

Abstract
The human small intestine is the key organ for absorption, metabolism, and excretion of orally administered drugs. To preclinically predict these reactions in drug discovery research, a cell model that can precisely recapitulate the in vivo human intestinal monolayer is desired. In this study, we developed a monolayer platform using human biopsy-derived duodenal organoids for application to pharmacokinetic studies. The human duodenal organoid-derived monolayer was prepared by a simple method in 3–8 days. It consisted of polarized absorptive cells and had tight junctions. It showed much higher cytochrome P450 (CYP)3A4 and carboxylesterase (CES)2 activities than did the existing models (Caco-2 cells). It also showed efflux activity of P-glycoprotein (P-gp) and inducibility of CYP3A4. Finally, its gene expression profile was closer to the adult human duodenum, compared to the profile of Caco-2 cells. Based on these findings, this monolayer assay system using biopsy-derived human intestinal organoids is likely to be widely adopted.
Keywords: intestinal organoids, small intestine, intestinal first-pass effect, monolayer culture, CYP3A4, P-glycoprotein, CES1, CES2, PEPT1, microarray
Graphical abstract
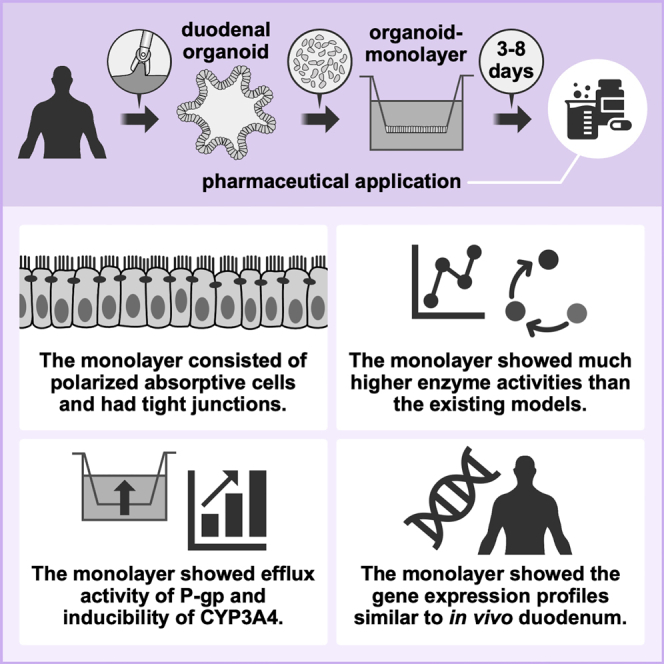
Mizuguchi and colleagues produced a monolayer from biopsy-derived human duodenal organoids by means of a simple and straightforward method. The monolayer recapitulates the nature of the in vivo small intestine, and therefore this monolayer can be a general platform for a wide range of applications including pharmaceutical research.
Introduction
The human small intestine plays a central role in the pharmacokinetics of orally administrated drugs, which account for around 70% of US Food and Drug Administration (FDA)-approved drugs (as of 2019). The process of absorbing, metabolizing, and excreting orally administered drugs begins in the small intestine. These series of reactions are called the first-pass effect and are an essential consideration in drug discovery research.1, 2, 3 An in vitro system to recapitulate the human in vivo drug absorption, metabolism, and excretion is crucially important for the structural design of compounds in the early stages of drug discovery research.
It has been difficult to culture primary human intestinal epithelial cells (iECs) for a long period without diminishing their functions and viability. Therefore, small intestinal tissues obtained from rodents or a human colon cancer-derived cell line, i.e., Caco-2 cells, have been broadly used and have made a major contribution to pharmacokinetic studies. Nonetheless, the former has the problem of species differences,4 and the latter has the problem that some of the drug-metabolizing enzymes and drug transporters are absent or poorly expressed in Caco-2 cells.5,6 To solve these problems, an alternative system is urgently needed.
Biopsy-derived intestinal organoids (also referred to as primary intestinal organoids, or enteroids) were first established in 2009 and 2011 from mouse and human intestinal tissues, respectively.7,8 These organoids can proliferate and they can be passaged, maintained for a long period in in vitro culture, and cryopreserved for a long period. Moreover, they have structural characteristics and mature functional capacities close to those of the original organs. Widespread use of the culture method for intestinal organoids has greatly contributed to the elucidation of the mechanisms of stem cell maintenance,9,10 stem cell differentiation,11 epithelial carcinogenesis,12 and inflammatory disease13 in the human intestine.
There have been few reports on the application of biopsy-derived human intestinal organoids for pharmacokinetic studies. This may be because the three-dimensional structure of them causes an inaccessible luminal compartment, which is the first part to interact with orally administrated drugs. Wang et al.14 have generated monolayer models by using intestinal organoids, and they applied the model to study effects of dietary compounds on epithelial proliferation. Similarly, Roodsant et al.15 have produced a human intestinal organoid-derived monolayer to elucidate intestinal host-pathogen interaction. Although these models are attractive, their applicability, especially in the field of pharmacokinetics, has not been evaluated.
In order to investigate the possibility of pharmacokinetic application of human intestinal organoids, we conducted this study. First, we established human biopsy-derived intestinal organoids using tissue specimens from the human duodenum, which is a relatively easy region of the human small intestinal tract to biopsy. Then, we produced the human duodenal organoid-derived monolayer by a simple method and applied it to pharmacokinetic studies.
Results
Establishment of the human duodenal organoid-derived monolayer
Duodenal biopsy tissues were taken from consenting patients, and human biopsy-derived duodenal organoids were established from them. Under endoscopic observation, there was no inflammation in the area of the intestinal tract from which the biopsies were taken (Figure 1A), and a sufficient volume of tissues could be obtained for the establishment operation (Figure 1B). When the crypts were isolated and cultured from the collected biopsy tissue according to the previously reported methods,7,8,16, 17, 18 the generated organoids were fully grown within 10 days and exhibited budding structures (Figure 1C, left and center). In addition, we were able to amplify the human duodenal organoids by passaging them (Figure 1C, right). After several passages, most of the human duodenal organoids began to exhibit a spherical structure. We confirmed that the established organoids could be cultured for more than 10 months (data not shown). Since a monolayer culture system is desirable for pharmacokinetic studies, we prepared a monolayer from the human duodenal organoids by dissociating them into single cells and seeding the cells on a cell culture insert. The gene expression level of a stem cell marker (leucine-rich repeat-containing G protein-coupled receptor 5 [LGR5]) in the seeded cells decreased immediately after seeding (Figure S1A), corresponding to the loss of proliferative potential after day 3 (data not shown). The gene expression levels of a drug-metabolizing enzyme (cytochrome P450 3A4 [CYP3A4]) and transporters (breast cancer resistance protein [BCRP], multiple drug resistance 1 [MDR1], and peptide transporter 1 [PEPT1]) increased over time (Figures S1B–S1E). In particular, the gene expression level of CYP3A4, a representative drug-metabolizing enzyme expressed in the human small intestine,19 was upregulated 1,000-fold during the monolayer culture (Figure S1B). Importantly, similar gene expression profiles were obtained for duodenal organoid cultures derived from different donors (Figure S2A). The gene expression level of an absorptive epithelial cell marker (villin 1 [VIL1]) was slightly decreased after day 10 (Figure S1F). The gene expression of a tight junction marker (claudin 3 [CLDN3]) and the level of transepithelial electrical resistance (TEER) in the seeded cells were increased through days 2–4 and plateaued thereafter (Figures S1G and S1H), indicating that the cells formed tight junctions after about 3 days. These results indicated that it is preferable to use the human duodenal organoid-derived monolayer at 3–8 days after seeding.
Figure 1.

Establishment of the human duodenal organoid-derived monolayer
(A) Image of the duodenal lumen obtained during upper gastrointestinal endoscopy. A biopsy from the non-inflammatory lamina propria mucosae of the duodenum was performed with the patient’s consent. (B) Collected specimens were in the low protein adsorption tube during the crypt isolation process. (C) Bright-field images of the isolated crypts (left panel) and established organoids (center panel, day 10 of establishment; right panel, day 5 of passage) are shown.
Growth factors known as niche factors enable the human intestinal organoids to proliferate, whereas withdrawal of several factors such as Wnt3A and nicotinamide promote their differentiation.8 Two types of media have widely been used, one containing all of the growth factors (maintenance media) and another lacking some of them (differentiation media). Although the differentiation medium was expected to improve the function of the human duodenal organoid-derived monolayer, it did not robustly alter the gene expression levels of major pharmacokinetic-related genes (Figure S3). Based on this result, we consistently used the maintenance medium for the culture of the human duodenal organoid-derived monolayer in the present report.
