

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by airway inflammation, small airway remodeling, and emphysema. Airway remodeling in patients with COPD involves both the airway epithelium and the subepithelial extracellular matrix (ECM). However, it is currently unknown how epithelial remodeling in COPD airways depends on the relative influence from inherent defects in the epithelial cells and alterations in the ECM. To address this, we analyzed global gene expression in COPD human bronchial epithelial cells (HBEC) and normal HBEC after repopulation on decellularized bronchial scaffolds derived from patients with COPD or donors without COPD. COPD HBEC grown on bronchial scaffolds showed an impaired ability to initiate ciliated-cell differentiation, which was evident on all scaffolds regardless of their origin. In addition, although normal HBEC were less affected by the disease state of the bronchial scaffolds, COPD HBEC showed a gene expression pattern indicating increased proliferation and a retained basal-cell phenotype when grown on COPD bronchial scaffolds compared with normal bronchial scaffolds. By using mass spectrometry, we identified 13 matrisome proteins as being differentially abundant between COPD bronchial scaffolds and normal bronchial scaffolds. These observations are consistent with COPD pathology and suggest that both epithelial cells and the ECM contribute to epithelial-cell remodeling in COPD airways.
Keywords: chronic obstructive pulmonary disease, extracellular matrix, airway, epithelium, remodeling
Clinical Relevance
We provide novel insight into how epithelial cells and the extracellular matrix contribute in different ways to central airway remodeling in chronic obstructive pulmonary disease. A better understanding of the pathological processes behind this remodeling has the potential to benefit the search for pathways and targets that can be harnessed for the development of novel chronic obstructive pulmonary disease therapies.
Chronic obstructive pulmonary disease (COPD) is characterized by chronic airway inflammation, small airway obstruction, and destruction of the lung parenchyma (1). Narrowing of airways and loss of lung tissue architecture lead to progressive airflow limitation and decreased lung function (2). Persistent exposure to cigarette smoke and other inhaled noxious agents enhances the risk of developing COPD and causes damage to the central airway epithelium, thereby compromising its function as a protective barrier against the external environment. Although the normal central airway epithelium has an inherent capacity to regenerate after injury, that process is impaired in patients with COPD (3, 4) and can lead to epithelial remodeling, including goblet-cell hyperplasia (5), squamous metaplasia (6), and decreased epithelial barrier function (7). Shortening of cilia (8) and decreased ciliary beating (9) have also been observed in the airways of patients with COPD. The damage and subsequent remodeling of the airway epithelium results in dysfunctional mucociliary clearance and leads to increased susceptibility to respiratory infections in patients with COPD (2).
The extracellular matrix (ECM) is a complex network of proteins and carbohydrates that provide organs with structural support, like tensile strength and elasticity. However, the ECM can also regulate cell function by harboring soluble mediators, like cytokines and growth factors (10–12), and mechanical properties of the ECM, like stiffness, have been suggested to dictate cell behavior (13). ECM remodeling in COPD airways has been reported in several studies (14–18), and we have previously shown that the bronchial ECM from patients with COPD modulates gene expression in normal human bronchial epithelial cells (HBEC) early during differentiation (19). However, it is not known how the impaired epithelial-cell function in COPD depends on the relative influence from pathological changes in the epithelial cells themselves and the underlying ECM. We hypothesized that HBEC derived from patients with COPD would display an aberrant phenotype when grown on a bronchial ECM and aimed to identify cellular functions affected by the disease state of the epithelial cells or the ECM. We therefore performed global transcriptomic profiling in COPD HBEC and normal HBEC after repopulation on decellularized bronchial scaffolds derived from patients with COPD and donors without COPD (19). In addition, the matrisome (20) composition of the bronchial scaffolds was characterized by using a proteomic approach based on mass spectrometry.
Here, we present data showing that gene expression in COPD airways depends on the disease states of both the epithelial cells and the ECM but that the two components affect gene expression in different ways. COPD HBEC showed an impaired ability to initiate ciliated-cell differentiation, which was induced by the bronchial scaffolds regardless of their origin. In addition, the gene expression pattern in COPD HBEC indicated increased proliferation and a retained basal-cell phenotype after repopulation on COPD bronchial scaffolds compared with normal bronchial scaffolds, suggesting that the proliferative capacity of epithelial cells in COPD airways is influenced by the ECM microenvironment. Portions of this article were included in the doctoral dissertation of the first author (21).
Methods
Tissue Acquisition
Lungs from patients with severe COPD (GOLD [Global Initiative for Chronic Obstructive Lung Disease] stage 4) and from organ donors without lung cancer or any chronic respiratory disease were acquired from Sahlgrenska University Hospital in Gothenburg, Sweden, and Skåne University Hospital in Lund, Sweden. All COPD samples were isolated from whole lungs of patients with COPD who had undergone transplantation. All non-COPD samples came from donated whole lungs deemed unsuitable for transplantation and are referred to as “normal” in the article. These studies were approved by the Swedish research ethical committees in Gothenburg and Lund. Informed consent was obtained from all subjects or their closest relatives. See the data supplement for more details.
Decellularization and Repopulation
Bronchial airways (second to fourth segment) were dissected and decellularized as described in our previous study, in which we demonstrated that decellularization preserved tissue morphology and major ECM components, whereas DNA content decreased drastically (19). Repopulation was performed as previously described (19), except the cells were in PneumaCult-Ex medium (Stemcell Technologies catalog number 05008) when seeded on the bronchial scaffolds and were in PneumaCult-ALI medium (Stemcell Technologies catalog number 05001) during the differentiation phase. HBEC and bronchial scaffolds were derived from different sets of patients with COPD and donors without COPD. The experimental design is described in detail in Figure 1 and in Table E1 in the data supplement. Differentiation was induced by switching to PneumaCult-ALI medium 4 days after addition of HBEC to the scaffolds, and this time point was defined as Day 0. The HBEC were also seeded in Transwell plates coated with bovine collagen I (Advanced Biomatrix catalog number 5005) for culture at the air–liquid interface (ALI). On Day 0, the apical medium was removed, and the basolateral PneumaCult-Ex medium was exchanged for the PneumaCult-ALI medium. Repopulated scaffolds for RNA sequencing (RNA-Seq) and protein extraction were collected and frozen in liquid nitrogen, which was followed by storage at −80°C. Repopulated scaffolds for histology and immunofluorescence were fixed in 4% formaldehyde for 20–24 hours at room temperature. ALI cultures were lysed in RLT buffer (Qiagen catalog number 79216), which was followed by freezing in liquid nitrogen and storage at −80°C.
Figure 1.

(A) The RNA-sequencing study followed an experimental design in which human bronchial epithelial cells (HBEC) from three patients with chronic obstructive pulmonary disease (COPD) and three donors without COPD were repopulated on bronchial scaffolds derived from a different set of three patients with COPD and three donors without COPD, resulting in four combinations of HBEC and bronchial scaffolds: COPD HBEC/COPD scaffolds, COPD HBEC/normal scaffolds, normal HBEC/COPD scaffolds, and normal HBEC/normal scaffolds (n = 9 repopulated scaffolds per combination and time point). The HBEC were also seeded on Transwell plates for culture at the air–liquid interface (ALI). Four days after repopulation, Day 0 samples were collected, and differentiation was induced for the remaining samples by switching to a differentiation medium. Day 7 samples were collected after 7 days of differentiation. The experimental design is further described in Table E1. (B) Before seeding of cells, the decellularized bronchial scaffolds were placed on sterile Whatman filters floating on medium in 6-well plates. The bronchial scaffold, medium, and Whatman filter are indicated by arrows. This setup allowed the cells to differentiate close to the ALI.
RNA-Seq
Repopulated bronchial scaffolds were disrupted and homogenized in QIAzol lysis reagent (Qiagen catalog number 79306) using a TissueLyser II bead mill (Qiagen). Phenol- and chloroform-based RNA extraction was performed by using the RNeasy 96 kit (Qiagen catalog number 74182), which was also used for ALI samples, including on-column DNase I digestion. RNA integrity was evaluated on the Fragment Analyzer Automated CE platform (Advanced Analytical Technologies), and quantification was performed by using the Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific catalog number R11490). RNA was diluted to 6 ng/μl and used as input to create cDNA libraries by using a TruSeq Stranded mRNA Library Preparation kit (Illumina catalog number RS-122-2103) with dual indexing and by following the manufacturer’s instructions. Libraries were validated on the Fragment Analyzer Automated CE platform (Advanced Analytical Technologies) by using the standard sensitivity NGS (Next Generation Sequencing) fragment analysis kit. Sample libraries were pooled in equimolar concentrations and diluted and denatured according to Illumina guidelines. RNA-Seq was performed by using a high-output 1 × 76–bp kit on an Illumina NextSeq 500 platform.
