD donor samples were observed. Erythroid cells derived from PKD HSPCs presented a significant reduction in the expression of endogenous RPK mRNA. Only erythroid cells derived from gene-edited PKD HSPCs expressed exogenous coRPK mRNA (Figure S6B). Furthermore, to study the recovery of the normal function in PKD edited cells, a functional analysis based on the quantification of ATP production of in vitro differentiated erythroid cells was performed in the four different PKD samples. Importantly, erythroid cells raised from PKD-CD34+ cells that had undergone gene editing with the RNP and therapeutic donor showed ATP production levels similar to healthy donor (HD) cells and higher than unedited cells from the same PKD patient, demonstrating the recovery of the normal function of the erythroid energetic pathway upon gene editing correction (Figure 6A; Figure S6C).
Figure 6.

Edited PKD-HSPCs engraft in immunodeficient mice and restore the energetic balance in erythroid derived cells
(A) ATP quantification of in vitro differentiated erythroid cells obtained from peripheral blood CD34+ cells of four different PKD patients. Cells were edited with the therapeutic AAV donor. Peripheral blood CD34+ cells from 6 different healthy donors were also in vitro differentiated and analyzed. Two replicates per healthy donor were performed. HD unedited, HSPCs from a healthy donor who had undergone in vitro erythroid differentiation; PKD control, HSPCs from a PKD patient who had undergone in vitro erythroid differentiation; PKD GFP or coRPK edited, HSPCs from a PKD patient who had undergone gene editing with either GFP-AAV or coRPK-AAV, respectively, and subsequent in vitro erythroid differentiation. Mean ± SD is shown. Dunn’s multiple comparison test was performed. The significance is represented by p values: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.001. (B) Percentage of human CD45+ cells in BM of immunodeficient NBSGW animals transplanted with mobilized peripheral blood CD34+ (PB-CD34+) cells with or without gene editing correction from a single PKD patient, analyzed 2 mpt. (C) Percentage of hCD34+ cells within the human population (hCD45+) in animals engrafted with PKD edited human cells. (D) Percentage of the hCD19+ cells within the human population (hCD45+) in animals engrafted with PKD edited human cells. (E) Percentage of the hCD33+ cells within the human population (hCD45+) in animals engrafted with PKD edited human cells. (F) Percentage of the hCD235a+ cells in mouse BM in animals engrafted with PKD edited human cells. (G) Representative agarose gel of 5′ and 3′ PCRs in samples pre-transplant (liquid culture), after erythroid differentiation (Ery diff), in mice receiving edited cells (1.1, 1.2, 1.3, 1.4, 1.5, and 1.6) and in mice receiving unedited cells (2.1, 2.2, 2.3, and 2.4). Mock, unedited cells; Mock+EP, unedited electroporated cells; coRPK, cells edited with therapeutic donor. Arrows indicate amplification of the band showing homologous directed integration. (H) Percentage of HDR analyzed by ddPCR in BM from primary NBSGW mice transplanted with unedited and edited PKD-HSPCs. (I) Percentage of correct HDR CFUs derived from hCD34+ cells, which were sorted from BM of individual NBSGW mice transplanted with mPB-CD34+ cells from a single PKD patient with (n = 5 mice) or without (n = 4 mice)gene editing correction. Cell sorting was performed 2 mpt. Sorted cells were cultured in methylcellulose for 2 weeks, and the percentage of HDR was assessed by specific PCR in 5′ and 3′ junctions. From 3 to 23 CFUs were analyzed per animal. (J) ATP quantification in hCD235a+ cells sorted from BM of individual NBSGW mice transplanted with mPB-CD34+ cells from a single PKD patient with (n = 5 mice) or without (n = 4 mice) gene editing correction. Cell sorting was performed 2 mpt. Kruskal-Wallis multiple comparison test was performed. The significance is represented by p values: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.001.