Characterization of the human duodenal organoid-derived monolayer
To morphologically characterize the human duodenal organoid-derived monolayer, we conducted various evaluations. Phase-contrast microscopy revealed that the human duodenal organoid-monolayer had a simple columnar epithelium-like structure (Figure 2A). Microvilli (Figure 2B, left) and a tight junction structure (Figure 2B, right) were observed by transmission electron microscopy. Immunostaining showed that E-cadherin (an epithelial cell marker) and zonula occludens-1 (ZO-1; a tight junction marker) were expressed in the intercellular space (Figure 2C, upper). It was also shown that villin (a brush border marker) and CYP3A4 (a typical small intestinal drug-metabolizing enzyme) were expressed in the apical membrane and the cytoplasm, respectively (Figure 2C, lower). Alkaline phosphatase staining showed that most of the cells constituting the monolayer had alkaline phosphatase activity (Figure 2D). TEER was reduced by 10 mM capric acid (C10), an absorption-enhancing agent (Figure 2E). Fluorescence-activated cell sorting (FACS) analysis showed that villin-positive cells constituted 75.2%–98.5% of total cells in the human duodenum organoid-derived monolayer (Figure S4). We also confirmed clear immunostaining of chromogranin A (CHGA), lysozyme (LYZ), and mucin 2 (MUC2), suggesting the existence of enteroendocrine, Paneth, and goblet cells, respectively (Figure S5). Collectively, these results suggested that the human duodenum-derived organoid monolayer has a polarized monolayer structure, that it consists of a high percentage of intestinal absorptive epithelial cells and other major iEC types, and that it possesses an integrated barrier function.
Figure 2.

Morphological analysis of the human duodenal organoid-derived monolayer
The human duodenal organoid-derived monolayer at 7 days after seeding was subjected to assays. (A) Phase-contrast image of the human duodenal organoid-derived monolayer. (B) Transmission electron micrographs of the monolayer. A brush border with microvilli (left) and tight junctions (right, black arrowhead) was observed. (C) The expression and localization of marker proteins for epithelial cells (E-cadherin), tight-junctions (ZO-1), brush borders (villin), and a drug-metabolizing enzyme (CYP3A4) were examined by immunostaining. Nuclei were counterstained with DAPI. (D) The alkaline phosphatase activity of the human duodenal organoid-derived monolayer was confirmed by alkaline phosphatase staining. (E) The barrier function of the organoid-derived monolayer in the presence or absence of 10 mM C10 was examined by TEER measurement. Data are expressed as means ± SD (n = 3). Statistical significance was evaluated by an unpaired two-tailed Student’s t test (∗∗p < 0.01).
Gene expression profile of the human duodenal organoid-derived monolayer
To characterize the human duodenal organoid-derived monolayer, gene expression levels were examined by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). Caco-2 cells, which are a typical intestinal cell model in pharmacokinetic studies, and adult duodenum (human duodenal total RNA was purchased from the BioChain Institute) were used for the comparison. The major intestinal CYP genes in the human duodenal organoid-derived monolayer were highly expressed compared to Caco-2 cells (Figure 3A; Figure S2A). In particular, the gene expression level of CYP3A4 in the human duodenal organoid-derived monolayer was comparable to that in the adult duodenum. It has been reported that carboxylesterase (CES)1 is more highly expressed than CES2 in the liver, while CES2 is more highly expressed than CES1 in the small intestine.20, 21, 22 Our qRT-PCR results indicated that Caco-2 cells and the human duodenal organoid-derived monolayer displayed hepatic and small intestinal CES expression profiles, respectively (Figure 3B; Figure S2A). The gene expression level of UDP-glucuronosyltransferase (UGT)1A1 in the human duodenum organoid-derived monolayer was similar to that in the adult duodenum, and higher than that in Caco-2 cells (Figure 3C; Figure S2A). The gene expression levels of intestinal apical transporters (BCRP, monocarboxylate transporter 1 [MCT1], MDR1, PEPT1) in the human duodenal organoid-derived monolayer were similar to or higher than those in Caco-2 cells (Figure 3D; Figure S2A). Alternatively, the gene expression levels of intestinal basolateral transporters (multidrug resistance protein [MRP]2, MRP3, organic cation transporter 1 [OCT1]) were lower than those in Caco-2 cells (Figure 3E; Figure S2A). These results suggest that the human duodenal organoid-derived monolayer has higher potential as a model for pharmacokinetic studies, and more characteristics similar to the original organ, i.e., the small intestine, compared to Caco-2 cells.
Figure 3.

Gene expression levels in the human duodenal organoid-derived monolayer
The gene expression profiles in the human duodenal organoid-derived monolayer (organoid-monolayer), Caco-2 cells (Caco-2), and adult duodenum (total RNA was purchased from the BioChain Institute) were characterized. The organoid monolayer at 8 days after seeding is shown. (A) Gene expression levels of CYP enzymes (CYP2C19, CYP2C9, CYP2J2, CYP2S1, CYP3A4). (B) Gene expression levels of CES enzymes (CES1, CES2). (C) Gene expression levels of UGT enzymes (UGT1A1, UGT2B17, UGT2B7). (D) Gene expression levels of transporters expressed on the apical side of the human small intestinal epithelium (BCRP, MCT1, MDR1, PEPT1). (E) Gene expression levels of transporters expressed on the basolateral side of the human small intestinal epithelium (MRP3, OCT1). Data are expressed as means ± SD (n = 3). The expression level of GAPDH in each group was taken as 1.0. Statistical significance was evaluated by one-way ANOVA followed by a Tukey’s post hoc test. Groups that do not share the same letter had significantly different results (p < 0.05).
Comparison of enzymatic and transporter activities with existing models
To compare the activities of drug-metabolizing enzymes and transporters in the human duodenal organoid monolayer with those of Caco-2 cells, we performed several activity measurements. The CYP3A4 activity measurement using the pro-luciferin substrate showed that the human duodenal organoid-derived monolayer had much higher CYP3A4 activity than did Caco-2 cells (Figure 4A). Higher CYP3A4 activity was also observed in the human duodenal organoid-derived monolayer from other donors (Figure S2B). The CES2 activity measurement using the pro-fluorescein substrate showed that the human duodenal organoid-derived monolayer also had higher CES2 activity than did Caco-2 cells (Figure 4B). Both the CYP3A4 and CES2 activities were efficiently inhibited by ketoconazole and loperamide, respectively, which are specific inhibitors of the respective activities (Figures 4A and 4B).
Figure 4.

Activities of drug-metabolizing enzymes or drug transporters in the human duodenal organoid-derived monolayer
The activities of drug-metabolizing enzymes or drug transporters in the human duodenal organoid-derived monolayer (organoid-monolayer) and Caco-2 cells (Caco-2) were characterized. The human duodenal organoid-derived monolayer at 3 days after seeding was subjected to the assay shown in Figure 4E. The human duodenal organoid-derived monolayer at 7 days after seeding was subjected to the assays shown in Figure 4A, B, C, D, and F. (A) CYP3A4 activity was examined by using a P450-Glo CYP3A4 assay kit. As an inhibitor of CYP3A4, 10 μM ketoconazole was used. (B) CES2 activity was measured by treatment with fluorescein diacetate (CES2 substrate) in the presence or absence of 1 mM loperamide (a CES2 inhibitor). (C) Metabolic pathways of dabigatran etexilate involving CES1 and CES2. (D) Detection of metabolites of dabigatran etexilate in the human duodenal organoid-derived monolayer. Dabigatran was added to the apical compartment of the monolayer and incubated at 37°C. At the indicated time points, samples were taken from the basal compartment of the monolayer. The metabolites were quantified using LC-MS/MS. (E) The apparent permeability coefficient (Papp) values of digoxin (substrate of P-gp) in the apical-to-basolateral (A to B, closed column) and basolateral-to-apical (B to A, open column) directions were evaluated. ER values (B to A/A to B) are indicated above the column. (F) The Papp values of glycylsarcosine (a substrate of PEPT1) in the apical-to-basolateral direction were evaluated. Data are expressed as means ± SD (n = 3). Statistical significance was evaluated by an unpaired two-tailed Student’s t test (∗∗p < 0.01).