Results
Bronchial Scaffolds Enhance Gene Expression Differences between COPD and Normal HBEC
Immunofluorescence demonstrated that the HBEC used for repopulation predominantly consisted of cells positive for the basal-cell marker TP63 (tumor protein p63) (see Figure E1 in the data supplement). Histology confirmed that COPD HBEC could repopulate both COPD bronchial scaffolds and normal bronchial scaffolds (Figure E2), and the morphology and distribution of cells were similar to those in our previously reported results for normal HBEC when using the same repopulation model (19). No difference in cell confluency was observed between COPD bronchial scaffolds and normal bronchial scaffolds. In the earlier study, this ex vivo model was shown to support differentiation of normal HBEC on COPD bronchial scaffolds and normal bronchial scaffolds for up to 35 days, and the strongest differential effects on gene expression were seen during early time points. On the basis of these findings, 0 days and 7 days were chosen as time points in the present study, which was designed to allow for exploration of the relative influence from both HBEC and bronchial scaffolds on global gene expression in COPD airways (Figure 1 and Table E1).
Principal component analysis (PCA) revealed that the RNA-Seq samples were clustered primarily on the basis of the differentiation time and on the basis of whether the HBEC had grown on bronchial scaffolds or at the ALI (Figure E3A). A large number of genes were differentially expressed between Day 0 and Day 7 (Figure E3B and Table E2), confirming an overall change in gene expression upon differentiation induction. Moreover, pronounced differences in gene expression were seen between HBEC grown on bronchial scaffolds and HBEC grown at the ALI (Figure E3C and Table E3), indicating a significant influence from the bronchial scaffolds. PCA performed separately on Day 0 and Day 7 samples showed that COPD and normal HBEC samples formed two different clusters at both time points (Figure 2A). Poor separation was seen between samples representing COPD HBEC and normal HBEC grown at the ALI, but the separation between COPD HBEC and normal HBEC samples was more prominent in the bronchial scaffold group, indicating that the bronchial scaffolds contributed to enhanced gene expression differences between COPD HBEC and normal HBEC. This was confirmed by a substantially larger number of genes differentially expressed between COPD HBEC and normal HBEC on bronchial scaffolds compared with the ALI environment (Figures 2B and 2C and Table E4). PCA showed no separation between samples representing COPD scaffolds and normal bronchial scaffolds on Day 0 (Figures 3A and 3B). However, for COPD HBEC on Day 7, PCA did show a separation between COPD bronchial scaffold samples and normal bronchial scaffold samples, whereas this was not seen for normal HBEC at the same time point (Figures 3C and 3D). In agreement with this observation, 1,694 genes were found to be differentially expressed in COPD HBEC on COPD scaffolds compared with normal scaffolds on Day 7, whereas the corresponding number for normal HBEC was considerably lower (Figures 3E and 3F and Table E5), demonstrating a fundamental difference between COPD HBEC and normal HBEC in terms of how gene expression is modulated by the bronchial scaffolds over time.
Figure 2.
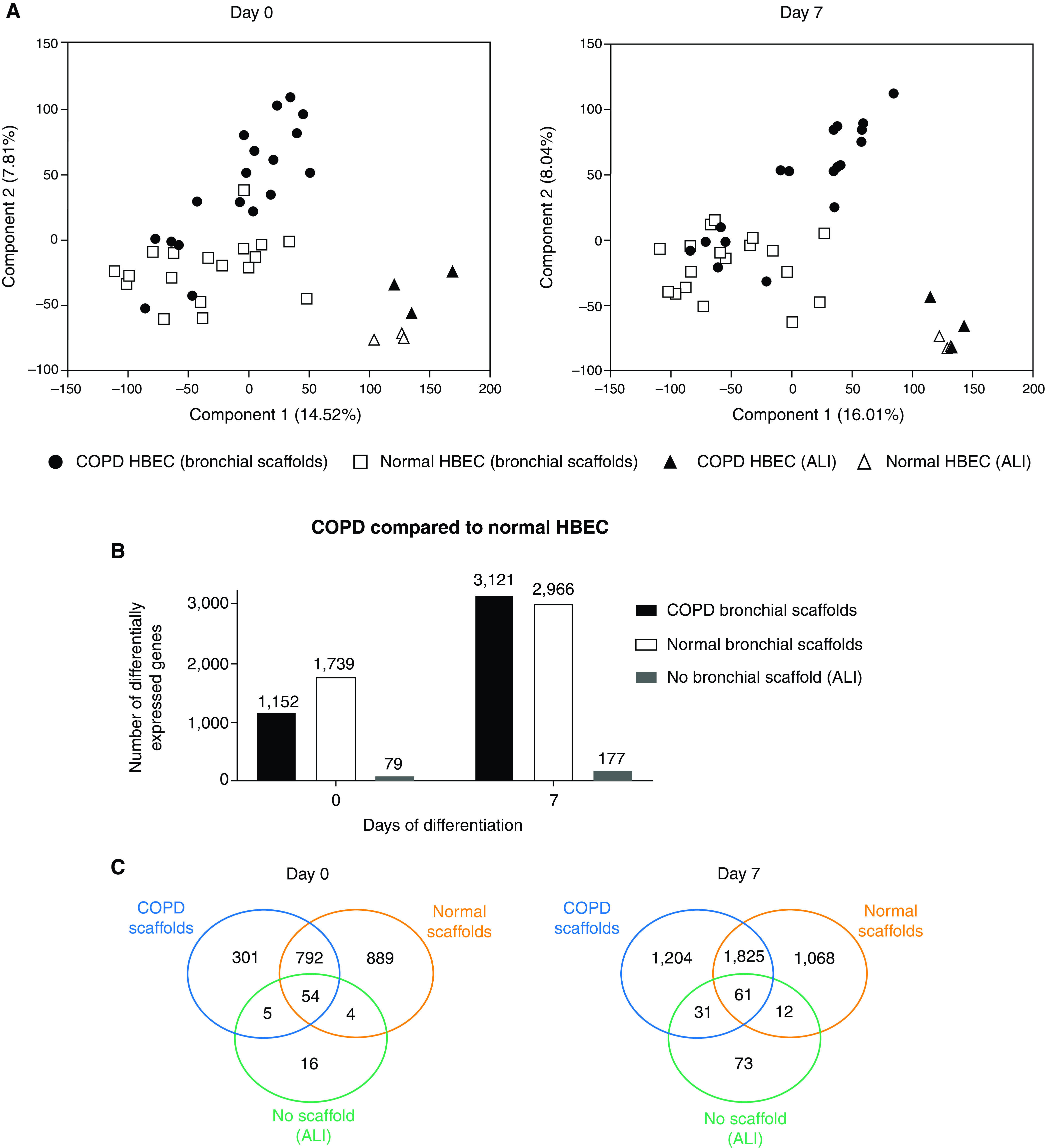
(A) Principal component analysis of RNA-sequencing data for samples representing 0 and 7 days of differentiation of COPD HBEC and normal HBEC repopulated on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination and time point) or grown at the ALI (n = 3 HBEC donors). Day 0 was defined as the day of differentiation induction, which was 4 days after the seeding of cells. (B) Number and (C) overlap of differentially expressed genes in COPD HBEC compared with normal HBEC when grown on COPD scaffolds, normal scaffolds, or at the ALI on Day 0 and Day 7. A gene was considered to be differentially expressed if the comparison resulted in a false discovery rate (FDR) < 0.05 using DESeq2 (see the data supplement) and the Benjamini-Hochberg method for multiple testing correction.
Figure 3.
