

Abstract
Metabolism is essential for a living organism to sustain life. It provides energy to a cell by breaking down compounds (catabolism) and supplies building blocks for the synthesis of macromolecules (anabolism). Signal transduction pathways tightly regulate mammalian cellular metabolism. Simultaneously, metabolism itself serves as a signaling pathway to control many cellular processes, such as proliferation, differentiation, cell death, gene expression, and adaptation to stress. Considerable progress in the metabolism field has come from understanding how cancer cells co-opt metabolic pathways for growth and survival. Recent data also show that several metabolic pathways may participate in the pathogenesis of lung diseases, some of which could be promising therapeutic targets. In this translational review, we will outline the basic metabolic principles learned from the cancer metabolism field as they apply to the pathogenesis of pulmonary arterial hypertension and fibrosis and will place an emphasis on therapeutic potential.
Keywords: metabolism, pulmonary arterial hypertension, pulmonary fibrosis, metformin
The traditional functions of metabolism are bioenergetics, the breakdown of nutrients to generate energy (catabolism); and biosynthesis of the macromolecules, such as proteins, lipids, and nucleic acids, necessary for cell growth (anabolism). An emerging function of metabolism is to control biology by regulating signaling pathways and factors that control gene expression (1). Previously, changes in metabolism were believed to be a consequence of alterations in signaling pathways or gene expression. In other words, metabolism is controlled by and reacts to a cell’s needs to conduct cellular processes that require ATP (i.e., bioenergetics and biosynthesis of macromolecules). Recent accumulating evidence, however, suggests that metabolism itself can regulate cellular signaling pathways and gene expression, playing a causal role in dictating many biological outcomes, such as proliferation, differentiation, cell death, and adaptation to stress (Figure 1). Intuitively, it makes sense that the interaction between metabolism and cellular processes is bidirectional, as it would be dangerous (or unnecessary) for the cell to commit to a certain cellular process when nutrients and metabolites are deficient (or in surplus). This relatively new perspective of metabolic signaling has been established by the growing evidence in the past two decades, especially in cancer metabolism. Cancer metabolism is based on the principle that altered metabolism in cancer cells, compared with metabolism in normal cells, is essential to acquire and maintain the malignancy (2). This knowledge has been successfully translated into effective therapy in some cases, as with, for example, the use of asparaginase for the treatment of acute lymphoblastic leukemia (3). In this review, we present the current evidence and lessons learned from cancer metabolism and how they relate to lung diseases such as pulmonary arterial hypertension (PAH) and idiopathic pulmonary fibrosis (IPF) (4, 5). This review is intended to paint a picture of the broad conceptual advances in metabolism with respect to anabolic diseases (cancer, PAH, and IPF) on the basis of in vivo findings. The regulation of intermediary metabolism pathways or mitochondria are beyond the scope of the review.
Figure 1.

Overview of intermediary metabolism. Upon changes in environmental conditions, cells respond by altering intermediary metabolism: the balance between AMPK-driven catabolism and mTORC1-driven anabolism. As a result of metabolism, H2O2 is generated that can control the cellular state and function. H2O2 can also have a detrimental consequence when it is converted into hydroxyl radicals (•OH) in the presence of ferrous iron (Fe2 +) by the Fenton reaction. Subsequently, •OH in the presence of PUFAs can generate lipid hydroperoxides that can induce cell death known as ferroptosis. AMPK = AMP-activated protein kinase; mTORC1 = mTOR complex I; PUFA = polyunsaturated fatty acid; TCA = tricarboxylic acid.
Basic Fundamentals of Intermediary Mammalian Metabolism
The roots of the cancer metabolism field are in the seminal observations made in the 1920 s by Otto Warburg that tumor tissues generate a copious amount of lactate compared with normal tissue, even in the presence of oxygen. This is referred to as the Warburg effect or aerobic glycolysis. Warburg went on to win the Nobel Prize for the discovery of cytochrome c oxidase, the final complex in the electron transport chain (ETC), which transfers electrons from cytochrome c to molecular oxygen. Warburg generated a logical explanation, at first glance, for aerobic glycolysis. He surmised that cancer cells acquire “an injury to respiration” as they go from a differentiated cellular state to a proliferative state, causing an increase in lactate production under normal oxygen conditions. Indeed, positron emission tomography (PET) imaging with fluorodeoxyglucose F 18 (18F-FDG) reveals that many tumors in patients have higher glucose uptake than the neighboring normal tissue. However, today we know that most cancer cells have an intact ETC and respire. A key lesson that has emerged from the field of cancer metabolism is that many proliferating cells (e.g., cancer cells, stem cells, and immune cells) increase uptake of extracellular nutrients (glucose, amino acids, and oxygen) to fuel enhanced flux through both glycolysis and mitochondrial tricarboxylic acid (TCA) cycle metabolism to supply precursors for the production of proteins, lipids, and nucleotides (6) (Figure 2).
Figure 2.