In order to profile the activity of CES enzymes in the human duodenal organoid-derived monolayer, we investigated the metabolism of dabigatran etexilate.23 The formation of the intermediate metabolite BIBR 1087 from the double prodrug dabigatran etexilate and the formation of BIBR 953 from the intermediate metabolite BIBR 951 are catalyzed by CES1. The formation of BIBR 951 from dabigatran etexilate and the formation of BIBR 953 from BIBR 1087 are catalyzed by CES2 (Figure 4C). In tissues and cells with predominant expression of CES1 (e.g., liver), BIBR 1087 should be the major metabolite; in tissues and cells with predominant expression of CES2 (e.g., small intestine), BIBR 951 should be the major metabolite. In the human duodenal organoid-derived monolayer, BIBR 951 was found to be the main metabolite (Figure 4D). Meanwhile, it has been reported that BIBR 1087 is the main metabolite in Caco-2 cells.23
The efflux activity of P-glycoprotein (P-gp; the product of MDR1), a major drug efflux transporter, was determined using its substrate, digoxin. The human duodenal organoid-derived monolayer showed an efflux ratio (ER) comparable to that in Caco-2 cells (Figure 4E). The activity of PEPT1, a major dipeptide transporter, was determined using its substrate, glycylsarcosine. The human duodenal organoid-derived monolayer showed higher PEPT1 activity compared to Caco-2 cells (Figure 4F). These results were generally consistent with those in the gene expression analysis (Figure 3).
In addition, we compared the human duodenal organoid-derived monolayer with human iPS cell-derived IECs (iPS-IECs), which are a relatively new model for pharmacokinetic studies. The results showed that the human duodenal organoid-derived monolayer had much higher CYP3A4 and P-gp activity than did human iPS-IECs (Figure S6). Taken together, these findings suggest that the human duodenal organoid-derived monolayer has mature functions that recapitulate the original organ, and that it would be more useful than the existing models for various pharmacokinetic studies.
CYP3A4 inducibility in the human duodenal organoid-derived monolayer
To examine the CYP3A4 inducibility in the human duodenal organoid-derived monolayer, we measured the gene expression levels of nuclear receptors and the CYP3A4 activities with or without inducer treatment. The gene expression levels of pregnane X receptor (PXR) and vitamin D receptor (VDR) in the human duodenal organoid-derived monolayer were higher than and the same as those of Caco-2 cells, respectively (Figure 5A). PXR and VDR contribute to rifampicin (RIF)- and vitamin D3 (VD3)-mediated CYP3A4 induction, respectively. Consistent with the results of qRT-PCR and the previous report on the CYP3A4 inducibility of Caco-2 cells,5 we found that RIF treatment induced CYP3A4 mRNA expression and activity only in the human duodenal organoid-derived monolayer, whereas VD3 treatment induced such expression/activity in both the human duodenal organoid-derived monolayer and Caco-2 cells (Figures 5B and 5C). We also confirmed similar trends in the induction of MDR1 mRNA expression and P-gp activity (Figure 5D; Figure S6B), and in the protein induction of CYP3A4 and P-gp in the human duodenal organoid-monolayer (Figure 5E). From these results, it was suggested that the human duodenal organoid-derived monolayer would be a useful tool to study the drug-mediated induction of pharmacokinetic-related factors.
Figure 5.

Induction potency in the human duodenal organoid-derived monolayer
Induction potency in the human duodenal organoid-derived monolayer (organoid-monolayer) and Caco-2 cells (Caco-2) was characterized. (A) Gene expression levels of nuclear receptors (pregnane X receptor [PXR], vitamin D receptor [VDR]). The organoid-monolayer at 8 days after seeding is shown. The expression level of GAPDH in each group was taken as 1.0. (B) The gene expression levels and (C) the activities of CYP3A4 were examined by real-time RT-PCR analysis and a P450-Glo CYP3A4 assay kit, respectively. (D) MDR1 mRNA induction was characterized by real-time RT-PCR analysis. The values in the solvent-treated cells were taken as 1.0. (E) CYP3A4 and P-gp protein inductions in the human duodenal organoid-monolayer were measured by LC-MS/MS. The values represent protein amounts of pooled triplicate cultures. The human duodenal organoid-derived monolayer at 7 days after seeding was subjected to induction assays. Data are expressed as means ± SD (n = 3) for (A)–(D). For (A), statistical significance was evaluated by one-way ANOVA followed by a Tukey’s post hoc test. Groups that do not share the same letter had significantly different results (p < 0.05). For (B)–(D), statistical significance was evaluated by an unpaired two-tailed Student’s t test (∗p < 0.05, ∗∗p < 0.01), and the cells were treated with 0.1% DMSO (solvent), 20 mM rifampicin (RIF), or 100 nM 1α,25-dihydroxyvitamin D3 (VD3) for 48 h at the end of the culture period. For (D), the cells were treated the inducer (RIF and VD3) for 48 h at the end of the culture period.
Transcriptomic analysis of the human duodenal organoid-derived monolayer
For further characterization of the human duodenal organoid-derived monolayer, we conducted a transcriptomic analysis. Human adult duodenal biopsies (Figure 6, adult duodenum), human duodenal organoids (Figure 6, organoid), the human duodenal organoid-derived monolayer (Figure 6, organoid-monolayer), and Caco-2 cells (Figure 6, Caco-2) were subjected to DNA microarray analysis. Hierarchical clustering revealed that the gene expression profiles of the human duodenal organoid-derived monolayer and Caco-2 cells were closest and farthest from those of the human adult duodenum, respectively (Figure 6A). In addition, the human duodenal organoid-derived monolayer showed a gene expression profile that was closer to the human adult duodenum than was the profile of the human duodenal organoids (Figure 6A). We grouped the genes with similar patterns of variation into six clusters, and the expression levels in each cluster are shown in Figure 6B. Gene clusters no. 1, 2, 4, and 6 had relatively low expression levels in Caco-2 cells, while cluster nos. 3 and 5 had relatively high-level expression in Caco-2 cells. In all clusters, the human duodenal organoid-derived monolayer showed higher expression levels than did the human duodenal organoids. To understand the functions of each cluster, we performed a Gene Ontology (GO) enrichment analysis (Table 1). Gene cluster no. 1 contained genes with GO terms such as “drug metabolic process,” no. 2 contained “intestinal epithelial cell differentiation,” no. 4 contained “cell-cell signaling,” and no. 6 contained “transmembrane transport” (Table 1). Gene cluster no. 3 contained genes with GO terms such as “regulation of muscle contraction,” and no. 5 contained genes with terms such as “calcium ion binding” (Table 1). Taken together, these results suggested that human duodenal organoids were successfully differentiated by monolayer culture, resulting in properties closer to the in vivo small intestine than the existing models.
Figure 6.

Comprehensive gene expression analysis
Human adult duodenal biopsies (five-donor pool, adult duodenum), the human duodenal organoid (organoid), the human duodenal organoid-derived monolayer at 8 days after seeding (organoid-monolayer), and Caco-2 cells (Caco-2) were subjected to DNA microarray analysis. (A) Heatmap and hierarchical clustering of gene expression levels obtained by microarray analysis. (B) Violin plots to visualize the expression levels of genes in each gene cluster. Statistical significance was evaluated by one-way ANOVA followed by a Tukey’s post hoc test. Groups that do not share the same letter had significantly different results (p < 0.05).
Table 1.