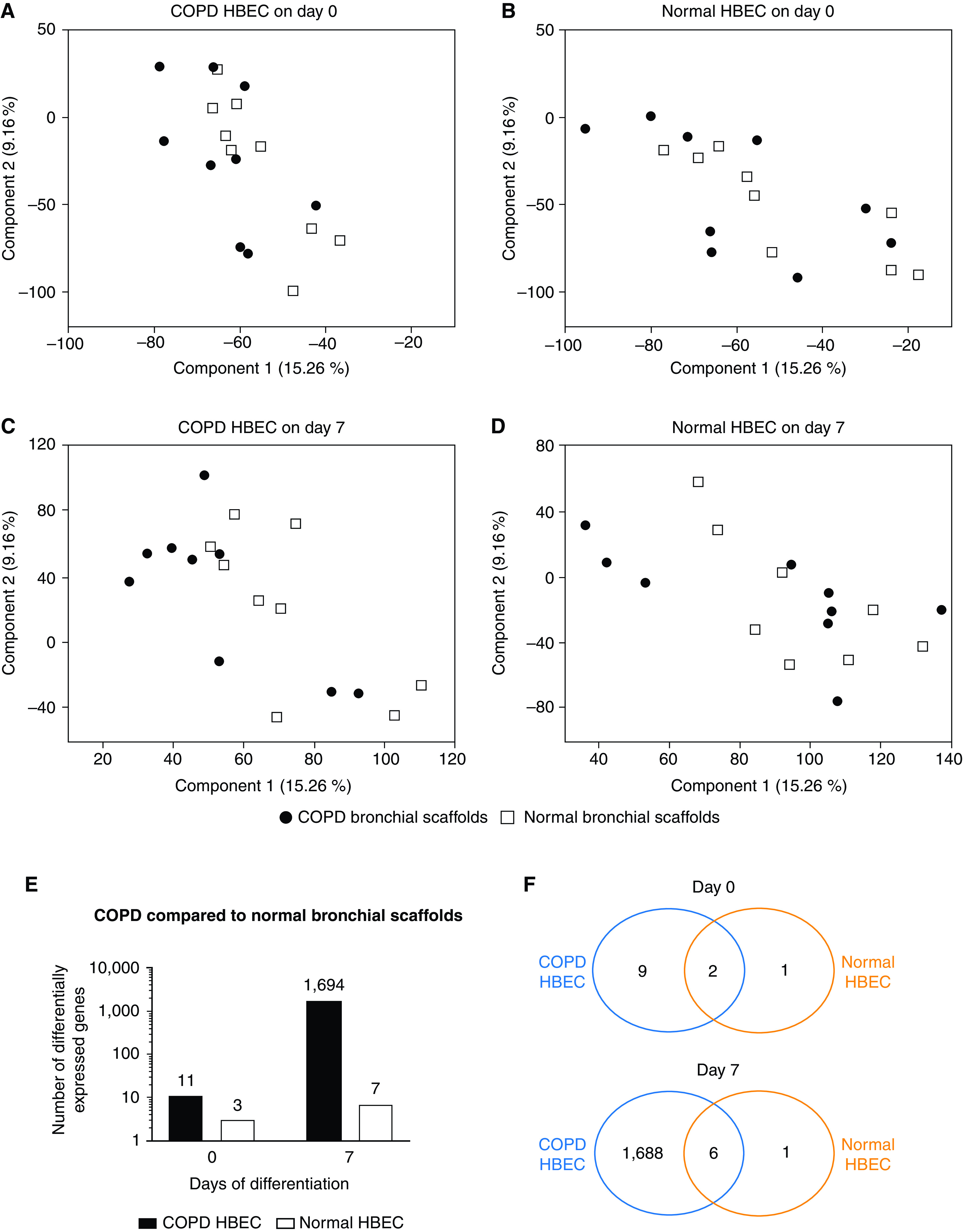
Principal component analysis of RNA-sequencing data for samples representing COPD HBEC and normal HBEC after (A and B) 0 days and (C and D) 7 days of differentiation on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination and time point). Day 0 was defined as the day of differentiation induction, which was 4 days after the seeding of cells. (E) Number and (F) overlap of genes differentially expressed in COPD HBEC and normal HBEC on COPD bronchial scaffolds compared with normal bronchial scaffolds. A gene was considered to be differentially expressed if the comparison resulted in an FDR < 0.05 using DESeq2 (see the data supplement) and the Benjamini-Hochberg method for multiple testing correction.
Differential Gene Expression Reveals Impaired Ciliary Development in COPD HBEC Grown on Bronchial Scaffolds
A Gene Ontology term enrichment search was performed for genes differentially expressed in COPD HBEC compared with normal HBEC on bronchial scaffolds on Day 0, showing enrichment for genes annotated with biological process terms such as “ECM organization” and “cell adhesion” as well as several terms related to development (Table E6). The temporal expression pattern of marker genes representing ciliated cells (FOXJ1 [forkhead box J1]) (22), goblet cells (MUC5AC), and basal cells (TP63 [tumor protein p63]) indicated successful differentiation induction (Figure 4A) and was consistent with our previous study using the model (19). Interestingly, the FOXJ1 expression on Day 7 was lower in COPD HBEC than in normal HBEC on bronchial scaffolds, regardless of scaffold origin, a finding that was also confirmed at the protein level (Figure 4B). TP63 expression was higher in COPD HBEC at the same time point, but no differences were seen for MUC5AC (Figure 4A). In agreement with the differential FOXJ1 expression, a Gene Ontology term enrichment search showed an overrepresentation of genes annotated with biological process terms related to ciliogenesis among the genes differentially expressed in COPD HBEC compared with normal HBEC on scaffolds on Day 7 (Tables 1 and 2). Numerous genes known to be involved in the development and assembly of cilia showed a consistent pattern of lower expression in COPD HBEC on both diseased and normal bronchial scaffolds, including ZMYND10 (zinc finger MYND type–containing 10) (23), DRC1 (dynein regulatory complex subunit 1) (24), DNAI2 (dynein axonemal intermediate chain 2) (25), ARMC4 (armadillo repeat–containing 4) (26), RSPH1 (radial spoke head component 1) (27), TMEM231 (transmembrane protein 231), B9D1 (B9 domain–containing 1), and CC2D2A (coiled-coil and C2 domain–containing 2A) (28) (Figure 5). Meanwhile, these differences were not seen in HBEC grown at the ALI, and none of the genes were induced between Day 0 and Day 7 in the absence of bronchial scaffolds. In contrast, all of the genes were induced over time in HBEC grown on bronchial scaffolds. Validation using real-time qRT-PCR confirmed the results (Figure E4). In summary, these results indicate that interactions with the bronchial scaffolds trigger an induction of ciliated-cell differentiation in HBEC, but COPD HBEC have an impaired ability to initiate this differentiation.
Figure 4.

(A) Expression of FOXJ1 (forkhead box J1) (ciliated-cell marker), MUC5AC (goblet-cell marker), and TP63 (tumor protein p63) (basal-cell marker) from RNA sequencing in COPD HBEC and normal HBEC repopulated on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination and time point) or grown at the ALI (n = 3 HBEC donors). Day 0 was defined as the day of differentiation induction, which was 4 days after the seeding of cells. FOXJ1 expression was higher in COPD HBEC than in normal HBEC on both COPD scaffolds and normal scaffolds on Day 0. FOXJ1 increased between Day 0 and Day 7 in HBEC repopulated on scaffolds (FDR < 0.001) but was not increased when HBEC were grown at the ALI. MUC5AC increased between Day 0 and Day 7 in HBEC repopulated on scaffolds and grown at the ALI (FDR < 0.001). TP63 decreased between Day 0 and Day 7 in HBEC repopulated on scaffolds and grown at the ALI (FDR < 0.001), except in COPD HBEC on COPD scaffolds. (B) Expression of FOXJ1 protein in COPD HBEC and normal HBEC after 7 days of differentiation on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination). FOXJ1 concentrations were normalized against GLB1 (β-galactosidase) to account for varying number of cells on the scaffolds. Means and SDs are indicated. *FDR < 0.05, **FDR < 0.01, and ***FDR < 0.001 using DESeq2 (see the data supplement) and the Benjamini-Hochberg method for multiple testing correction in A. *P < 0.05 and **P < 0.01 using the Mann-Whitney test in B.
Table 1.