To assay maintenance of long-term correction, unedited or edited mobilized peripheral blood CD34+ (PB-CD34+) cells from a PKD patient were transplanted into immunodeficient NOD.Cg-KitW-41JTyr+PrkdcscidIl2rgtm1Wjl/ThomJ (NBSGW) mice, which allow human erythroid differentiation in vivo, to analyze engraftment ability and restoration of erythroid ATP production in the long term. Level of human hematopoietic chimerism was analyzed 60 days post-transplant. High percentages of human engraftment (95.5% ± 1.1% and 90.0% ± 6.8% hCD45+ cells from unedited and edited PKD HSPCs, respectively) were obtained (Figure 6B). We detected differences in the percentage of the different lineages (Figures 6C–6F), contrary to expectations from previous experiments with HD cells in NSG mice. A greater amount of hematopoietic progenitors (hCD34+) and a lower contribution to the myeloid cells in the hematopoietic engraftment were observed in mice transplanted with edited cells (Figures 6C and 6D). However, human erythroid percentages in mouse BM were similar between both groups (Figure 6F). These data indicate that gene editing in PKD HSPCs is feasible and does not hamper engraftment potential.
Analysis of specific integration of the therapeutic donor was assessed in pre-transplant samples and in BM of transplanted mice. HDR efficiency in pre-transplanted cells was assessed in a liquid culture 10 dpt (13.0% HDR) and in edited cells differentiated toward erythroid lineage (19.3% HDR) by digital droplet PCR (ddPCR) (Figure S6E). Expected bands showing specific editing in 5′ and 3′ junctions were only observed in both samples (Figure 6G). We assessed the percentage of gene targeting in total BM of transplanted animals with two different methods. First, we quantified HDR in the BM of mice transplanted with these cells 60 days post-transplant using ddPCR. An average of 6.8% ± 1.8% HDR was detected (Figure 6H). In parallel, we performed specific 5′ and 3′ PCRs of individual colonies obtained after CFU assay of sorted hCD34+ cells from the BM of each mouse transplanted with or without gene-edited mPB-CD34+ cells from the PKD patient. Up to 61 individual colonies from 5 different transplanted mice were analyzed. Among colonies positive for the Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) housekeeping PCR (51 colonies in total), 12 presented the expected integration of coRPK sequences at both 5′ and 3′ junctions, indicating an efficiency of 25.2% ± 7.3% HDR in PKD patient HSCs (Figure 6I). As expected, no colonies from mock-edited CD34+ cells showed coRPK integration. In addition, the specific integration was also detected in either total BM (Figure 6G) or hCD19+ and hCD33+ sorted populations from the transplanted NBSGW mouse BM (Figure S6D), confirming that editing had occurred in multipotent HSCs.
Finally, to assess the phenotypic correction in erythroid cells generated in vivo from gene-edited PKD cells, we sorted human erythroid cells derived from the transplanted PKD mPB-CD34+ cells 2 months after transplant, and ATP level was evaluated. Importantly, ATP level in in vivo-generated erythroid cells from gene-edited PKD HSPCs was increased in comparison with their counterparts without gene correction, similar to the increase observed in the in vitro differentiated erythroid cells (Figure 6J; Figure S6F). Altogether, these results show the potential use of our gene editing approach to correct long-term HSPC from PKD patients.
Discussion
PKD is nowadays considered a good candidate for gene therapy, supported by key factors such as the monogenic origin of the disease and the confirmation of the efficacy of allogenic HSCT as a curative treatment for some PKD patients.11 In fact, an international multicentric clinical trial for the treatment of PKD patients is currently on-going (NCT04105166), and has already shown first evidence of therapeutic efficacy.19 Despite this promising therapy for PKD patients, the emergence of programmable nucleases has revolutionized the gene therapy field, making specific driven integration gene therapy an applicable clinical option.34 The feasibility of knocking in a cDNA immediately after the start codon of the gene has been recently demonstrated.24,39,40 This approach allows the restoration of the specific gene functionality without compromising the endogenous regulatory control of the gene expression. In the case of PKD, this strategy would be applicable to most PKD patients, with the exception of those carrying mutations in the promoter or regulatory regions (only 1.1% of the identified7). Here, we have addressed the development of a CRISPR/Cas9 + donor AAV6 delivery strategy to knock in a healthy version of the RPK gene at the PKLR locus to correct PKD.