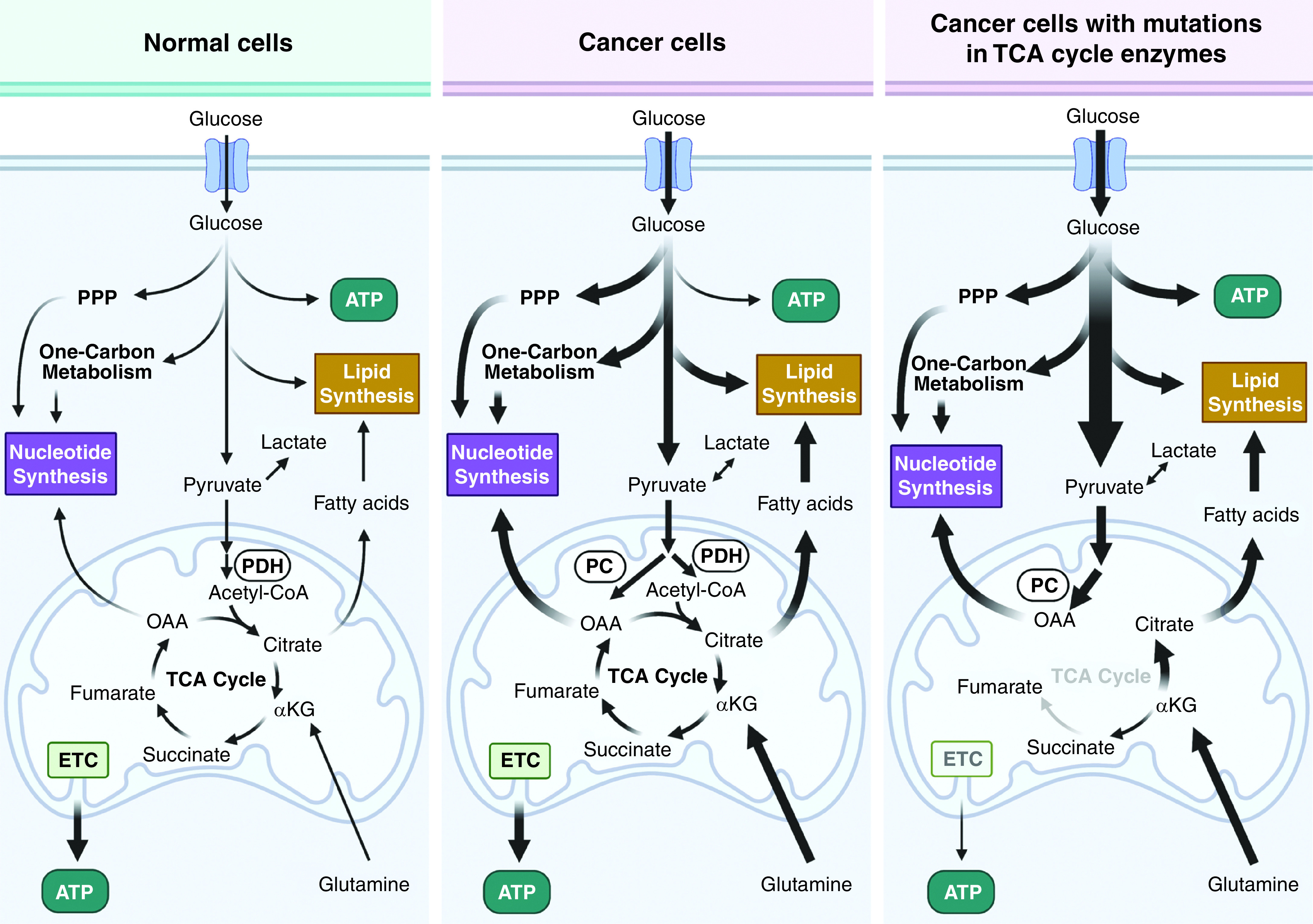
Metabolic heterogeneity among cancer cells. Most cancer cells exhibit robust flux through glycolysis and TCA cycle metabolism as well as their branched pathways. For tumor cells with mutations in TCA-cycle enzymes, tumor growth is still supported by alternative pathways such as glutamine-dependent reductive carboxylation or PC. αKG = α-ketoglutarate; ETC = electron transport chain; OAA = oxaloacetate; PC = pyruvate carboxylase; PDH = pyruvate dehydrogenase; PPP = pentose phosphate pathway.
Glycolytic intermediates funnel into various anabolic pathways, including the pentose phosphate pathway, hexosamine pathway, and de novo serine synthesis–dependent one-carbon metabolism (2). The TCA cycle intermediates citrate, succinyl-CoA, and oxaloacetate are precursors for the synthesis of lipids, heme, and nucleotides, respectively (2). Glutamine and other amino acids replenish TCA cycle intermediates (i.e., anaplerosis) to support anabolism. Although the vast majority of tumors respire, there are rare cancers that are largely dependent on glycolysis for ATP generation because of mutations in TCA cycle enzymes (SDH [succinate dehydrogenase] or FH [fumarate hydratase]). However, these cancers are able to generate the TCA-cycle intermediates citrate and oxaloacetate through glutamine-dependent reductive carboxylation and PC (pyruvate carboxylase), respectively (Figure 2). The mutations in SDH or FH provide the metabolites succinate and fumarate, which maintain the malignant phenotype through epigenetic regulation of certain cancers as discussed below.
It should be emphasized that there is considerable metabolic heterogeneity within tumor cells in vivo during tumorigenesis and metastasis (Figure 2). However, no pathogenic mutations that completely disable glycolysis or respiration have been reported as being linked to tumor progression. Pathogenic mutations that result in complete loss of ETC function are probably selected against for tumor progression (7). Partial inhibition of mitochondrial respiration is not pathogenic for tumor growth because mitochondria still can generate TCA-cycle metabolites as in the above examples. There is a gain of function by decreasing respiration: primarily the generation of oncometabolites, including D(R)-2-hydroxyglutarate [D(R)-2HG], fumarate, or succinate, to promote tumorigenesis, as well as an increase in mitochondrial reactive oxygen species (ROS) to stimulate metastasis (8). In addition, a recent functional genomic-screen study revealed that targeting mitochondrial metabolism in metastatic lung cancer cells was efficacious, even though those cells had diminished respiration (9). Finally, slow-cycling tumor cells that emerge from a standard-of-care therapy tend to be more dependent on oxidative phosphorylation for survival than the more quickly proliferating cancer cells that use a robust amount of glycolysis (10).
The increase in oxidative metabolism observed in most cancer cells results in the production of high levels of ROS (11). The mitochondrial ETC, as well as the plasma membrane and intracellular NOX (nicotinamide adenine dinucleotide [NAD] phosphate oxidase) enzymes, produce ROS in the form of superoxide, which is quickly converted into hydrogen peroxide (H2O2) by SOD1 (superoxide dismutase 1) or SOD2. Cancer cells express high levels of SOD1 or SOD2 because superoxide can be a toxic molecule (12). Subsequently, H2O2 can oxidize specific cysteine residues within proteins to alter their function, impacting a range of biological processes, including cell proliferation, survival, and metabolic adaptation to stress. Pathways known to be positively regulated by H2O2 include those of HIFs (hypoxia-inducible factors), NF-κB, PI3K, and MAP (mitogen-activated protein) kinases. A detrimental consequence of H2O2 is its conversion into hydroxyl radicals in the presence of ferrous iron (Fe2 + ) by the Fenton reaction. The presence of hydroxyl radicals can subsequently lead to the generation of lipid hydroperoxides in the presence of polyunsaturated fatty acids, which can cause a type of cell death known as ferroptosis. Consequently, cancer cells run the risk of ferroptosis because of the high levels of H2O2 and polyunsaturated fatty acids, and they thus overexpress enzymes that detoxify lipid hydroperoxides into lipid alcohols such as GPX4 (glutathione peroxidase 4) and FSP1 (fibroblast-specific protein 1). Interestingly, dietary antioxidants can detoxify lipid hydroperoxides to prevent ferroptosis during tumor progression, resulting in increased tumor growth (13). This likely explains why dietary antioxidants promoted lung cancer in clinical trials. It is important to note that the steady-state level of ROS in tumor cells can potentially be increased, decreased, or the same, compared with that of normal cells. But once antioxidant capacity is inhibited in both cell types, tumor cells show much higher levels of ROS than normal cells (14). This indicates that the rate of ROS production in cancer cells is higher than that in normal cells. Collectively, these studies indicate that cancer cells use localized pools of H2O2 to activate protumorigenic pathways while limiting the generation of lipid hydroperoxides to prevent ferroptosis.