Significantly enriched GO terms detected by GO enrichment analysis of each gene cluster
| ID | Term | p Value |
|---|---|---|
| Gene cluster no. 1 | ||
| GO:0070062 | extracellular exosome | 4.88E−08 |
| GO:0072562 | blood microparticle | 5.87E−08 |
| GO:0006955 | immune response | 4.04E−07 |
| GO:0019825 | oxygen binding | 1.78E−06 |
| GO:0009897 | external side of plasma membrane | 2.82E−06 |
| GO:0009986 | cell surface | 4.52E−06 |
| GO:0031295 | T cell costimulation | 6.21E−06 |
| GO:0042613 | MHC class II protein complex | 6.36E−06 |
| GO:0006954 | inflammatory response | 6.64E−06 |
| GO:0020037 | heme binding | 8.68E−06 |
| GO:0008202 | steroid metabolic process | 1.56E−05 |
| GO:0017144 | drug metabolic process | 1.97E−05 |
| GO:0042737 | drug catabolic process | 3.86E−05 |
| GO:0003823 | antigen binding | 4.49E−05 |
| GO:0002504 | antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | 5.17E−05 |
| Gene cluster no. 2 | ||
| GO:0007165 | signal transduction | 1.07E−05 |
| GO:0006955 | immune response | 1.77E−04 |
| GO:0005102 | receptor binding | 7.54E−04 |
| GO:0070062 | extracellular exosome | 1.19E−03 |
| GO:0009887 | organ morphogenesis | 1.76E−03 |
| GO:0005615 | extracellular space | 1.78E−03 |
| GO:0060575 | intestinal epithelial cell differentiation | 1.79E−03 |
| GO:0005164 | tumor necrosis factor receptor binding | 2.83E−03 |
| GO:0006954 | inflammatory response | 3.17E−03 |
| GO:0006935 | chemotaxis | 5.94E−03 |
| GO:0006805 | xenobiotic metabolic process | 6.28E−03 |
| GO:1902166 | negative regulation of intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator | 6.41E−03 |
| GO:0050892 | intestinal absorption | 7.43E−03 |
| GO:0005125 | cytokine activity | 7.92E−03 |
| GO:0006366 | transcription from RNA polymerase II promoter | 9.30E−03 |
| Gene cluster no. 3 | ||
| GO:0005515 | protein binding | 5.01E−03 |
| GO:0031014 | troponin T binding | 7.09E−03 |
| GO:0014883 | transition between fast and slow fiber | 1.33E−02 |
| GO:0005861 | troponin complex | 1.44E−02 |
| GO:0016540 | protein autoprocessing | 2.45E−02 |
| GO:0006937 | regulation of muscle contraction | 2.64E−02 |
| GO:0050750 | low-density lipoprotein particle receptor binding | 2.81E−02 |
| GO:0003009 | skeletal muscle contraction | 4.48E−02 |
| GO:0000122 | negative regulation of transcription from RNA polymerase II promoter | 4.66E−02 |
| GO:0030049 | muscle filament sliding | 7.00E−02 |
| GO:0007154 | cell communication | 7.00E−02 |
| GO:0030326 | embryonic limb morphogenesis | 7.35E−02 |
| GO:0043621 | protein self-association | 7.87E−02 |
| GO:0009267 | cellular response to starvation | 8.59E−02 |
| GO:0042277 | peptide binding | 9.98E−02 |
| Gene cluster no. 4 | ||
| GO:0005615 | extracellular space | 1.56E−04 |
| GO:0005576 | extracellular region | 2.73E−03 |
| GO:0005887 | integral component of plasma membrane | 4.59E−03 |
| GO:0060326 | cell chemotaxis | 8.09E−03 |
| GO:0070062 | extracellular exosome | 1.34E−02 |
| GO:0005578 | proteinaceous extracellular matrix | 1.45E−02 |
| GO:0007267 | cell-cell signaling | 1.57E−02 |
| GO:0005886 | plasma membrane | 1.58E−02 |
| GO:0007165 | signal transduction | 3.11E−02 |
| GO:0055114 | oxidation-reduction process | 3.37E−02 |
| GO:0005520 | insulin-like growth factor binding | 3.57E−02 |
| GO:0008285 | negative regulation of cell proliferation | 4.89E−02 |
| GO:0043066 | negative regulation of apoptotic process | 6.83E−02 |
| GO:0008284 | positive regulation of cell proliferation | 7.23E−02 |
| GO:0043407 | negative regulation of MAP kinase activity | 7.24E−02 |
| Gene cluster no. 5 | ||
| GO:0005576 | extracellular region | 4.11E−08 |
| GO:0005615 | extracellular space | 6.28E−07 |
| GO:0070062 | extracellular exosome | 1.04E−06 |
| GO:0030198 | extracellular matrix organization | 7.02E−06 |
| GO:0072562 | blood microparticle | 7.90E−06 |
| GO:0010951 | negative regulation of endopeptidase activity | 1.74E−05 |
| GO:0001523 | retinoid metabolic process | 5.56E−05 |
| GO:0030195 | negative regulation of blood coagulation | 6.25E−05 |
| GO:0005509 | calcium ion binding | 1.16E−04 |
| GO:0034364 | high-density lipoprotein particle | 4.01E−04 |
| GO:0006810 | Transport | 5.73E−04 |
| GO:0033344 | cholesterol efflux | 6.13E−04 |
| GO:0002576 | platelet degranulation | 6.54E−04 |
| GO:0042632 | cholesterol homeostasis | 9.05E−04 |
| GO:2000096 | positive regulation of Wnt signaling pathway, planar cell polarity pathway | 9.22E−04 |
| Gene cluster no. 6 | ||
| GO:0070062 | extracellular exosome | 1.93E−07 |
| GO:0005615 | extracellular space | 2.82E−04 |
| GO:0005215 | transporter activity | 5.79E−04 |
| GO:0031526 | brush border membrane | 2.04E−03 |
| GO:0016021 | integral component of membrane | 3.21E−03 |
| GO:0005903 | brush border | 3.25E−03 |
| GO:0031528 | microvillus membrane | 3.91E−03 |
| GO:0005385 | zinc ion transmembrane transporter activity | 5.55E−03 |
| GO:0055085 | transmembrane transport | 8.34E−03 |
| GO:0090675 | intermicrovillar adhesion | 1.02E−02 |
| GO:0005576 | extracellular region | 1.20E−02 |
| GO:0005886 | plasma membrane | 1.28E−02 |
| GO:0016324 | apical plasma membrane | 1.46E−02 |
| GO:0044214 | spanning component of plasma membrane | 1.47E−02 |
| GO:0030199 | collagen fibril organization | 1.70E−02 |
GO enrichment analysis was performed based on the functions of each of the gene clusters obtained by hierarchical clustering (Figure 6A). The list of genes contained in each cluster was input into DAVID (https://david.ncifcrf.gov/summary.jsp), and 15 of the outputs are shown in this table. MHC, major histocompatibility complex; MAP, mitogen-activated protein.
The gene set enrichment analysis (GSEA) of the DNA microarray data showed that the expression levels of genes involved in drug and xenobiotic metabolism were upregulated in the human duodenal organoid-derived monolayer compared to Caco-2 cells (Figure S7). This result suggests that the organoid monolayer could solve the problems associated with the use of Caco-2 cells in pharmacokinetic studies.
Discussion
In this study, we successfully produced a monolayer from biopsy-derived human duodenal organoids by means of a very simple and straightforward method. The human duodenal organoid-derived monolayer can be generated from the organoids in 3–8 days by using a single medium without complicated procedures. The monolayer showed higher expressions and functions of major pharmacokinetic-related enzymes and transporters than did Caco-2 cells and human iPS-IECs, and these profiles in the monolayer were close to those of the human intestine in general. In addition, the human duodenal organoid-derived monolayer exhibited gene expression profiles similar to those of the adult small intestine. It was suggested that the human duodenal organoid-derived monolayer can recapitulate the characteristics of the in vivo small intestine, and therefore this monolayer could be a powerful tool to predict the pharmacokinetics of orally administered drugs.
In our model, some of the pharmacokinetic-related genes were upregulated and then maintained at these higher levels for at least 5 days of the monolayer culture (from day 3 to 8). This suggests that the monolayer is able to remain functional for 5 days, which is a sufficient time window for various pharmacokinetic studies. Thus, the human duodenal organoid-derived monolayer can be used for a wide range of applications, in addition to conventional pharmacokinetic testing, such as for tests of intestinal toxicity from long-term drug exposure and intestinal induction of enzymes or transporters by habitual drug use.
There are few reports using human biopsy-derived intestinal organoids for in vitro compound testing. Dekkers et al.24 used rectal organoids of patients suffering from cystic fibrosis and demonstrated the usefulness of human biopsy-derived intestinal organoids for drug screening. Kozuka et al.25 made a functional monolayer from ileum organoids by optimizing the culture medium. The organoid-derived monolayer was capable of hormone production and ion transport in response to external stimuli from the compounds. Kasendra et al.26,27 combined the duodenal organoids and a microfluidic device for pharmacokinetic studies. The device was made from polydimethylsiloxane (PDMS) and enabled cell culture with physiological fluid flow and peristalsis-like mechanical motion using a vacuum pump controlled by an electronic regulator. Alternatively, in the present study, we used cell culture plates that have been widely used and one type of medium that is widely known. Therefore, it is considered that our protocol can be easily performed at any laboratories. In addition, to the best of our knowledge, this is the first report to describe a practical method for generating a human intestinal organoid-derived monolayer for pharmacokinetic applications.