To assess the safety of the designed PKLR guides, we have used a robust technique, such as GUIDE-seq, which ensured the safety of the PKLR SG1 regarding its OT effect (Figure 1). This strict requirement of little OT activity is crucial when a multiclonal cell population is targeted, as is the case for HSPC gene therapy. Additionally, to deliver HDR donors we selected the AAV6 platform, which has been widely used to edit human HSPCs. A therapeutic AAV6 donor carrying the coRPK cDNA and no promoter regions was synthesized. In parallel, a reporter donor was also generated. Both therapeutic and reporter donors’ cDNA were diverged to prevent re-cutting of the SG1-RNP after gene editing had occurred. The combination of SG1-RNP and either therapeutic or reporter donor AAV6 boosted the efficacy of HDR at PKLR locus in human hematopoietic cells. No toxic effects linked to the protocol were found in hematopoietic progenitors (Figure 2). When transduced with the reporter donor, 21.1% ± 17.4% of CFUs were GFP+ (Figure S3B). Additionally, 35.5% ± 4.7% and 38.5% ± 7.4% of the analyzed hematopoietic colonies presented the specific integration of the reporter donor or the therapeutic donor, respectively (Figure 2D). The discrepancy between the fluorescence analyses and the molecular characterization could be attributed to differences in the sensitivity in the GFP+ quantification, since the monoallelic integration could minimize the fluorescence intensity. Furthermore, edited cells, when transplanted into immunodeficient mice, were able to repopulate primary and secondary recipients very efficiently (Figures 3 and 4). Edited cells were able to give rise to different hematopoietic lineages, evidencing the successful editing of HSCs. However, the percentage of gene editing within the human cells in mice was below 5% (Figure 3C and Figure 4D), not feasible for a potential treatment to solve PKD. Consequently, we attempted the optimization of the protocol. We observed that increasing the vector concentration 5-fold allowed us to achieve higher percentages of gene editing with the reporter donor in committed progenitors and in xenotransplant experiments, without compromising human cell viability or stem potential (Figure 5). More importantly, when we applied these optimizations to HSPCs coming from the PKD patient, we observed that edited PKD-HSPCs were able to long-term reconstitute the BM of NBSGW mice efficiently (Figures 6B and 6G). Although the percentage of gene editing in human cells in the BM of these mice, assessed by ddPCR, was 6.8% ± 1.9% (Figure 6H), we corroborated a 25.2% ± 7.2% HDR by specific in-out PCRs in hematopoietic colonies obtained from hCD34+ cells of the BM of transplanted mice (Figure 6I), a value that is within the therapeutic window for PKD correction. The discrepancy between estimated gene editing efficiency in total human engraftment and in HSPCs present in the BM of the animals may be due to high transduction efficiency in human progenitors or to an over-representation of human lymphoid cells in this animal model. Additionally, technical difficulties linked to the ddPCR design could also account for the lower efficiencies detected. Furthermore, erythroid cells generated both in vitro and in vivo from edited PKD-HSPCs restored their ATP level after gene editing to values similar to those obtained from HD cells (Figures 6A and 6J). Taking into account in vitro data and previous results in the PKD animal model, we have observed that this almost complete recovery of the ATP levels is reached when 20%–30% correction is achieved, a value similar to that obtained with the analysis performed in HSPCs. Although the in vivo differentiation of corrected erythroid cells from gene-edited PKD HSPCs should be confirmed when more patient samples are available, our data suggest a long-term restoration of ATP levels in PKD patient-derived erythrocytes after gene editing.