The growth of cancer cells is supported by increased flux through anabolic pathways, which are transcriptionally activated by the deregulation of oncogenes (e.g., MYC) and tumor suppressors (e.g., p53). Furthermore, cancer cells can activate metabolic pathways through enzyme phosphorylation, without growth factor stimulation. In normal cells, growth factors activate PI3K, which subsequently recruits the serine/threonine-specific protein kinase, AKT, that activates mTORC1 (mTOR complex I). In the presence of nutrients, the activation of the PI3K–AKT–mTORC1 network promotes phosphorylation of enzymes involved in de novo lipid and nucleotide synthesis pathways, which is associated with decreased autophagy. Tumor cells often have mutations that promote the PI3K–AKT–mTOR network without extrinsic stimulation by growth factors (2). The protooncogene MYC transcriptionally upregulates glycolytic enzymes and genes involved in ETC function. During the progression of tumor growth, cancer cells encounter nutrient deprivation as metabolic demand exceeds the nutrient supply because of insufficient angiogenesis. Thus, cancer cells activate AMPK (AMP-activated protein kinase), which senses changes in the ATP/AMP ratio, resulting in diminished anabolism through decreased mTORC1 activity and de novo lipogenesis. In addition, AMPK activation leads to an increase in catabolism through stimulation of autophagy and fatty acid oxidation to maintain the ATP/ADP ratio for survival.
An exciting theme that has emerged from cancer metabolism is the appreciation that metabolites can modulate protein function to change the cell fate/function by altering gene expression (15) (Figure 3). For example, ACLY (ATP-citrate lyase) uses citrate to generate acetyl-CoA that can be used to acetylate histones at certain markers (e.g., H3K27), which is associated with active gene expression. Acetate (16) or nuclear PDH (pyruvate dehydrogenase) (17) can also generate acetyl-CoA. Methylation of H3K4 is also associated with actively transcribed genes. The methyl group comes from S-adenosyl methionine that is generated by the methionine cycle, supported by one-carbon metabolism. Demethylation of histones, DNA, and RNA is controlled by a family of α-ketoglutarate (α-KG)–dependent dioxygenases. Prolyl hydroxylases that negatively regulate HIFs are also members of this α-KG–dependent dioxygenases family. The α-KG–dependent dioxygenases can be inhibited by succinate, fumarate, and D(R)- or L(S)-2HG. D- and L-2HG are reduced forms of α-KG. D-2HG is a well-recognized oncometabolite and markedly accumulates in tumors with mutations in IDH1 (isocitrate dehydrogenase 1) or IDH2 that convert α-KG to D-2HG (18). Cells with high D-2HG levels consequently fail to activate differentiation programs and instead maintain a proliferative phenotype, in part because of DNA and histone hypermethylation. Currently, D-2HG is used as a biomarker to monitor disease activity for acute myeloid leukemia (AML) and solid tumors like glioma. Recently, drugs targeting mutant IDH1/2 have shown efficacy in AML by diminishing D-2HG levels. On the other hand, the enantiomer L-2HG is not produced by IDH1/2 mutations but rather by LDHA (lactate dehydrogenase A)/LDHC (lactate dehydrogenase C) and MDH1 (malate dehydrogenase 1)/MDH2. L-2HG is substantially generated under conditions when the NADH/NAD+ ratio is elevated, such as during hypoxia or ETC dysfunction (19), as well in conditions with low levels of L-2HGDH (L-2HG dehydrogenase), an enzyme that converts L-2HG back to α-KG. VHL (Von Hippel Lindau)–null renal cancer cells display high levels of L-2HG, which is associated with DNA hypermethylation and maintenance of a malignant phenotype. In addition, cancers that harbor SDH or FH mutations, such as paragangliomas, pheochromocytomas, gastrointestinal stromal tumors, and renal-cell carcinomas, have high levels of succinate or fumarate, respectively. They display increased levels of HIF activation under normal oxygen conditions and interfere with the activity of α-KG–dependent dioxygenases, leading to hypermethylation of histones, DNA, and RNA to maintain malignant phenotypes (20).
Figure 3.
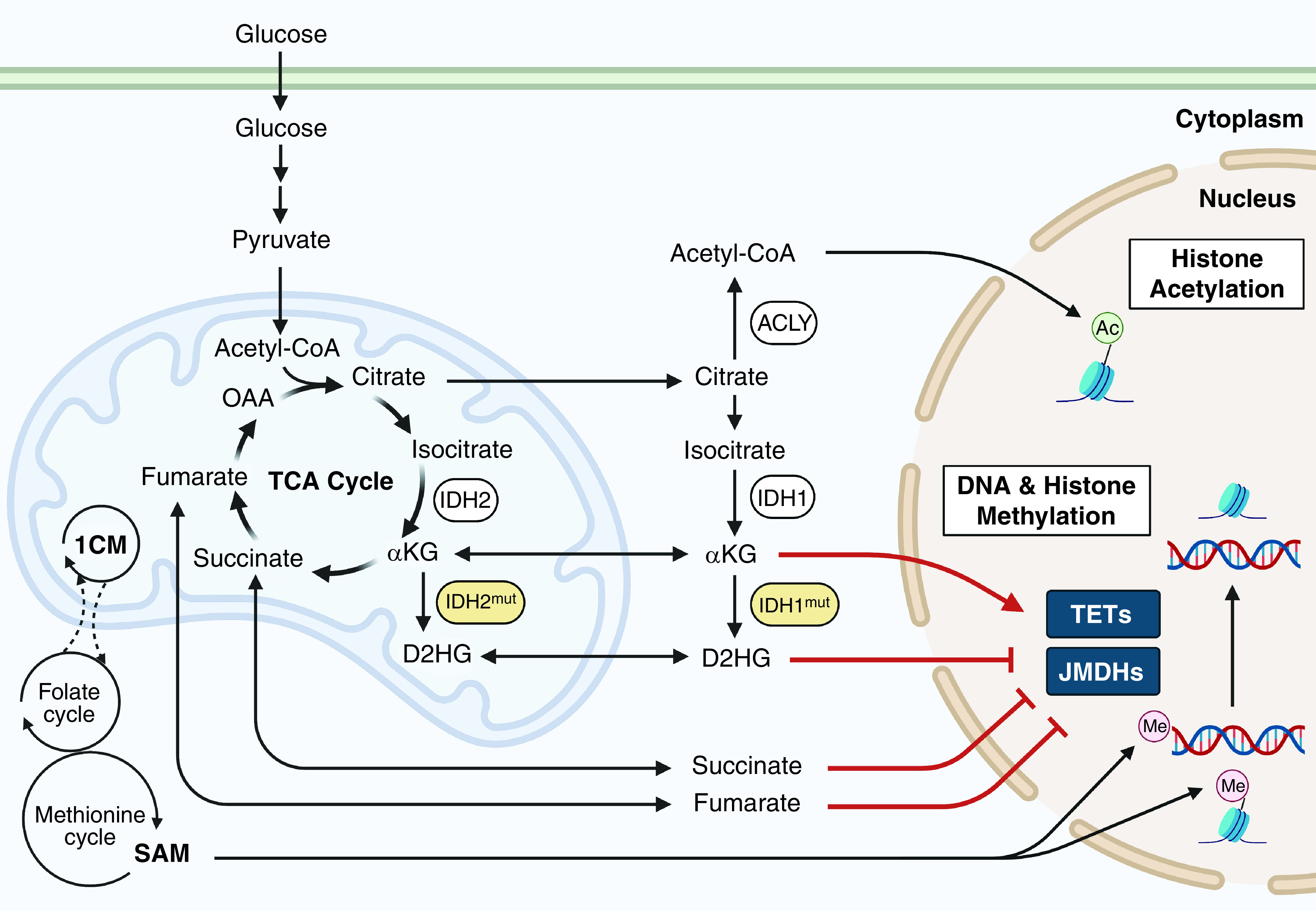
Metabolic intermediates as epigenetic regulators. TCA-cycle metabolites such as succinate, fumarate, and αKG control DNA and histone methylation by regulating αKG-dependent dioxygenases. D2HG, which is increased with mutations in IDH1 (isocitrate dehydrogenase 1) or IDH2, inhibits αKG-dependent dioxygenases. 1CM = one-carbon metabolism; ACLY = ATP-citrate lyase; D2HG = D-2-hydroxyglutarate; JMDH = Jumonji-c domain histone demethylase; SAM = S-adenosyl methionine; TET = ten–eleven translocation methylcytosine dioxygenase.