Intestinal organoids derived from human iPS cells,28,29 which are referred to as induced human intestinal organoids (iHIOs) or simply HIOs, are also starting to be applied for pharmacokinetic testing. The iHIOs can be subjected to the evaluation of efflux transport activity through P-gp and the induction of CYP3A4 in the three-dimensional state.30 iHIO-derived monolayers can be produced by sorting and seeding epithelial cell adhesion molecule (EpCAM)-positive cells, and these monolayers are useful as a cell model to predict drug absorption and metabolism in the small intestine.31,32 However, several assays using the intestinal organoids in a three-dimensional state and the sorting operations necessary to produce the monolayer are relatively complicated and may not be suitable for large-scale testing. In any case, it will be necessary to conduct a detailed comparison between iHIOs and human biopsy-derived intestinal organoids in order to elucidate which type of organoid is more suitable for pharmacokinetic study.
Comprehensive gene expression analysis using a DNA microarray provided us with interesting insights from viewpoints beyond the pharmacokinetics. GO enrichment analysis showed that genes with GO terms such as “immune response,” “T cell costimulation,” “MHC class II protein complex” and “inflammatory response” had higher expression levels in human duodenal organoid-derived monolayer than in Caco-2 cells. Moreover, GSEA showed that genes belonging to lists such as “intestinal immune network for IgA production,” “antigen processing and presentation,” “B cell receptor signaling pathway,” and “natural killer cell-mediated cytotoxicity” had higher expression levels in the human duodenal organoid-derived monolayer than in Caco-2 cells. These results suggest that our human duodenal organoid-derived monolayer may retain its functions related to the immune system. Although biopsy-derived intestinal organoids in a basically three-dimensional state have been used in basic studies of the intestinal immune system against parasites,33 bacteria,34 and viruses,35 our results suggest that it will also be possible to use these organoids in a monolayer state in the near future.
Interestingly, monolayerization itself caused a 3-log order increase of CYP3A4 expression level compared to that in the organoid and promoted cell differentiation, while the use of differentiation media did not appear to have any major effects on the maturation. If the mechanism behind monolayerization-mediated maturation can be elucidated, it may become possible to improve the functions of not only organoid monolayers, but also iPS-IECs. Although monolayerization might have a larger effect on the cellular maturation, investigations to optimize the culture conditions (extracellular matrix and medium composition) for human intestinal organoids are very important and are underway. Gjorevski et al.36 have successfully developed a chemically defined extracellular matrix as an alternative to Matrigel for intestinal organoid cultures. Fujii et al.37 have established a refined medium condition that allows human intestinal organoids to concurrently undergo multi-differentiation and self-renewal. In the present study, to prepare the human intestinal organoid-derived monolayer, we used the simplest and most practical of all the feasible methods. Therefore, it might be possible to further improve the culture conditions for the human intestinal organoid-derived monolayer.
Although Caco-2 cells are derived from human colon cancer, they have been used as a golden standard in conventional pharmacokinetic studies because they are known to exhibit small intestinal epithelial-like properties after 21 days of monolayer culture.5 Therefore, in the present study, we compared the functions of our model (human duodenal organoid-derived monolayer) with those of Caco-2 cells. However, there is a common understanding that the organoids model should be better than Caco-2 cells. In vivo human small intestine should be used to compare with the human duodenal organoid-derived monolayer from the scientific perspective. Currently, biological assay systems using primary human small iECs are very difficult because of the difficulty in culturing them. We would like to obtain the data somehow and perform a true comparative study in the future.
Currently, many kinds of research combining organoids with cutting-edge technologies such as genome editing are accelerating. However, assays using organoids are often hampered by technical problems that arise from the three-dimensional structure of the organoids. We hope that our simple method for generating an intestinal organoid-derived monolayer will offer a solution to this problem and will provide a general platform for a wide range of applications.
Materials and methods
Ethics statement
This study was approved by the Ethics Committees of Sapporo Medical University, Osaka University, and Nippon Boehringer Ingelheim. All experiments were performed in accordance with the ethical standards of the institutional and national research committees, and with the Declaration of Helsinki. Written informed consent was obtained from all participants.
Duodenal biopsy
The human duodenal biopsy was performed during upper gastrointestinal endoscopy in patients from Sapporo Medical University Hospital. Under endoscopic observation, two to four biopsy samples were obtained from the noninflammatory lamina propria mucosae of the duodenum. The obtained samples were stored in ice-cold PBS containing 1× antibiotic-antimycotic (15240062; Thermo Fisher Scientific, MA, USA) until the procedures to establish the organoids.
Establishment of human duodenal organoids
We established human duodenal organoids based on the methods described in several previous reports with some modifications.7,8,16, 17, 18 Briefly, obtained samples were incubated in 2.5 mM EDTA at 4°C for 30 min and vigorously pipetted up and down to isolate crypts. The crypts were resuspended in Matrigel (354230; Corning, NY, USA), and 25-40 μL of the crypt suspension was applied to the center of each well of a 24-well plate (142475; Thermo Fisher Scientific). The Matrigel was polymerized for 10 min at 37°C, and 500 μL/well of the organoid culture medium (IntestiCult organoid growth medium (human), ST-06010; STEMCELL Technologies, BC, Canada) containing 1× antibiotic-antimycotic was overlaid. According to the manufacturer, the composition of the organoid culture medium is based on two reports.8,38
Maintenance of human duodenal organoids
In order to maintain human duodenal organoids, the medium was changed every 2 days and organoids were passaged at 1:3 to 1:10 every week. The procedure for passaging human duodenal organoids was mainly based on the report of Miyoshi and Stappenbeck16 with some modifications. Each well was washed with 0.5 mM EDTA, and the organoid cultures were scraped and suspended in TrypLE select (12563029; Thermo Fisher Scientific). The suspension was incubated at 37°C for 5–7 min to dissolve the Matrigel. After incubation, the organoids were pipetted up and down two to five times, then resuspended with Matrigel to the desired concentration. Then, 25–40 μL of the organoid suspension was applied to the center of each well of a 24-well plate. The Matrigel was polymerized for 10 min at 37°C, and 500 μL/well of the organoid culture medium containing 1× antibiotic-antimycotic was overlaid.
Human duodenal organoid-derived monolayer
To maximize seeding efficiency, a Transwell filter insert (24-well, 0.4-μm pore size, 353095; Corning) was coated with 250 μL of 2% (v/v) Matrigel in advanced DMEM/F-12 (12634010; Thermo Fisher Scientific) for 1 h at 37°C. Organoids were collected and incubated in TrypLE select following the passaging protocol described above. After incubation, the organoids were pipetted up and down to dissociate organoids into single cells. The cells were resuspended with the organoid culture medium containing 10 μM Y-27632 (036-24023; Fujifilm Wako Pure Chemical Industries, Osaka, Japan). The cell suspension was filtered through a 40-μm cell strainer (352340; Corning) and applied to the Matrigel-coated Transwell filter insert to be 5.0 × 105 to 1.0 × 106 cells/cm2. The culture medium was added to only the apical side and replaced with the organoid culture medium every 1–2 days until the assay. Note that we tried seeding the cells as a fragment to establish the monolayer, but the adhesion efficiency was poor and any confluent monolayer could not be obtained. Alternatively, single-cell seeding resulted in a confluent monolayers that could be applied to absorption tests. Although this result may seem to contradict a report from another group,26 it is considered that the adhesion efficiency can vary depending on the coating or the material of the plate, the site of origin of the intestinal organoids, and the culture conditions.
Caco-2 cells
Caco-2 cells (HTB-37; ATCC, VA, USA) were cultured with minimum essential medium (MEM, M4655; Merck, Hessen, Germany) containing 10% fetal bovine serum, 1× MEM-non-essential amino acid (NEAA) solution (11140050; Thermo Fisher Scientific), 1× GlutaMAX-I (35050061; Thermo Fisher Scientific), and 1× antibiotic-antimycotic. For differentiation of Caco-2 cells, the cells were cultured on a Transwell filter insert for 21 days after they reached confluence.
Phase-contrast images
The human duodenal organoid-derived monolayer was observed with an inverted phase-contrast microscope (CKX41; Olympus, Tokyo, Japan). The image was captured with a computer-assisted digital camera (DP21; Olympus).