This knockin strategy represents the most promising gene editing approach in PKLR gene so far. However, the use of nucleases targeting areas near the start site of genes might alter the expression of those genes by introducing indels. On the other hand, the addition of HDR donors, able to lead normal gene expression, might compensate this potential risk by reducing the presence of indels and restoring normal gene regulation through the HDR donors. Consequently, a high frequency of HDR at the expense of non-homologous end joining (NHEJ) might balance DNA repair to prevent harmful events.
Optimizations in vector concentration have improved the outcome of the protocol, but transduction efficiency in the most primitive compartment (HSCs) is still in the limits of the therapeutic application. We have considered the use of different editing enhancers, such as small molecules or specific microRNAs, to enhance the knockin in HSPCs. Although in previous gene editing experiments focused on the knockin in the second intron of the gene36 we observed that dimethyl prostaglandin E2 did not enhance the procedure, it may be worth testing it in this new location, since it was reported to enhance gene editing in similar contexts.23 Besides, other molecules such as Scr7 have been shown to affect NHEJ pathway by the inhibition of DNA ligase IV, a key enzyme for this pathway. Thus, the downregulation of NHEJ increased the efficiency of HDR in mammalian cells.41,42 In the same direction, 53BP1 inhibition facilitates HDR in human HSPCs.43,44 Other groups have reported the use of SR-1 molecule as an HDR pathway enhancer in TALEN and CRISPR/Cas9-mediated editing systems.45 Aside from small molecules, it has been recently reported that inhibition of p53 increases the rate of HDR.46 Furthermore, Schiroli et al. reported that AAV6-mediated gene editing aggravates p53 activation and delays HSPCs proliferation,37 which can explain our limited levels of HDR in long-term HSCs. They also claimed that transient p53 inhibition alleviated repopulating defects in edited HSPCs. Either way, inhibiting p53 is a risky approach, since it is widely known that stable inactivation of p53 pathway can lead to development of malignancies. However, a recent study in mice reported no increases in mutational load upon stable p53 genetic inactivation in HSCs,47 opening the door to the possible regulation of p53 during the gene editing procedure in order to increase the yield of edited HSPCs and ensure rapid establishment of therapeutic benefit. Additional experiments using some of the above-mentioned HDR enhancers will be performed to further improve gene editing for PKD.
In summary, the present study demonstrates that a gene editing approach based on RNP electroporation and donor rAAV6 transduction in PKD HSCs is safe and efficient to correct PKD. Moreover, the levels of gene editing in HSPCs achieved with the proposed strategy are in the range required to be of clinical relevance for the treatment of PKD patients.
Materials and methods
Human cells
The K562 cell line (chronic myelogenous leukemia; ATCC, CCL-243) was cultured in Iscove’s modified Dulbecco’s medium (IMDM; Gibco), 20% HyClone (GE Healthcare), and 1% penicillin-streptomycin (P/S) solution (Gibco). Cells were maintained at 5 × 105 to 1 × 106 cells/mL.
Umbilical CB samples from healthy donors were provided by Centro de Transfusiones de la Comunidad de Madrid, and samples from PKD patients were provided by Hospital Universitario Fundación Jiménez Díaz, Hospital Infantil Universitario Niño Jesús, and Ospedale Maggiore Policlinico. All samples were collected under written consent from the donors and Centro de Transfusiones de la Comunidad de Madrid’s institutional review board agreement (SAF2017-84248-P), in accordance with the Helsinki Declaration of 1975, revised in 2000. Purified CD34+ cells were obtained by immunoselection with the CD34 Micro-Bead kit (MACS; Miltenyi Biotech).
Cells were grown in StemSpan (STEMCELL Technologies) supplemented with 0.5% P/S, 100 ng/mL human stem cell factor (SCF), 100 ng/mL human thrombopoietin (TPO), 100 ng/mL human FMS-like tyrosine kinase 3 ligand (Flt3), and 100 ng/mL human interleukin 6 (hIL-6) (all obtained from EuroBioSciences) and 35 nM UM171 molecule (STEMCELL Technologies). Cells were cultured under normoxic conditions: 37°C, 21% O2, 5% CO2, and 95% relative humidity.
rAAV/Cas9 gene editing of human hematopoietic progenitors
To assemble RNPs, 6 μg of Alt-R S.p. Cas9 Nuclease V3 (IDT) was combined with 3.2 μg of synthetic single guide RNAs (sgRNAs) (Synthego) at room temperature (RT) for 10 min prior to being used.