In summary, four important insights learned from cancer metabolism are 1) both TCA cycle- and glycolytic intermediates support macromolecule synthesis; 2) ROS in the form of H2O2 can support tumor progression, but ROS in the form of lipid hydroperoxides can limit tumor growth by inducing ferroptosis; 3) mTORC1 and AMPK signaling pathways can control the balance between anabolism and catabolism, respectively; and 4) TCA cycle metabolites sustain histone acetylation and inhibit DNA, RNA, and histone demethylases. Thus, cancer cells use metabolism to maintain their malignant phenotype and support tumor progression by promoting anabolism under nutrient-replete conditions and by promoting catabolism under nutrient-depleted conditions (Figure 1).
In Vivo Veritas: Intermediary Metabolism Differs between Cell Culture versus In Vivo Settings
After the establishment of the first cell lines in the 1940s, there was a concerted effort to develop media that would efficiently support cell proliferation in culture. In the 1950s, Harry Eagle developed media containing excessive amounts of nutrients (glucose and amino acids) that were optimal for cell proliferation. Of note, these media were not designed to mimic in vivo physiologic nutrient levels. The high levels of glucose found in DMEM (25mM) and RPMI (11mM) compared with normal plasma glucose levels (5–5.5mM) invoke a robust production of lactate at normal oxygen levels (i.e., aerobic glycolysis) in both proliferating cancer and T cells in vitro. By contrast, in vivo 13C-glucose isotope tracing in mouse models of cancer and proliferating T cells, as well as in human lung and glioblastoma cancers in patients, demonstrates that a large fraction of glucose enters into the mitochondrial TCA cycle. Thus, the fate of glucose in vitro culture is distinct from its fate in vivo. Interestingly, 13C-lactate isotope tracing in human lung cancer demonstrates that a large fraction of lactate, converted to pyruvate, enters into the TCA cycle as well. A notable exception is in VHL-null cancers that display high levels of HIFs. HIFs target PDKs (PDH kinases), negative regulators of PDH, and thus decrease PDH activity and subsequently suppress oxidation of pyruvate to acetyl-CoA. These cells display robust lactate production (i.e., the Warburg effect) and rely on glutamine to fuel the TCA cycle. The recent advent of human plasma–like media (21) was an attempt to define media that represent plasma nutrient levels, and these media should be used for in vitro experiments. Ultimately, any in vitro finding in metabolism should be confirmed in vivo, as the availability of nutrients and the cellular microenvironment differ significantly during the course of pathology between in vitro and in vivo conditions (22).
Metabolism Alterations Linked to PAH
PAH is a vascular remodeling disease characterized by high pressure in the pulmonary artery that leads to right heart failure and premature death. Patients with PAH display intimal and medial thickening of the pulmonary arteries. Furthermore, the lungs of patients with PAH contain plexogenic lesions with proliferating pulmonary arterial smooth muscle cells (PASMCs), pulmonary arterial endothelial cells (PAECs), and fibroblasts in and around occluded pulmonary arteries. In addition, accumulating evidence indicates infiltration of inflammatory cells within the vascular wall. Consequently, the vascular remodeling in PAH results in the narrowing of the vascular lumen. The in vivo pathologic features of PAH are reminiscent of cancer, and both diseases can be simply thought of as dysregulated anabolism leading to a cellular proliferative disorder (Figure 4). In both human and animal models of PAH, PASMCs show enhanced glycolysis, activation of HIF-1 even under normoxia, and upregulation of oncogenes (23, 24). Furthermore, PET scans with 18F-FDG revealed significantly higher uptake in PAH lungs both in human and rodent models than in control lungs. This suggests that an enhanced glycolytic rate supports the anabolic phenotype of proliferating cells in vivo in PAH, which is similarly observed in cancer (25, 26). However, the data with respect to the role of mitochondrial metabolism in PAH are more complex. PAECs isolated from patients with PAH display a modest increase in glycolytic rates compared with PAECs isolated from healthy control subjects, which is concomitant with a slight decrease in oxygen consumption (26). One caution about the studies using PAECs or PASMCs isolated from patients with PAH is that these studies were done in high-glucose media without the substrates that fuel the mitochondrial TCA cycle (e.g., lactate and alanine) that are present in human plasma. Evidence for glycolysis contributing to the development of PAH comes in part from the observation that pharmacologic or genetic inhibition of the positive glycolytic regulator PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3) ameliorates the development of pulmonary hypertension (PH) in rodent models (27). Furthermore, PDH is inhibited by HIFs and TNF-α in PAH PASMCs, decreasing the conversion of pyruvate into acetyl-CoA in the mitochondrial matrix (28). The importance of suppressed PDH for the pathogenesis of PAH in vivo is supported by the observation that a PDK inhibitor, dichloroacetate, ameliorates or reverses PH in animal models of PAH (29, 30) and impressively improves mean pulmonary arterial pressures (>5 mm Hg) in a small number of patients with PAH (31). Surprisingly, there have been no follow-up investigational human studies or clinical trials reported in clinicaltrials.gov or peer-reviewed papers using dichloroacetate.
Figure 4.