Transmission electron microscopy (TEM) images
The human duodenal organoid-derived monolayer was fixed in phosphate-buffered 2% glutaraldehyde. Post-fixation, dehydration, embedding, ultrathin sectioning, staining, and observation were performed at Hanaichi Ultrastructure Research Institute (Aichi, Japan). Briefly, the specimen was post-fixed in 2% osmium tetroxide for 3 h in an ice bath. Then, it was dehydrated in graded ethanol and embedded in the epoxy resin. Ultrathin sections were obtained by the ultramicrotome technique. Ultrathin sections, which were stained with uranyl acetate for 15 min and lead staining solution for 5 min, were submitted to TEM observation (H-7600; Hitachi, Tokyo, Japan) at 100 kV.
Alkaline phosphatase staining
Alkaline phosphatase staining was performed by using a red-color alkaline phosphatase (AP) staining kit (AP100R-1; System Biosciences, CA, USA) according to the manufacturer’s instructions. The stained specimen was observed with an inverted phase-contrast microscope, and the image was captured with a computer-assisted digital camera.
Immunostaining
The human duodenal organoid-derived monolayer was fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. After blocking the monolayer with the blocking buffer, which contains PBS containing 2% bovine serum albumin (01863-35; Nacalai Tesque, Kyoto, Japan) and 0.2% Triton X-100 (X100-100ML; Merck), for 15 min at room temperature, the cells were incubated with the blocking buffer containing a primary antibody (described in Table S2) at 4°C overnight, and finally with the blocking buffer containing a secondary antibody (described in Table S2) for 1 h at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 11034-56; Nacalai Tesque). The images were acquired with a confocal laser scanning microscope (FV10i; Olympus) and fluorescence microscope (BZ-X800; Keyence, Osaka, Japan).
TEER measurement
TEER values of the human duodenal organoid-derived monolayer were measured by using a Millicell ERS-2 voltohmmeter with an STX01 electrode (MERS00002; Merck). The raw data were converted to Ω × cm2 based on the culture insert area. The blank resistance was then subtracted from the measured resistance to obtain the effective TEER. As an absorption-enhancing agent, 10 mM capric acid was used.
P-gp activity
To measure the P-gp activity, digoxin was used in the bi-directional transporter assay as described previously.39 The concentrations of digoxin in samples were determined with ultra high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). In brief, the solutions were mixed with a 3-fold volume of acetonitrile/0.2% formic acid containing 100 nM hesperetin as an internal standard. Mixed solutions were centrifuged for 5 min at 1,000 × g. Then, the supernatant was analyzed by UPLC-MS/MS to measure the concentration of digoxin according to a standard curve. UPLC analysis was performed using an ExionLC (AB Sciex, CA, USA), and MS/MS was performed on a 5500 QTRAP (AB Sciex). The mass spectrometer was set to the multiple-reaction monitoring (MRM) mode and was operated with the electrospray ionization source in the negative ion mode. The MRM transition (m/z of precursor ion/m/z of product ion) for digoxin was 825.3/779.4. For transition, the ion spray voltage and collision energy were set at −4,500 V and −30 eV. The dwell time for each MRM transition was set at 50 ms. LC separations were carried out at 40°C with an Atlantis T3 column (3 μm, 2.1 × 50 mm; Waters, MA, USA). The mobile phase was delivered at a flow rate of 0.4 mL/min using a gradient elution profile consisting of solvent A (0.1% formic acid/distilled water) and solvent B (acetonitrile). The initial composition of the binary solvent was 3% solvent B. Solvent B was increased from 3% to 95% during 2.6 min. The solvent composition remained at 100% Solvent B for 0.9 min. 2 μL of sample solution was injected into the column.
PEPT1 activity
The cells were cultured on the cell culture inserts, then washed with Hanks’ balanced salt solution (HBSS, 14025092; Thermo Fisher Scientific). HBSS containing 30 mM glycylsarcosine (G3127; Merck) was added to the apical side, and HBSS was also added to the basolateral side. After 90 min of incubation at 37°C, the solution was collected from the basolateral side. The solutions were mixed with a 2-fold volume of acetonitrile, then centrifuged for 10 min at 20,000 × g. The supernatants were mixed with a 5.6-fold volume of water and centrifuged for 10 min at 20,000 × g, and then the supernatant was analyzed by UPLC-MS/MS to measure the concentration of glycylsarcosine according to a standard curve. UPLC analysis was performed using an Acquity UPLC (Waters), and MS/MS was performed on a Q-Premier/XE (Waters). The mass spectrometer was set to the MRM mode and was operated with the electrospray ionization source in the positive ion mode. The MRM transition (m/z of precursor ion/m/z of product ion) for glycylsarcosine was 146.92/89.90. For transition, the cone voltage and collision energy were set at 18 V and 12 eV. The dwell time for each MRM transition was set at 100 ms. LC separations were carried out at 40°C with an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm; Waters). The mobile phase was delivered at a flow rate of 0.5 mL/min using a gradient elution profile consisting of solvent A (0.1% formic acid/distilled water) and solvent B (acetonitrile). The initial composition of the binary solvent was 0% solvent B from 0 to 1.0 min. Solvent B was increased from 0% to 100% during 1.5 min. The solvent composition remained at 100% solvent B for 1.0 min. 10 μL of sample solution was injected into the column.
qRT-PCR
Total RNA was isolated using ISOGEN (319-90211; Nippon Gene, Tokyo, Japan). Using 500 ng of the total RNA, cDNA was synthesized with a SuperScript VILO cDNA synthesis kit (11754250; Thermo Fisher Scientific). qRT-PCR was performed with Fast SYBR Green master mix (4385614; Thermo Fisher Scientific) using a StepOnePlus real-time PCR system (4376592; Thermo Fisher Scientific). The Ct values of the target genes were normalized by those of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and the 2−ΔΔCt method was adopted for the relative quantification. PCR primer sequences (described in Table S3) were obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/).40, 41, 42 Human duodenal total RNA (R1234101-50) was purchased from the BioChain Institute (CA, USA).
CYP3A4 activity
To measure the CYP3A4 activity, we performed lytic assays by using a P450-Glo CYP3A4 assay kit (V9001; Promega, WI, USA). Luciferin-IPA was used for the CYP3A4 substrate. We measured the fluorescence activity with a luminometer (Lumat LB 9507; Berthold Technologies, Baden-Wurttemberg, Germany) according to the manufacturer’s instructions. The CYP3A4 activities were normalized with the protein content per well by using a Pierce bicinchoninic acid (BCA) protein assay kit (23227; Thermo Fisher Scientific) according to the manufacturer’s instructions. As an inhibitor of CYP3A4, 10 μM ketoconazole (116-00551; Fujifilm Wako Pure Chemical Industries) was incubated with the substrate.
CES2 activity
To measure the CES2 activity, the cells were homogenized with a lysis buffer consisting of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl (191-01665; Fujifilm Wako Pure Chemical Industries), 0.5% Triton X-100, and 1 mM EDTA for 20 min on ice. The homogenates were centrifuged at 15,000 × g at 4°C for 15 min, and the supernatants were collected into a new tube. 1 mL of the supernatant containing 20 μg protein was incubated with 1 mM of a CES2 inhibitor, loperamide (129-05721; Fujifilm Wako Pure Chemical Industries), at 37°C for 5 min, and then treated with 10 μM of a CES2 substrate, fluorescein diacetate (069-05451; Fujifilm Wako Pure Chemical Industries). After 15 min of incubation, the reaction was terminated by the addition of an equal volume of ice-cold acetonitrile (012-19851; Fujifilm Wako Pure Chemical Industries). After centrifugation, the fluorescence intensity was measured with a multimode microplate reader (TriStar LB941; Berthold Technologies) using wavelengths of 485 nm for excitation and 535 nm for emission.
CES-mediated metabolism of dabigatran etexilate
Using a human duodenal organoid-derived monolayer at day 7 grown on Transwell filter inserts, CES-mediated metabolism of dabigatran etexilate was measured as described previously.23 In the experiments with Transwell filter inserts, dabigatran etexilate (10 μM) was given to the apical compartments, and metabolites formed in the basal (receiver) compartments were measured. Concentrations of three metabolites were determined by HPLC-MS/MS simultaneously using same internal standard and expressed as a ratio.