For nucleofection of RNP, the P3 Primary Cell 4D Nucleofector X Kit for Amaxa 4D device (Lonza, Basel, Switzerland) was used. Two hundred thousand cells were pre-stimulated for 24 h or 48 h, resuspended in 20 μL of nucleofection solution, and nucleofected with the DZ-100 program. After the pulse, nucleofected HSPCs were incubated for 10 min at 37°C. Then, 180 μL of pre-warmed medium was added, and cells were transferred to a 96-well cell culture plate.
Nucleofected cells were immediately transduced with the corresponding AAV at different concentrations in a 96-well culture plate in a final volume of 200 μL. Twenty-four hours after transduction, 100 μL of pre-warmed medium was added.
Human hematopoiesis in immunodeficient mice
All mice were kept under standard pathogen-free conditions in the animal facility of CIEMAT. All animal experiments were performed in compliance with European and Spanish legislation and institutional guidelines and following ARRIVE guidelines (https://arriveguidelines.org/arrive-guidelines). The protocol was approved by “Consejeria de Medio Ambiente y Ordenación del Territorio” (Protocol number PROEX 073/15).
Human CD34+ cells were administered through the tail vein of 6- to 8-week-old female NSG or 6- to 8-week-old female NBSGW mice sub-lethally irradiated the day before transplant with 1.5 Gy or 1 Gy, respectively. Together with these cells, 1 × 106 hCD34− cells (negative fraction of the CD34+ purification) irradiated with 20 Gy were transplanted as support population. Human engraftment follow-up was conducted at 30 and 90 days post-transplantation. To evaluate long-term engraftment, BM cells from primary NSG mice were transplanted into secondary NSG recipients, and human engraftment was analyzed 30 and 90 days post-transplant.
Genome targeting and quantification
Frequency of gene editing in HSPCs, was assessed by specific integration in CFUs. Two days after the gene editing protocol, hCD34+ cells were resuspended in enriched methylcellulose medium (StemMACS HSC-CFU complete with Epo, Miltenyi Biotec). Fourteen days afterward, colonies were counted and CFU-GMs (granulocyte-macrophage colony forming units), BFU-Es (erythroid burst forming units) and CFU-GEMMs (granulocyte-erythroid-macrophage-megakaryocyte colony forming units) were scored. Individual colonies were picked in 100 μL of PBS. Genomic DNA from colonies was extracted as previously described.48 Specific in and out primers (donorGFP-LHA/RHA and donorRPK-LHA/RHA) (see Table S7) were designed to amplify the regions of junction between the endogenous and exogenous DNA using Herculase II Fusion High-Fidelity DNA Polymerase (Agilent Technologies) (as shown in Figure S2C). PCR products were verified in 1% agarose gels. To analyze gene editing levels in cells engrafted in immunodeficient mice or erythroid differentiation experiments, cell DNA was purified with the DNeasy Blood & Tissue Kit (QIAGEN) and the previously described in-out PCR strategy was performed. In some cases, HDR was analyzed by ddPCR (QX200 Droplet Digital PCR System, Bio-Rad, Hercules, CA, USA), using ddPCR Supermix for Probes (no dUTP) (Bio-Rad, Hercules, CA, USA). To quantify gene editing in PKLR locus, three primers (common Fwd, WT Rev, and HR Rev) and two probes (Ref and HR) were used in one PCR reaction (Table S8). Droplets were generated and analyzed according to manufacturer’s instructions with the QX200 system (Bio-Rad). ddPCR cycling conditions were as follows: 37°C (10 min) for genomic DNA digestion with HindII; followed by 95°C (10 min) and 50 cycles of 94°C (30 s), 57.4°C (30 s), and 72°C (2 min); and then 98°C (10 min).