GO Term Enrichment for Genes Differentially Expressed on Day 7 in COPD HBEC Compared with Normal HBEC after Repopulation on COPD Bronchial Scaffolds
| GO Biological Process | Fold Enrichment | FDR |
|---|---|---|
| Cilium organization (GO:0044782) | 2.7 | 1.4 × 10−16 |
| Cilium assembly (GO:0060271) | 2.6 | 5.6 × 10−15 |
| Plasma membrane–bounded cell projection assembly (GO:0120031) | 2.3 | 2.4 × 10−13 |
| Cell projection assembly (GO:0030031) | 2.3 | 3.6 × 10−13 |
| Cell projection organization (GO:0030030) | 1.6 | 2.1 × 10−7 |
| Microtubule-based process (GO:0007017) | 1.8 | 2.8 × 10−7 |
| Microtubule bundle formation (GO:0001578) | 3.5 | 3.5 × 10−7 |
| Plasma membrane–bounded cell projection organization (GO:0120036) | 1.6 | 6.4 × 10−7 |
| Organelle assembly (GO:0070925) | 1.7 | 7.5 × 10−7 |
| Axoneme assembly (GO:0035082) | 4.1 | 8.1 × 10−7 |
Definition of abbreviations: COPD = chronic obstructive pulmonary disease; FDR = false discovery rate; GO = Gene Ontology; HBEC = human bronchial epithelial cells; RNA-Seq = RNA sequencing.
Shown are the GO biological process terms for which there is an overrepresentation among the differentially expressed genes relative to the reference list, which consisted of all GO-annotated genes that were detected in at least one RNA-Seq sample. Shown are the 10 terms with the lowest FDR for each analysis.
Table 2.
GO Term Enrichment for Genes Differentially Expressed on Day 7 in COPD HBEC Compared with Normal HBEC after Repopulation on Normal Bronchial Scaffolds
| GO Biological Process | Fold Enrichment | FDR |
|---|---|---|
| Cilium organization (GO:0044782) | 2.6 | 5.7 × 10−15 |
| Cilium assembly (GO:0060271) | 2.6 | 7.2 × 10−14 |
| Plasma membrane–bounded cell projection assembly (GO:0120031) | 2.3 | 2.1 × 10−12 |
| Cell projection assembly (GO:0030031) | 2.3 | 3.0 × 10−12 |
| Cell projection organization (GO:0030030) | 1.7 | 3.2 × 10−11 |
| Plasma membrane–bounded cell projection organization (GO:0120036) | 1.7 | 1.6 × 10−10 |
| Microtubule bundle formation (GO:0001578) | 3.9 | 3.7 × 10−9 |
| Microtubule-based process (GO:0007017) | 1.8 | 1.0 × 10−8 |
| Axoneme assembly (GO:0035082) | 4.5 | 2.9 × 10−8 |
| Cilium movement (GO:0003341) | 4.5 | 5.7 × 10−8 |
Shown are the GO biological process terms for which there is an overrepresentation among the differentially expressed genes relative to the reference list, which consisted of all GO-annotated genes that were detected in at least one RNA-Seq sample. Shown are the 10 terms with the lowest FDR for each analysis.
Figure 5.
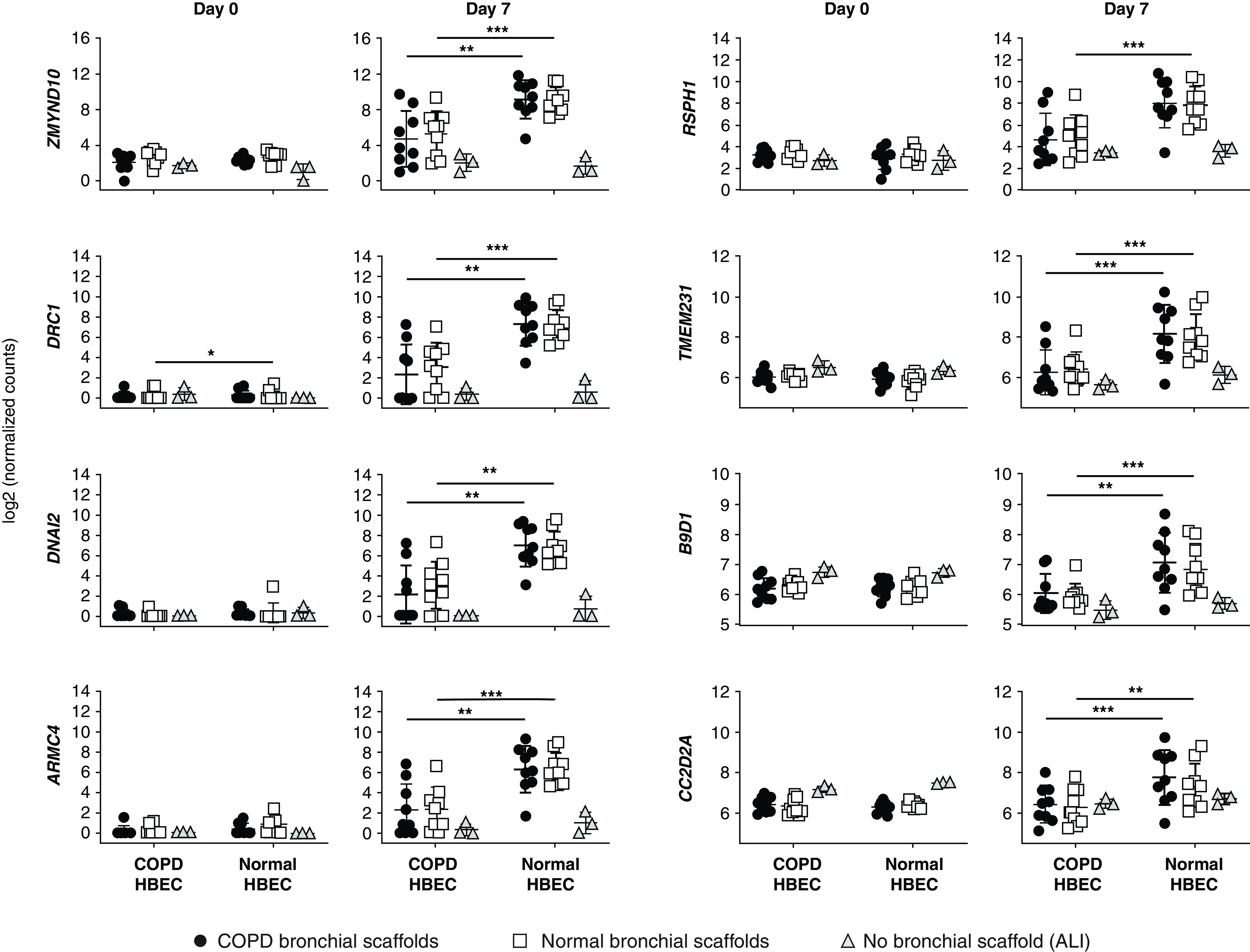
RNA-sequencing data for genes involved in ciliary development and assembly in COPD HBEC and normal HBEC after 0 and 7 days of differentiation on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination and time point) or at the ALI (n = 3 HBEC donors). Day 0 was defined as the day of differentiation induction, which was 4 days after the seeding of cells. DRC1 (dynein regulatory complex subunit 1) expression was higher in COPD HBEC than in normal HBEC on normal scaffolds on Day 0. No induction was seen between Day 0 and Day 7 for any of the genes in HBEC grown at the ALI, whereas all genes increased significantly over time in normal HBEC grown on bronchial scaffolds. In COPD HBEC grown on bronchial scaffolds, the following genes increased between Day 0 and Day 7: ZMYND10, ARMC4, and RSPH1 (both scaffold types) and DRC1, DNAI2, and TMEM231 (normal scaffolds only). Means and SDs are indicated. *FDR < 0.05, **FDR < 0.01, and ***FDR < 0.001 for COPD HBEC compared with normal HBEC grown on bronchial scaffolds using DESeq2 (see the data supplement) and the Benjamini-Hochberg method for multiple testing correction. ARMC4 = armadillo repeat–containing 4; B9D1 = B9 domain–containing 1; CC2D2A = coiled-coil and C2 domain–containing 2A; DNAI2 = dynein axonemal intermediate chain 2; RSPH1 = radial spoke head component 1; TMEM231 = transmembrane protein 231; ZMYND10 = zinc finger MYND type–containing 10.