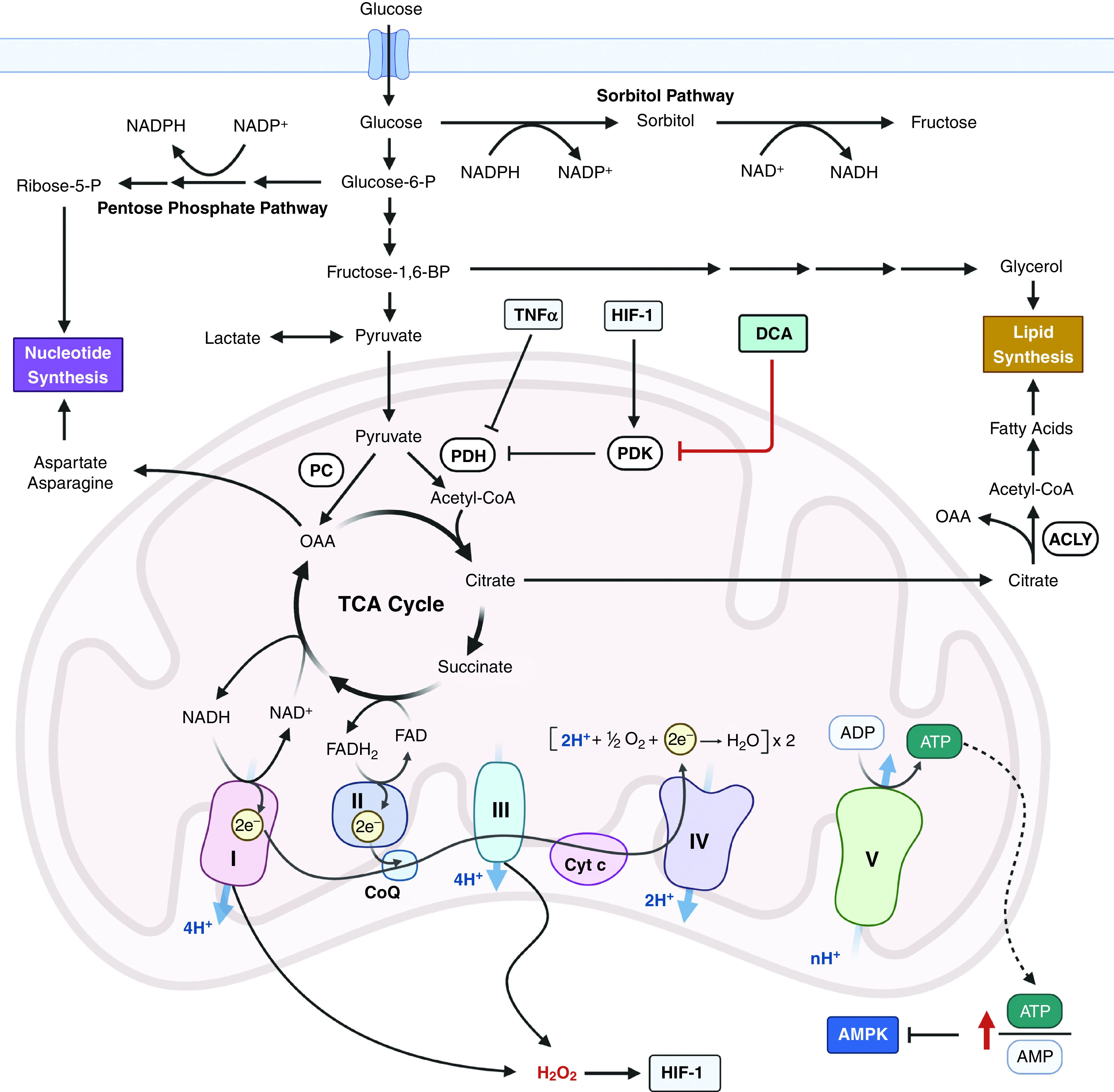
Metabolic pathways involved in the pathology of pulmonary arterial hypertension (PAH). Dysregulated anabolism, characterized by enhanced glycolysis and TCA intermediates, is central to the pathogenesis of PAH. Targeting PDH in animal models showed promise for PAH therapy (DCA), indicating that suppressed PDH activity is important in PAH pathology. Cyt c = cytochrome c; DCA = dichloroacetate; e− = electron; FAD = flavin adenine dinucleotide; FADH2 = reduced form of FAD; Fructose-1,6-BP = fructose-1,6-bisphosphate; Glucose-6-P = glucose-6-phosphate; HIF-1 = hypoxia-inducible factor; PDK = PDH kinase; Ribose-5-P = ribose-5-phosphate.
It is important to note that the decrease in pyruvate oxidation during the progression of PAH causes an increased reliance on mitochondrial fatty acid oxidation to generate mitochondrial acetyl-CoA to support pulmonary vascular remodeling and PAH in mice (32). Fatty acid oxidation burns fatty acids into CO2 that cannot generate any net carbons for macromolecular synthesis. Thus, there must be other fuels in the TCA cycle to support growth during the development of PAH. One possibility is that glutaminolysis generates α-KG within the TCA cycle to provide net carbons for growth. Indeed, pharmacologic inhibition of GLS1 (glutaminase 1) attenuated rodent PAH (33). A study that provides some insight into the role of TCA cycle metabolism in patients with PAH comes from metabolite profiling of lung samples obtained from patients with PAH at the time of lung transplantation compared with normal lung tissues obtained from patients with cancer undergoing surgical lobectomy (34). Principal component analysis from PAH lung tissues exhibited a distinct metabolic signature compared with the normal lung tissue. Notably, increased glucose levels were accompanied by elevated sorbitol levels, whereas lactate levels remained unchanged. Interestingly, glycolytic intermediates upstream of fructose-1,6-bisphosphate were increased, indicating that excess glucose uptake in PAH lungs is funneled into the sorbitol and pentose phosphate pathways, the latter of which would be used to generate ribose for de novo nucleotide production. The TCA-cycle intermediates citrate and succinate were increased in PAH lung tissues compared with normal lung tissues. These data are consistent with the cancer metabolic phenotype characterized by increased production of glycolytic and TCA-cycle intermediates to fuel anabolism. Going forward, it would be important to perform stable isotope tracing with different carbon sources (e.g., glucose, glutamine, and lactate) in rodent PAH models and perform PET scans using different tracers in patients with PAH to determine the contribution of different carbon molecules to fueling the TCA cycle during PAH pathology in vivo. This could lead to the development of new, targeted metabolic therapies for PAH. For example, if glutamine were found to be a significant fuel for TCA-cycle intermediates, perhaps GLS inhibitors (currently in phase 2 clinical trials for certain cancers) could be used to treat PAH.