Acknowledgments
The authors would like to thank Miguel A. Martin for the careful maintenance of PKD deficient mice, and Mrs. Aurora de la Cal, María del Carmen Sánchez, Soledad Moreno, Nadia Abu-sabha, Montserrat Aldea, and Mr. Sergio Losada for their dedicated administrative help. This work was supported by grants from “Ministerio de Economía, Comercio y Competitividad y Fondo Europeo de Desarrollo Regional (FEDER)” (SAF2017-84248-P), “Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III” (Red TERCEL; RD16/0011/0011) and Comunidad de Madrid (AvanCell, B2017/BMD-3692). The authors also thank Fundación Botín for promoting translational research at the Hematopoietic Innovative Therapies Division of the CIEMAT. CIBERER is an initiative of the “Instituto de Salud Carlos III” and “Fondo Europeo de Desarrollo Regional (FEDER).”
Author contributions
S.F.-B. and O.Q.-B. designed and performed the experiments and wrote the manuscript; O.A., R.S., M.D.-R., and I.O.-P. helped in the experimental procedures; D.P.D. and J.C. contributed in the experimental design; M.S.S., R.T., and M.A.B. performed and analyzed experiments; J.L.L.-L. and P.B. provided samples; J.A.B. suggested procedures; M.P. contributed to the experimental design and suggested procedures; J.-C.S. designed the experiments, wrote the manuscript, and provided grant support.
Declaration of interests
J.-C.S. and J.A.B. are consultants and hold shares and receive funding from Rocket Pharma. M.P. serves on the scientific advisory board of CRISPR Therapeutics and Graphite Bio. D.P.D. is employed by Graphite Bio. R.T., M.S.S., and M.A.B. are employed by Integrated DNA Technologies, Inc (IDT), which manufactures reagents similar to some described in the manuscript. R.T. and M.A.B. own equity in DHR, the parent company of IDT. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.05.001.
Contributor Information
Oscar Quintana-Bustamante, Email: oscar.quintana@ciemat.es.
Jose-Carlos Segovia, Email: jc.segovia@ciemat.es.
Supplemental information
References
- 1.Canu G., De Bonis M., Minucci A., Capoluongo E. Red blood cell PK deficiency: An update of PK-LR gene mutation database. Blood Cells Mol. Dis. 2016;57:100–109. doi: 10.1016/j.bcmd.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Grace R.F., Bianchi P., van Beers E.J., Eber S.W., Glader B., Yaish H.M., Despotovic J.M., Rothman J.A., Sharma M., McNaull M.M. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131:2183–2192. doi: 10.1182/blood-2017-10-810796. [DOI] [PubMed] [Google Scholar]
- 3.Meza N.W., Alonso-Ferrero M.E., Navarro S., Quintana-Bustamante O., Valeri A., Garcia-Gomez M., Bueren J.A., Bautista J.M., Segovia J.C. Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Mol. Ther. 2009;17:2000–2009. doi: 10.1038/mt.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grace R.F., Barcellini W. Management of pyruvate kinase deficiency in children and adults. Blood. 2020;136:1241–1249. doi: 10.1182/blood.2019000945. [DOI] [PubMed] [Google Scholar]
- 5.Christensen R.D., Eggert L.D., Baer V.L., Smith K.N. Pyruvate kinase deficiency as a cause of extreme hyperbilirubinemia in neonates from a polygamist community. J. Perinatol. 2010;30:233–236. doi: 10.1038/jp.2009.118. [DOI] [PubMed] [Google Scholar]
- 6.Muir W.A., Beutler E., Wasson C. Erythrocyte pyruvate kinase deficiency in the Ohio Amish: origin and characterization of the mutant enzyme. Am. J. Hum. Genet. 1984;36:634–639. [PMC free article] [PubMed] [Google Scholar]