COPD HBEC Gene Expression Indicates Dysregulated Cell Cycle Progression Induced by the Bronchial Scaffolds
A Gene Ontology term enrichment search was performed for the 1,694 genes differentially expressed in COPD HBEC on COPD bronchial scaffolds compared with normal bronchial scaffolds on Day 7. Enrichment was seen for genes annotated with biological process terms such as “chromosome segregation,” “cell proliferation,” “DNA replication,” and “regulation of cell cycle” (Table 3). Moreover, the gene expression pattern indicated that several mediators known to promote cell growth or cell cycle progression were predicted to have increased activity in COPD HBEC on COPD scaffolds compared with normal scaffolds (Table 4). These mediators included MYC (MYC protooncogene, bHLH transcription factor) (29), ERBB2 (Erb-b2 receptor tyrosine kinase 2) (30), and the E2F transcription factors (31). Meanwhile, proteins that negatively regulate cell cycle progression were predicted to have decreased activity on COPD scaffolds, and these included proteins such as p53 (32), CDKN1A (cyclin-dependent kinase inhibitor 1A [p21]) (33), CDKN2A (cyclin-dependent kinase inhibitor 2A [p16-INK4A/p14-ARF]) (34), and RB1 (retinoblastoma protein transcriptional corepressor 1) (35) (Table 4). Several genes known to promote cell cycle progression or cell division had a higher expression level on COPD scaffolds on Day 7, including MKI67 (36), E2F2 (31), TTK (TTK protein kinase) (37), MCM10 (minichromosome maintenance 10 replication initiation factor) (38), ANLN (anillin actin binding protein) (39), CCNB1 (cyclin B1) (40), CCNA2 (cyclin A2) (41), and CDK1 (cyclin-dependent kinase 1) (40), whereas CDKN1A and CDKN2A were both downregulated on COPD scaffolds (Figure 6A). In addition, the basal-cell markers TP63, KRT5 (keratin 5), and KRT14 (keratin 14) (42) were all increased on COPD bronchial scaffolds relative to normal bronchial scaffolds in COPD HBEC at the same time point (Figure 6B). Results from validation with real-time qRT-PCR were largely consistent with the RNA-Seq data (Figure E5). Immunofluorescence confirmed expression of MKI67 and TP63 in COPD HBEC on COPD scaffolds and normal scaffolds on Day 7 (Figure E6), showing that TP63+ cells were predominantly present on the basolateral side of the repopulated epithelium. Finally, differential expression of genes associated with integrin signaling suggested that the cell–ECM interaction between COPD HBEC and the scaffolds depended on the scaffold disease state (Table E7). Taken together, these results indicate enhanced cell cycle progression and increased proliferation of COPD HBEC when cultured on COPD bronchial scaffolds compared with normal bronchial scaffolds.
Table 3.
GO Term Enrichment for Genes Differentially Expressed in COPD HBEC on COPD Bronchial Scaffolds Compared with Normal Bronchial Scaffolds after 7 Days of Differentiation
| GO-Slim Biological Process | Fold Enrichment | FDR |
|---|---|---|
| Chromosome segregation (GO:0007059) | 3.4 | 4.2 × 10−5 |
| DNA metabolic process (GO:0006259) | 2.1 | 8.9 × 10−5 |
| DNA replication (GO:0006260) | 2.7 | 1.2 × 10−4 |
| Regulation of cell cycle (GO:0051726) | 2.5 | 2.0 × 10−4 |
| Cellular process (GO:0009987) | 1.1 | 5.9 × 10−4 |
| Cell cycle (GO:0007049) | 1.6 | 6.5 × 10−4 |
| Metabolic process (GO:0008152) | 1.2 | 4.4 × 10−3 |
| Translation (GO:0006412) | 2.0 | 9.1 × 10−3 |
| Cell proliferation (GO:0008283) | 2.8 | 1.6 × 10−2 |
| DNA recombination (GO:0006310) | 3.1 | 1.6 × 10−2 |
| Biosynthetic process (GO:0009058) | 1.3 | 1.9 × 10−2 |
| Phosphate-containing compound metabolic process (GO:0006796) | 1.3 | 2.1 × 10−2 |
| DNA repair (GO:0006281) | 2.0 | 2.9 × 10−2 |
| Mitosis (GO:0007067) | 1.8 | 3.8 × 10−2 |
| Meiosis (GO:0007126) | 2.6 | 3.9 × 10−2 |
Shown are the GO-slim biological process terms for which there is an overrepresentation among the differentially expressed genes relative to the reference list, which consisted of all GO-annotated genes that were detected in at least one RNA-Seq sample. Shown are the 15 terms with the lowest FDR.
Table 4.
Upstream Mediators Predicted to Have a Changed Activity in COPD HBEC on COPD Bronchial Scaffolds Compared with Normal Bronchial Scaffolds after 7 Days of Differentiation, Based on the Expression Pattern of Differentially Expressed Genes
| Upstream Mediator | Predicted Activity | Activation Z-Score | P Value |
|---|---|---|---|
| MYC | Increased | 7.8 | 4.4 × 10−34 |
| MYCN | Increased | 7.3 | 1.3 × 10−25 |
| ESR1 | Increased | 6.4 | 5.1 × 10−27 |
| RABL6 | Increased | 5.8 | 2.0 × 10−26 |
| E2F family | Increased | 4.3 | 1.8 × 10−26 |
| β-Estradiol | Increased | 4.1 | 4.5 × 10−32 |
| HGF | Increased | 3.9 | 1.1 × 10−32 |
| E2F1 | Increased | 3.8 | 2.3 × 10−28 |
| VEGF family | Increased | 3.7 | 3.0 × 10−25 |
| E2F3 | Increased | 3.4 | 3.8 × 10−26 |
| ERBB2 | Increased | 2.6 | 1.0 × 10−56 |
| FOXO3 | Decreased | −2.3 | 5.4 × 10−23 |
| CDKN1A | Decreased | −3.2 | 1.8 × 10−40 |
| RB1 | Decreased | −3.8 | 1.9 × 10−28 |
| MIRLET7I | Decreased | −4.6 | 1.9 × 10−27 |
| CDKN2A | Decreased | −5.8 | 3.0 × 10−32 |
| TP53 | Decreased | −6.0 | 1.3 × 10−68 |
| TNFα | Decreased | −6.3 | 1.0 × 10−25 |
| NUPR1 | Decreased | −7.2 | 5.2 × 10−29 |
| TGFB1 | Decreased | −8.1 | 5.8 × 10−46 |
Definition of abbreviations: CDKN1A = cyclin-dependent kinase inhibitor 1A (p21); CDKN2A = cyclin-dependent kinase inhibitor 2A (p16-INK4A/p14-ARF); COPD = chronic obstructive pulmonary disease; ERBB2 = Erb-b2 receptor tyrosine kinase 2; ESR1 = estrogen receptor 1; FOXO3 = forkhead box O3; HBEC = human bronchial epithelial cells; HGF = hepatocyte growth factor; MIRLET7I = microRNA let-7i (let-7); MYC = MYC protooncogene, bHLH transcription factor; MYCN = MYCN protooncogene, bHLH transcription factor; NUPR1 = nuclear protein 1, transcriptional regulator; RABL6 = RAB, member RAS oncogene family–like 6; RB1 = retinoblastoma protein transcriptional corepressor 1; TGFB1 = transforming growth factor β1; TP53 = tumor protein p53; VEGF = vascular endothelial growth factor.
Positive and negative activation z-scores indicate increased and decreased activity, respectively, on COPD scaffolds compared with normal scaffolds. The analysis was performed by using Ingenuity Pathway Analysis. Shown are the 20 upstream mediators with the lowest P values sorted by the activation z-score.
Figure 6.