Metabolic Requirements for Progression of Pulmonary Fibrosis
IPF is a progressive fibrotic lung disease related to the aging process, which also displays an anabolic metabolic phenotype (Figure 5). It is characterized by alveolar epithelial cell damage and accumulated fibroblasts that undergo transdifferentiation into myofibroblasts secreting collagen and other proteins into the extracellular matrix. TGF-β (transforming growth factor β) has been implicated as the major driver of collagen production in fibroblasts during the pathogenesis of IPF. The production of collagen requires the amino acid glycine, which is generated from serine in the cytosol or mitochondria by the one-carbon metabolism enzymes SHMT1 (serine hydroxymethyltransferase 1) or SHMT2, respectively. TGF-β induces the mRNA expression of the de novo serine synthesis pathway enzymes PHGDH (phosphoglycerate dehydrogenase) and PSAT1 (phosphoserine aminotransferase 1), resulting in increased flux from the glycolytic intermediate 3-phosphoglycerate to serine. Importantly, inhibition of PHGDH attenuates TGF-β–induced collagen synthesis in human lung fibroblasts (35) and attenuates bleomycin-induced lung fibrosis in mice (36). TGF-β also increases glucose uptake and glycolysis to generate 3-phosphoglycerate to support de novo serine and glycine synthesis. In animal models of lung fibrosis, enhanced glycolysis promotes myofibroblast differentiation, whereas inhibiting glycolysis by targeting GLUT1 (glucose transporter 1) or PFKFB3 attenuates lung fibrosis (37, 38). These results are supported by the observation that patients with IPF exhibit an increased 18F-FDG PET signal in their normal-appearing lung regions on high-resolution computed tomography images compared with normal areas of lungs in patients without lung disease (39). Furthermore, IPF lung tissues, compared with normal lung tissues, demonstrate increases in the product-to-substrate ratios of glucose-6-phosphate to glucose and lactic acid to pyruvic acid, suggesting an increase in glycolytic flux (40). Lactic acid levels are increased in IPF lung tissue compared with tissue from healthy control subjects, which promotes TGF-β–induced myofibroblast differentiation (41, 42). Collectively, these results suggest that IPF is characterized by increased glucose uptake that generates the glycolytic intermediate 3-phosphoglycerate, supporting de novo serine and glycine production, which is necessary for collagen generation in fibroblasts (Figure 5).
Figure 5.
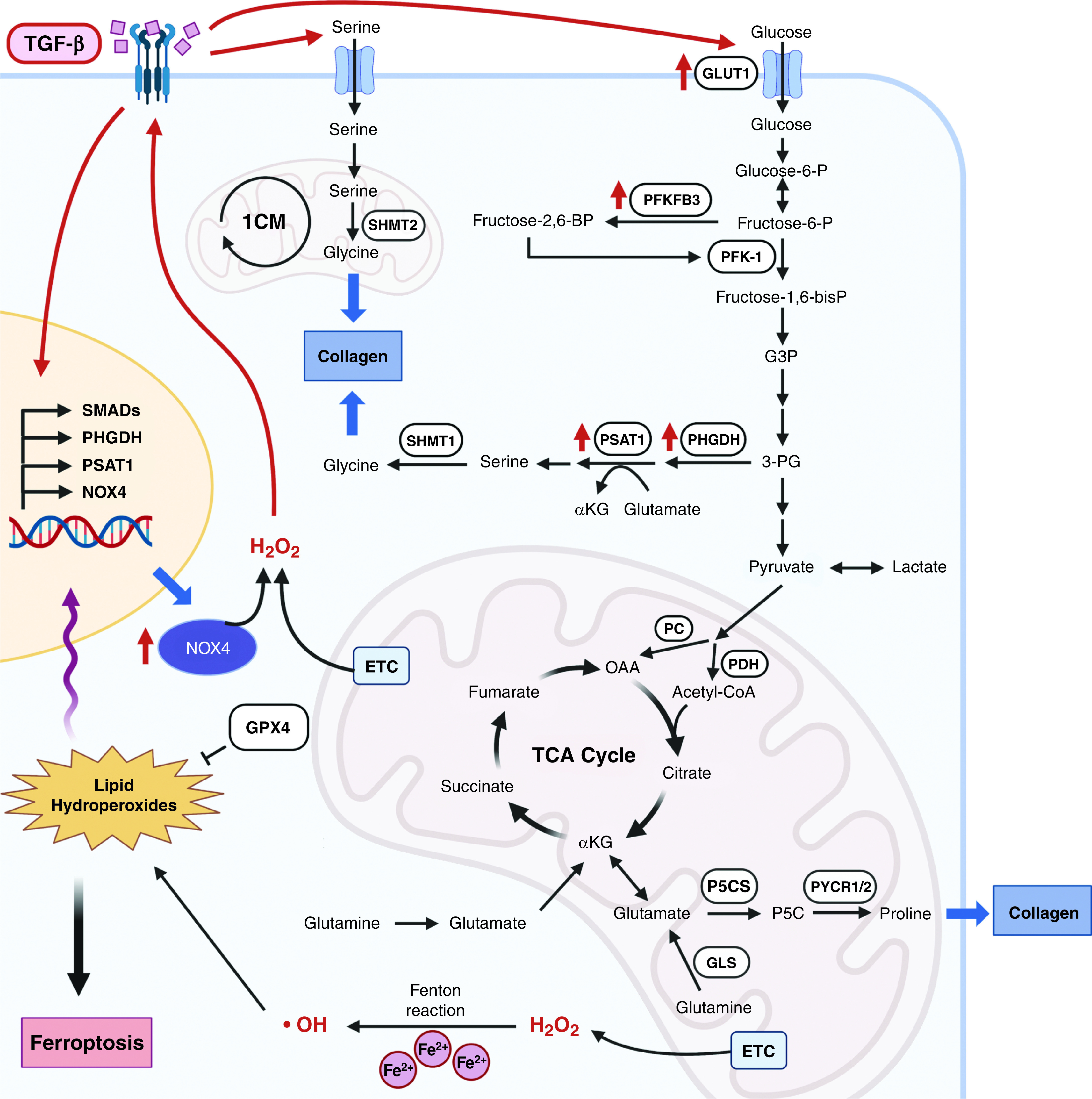
Signaling pathways that regulate the pathogenesis of pulmonary fibrosis. During the pathogenesis of idiopathic pulmonary fibrosis, TGF-β modulates metabolic signaling pathways by regulating redox signaling, enhancing serine and glycine synthesis by increased glucose influx, and promoting proline synthesis in mitochondria, which result in aberrant production of collagen. Fructose-2,6-BP = fructose-2,6-bisphosphate; Fructose-6-P = fructose-6-phosphate; G3P = glyceraldehyde 3-phosphate; GLS = glutaminase; GLUT1 = glucose transporter 1; GPX4 = glutathione peroxidase 4; NOX4 = NADP oxidase 4; P5CS = Δ1‐pyrroline‐5‐carboxylate synthase; PFKFB3 = 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PHGDH = phosphoglycerate dehydrogenase; PSAT1 = phosphoserine aminotransferase 1; PYCR1/2 = pyrroline‐5‐carboxylate reductase family member 1 or 2; SHMT = serine hydroxymethyltransferase; TGF-β = transforming growth factor-β.
In addition to glycine, proline is another key amino acid that is necessary for collagen production. Proline levels are elevated in IPF lung tissue compared with normal control lungs (40). Proline is produced in mitochondria from glutamine through a two-step process involving P5CS (delta‐1‐pyrroline‐5‐carboxylate synthase), PYCR1 (pyrroline‐5‐carboxylate reductase family member 1), and PYCR2 (Figure 5). Genetic deletion of P5CS in human fibroblasts abrogates TGF-β–induced collagen production, and TGF-β induces the biosynthesis of proline from glutamine in fibroblasts by upregulating enzymes in this pathway (43, 44). In vitro studies demonstrate that glutamine conversion to glutamate by GLS1 is required for TGF-β1–induced myofibroblast differentiation and collagen production (45, 46). Going forward, it would be important to use glutamine-based PET scans in patients with IPF and test GLS inhibitors in clinical trials to determine whether glutamine-dependent proline production in the mitochondria fuels collagen production, promoting the progression of IPF pathology.