- 7.Zanella A., Fermo E., Bianchi P., Chiarelli L.R., Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21:217–231. doi: 10.1016/j.blre.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Rab M.A.E., Van Oirschot B.A., Kosinski P.A., Hixon J., Johnson K., Chubukov V., Dang L., Pasterkamp G., Van Straaten S., Van Solinge W.W. AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes. Haematologica. 2021;106:238–249. doi: 10.3324/haematol.2019.238865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henig I., Zuckerman T. Hematopoietic stem cell transplantation-50 years of evolution and future perspectives. Rambam Maimonides Med. J. 2014;5:e0028. doi: 10.5041/RMMJ.10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smetsers S.E., Smiers F.J., Bresters D., Sonnevelt M.C., Bierings M.B. Four decades of stem cell transplantation for Fanconi anaemia in the Netherlands. Br. J. Haematol. 2016;174:952–961. doi: 10.1111/bjh.14165. [DOI] [PubMed] [Google Scholar]
- 11.van Straaten S., Bierings M., Bianchi P., Akiyoshi K., Kanno H., Serra I.B., Chen J., Huang X., van Beers E., Ekwattanakit S. Worldwide study of hematopoietic allogeneic stem cell transplantation in pyruvate kinase deficiency. Haematologica. 2018;103:e82–e86. doi: 10.3324/haematol.2017.177857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 13.Bueren J.A., Quintana-Bustamante O., Almarza E., Navarro S., Río P., Segovia J.C., Guenechea G. Advances in the gene therapy of monogenic blood cell diseases. Clin. Genet. 2020;97:89–102. doi: 10.1111/cge.13593. [DOI] [PubMed] [Google Scholar]
- 14.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.A., Hongeng S., Magrin E., Schiller G.J., Payen E., Semeraro M. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 2018;378:1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 15.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marktel S., Scaramuzza S., Cicalese M.P., Giglio F., Galimberti S., Lidonnici M.R., Calbi V., Assanelli A., Bernardo M.E., Rossi C. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019;25:234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 17.Ribeil J.A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Gomez M., Calabria A., Garcia-Bravo M., Benedicenti F., Kosinski P., López-Manzaneda S., Hill C., Del Mar Mañu-Pereira M., Martín M.A., Orman I. Safe and efficient gene therapy for pyruvate kinase deficiency. Mol. Ther. 2016;24:1187–1198. doi: 10.1038/mt.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López Lorenzo J.L., Navarro S., Shah A.J., Roncarolo M.G., Sevilla J., Llanos L., Pérez Camino de Gaisse B., Sanchez S., Glader B., Chien M. Lentiviral Mediated Gene Therapy for Pyruvate Kinase Deficiency: A Global Phase 1 Study for Adult and Pediatric Patients. Blood. 2020;136:47. [Google Scholar]
- 20.Bonafont J., Mencía Á., García M., Torres R., Rodríguez S., Carretero M., Chacón-Solano E., Modamio-Høybjør S., Marinas L., León C. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019;27:986–998. doi: 10.1016/j.ymthe.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zabaleta N., Barberia M., Martin-Higueras C., Zapata-Linares N., Betancor I., Rodriguez S., Martinez-Turrillas R., Torella L., Vales A., Olagüe C. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun. 2018;9:5454. doi: 10.1038/s41467-018-07827-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:16007. doi: 10.1038/ncomms16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 27.De Ravin S.S., Reik A., Liu P.Q., Li L., Wu X., Su L., Raley C., Theobald N., Choi U., Song A.H. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016;34:424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diez B., Genovese P., Roman-Rodriguez F.J., Alvarez L., Schiroli G., Ugalde L., Rodriguez-Perales S., Sevilla J., Diaz de Heredia C., Holmes M.C. Therapeutic gene editing in CD34+ hematopoietic progenitors from Fanconi anemia patients. EMBO Mol. Med. 2017;9:1574–1588. doi: 10.15252/emmm.201707540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rio P., Baños R., Lombardo A., Quintana-Bustamante O., Alvarez L., Garate Z., Genovese P., Almarza E., Valeri A., Díez B. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol. Med. 2014;6:835–848. doi: 10.15252/emmm.201303374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. A erythroid enhancer of BCL11A subject to genetic variation. Science. 