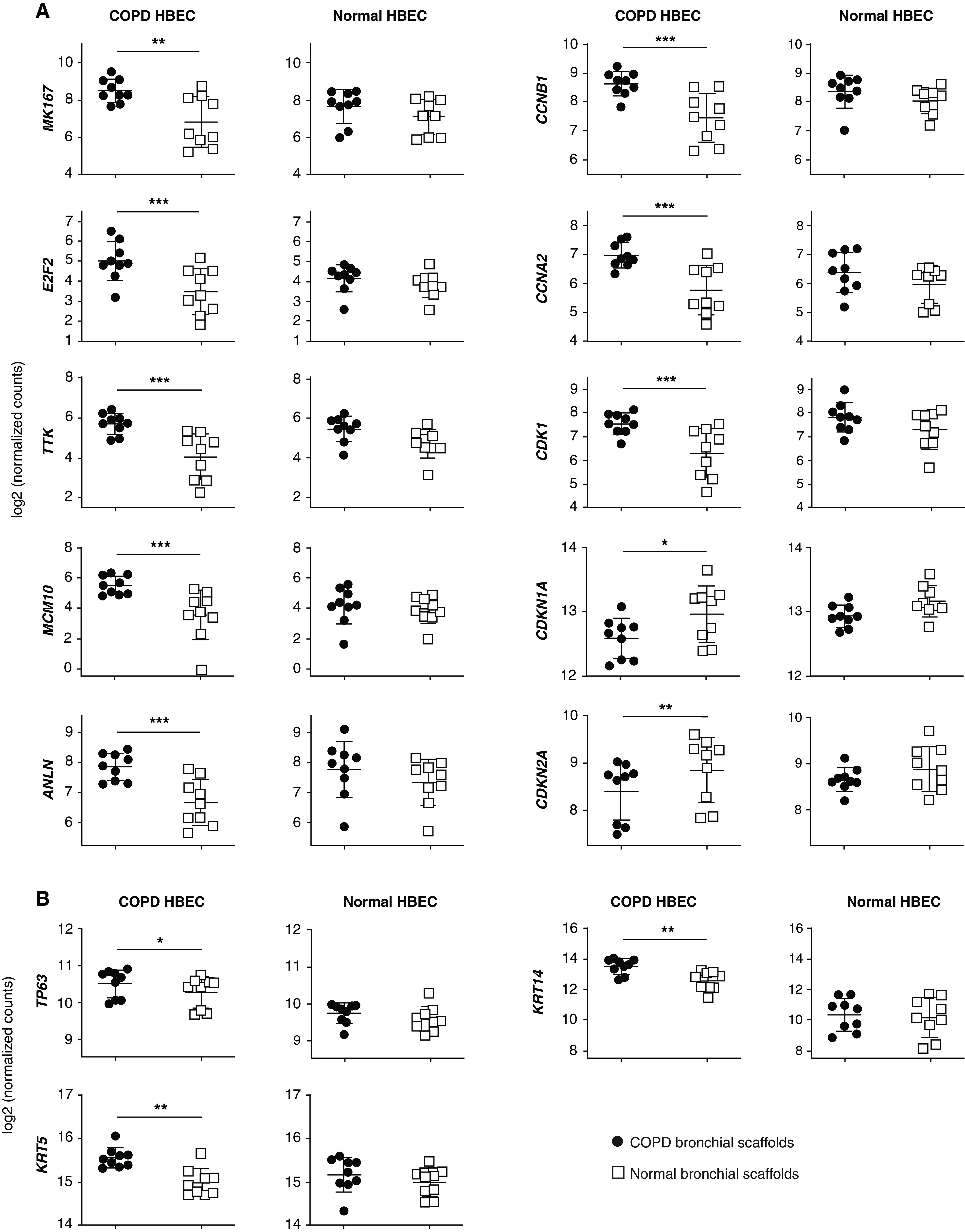
RNA-sequencing data for (A) genes involved in cell cycle regulation and cell division and (B) basal-cell marker genes in COPD HBEC and normal HBEC after 7 days of differentiation on COPD bronchial scaffolds or normal bronchial scaffolds (n = 9 repopulated scaffolds per combination and time point). Means and SDs are indicated. *FDR < 0.05, **FDR < 0.01, and ***FDR < 0.001 using DESeq2 (see the data supplement) and the Benjamini-Hochberg method for multiple testing correction. ANLN = anillin actin binding protein; CCNA2 = cyclin A2; CCNB1 = cyclin B1; CDK1 = cyclin-dependent kinase 1; CDKN1A = cyclin-dependent kinase inhibitor 1A (p21); CDKN2A = cyclin-dependent kinase inhibitor 2A (p16-INK4A/p14-ARF); KRT14 = keratin 14; KRT5 = keratin 5; MCM10 = minichromosome maintenance 10 replication initiation factor; TTK = TTK protein kinase.
COPD Bronchial Scaffolds Show an Altered ECM Composition
In total, 3,340 proteins were detected in the bronchial scaffolds, 384 of which belonged to the matrisome as defined by Naba and colleagues (20). Many core matrisome components, such as ECM glycoproteins and collagens, were identified, but matrisome-associated proteins, like ECM regulators and secreted factors (Figures E7A and E7B), were also identified. Thirteen matrisome proteins were differentially abundant in COPD scaffolds compared with normal scaffolds (Figure E8). LOXL1 (lysyl oxidase–like 1), FBLN5 (fibulin 5), EFEMP1 (EGF-containing fibulin ECM protein 1), MMP12 (matrix metallopeptidase 12), and the C1QA (complement C1q A), C1QB (complement C1q B), and C1QC (complement C1q C) chains were all increased in COPD scaffolds. Proteins decreased in COPD scaffolds included IGFBP2 (insulin-like growth factor binding protein 2), S100A8 (S100 calcium binding protein A8), and S100A9 (S100 calcium binding protein A9) as well as the FGA (fibrinogen α), FGB (fibrinogen β), and FGG (fibrinogen γ) chains.
Discussion
We demonstrate that gene expression in COPD bronchial airways depends both on inherent defects in the epithelial cells and on the disease state of the bronchial ECM. Bronchial scaffolds induced ciliated-cell differentiation in HBEC, but COPD HBEC showed an impaired ability to initiate this differentiation. Furthermore, COPD HBEC were considerably more affected than normal HBEC by the disease state of the bronchial scaffolds, showing a gene expression pattern indicating increased proliferation and a retained basal-cell phenotype when grown on COPD scaffolds compared with normal scaffolds.
Many of the pathological changes observed in COPD airways have been linked to cigarette smoke, which affects the structure and function of the airway epithelium as well as its regenerative potential (43). Basal cells have an inherent capacity as progenitors to regenerate a damaged airway epithelium by differentiating into ciliated cells and goblet cells, and smoking is known to have a strong influence on the basal-cell transcriptome, affecting genes associated with the risk of developing COPD (44). Functionally, cigarette smoke has a detrimental effect on ciliary development (45–47) and is also known to induce epigenetic alterations in human airway epithelial cells (48). In the present study, all COPD HBEC came from smokers or ex-smokers, which may suggest that the impaired induction of ciliary genes in these cells is a result of epigenetic alterations caused by cigarette smoke exposure. Moreover, the gene expression pattern showing impaired ciliated-cell differentiation in COPD HBEC became evident only after repopulation on bronchial scaffolds, and not at the ALI (Figure 5), suggesting that signals from components of the bronchial ECM contribute to the differentiation process. Shojaie and colleagues (49) showed that murine endodermal progenitor cells could be differentiated into proximal airway epithelial cells, including ciliated cells, when cultured on decellularized rat lung scaffolds, and pretreatment of the scaffolds with heparinase demonstrated that heparan sulfate and its associated factors were essential for supporting the observed differentiation. This suggests that heparan sulfate proteoglycans in the bronchial scaffolds may have played a role in promoting ciliated-cell differentiation in the present study. Importantly, MUC5AC was not differentially expressed between COPD HBEC and normal HBEC, and, in contrast to FOXJ1, it was induced between Day 0 and Day 7 in HBEC grown at the ALI (Figure 4A), indicating that differentiation toward the goblet-cell lineage is less dependent on signals from the ECM.