CYP3A4 and P-gp protein quantification
Protein quantification of CYP3A4 and P-gp in the microsomal fraction of the human duodenal organoid-derived monolayer was conducted according to the previously reported methods43 with minor modification. In brief, cells cultured on a Transwell filter insert were detached by sonification and disrupted by a Bioruptor (Cosmo Bio, Tokyo, Japan). Homogenates were subjected to several centrifugation steps to collect a microsomal fraction. The resulting microsomal pellet was solubilized using the membrane protein extraction phase-transfer surfactant (MPEX-PTS) reagent kit (GL Science, Tokyo, Japan) following the assay kit protocol. Total protein content of enriched microsomal proteins was determined using a 3-(4-carboxybenzoyl)-quinolone-2-carboxaldehyde protein quantitation kit (Thermo Fisher Scientific). A subsequent protein quantification method using HPLC-MS/MS applied in this study was adapted on the basis of techniques developed at Tohoku University (Miyagi, Japan). In brief, microsomal proteins solubilized in MPEX-PTS reagent were subjected to sequential proteolytic digestion with sequencing-grade l-(tosylamido-2-phenyl)ethylchloromethyl ketone-treated trypsin (Promega) and lysyl endopeptidase (FUJIFILM Wako Pure Chemical Industries) after reduction and alkylation. Digested samples were spiked with SIL peptides (Proteomedix Frontiers, Miyagi, Japan) as internal standards (ISs) prior to liquid-liquid extraction to remove the detergents in MPEX-PTS reagent. Recovered peptides from the aqueous phase were desalted using GL-Tips GC (GL Sciences), and the dried residue was dissolved in 0.1% formic acid. Finally, surrogate peptides of CYP3A4 and P-gp were simultaneously detected by multiplexed selected reaction monitoring (SRM)/MRM analysis using HPLC-MS/MS. HPLC-MS/MS analysis was performed with a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an Ultimate 3000 (Dionex, Amsterdam, the Netherlands) nano-LC system consisting of a loading column (Acclaim PepMap 100; Thermo Fisher Scientific) and an analytical nano-HPLC column (NTCC-360/100; Nikkyo Technos, Tokyo, Japan). The mobile phase was delivered at a flow rate of 0.5 μL/min using a gradient elution profile consisting of solvent A (0.1% formic acid) and solvent B (95% acetonitrile with 0.1% formic acid). The initial composition of the binary solvent was 4% solvent B. Solvent B was increased from 4% to 30% from 7 to 35 min, and from 30% to 95% from 35 to 36 min. The composition of solvent remained for 4 min at 95% solvent B. The MRM transition (m/z of precursor ion/m/z of product ion) of CYP3A4 was 684.9/242.1 and 684.9/179.6. The MRM transition of P-gp was 455.7/243.2 and 455.7/138.1. Optimized parameters for SRM/MRM analysis of target peptides were applied. Each target peptide level was quantified based on sample peaks identified at the same retention time as the corresponding SIL peptides, and calibration curves were generated for every transition. Concentrations were calculated as the mean of quantitative values derived from two MRM transitions.
DNA microarray
Human adult duodenal biopsies (five-donor pool, excluding the material used to make the organoids), human duodenal organoids, the human duodenal organoid-derived monolayer, and Caco-2 cells were subjected to DNA microarray analysis. Total RNA was isolated using an RNeasy mini kit (74106; QIAGEN, Nordrhein-Westfalen, Germany). Then, cRNA amplifying, labeling, hybridizing, and analyzing were performed at Takara Bio (Shiga, Japan) using a SurePrint G3 Human Gene Expression 8×60K v3 microarray (G4851C; Agilent Technologies, CA, USA). The Gene Expression Omnibus (GEO) accession number for the microarray analysis is GEO: GSE160695. The quality of the obtained data was checked using the values “gSigEval,” which shows the results of the signal evaluation (2 for transcripts detected, 1 for transcripts detected at a low level, 0 for transcripts that were not detected), and signals considered to be noise (gSigEval of 1 or 0) were not used for further analysis.
GSEA was performed with GSEA software (v4.0.3; UC San Diego, CA, USA and Broad Institute, MA, USA; https://www.gsea-msigdb.org/gsea/index.jsp) using all signals obtained from the DNA microarray in the human duodenal organoid-derived monolayer and Caco-2 cells. The pre-defined MSigDB gene set “KEGG subset of CP” was used to determine which pathway is involved.44,45
For hierarchical clustering and GO enrichment analysis, we used the maximum/minimum (Max/Min) method to extract 1,000 genes with the largest expression change. Hierarchical clustering was performed with freely available software, that is, TM4 MeV Stand-Alone Client (v4.9.0; Dana-Farber Cancer Institute, MA, USA, http://mev.tm4.org). The parameters for “linkage method selection” and “distance metric selection” were set to “average linkage clustering” and “Euclidean distance,” respectively.46 GO enrichment analysis of the significantly changed genes was conducted using the DAVID tool (v6.8; LHRI, MD, USA; https://david.ncifcrf.gov/home.jsp). The annotation categories “GOTERM_BP_DIRECT,” “GOTERM_CC_DIRECT,” and “GOTERM_MF_DIRECT” were used as inputs.47,48 For basic statistical analysis and illustrations, Microsoft Excel (v2019; Microsoft, WA, USA), GraphPad Prism (v5; GraphPad, CA, USA), and PlotsOfData (https://huygens.science.uva.nl/PlotsOfData/) were used.49
Acknowledgments
We would like to thank Junko Tanba and Hideyuki Koide of the Department of Gastroenterology and Hepatology, School of Medicine, Sapporo Medical University for excellent technical support. We wish to acknowledge Yuki Fukuda of the Laboratory of Applied Environmental Biology, Graduate School of Pharmaceutical Sciences, Osaka University for valuable cooperation in our experiments. We also thank Dr. Kazuo Takayama of the Center for iPS Cell Research and Application, Kyoto University for helpful discussions. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant nos. 20K20381 and 19J21382) and by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from Japan Agency for Medical Research and Development (AMED, grant no. JP21am0101084). T.Y. is supported by a Grant-in-Aid for JSPS Fellows.
Author contributions
The author contributions are presented using the CRediT taxonomy.50 Conceptualization, W.K., H.N., and H.M.; methodology, T.Y., K.K., G.M., M.T., Y.C., S.R., and W.K.; validation; T.Y., T.I., K.K., G.M., M.T., D.H., J.Y., R.N., K.I., Y.C., S.R., and K.H.; formal analysis, T.Y.; investigation, T.Y. and K.K.; resources, K.K., D.H., W.K., H.N., and H.M.; data curation, T.Y.; writing – original draft, T.Y. and H.M.; writing – review & editing, T.Y., T.I., K.K., G.M., M.T., D.H., J.Y., R.N., K.I., K.H., W.K., H.N., and H.M.; visualization, T.Y.; supervision, W.K., H.N., and H.M.; project administration, W.K., H.N., and H.M.; funding acquisition, W.K., H.N., and H.M.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.05.005.