TGF-β binding to its receptor induces mitochondrial ROS, which augments TGF-β transcriptional responses, including those involving NOX4. As the generation of ROS by NOX4 is transcriptionally regulated, this sets up a positive feedback loop of ROS signaling that drives fibrosis. Mitochondrial ROS is not required for SMAD phosphorylation or nuclear translocation (47), suggesting that TGF-β activation of noncanonical pathways might drive TGF-β target gene expression. The incidence of IPF increases with age. In a mouse model of bleomycin-induced fibrosis, both young and aged mice exhibited similar severity at their peak lung fibrosis stage; however, only aged mice displayed persistent and unresolved fibrosis (48). Aged mice had reduced levels of NRF2-dependent antioxidant target genes, concomitant with elevated levels of NOX4. Importantly, inhibition of NOX4 in aged mice partially restored fibrosis resolution. Likewise, human IPF studies have demonstrated a markedly elevated oxidant burden with a perturbed balance between oxidants and antioxidants. There is also an age-related decrease in mitochondrial respiration in epithelial cells that likely causes an increase in mitochondrial ROS (49). For example, glutathione levels, as well as the levels of enzymes responsible for glutathione synthesis, are lower in IPF lungs than in normal control lungs (50). These findings are reminiscent of cancer cells, which show an increased rate of ROS production from NOX enzymes and mitochondria, as is necessary for activating a variety of protumorigenic signaling (e.g., HIFs). However, IPF diverges from cancer with respect to lipid hydroperoxide biology. Cancer cells generate high levels of lipid hydroperoxides that can induce ferroptosis; thus, cancer cells upregulate GPX4 to convert lipid hydroperoxides to lipid alcohols. In fact, inhibition of GPX4 in various cancer cells invokes ferroptosis both in vitro and in vivo. By contrast, heterozygous GPX4-deficient mice showed enhanced bleomycin-induced lung fibrosis even though lipid peroxidation was increased, whereas overexpression of GPX4 attenuated fibrosis (51). Inhibition of GPX4 increased TGF-β–induced myofibroblast differentiation concomitant with enhanced lipid peroxidation and SMAD2/3 signaling. Thus, in the lung fibrosis setting, lipid hydroperoxides act as signaling molecules rather than as damaging molecules. It is likely that the levels of lipid hydroperoxides in lung fibrosis are markedly lower than those in cancer cells. Nevertheless, these findings suggest that scavenging lipid hydroperoxides might be beneficial in slowing the progression of IPF.
Targeting Metabolism for PAH and IPF: Metformin
A few inhibitors targeting metabolism have made it into advanced clinical trials in cancer. Notably, inhibitors targeting mutant IDH1 or IDH2 have been successful in treating AML in clinical trials. However, these mutations have not been observed in patients with PAH or IPF; thus, these inhibitors are not likely to show efficacy in these patients. GLS inhibitors have shown early promise in patients with KEAP1 (Kelch-like ECH–associated protein 1) mutant non–small cell lung cancer (52). Patients with PAH and/or IPF might benefit from GLS inhibitors because they would decrease anaplerosis, as discussed above. The one drug that has garnered attention in cancer and lung diseases is metformin, a widely used antidiabetic drug. Epidemiologic retrospective studies in individuals with diabetes receiving metformin reported an association with reduced cancer incidence and mortality. Investigational studies in murine cancer models also concluded that metformin acts as an anticancer agent (53). Metformin combined with standard EGFR (epidermal growth factor receptor)–tyrosine kinase inhibitor therapy significantly improved both progression-free survival and overall survival in patients with advanced lung adenocarcinoma (54). In addition, a phase 3 clinical trial in 2021 will report their results on whether metformin (1,750 mg/d) is a viable therapeutic strategy against breast cancer (NCT 01101438). However, although a handful of clinical trials have reported some efficacy of metformin in various cancers, others have not observed a robust anticancer efficacy. This is likely due to the mechanism by which metformin exerts its antitumor effects.
There are two different widely accepted mechanisms by which metformin exerts its antitumor effects: 1) it reduces circulating insulin levels, a known mitogen for tumors, by improving hyperglycemia, and 2) it inhibits mitochondrial complex I within cancer cells, which decreases the TCA-cycle intermediates necessary for tumor growth (Figure 6) (53). There is considerable heterogeneity in the expression of insulin and/or insulin growth factor receptor among different tissues and cell types. Therefore, metformin-mediated reduction of circulating insulin levels in non–insulin-responsive cancers would be irrelevant to any potential antitumor effect. In addition, metformin requires OCTs (organic cation transporters) to enter cells (Figure 6). Normal tissues that express OCTs include the kidney, gut, and liver. Cancer cells that do not express OCTs are therefore refractory to metformin-induced inhibition of complex I and its antitumor effect (53). Heterogeneity in OCT expression might explain the variability in metformin’s antitumor efficacy in clinical trials. Going forward, the identification of OCT-expressing tumors may be used as a screening strategy to identify appropriate candidates for metformin therapy. Furthermore, an underappreciated aspect of metformin is its ability to modulate physiology and ameliorate disparate pathologies through its effects on inflammation. For example, metformin can inhibit IL-6 production, which is a cytokine associated with diverse lung pathologies, including cancer, PAH, IPF, and acute respiratory distress syndrome. Future studies should focus on how metformin modulates inflammation.
Figure 6.