We did not observe differential expression of genes involved in proliferation between COPD HBEC and normal HBEC grown at the ALI or on bronchial scaffolds. However, our data indicate increased proliferation of COPD HBEC when grown on COPD bronchial scaffolds compared with normal bronchial scaffolds (Figure 6A and Table 4), whereas no such effect was seen for normal HBEC. Interestingly, COPD HBEC also displayed higher expression of basal-cell markers on COPD scaffolds than on normal scaffolds (Figure 6B), and expression of the proliferation marker MKI67 decreased between Day 0 and Day 7 for all repopulated HBEC except COPD HBEC on COPD bronchial scaffolds (Figure E9). Taken together, these results suggest that normal HBEC have the ability to differentiate in a similar way on both COPD bronchial scaffolds and normal bronchial scaffolds, resulting in a lower proliferation rate over time, whereas COPD HBEC retain basal-cell properties and a higher proliferation rate, but only on COPD scaffolds, which also suggests that the normal scaffolds may have a normalizing effect on the COPD HBEC. Our observations indicate that the bronchial ECM in COPD airways limits differentiation and promotes proliferation of airway epithelial cells, which is in line with observations of basal-cell hyperplasia (50) and increased lung cancer risk (51) in patients with COPD. These findings may be related to differences in adhesion to COPD scaffolds compared with normal scaffolds. CDK1 has been shown to mediate a direct link between cell cycle regulation and cell adhesion (52). The activity of CDK1, and its interaction with CCNB1 and CCNA2, was shown to regulate the cell adhesion complex area during cell cycle progression. In the present study, all three of these genes were upregulated on COPD scaffolds compared with normal scaffolds in COPD HBEC (Figure 6A), suggesting that differential adhesion to diseased bronchial scaffolds compared with normal bronchial scaffolds may have influenced the cell cycle machinery in the repopulated COPD HBEC. There is support in our data for differential adhesion of the COPD HBEC to the bronchial scaffolds, as several genes related to integrin signaling display a lower expression level when the cells are grown on COPD scaffolds compared with normal scaffolds (Table E7).
The bronchial scaffolds were analyzed with mass spectrometry proteomics to identify disease-specific alterations in the bronchial ECM. The ECM was particularly well preserved with respect to ECM glycoproteins, proteoglycans, and collagens (Figure E7B) on the basis of the definition of the matrisome by Naba and colleagues (20). Consistent with other proteomic studies performed on decellularized lung tissue (53, 54), nonmatrisome proteins were also identified in the scaffolds. Thirteen matrisome proteins showed an altered abundance in COPD scaffolds compared with normal scaffolds (Figure E8), including LOXL1 (55), FBLN5 (56), EFEMP1 (57), and MMP12 (58), which are all known to bind tropoelastin or to be directly involved in regulation of elastic fiber integrity. Their increased abundance in COPD bronchial scaffolds may be a result of dysregulated elastic fiber homeostasis in COPD lungs, which show decreased elastin content not only in the alveolar parenchyma but also in the airway walls (17). By using proteomics on nondecellularized peripheral lung tissue, we have previously shown that MMP12 has an altered solubility profile in COPD lungs, although the total amounts were unchanged compared with those of controls (59). This suggests that the solubility, distribution, and abundance of MMP12 vary among the different compartments of COPD lungs. To our knowledge, the matrisome proteins we identify as being differentially abundant have not yet been associated with proliferation or cell cycle regulation in COPD. Furthermore, there could also be other ECM alterations in the COPD bronchial scaffolds that influenced gene expression in the repopulated HBEC but were not recorded by the mass spectrometry setup, including changes in biomechanical properties, like tissue stiffness and compliance, as well as posttranslational modifications of proteins, such as glycosylations. More studies are needed to clarify the mechanistic links between bronchial ECM alterations and the epithelial-cell phenotype in COPD.
In recent years, there have been advances in organ bioengineering made with the aim of mitigating the donor lung shortage for patients with end-stage chronic lung disease (60). One potential approach is to use decellularized organ scaffolds repopulated with patient-derived stem or progenitor cells, including HBEC, to generate functional organs (61, 62). In this study, we provide evidence that not only the origin of the cells and the scaffolds, but also how the cells interact with specific scaffolds to generate a functional bronchial epithelium, need to be considered. We show that the culture conditions have a large impact on the behavior of HBEC and that results from studies on epithelial cells grown on cell culture plastic can be difficult to compare with data from studies on epithelial cells grown on biological scaffolds. Our data demonstrate that the underlying ECM induces gene expression profiles that differ from those seen in HBEC cultured at the ALI and that differences between diseased and normal HBEC increase further when the cells are subjected to differentiation signals. Because of the limited supply of lung tissue, we were not able to match COPD and control groups with respect to sex, age, and smoking status, and it cannot therefore be fully excluded that these factors may have influenced the results to some extent.
In conclusion, we show that bronchial scaffolds induce ciliated-cell differentiation in primary HBEC and that COPD HBEC display an impaired ability to initiate this differentiation. The gene expression pattern in COPD HBEC also indicates increased proliferation and a retained basal-cell phenotype after repopulation on COPD scaffolds compared with normal scaffolds, suggesting that interactions between epithelial cells and the ECM modulate epithelial-cell proliferation and differentiation in COPD bronchial airways. Our findings provide novel insight into the importance of the bronchial ECM and its influence on the bronchial epithelial-cell phenotype in COPD and suggest that the cell–ECM interaction needs to be included in the experimental design of studies focused on the molecular mechanisms behind epithelial-cell dysfunction in respiratory diseases.
Acknowledgments
Acknowledgment
The authors thank the Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University, for performing the quantitative proteomic analysis and the Inga Britt and Arne Lundberg Research Foundation for donating the Orbitrap Fusion Tribrid Mass Spectrometry instrument. They thank Karolina Öst for characterizing the human bronchial epithelial cells by using the air–liquid interface model and thank Dr. Kristofer Thörn for helpful guidance around how to make cDNA libraries for RNA sequencing and for performing the sequencing step. They also thank Manasa Surakala for running the bcbio-nextgen pipeline and Dr. Graham Belfield for valuable input on the experimental design of the RNA-sequencing study. Finally, they thank all patients and donors for donating their organs as well as Skåne University Hospital in Lund and the Department of Cardiothoracic Surgery at Sahlgrenska University Hospital in Gothenburg.
Footnotes
Supported by AstraZeneca, The Swedish Research Council (DNR 2016-01190), The Swedish Foundation for Strategic Research, The Swedish Heart-Lung Foundation, ALF grants from the Swedish government, The Greta & Johan Kock Foundation, The Alfred Österlund Foundation, The Anna-Greta Crafoord Foundation, The Konsul Th C Bergh Foundation, King Gustav V’s and Queen Victoria’s Freemason Foundation, The Royal Physiographic Society of Lund, and The Medical Faculty of Lund University.