Supplemental information
References
- 1.Shen D.D., Kunze K.L., Thummel K.E. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Adv. Drug Deliv. Rev. 1997;27:99–127. doi: 10.1016/s0169-409x(97)00039-2. [DOI] [PubMed] [Google Scholar]
- 2.Lin J.H., Chiba M., Baillie T.A. Is the role of the small intestine in first-pass metabolism overemphasized? Pharmacol. Rev. 1999;51:135–158. [PubMed] [Google Scholar]
- 3.Kato M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab. Pharmacokinet. 2008;23:87–94. doi: 10.2133/dmpk.23.87. [DOI] [PubMed] [Google Scholar]
- 4.Martignoni M., Groothuis G.M.M., de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006;2:875–894. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 5.Sun H., Chow E.C., Liu S., Du Y., Pang K.S. The Caco-2 cell monolayer: Usefulness and limitations. Expert Opin. Drug Metab. Toxicol. 2008;4:395–411. doi: 10.1517/17425255.4.4.395. [DOI] [PubMed] [Google Scholar]
- 6.Balimane P.V., Chong S. Cell culture-based models for intestinal permeability: A critique. Drug Discov. Today. 2005;10:335–343. doi: 10.1016/S1359-6446(04)03354-9. [DOI] [PubMed] [Google Scholar]
- 7.Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E., van Es J.H., Abo A., Kujala P., Peters P.J., Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 8.Sato T., Stange D.E., Ferrante M., Vries R.G.J., Van Es J.H., Van den Brink S., Van Houdt W.J., Pronk A., Van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 9.Sato T., van Es J.H., Snippert H.J., Stange D.E., Vries R.G., van den Born M., Barker N., Shroyer N.F., van de Wetering M., Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buczacki S.J.A., Zecchini H.I., Nicholson A.M., Russell R., Vermeulen L., Kemp R., Winton D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 11.VanDussen K.L., Carulli A.J., Keeley T.M., Patel S.R., Puthoff B.J., Magness S.T., Tran I.T., Maillard I., Siebel C., Kolterud Å. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matano M., Date S., Shimokawa M., Takano A., Fujii M., Ohta Y., Watanabe T., Kanai T., Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 13.Kakiuchi N., Yoshida K., Uchino M., Kihara T., Akaki K., Inoue Y., Kawada K., Nagayama S., Yokoyama A., Yamamoto S. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature. 2020;577:260–265. doi: 10.1038/s41586-019-1856-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., DiSalvo M., Gunasekara D.B., Dutton J., Proctor A., Lebhar M.S., Williamson I.A., Speer J., Howard R.L., Smiddy N.M. Self-renewing monolayer of primary colonic or rectal epithelial cells. Cell. Mol. Gastroenterol. Hepatol. 2017;4:165–182.e7. doi: 10.1016/j.jcmgh.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roodsant T., Navis M., Aknouch I., Renes I.B., van Elburg R.M., Pajkrt D., Wolthers K.C., Schultsz C., van der Ark K.C.H., Sridhar A., Muncan V. A human 2D primary organoid-derived epithelial monolayer model to study host-pathogen interaction in the small intestine. Front. Cell. Infect. Microbiol. 2020;10:272. doi: 10.3389/fcimb.2020.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyoshi H., Stappenbeck T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanDussen K.L., Marinshaw J.M., Shaikh N., Miyoshi H., Moon C., Tarr P.I., Ciorba M.A., Stappenbeck T.S. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64:911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugimoto S., Sato T. Establishment of 3D intestinal organoid cultures from intestinal stem cells. Methods Mol. Biol. 2017;1612:97–105. doi: 10.1007/978-1-4939-7021-6_7. [DOI] [PubMed] [Google Scholar]
- 19.Paine M.F., Hart H.L., Ludington S.S., Haining R.L., Rettie A.E., Zeldin D.C. The human intestinal cytochrome P450 “pie”. Drug Metab. Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai T., Imoto M., Sakamoto H., Hashimoto M. Identification of esterases expressed in Caco-2 cells and effects of their hydrolyzing activity in predicting human intestinal absorption. Drug Metab. Dispos. 2005;33:1185–1190. doi: 10.1124/dmd.105.004226. [DOI] [PubMed] [Google Scholar]
- 21.Imai T., Taketani M., Shii M., Hosokawa M., Chiba K. Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug Metab. Dispos. 2006;34:1734–1741. doi: 10.1124/dmd.106.009381. [DOI] [PubMed] [Google Scholar]
- 22.Ishiguro N., Kishimoto W., Volz A., Ludwig-Schwellinger E., Ebner T., Schaefer O. Impact of endogenous esterase activity on in vitro P-glycoprotein profiling of dabigatran etexilate in Caco-2 monolayers. Drug Metab. Dispos. 2014;42:250–256. doi: 10.1124/dmd.113.053561. [DOI] [PubMed] [Google Scholar]
- 23.Cui Y., Claus S., Schnell D., Runge F., MacLean C. In-depth characterization of EpiIntestinal microtissue as a model for intestinal drug absorption and metabolism in human. Pharmaceutics. 2020;12:405. doi: 10.3390/pharmaceutics12050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dekkers J.F., Wiegerinck C.L., de Jonge H.R., Bronsveld I., Janssens H.M., de Winter-de Groot K.M., Brandsma A.M., de Jong N.W.M.M., Bijvelds M.J.C.C., Scholte B.J. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013;19:939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 25.Kozuka K., He Y., Koo-McCoy S., Kumaraswamy P., Nie B., Shaw K., Chan P., Leadbetter M., He L., Lewis J.G. Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Reports. 2017;9:1976–1990. doi: 10.1016/j.stemcr.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasendra M., Tovaglieri A., Sontheimer-Phelps A., Jalili-Firoozinezhad S., Bein A., Chalkiadaki A., Scholl W., Zhang C., Rickner H., Richmond C.A. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018;8:2871. doi: 10.1038/s41598-018-21201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasendra M., Luc R., Yin J., Manatakis D.V., Kulkarni G., Lucchesi C., Sliz J., Apostolou A., Sunuwar L., Obrigewitch J. Duodenum Intestine-Chip for preclinical drug assessment in a human relevant model. eLife. 2020;9:e50135. doi: 10.7554/eLife.50135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spence J.R., Mayhew C.N., Rankin S.A., Kuhar M.F., Vallance J.E., Tolle K., Hoskins E.E., Kalinichenko V.V., Wells S.I., Zorn A.M. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCracken K.W., Howell J.C., Wells J.M., Spence J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011;6:1920–1928. doi: 10.1038/nprot.2011.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onozato D., Yamashita M., Nakanishi A., Akagawa T., Kida Y., Ogawa I., Hashita T., Iwao T., Matsunaga T. Generation of intestinal organoids suitable for pharmacokinetic studies from human induced pluripotent stem cells. Drug Metab. Dispos. 2018;46:1572–1580. doi: 10.1124/dmd.118.080374. [DOI] [PubMed] [Google Scholar]
- 31.Akazawa T., Yoshida S., Ohnishi S., Kanazu T., Kawai M., Takahashi K. Application of intestinal epithelial cells differentiated from human induced pluripotent stem cells for studies of prodrug hydrolysis and drug absorption in the small intestine. Drug Metab. Dispos. 2018;46:1497–1506. doi: 10.1124/dmd.118.083246. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida S., Miwa H., Kawachi T., Kume S., Takahashi K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci. Rep. 2020;10:5989. doi: 10.1038/s41598-020-63151-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerbe F., Sidot E., Smyth D.J., Ohmoto M., Matsumoto I., Dardalhon V., Cesses P., Garnier L., Pouzolles M., Brulin B. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529:226–230. doi: 10.1038/nature16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson S.S., Tocchi A., Holly M.K., Parks W.C., Smith J.G. A small intestinal organoid model of non-invasive enteric pathogen-epithelial cell interactions. Mucosal Immunol. 2015;8:352–361. doi: 10.1038/mi.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J., Li C., Liu X., Chiu M.C., Zhao X., Wang D., Wei Y., Lee A., Zhang A.J., Chu H. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020;26:1077–1083. doi: 10.1038/s41591-020-0912-6. [DOI] [PubMed] [Google Scholar]
- 36.Gjorevski N., Sachs N., Manfrin A., Giger S., Bragina M.E., Ordóñez-Morán P., Clevers H., Lutolf M.P. Designer matrices for intestinal stem cell and organoid culture. Nature. 2016;539:560–564. doi: 10.1038/nature20168. [DOI] [PubMed] [Google Scholar]
- 37.Fujii M., Matano M., Toshimitsu K., Takano A., Mikami Y., Nishikori S., Sugimoto S., Sato T. Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell. 2018;23:787–793.e6. doi: 10.1016/j.stem.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 38.Jung P., Sato T., Merlos-Suárez A., Barriga F.M., Iglesias M., Rossell D., Auer H., Gallardo M., Blasco M.A., Sancho E. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 39.Kishimoto W., Ishiguro N., Ludwig-Schwellinger E., Ebner T., Schaefer O. In vitro predictability of drug-drug interaction likelihood of P-glycoprotein-mediated efflux of dabigatran etexilate based on [I]2/IC50 threshold. Drug Metab. Dispos. 2014;42:257–263. doi: 10.1124/dmd.113.053769. [DOI] [PubMed] [Google Scholar]
- 40.Wang X., Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spandidos A., Wang X., Wang H., Dragnev S., Thurber T., Seed B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics. 2008;9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spandidos A., Wang X., Wang H., Seed B. PrimerBank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaefer M., Morinaga G., Matsui A., Schänzle G., Bischoff D., Süssmuth R.D. Quantitative expression of hepatobiliary transporters and functional uptake of substrates in hepatic two-dimensional sandwich cultures: A comparative evaluation of upcyte and primary human hepatocytes. Drug Metab. Dispos. 2018;46:166–177. doi: 10.1124/dmd.117.078238. [DOI] [PubMed] [Google Scholar]
- 44.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 45.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saeed A.I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 47.Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 49.Postma M., Goedhart J. PlotsOfData—A web app for visualizing data together with their summaries. PLoS Biol. 2019;17:e3000202. doi: 10.1371/journal.pbio.3000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand A., Allen L., Altman M., Hlava M., Scott J. Beyond authorship: Attribution, contribution, collaboration, and credit. Learn. Publ. 2015;28:151–155. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.