2014;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang K.H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F., Sun C. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Psatha N., Reik A., Phelps S., Zhou Y., Dalas D., Yannaki E., Levasseur D.N., Urnov F.D., Holmes M.C., Papayannopoulou T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018;10:313–326. doi: 10.1016/j.omtm.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto R., Clement M.K., Cole M.A., Luk K., Baricordi C. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2020;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 35.Garate Z., Quintana-Bustamante O., Crane A.M., Olivier E., Poirot L., Galetto R., Kosinski P., Hill C., Kung C., Agirre X. Generation of a High Number of Healthy Erythroid Cells from Gene-Edited Pyruvate Kinase Deficiency Patient-Specific Induced Pluripotent Stem Cells. Stem Cell Reports. 2015;5:1053–1066. doi: 10.1016/j.stemcr.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quintana-Bustamante O., Fañanas-Baquero S., Orman I., Torres R., Duchateau P., Poirot L., Gouble A., Bueren J.A., Segovia J.C. Gene editing of PKLR gene in human hematopoietic progenitors through 5 ’ and 3 ’ UTR modified TALEN mRNA. PLoS One. 2019;14:e0223775. doi: 10.1371/journal.pone.0223775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro J., Iancu O., Jacobi A.M., McNeill M.S., Turk R., Rettig G.R., Amit I., Tovin-Recht A., Yakhini Z., Behlke M.A., Hendel A. Increasing CRISPR Efficiency and Measuring Its Specificity in HSPCs Using a Clinically Relevant System. Mol. Ther. Methods Clin. Dev. 2020;17:1097–1107. doi: 10.1016/j.omtm.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard N., Hagin D., Sommer K., Song Y., Khan I., Clough C., Ochs H.D., Rawlings D.J., Scharenberg A.M., Torgerson T.R. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127:2513–2522. doi: 10.1182/blood-2015-11-683235. [DOI] [PubMed] [Google Scholar]
- 40.Voit R.A., Hendel A., Pruett-Miller S.M., Porteus M.H. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014;42:1365–1378. doi: 10.1093/nar/gkt947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin C., Li H., Hao M., Xiong D., Luo Y., Huang C., Yuan Q., Zhang J., Xia N. Increasing the Efficiency of CRISPR/Cas9-mediated Precise Genome Editing of HSV-1 Virus in Human Cells. Sci. Rep. 2016;6:34531. doi: 10.1038/srep34531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maruyama T., Dougan S.K., Truttmann M., Bilate A.M., Ingram J.R., Ploegh H.L. Inhibition of non-homologous end joining increases the efficiency of CRISPR/Cas9-mediated precise genome editing. Nature. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Ravin S.S., Brault J., Meis R.J., Liu S., Li L., Pavel-Dinu M., Lazzarotto C.R., Liu T.Q., Koontz S., Choi U. Enhanced Homology-directed Repair for Highly Efficient Gene Editing in Hematopoietic Stem/Progenitor Cells. Blood. 2021;137:2598–2608. doi: 10.1182/blood.2020008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweeney C.L., Pavel-Dinu M., Choi U., Brault J., Liu T., Koontz S., Li L., Theobald N., Lee J., Bello E.A. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther. 2021 doi: 10.1038/s41434-021-00251-z. Published online March 12, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016;7:10548. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 47.Garaycoechea J.I., Crossan G.P., Langevin F., Mulderrig L., Louzada S., Yang F., Guilbaud G., Park N., Roerink S., Nik-Zainal S. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature. 2018;553:171–177. doi: 10.1038/nature25154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charrier S., Ferrand M., Zerbato M., Précigout G., Viornery A., Bucher-Laurent S., Benkhelifa-Ziyyat S., Merten O.W., Perea J., Galy A. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther. 2011;18:479–487. doi: 10.1038/gt.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.