Author Contributions: U.H. designed and performed all experiments except the mass spectrometry analysis, analyzed the data, prepared the figures, and wrote the manuscript. X.Z. conceived the overall research plan. G.W.-T., O.H., and X.Z. supervised the study and contributed to the experimental design, interpretation of the data, and writing of the manuscript. L.Ö. provided input for the experimental design and performed bioinformatic analyses of the RNA-sequencing data. O.V. provided intellectual input for the interpretation of data. G.D., M.S., and L.B. were responsible for surgical procedures and collection of samples. All authors reviewed the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0395OC on April 21, 2021
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365:1567–1575. doi: 10.1056/NEJMoa1106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35:71–86. doi: 10.1016/j.ccm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 3. Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:726–733. doi: 10.1513/pats.200605-126SF. [DOI] [PubMed] [Google Scholar]
- 4. Perotin JM, Adam D, Vella-Boucaud J, Delepine G, Sandu S, Jonvel AC, et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. 2014;15:151. doi: 10.1186/s12931-014-0151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC, et al. Epithelial mucin stores are increased in the large airways of smokers with airflow obstruction. Chest. 2006;130:1102–1108. doi: 10.1378/chest.130.4.1102. [DOI] [PubMed] [Google Scholar]
- 6. Rigden HM, Alias A, Havelock T, O’Donnell R, Djukanovic R, Davies DE, et al. Squamous metaplasia is increased in the bronchial epithelium of smokers with chronic obstructive pulmonary disease. PLoS One. 2016;11:e0156009. doi: 10.1371/journal.pone.0156009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hessel J, Heldrich J, Fuller J, Staudt MR, Radisch S, Hollmann C, et al. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One. 2014;9:e85453. doi: 10.1371/journal.pone.0085453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yaghi A, Zaman A, Cox G, Dolovich MB. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir Med. 2012;106:1139–1147. doi: 10.1016/j.rmed.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 10. Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC, et al. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-beta) by modulating assembly of latent TGF-beta-binding protein-1. J Biol Chem. 2007;282:26418–26430. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- 11. Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of protein function by glycosaminoglycans: as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 12. Kresse H, Schönherr E. Proteoglycans of the extracellular matrix and growth control. J Cell Physiol. 2001;189:266–274. doi: 10.1002/jcp.10030. [DOI] [PubMed] [Google Scholar]
- 13. Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Annoni R, Lanças T, Yukimatsu Tanigawa R, de Medeiros Matsushita M, de Morais Fernezlian S, Bruno A, et al. Extracellular matrix composition in COPD. Eur Respir J. 2012;40:1362–1373. doi: 10.1183/09031936.00192611. [DOI] [PubMed] [Google Scholar]
- 15. Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126:725–735. doi: 10.1309/jc477fael1ykv54w. [DOI] [PubMed] [Google Scholar]
- 16. Harju T, Kinnula VL, Pääkkö P, Salmenkivi K, Risteli J, Kaarteenaho R. Variability in the precursor proteins of collagen I and III in different stages of COPD. Respir Res. 2010;11:165. doi: 10.1186/1465-9921-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eurlings IM, Dentener MA, Cleutjens JP, Peutz CJ, Rohde GG, Wouters EF, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. 2014;14:90. doi: 10.1186/1471-2466-14-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hogg JC, McDonough JE, Gosselink JV, Hayashi S. What drives the peripheral lung-remodeling process in chronic obstructive pulmonary disease? Proc Am Thorac Soc. 2009;6:668–672. doi: 10.1513/pats.200907-079DP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hedström U, Hallgren O, Öberg L, DeMicco A, Vaarala O, Westergren-Thorsson G, et al. Bronchial extracellular matrix from COPD patients induces altered gene expression in repopulated primary human bronchial epithelial cells. Sci Rep. 2018;8:3502. doi: 10.1038/s41598-018-21727-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedström U. Lund, Sweden: Lund University; 2018. https://portal.research.lu.se/portal/files/54114677/Doctoral_thesis_UH.pdf [Google Scholar]
- 22. Yu X, Ng CP, Habacher H, Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 23. Zariwala MA, Gee HY, Kurkowiak M, Al-Mutairi DA, Leigh MW, Hurd TW, et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am J Hum Genet. 2013;93:336–345. doi: 10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, et al. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hjeij R, Lindstrand A, Francis R, Zariwala MA, Liu X, Li Y, et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am J Hum Genet. 2013;93:357–367. doi: 10.1016/j.ajhg.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kott E, Legendre M, Copin B, Papon JF, Dastot-Le Moal F, Montantin G, et al. Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am J Hum Genet. 2013;93:561–570. doi: 10.1016/j.ajhg.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 29. Bretones G, Delgado MD, León J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849:506–516. doi: 10.1016/j.bbagrm.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 30. Li LY, Chen H, Hsieh YH, Wang YN, Chu HJ, Chen YH, et al. Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 2011;71:4269–4279. doi: 10.1158/0008-5472.CAN-10-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 33. Georgakilas AG, Martin OA, Bonner WM. p21: A two-faced genome guardian. Trends Mol Med. 2017;23:310–319. doi: 10.1016/j.molmed.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 35. Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–1502. doi: 10.1101/gad.282145.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cuylen S, Blaukopf C, Politi AZ, Müller-Reichert T, Neumann B, Poser I, et al. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature. 2016;535:308–312. doi: 10.1038/nature18610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, et al. Mps1 phosphorylates borealin to control aurora B activity and chromosome alignment. Cell. 2008;132:233–246. doi: 10.1016/j.cell.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 38. Baxley RM, Bielinsky AK. Mcm10: a dynamic scaffold at eukaryotic replication forks. Genes (Basel) 2017;8:73. doi: 10.3390/genes8020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao WM, Fang G. Anillin is a substrate of anaphase-promoting complex/cyclosome (APC/C) that controls spatial contractility of myosin during late cytokinesis. J Biol Chem. 2005;280:33516–33524. doi: 10.1074/jbc.M504657200. [DOI] [PubMed] [Google Scholar]
- 40. Lindqvist A. Cyclin B-Cdk1 activates its own pump to get into the nucleus. J Cell Biol. 2010;189:197–199. doi: 10.1083/jcb.201003032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hein JB, Nilsson J. Interphase APC/C-Cdc20 inhibition by cyclin A2-Cdk2 ensures efficient mitotic entry. Nat Commun. 2016;7:10975. doi: 10.1038/ncomms10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang H, Liu X, Umino T, Sköld CM, Zhu Y, Kohyama T, et al. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am J Respir Cell Mol Biol. 2001;25:772–779. doi: 10.1165/ajrcmb.25.6.4458. [DOI] [PubMed] [Google Scholar]
- 44. Ryan DM, Vincent TL, Salit J, Walters MS, Agosto-Perez F, Shaykhiev R, et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS One. 2014;9:e88051. doi: 10.1371/journal.pone.0088051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brekman A, Walters MS, Tilley AE, Crystal RG. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am J Respir Cell Mol Biol. 2014;51:688–700. doi: 10.1165/rcmb.2013-0363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, Eickelberg O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep. 2015;5:8163. doi: 10.1038/srep08163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Simet SM, Sisson JH, Pavlik JA, Devasure JM, Boyer C, Liu X, et al. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am J Respir Cell Mol Biol. 2010;43:635–640. doi: 10.1165/rcmb.2009-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010;29:3650–3664. doi: 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shojaie S, Ermini L, Ackerley C, Wang J, Chin S, Yeganeh B, et al. Acellular lung scaffolds direct differentiation of endoderm to functional airway epithelial cells: requirement of matrix-bound HS proteoglycans. Stem Cell Reports. 2015;4:419–430. doi: 10.1016/j.stemcr.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11:S252–S258. doi: 10.1513/AnnalsATS.201402-049AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer. 2013;13:233–245. doi: 10.1038/nrc3477. [DOI] [PubMed] [Google Scholar]
- 52. Jones MC, Askari JA, Humphries JD, Humphries MJ. Cell adhesion is regulated by CDK1 during the cell cycle. J Cell Biol. 2018;217:3203–3218. doi: 10.1083/jcb.201802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wagner DE, Bonenfant NR, Parsons CS, Sokocevic D, Brooks EM, Borg ZD, et al. Comparative decellularization and recellularization of normal versus emphysematous human lungs. Biomaterials. 2014;35:3281–3297. doi: 10.1016/j.biomaterials.2013.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garlikova Z, Silva AC, Rabata A, Potesil D, Ihnatova I, Dumkova J, et al. Generation of a close-to-native in vitro system to study lung cells–extracellular matrix crosstalk. Tissue Eng Part C Methods. 2018;24:1–13. doi: 10.1089/ten.tec.2017.0283. [DOI] [PubMed] [Google Scholar]
- 55. Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 56. Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, et al. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 57. Kobayashi N, Kostka G, Garbe JH, Keene DR, Bächinger HP, Hanisch FG, et al. A comparative analysis of the fibulin protein family: biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- 58. Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon VK, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. 2012;55:1965–1975. doi: 10.1002/hep.25567. [DOI] [PubMed] [Google Scholar]
- 59. Åhrman E, Hallgren O, Malmström L, Hedström U, Malmström A, Bjermer L, et al. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J Proteomics. 2018;189:23–33. doi: 10.1016/j.jprot.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 60. Gilpin SE, Wagner DE. Acellular human lung scaffolds to model lung disease and tissue regeneration. Eur Respir Rev. 2018;27:180021. doi: 10.1183/16000617.0021-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gilpin SE, Charest JM, Ren X, Tapias LF, Wu T, Evangelista-Leite D, et al. Regenerative potential of human airway stem cells in lung epithelial engineering. Biomaterials. 2016;108:111–119. doi: 10.1016/j.biomaterials.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gilpin SE, Ren X, Okamoto T, Guyette JP, Mou H, Rajagopal J, et al. Enhanced lung epithelial specification of human induced pluripotent stem cells on decellularized lung matrix. Ann Thorac Surg. 2014;98:1721–1729, discussion 1729. doi: 10.1016/j.athoracsur.2014.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]