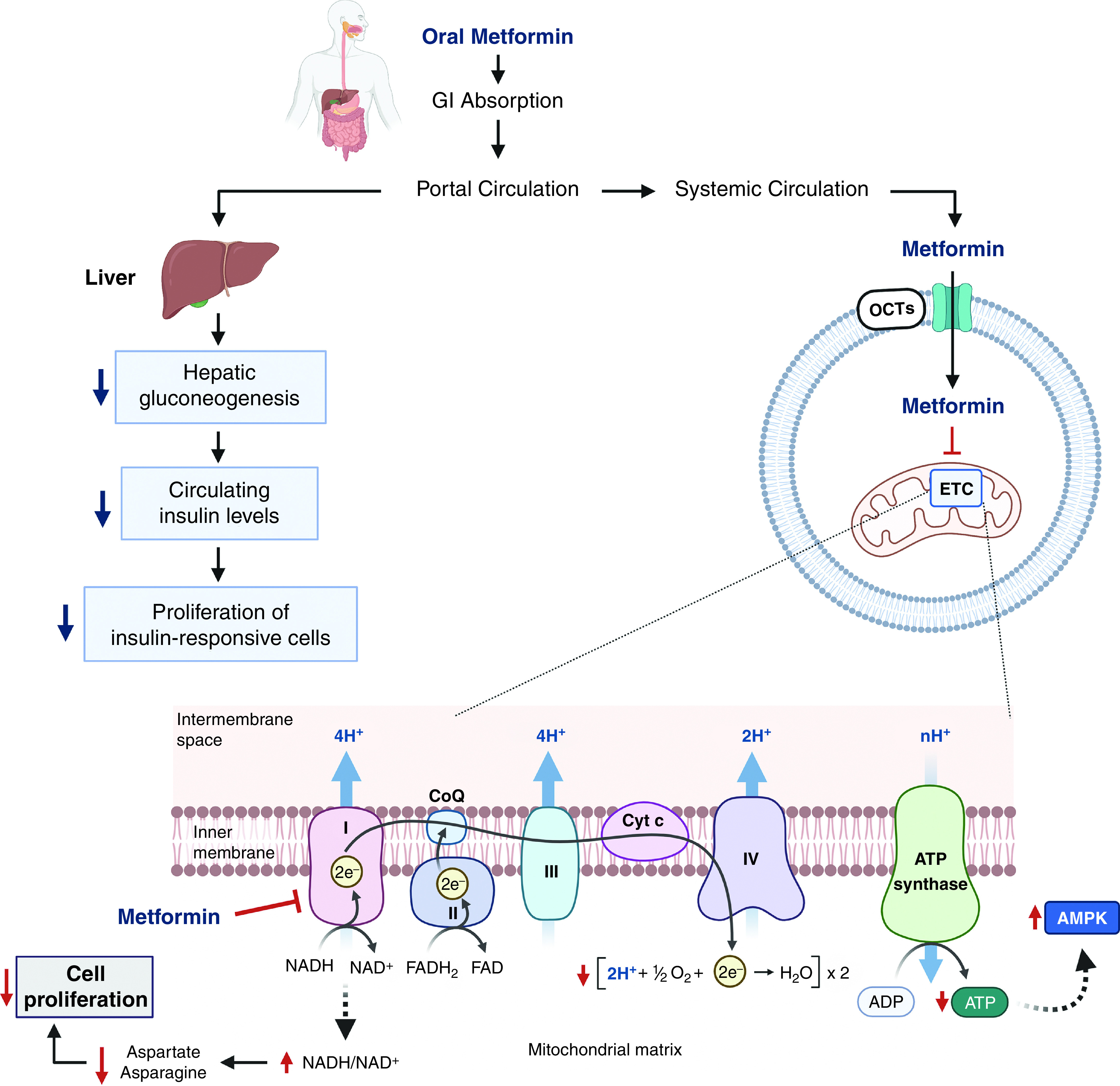
Cellular and molecular mechanisms of metformin. Oral metformin is absorbed in the GI tract and enters portal circulation to decrease hepatic glucose production. Improved hyperglycemia subsequently decreases circulating insulin levels and thus decreases the proliferation of insulin-responsive cells. On the other hand, metformin directly interacts with cells that express OCTs (organic cation transporters). Once metformin is transported into a cell via OCTs, it inhibits mitochondrial respiratory chain complex I. Mild inhibition of complex I induced by metformin results in an increase of the NADH/NAD+ ratio and reduced electron transfer. Consequently, cellular ATP levels decrease, and AMP accumulates, which leads to the activation of the AMPK signaling pathway. In addition, metformin limits the synthesis of aspartate and asparagine and thus inhibits cell proliferation (82). GI = gastrointestinal.
Evidence shows that metformin may be beneficial to patients with PH and/or IPF. Metformin attenuates PH in chronic hypoxia– and monocrotaline-induced rodent models of PAH (55–60) and may also reverse PH in a Sugen/hypoxic rat model of PAH (61), although the finding was not replicated in another study (62). Rather, it looks more promising in PH due to left heart disease (group 2) related to metabolic syndrome. Metformin improves both PH and metabolic syndrome–related outcomes in a Sugen/obese ZSF1 rat model of PH with heart failure with preserved ejection fraction (PH-HFpEF) (63, 64) and in a supracoronary aortic banding/high-fat diet/olanzapine rat model of PH-HFpEF (65). The effect seems to be mediated by IL-6 inhibition. In vitro studies showed that metformin decreases the proliferation of both rat and human PASMCs (66, 67). A human study showed that combination therapy with metformin and bosentan increased the 6-minute-walk distance in patients with a congenital heart defect associated PAH, compared with bosentan monotherapy (68). A phase 2 trial of metformin for PH-HFpEF is ongoing (NCT 03629340). Further studies are necessary to elucidate the mechanism by which metformin exerts its beneficial effects on PH, especially related to metabolic syndrome.
A molecular structure study showed that metformin may directly interact with TGF-β1 and inhibit downstream signaling (69). Metformin inhibits TGF-β1–induced myofibroblast differentiation and suppresses NOX4 expression in human lung fibroblasts in vitro (70) and attenuates bleomycin-induced lung fibrosis in murine (70–75) and rat (76) models. In addition, treatment with metformin of human fibroblasts isolated from patients with IPF induced significant upregulation of the lipogenic markers, suggesting that metformin induces lipogenic differentiation of lung fibroblasts (73). On the basis of the data so far, it seems clear that metformin attenuates pulmonary fibrosis, at least in a murine bleomycin-induced fibrosis model. However, the mechanism of action and target cell types expressing OCTs in the lung (77) remain uncertain. One proposed mechanism is through the activation of AMPK, as the beneficial effect of metformin on bleomycin-induced pulmonary fibrosis was abrogated in AMPKα1-deficient mice (71). Metformin induces mild and specific inhibition of mitochondrial respiratory chain complex I (53) independent of AMPK activation (78). Metformin-induced AMPK activation results from changes in ATP/ADP and ATP/AMP ratios caused by decreased cellular energy from complex I inhibition (79). It would be interesting to see whether the antifibrotic effect of metformin in vivo is dependent on its ability to inhibit complex I. On the other hand, human data have not shown a consistent benefit of metformin use in patients with IPF to date. Post hoc analysis of the pirfenidone trial on patients with IPF who received a placebo showed no difference in clinical outcomes in metformin users compared with non–metformin users (80, 81). Well-designed prospective studies would be necessary to evaluate the antifibrotic effect of metformin in human IPF.
Conclusions
In summary, metabolic homeostasis plays an important role in regulating physiologic and pathologic cellular processes that are integral to the pathogenesis of many lung diseases. A conceptual advance in the past 20 years is that intermediary metabolism is not only used for generation of ATP or metabolites for growth; but metabolites can serve as important signaling molecules to modulate physiology and pathology. We are beginning to understand the role of metabolic dysregulation in disease progression, and further study of this subject has the potential to yield therapeutic targets in many lung diseases for which we do not currently have effective therapies. However, the key to moving forward in modulating metabolism in the context of normal lung physiology and pathologies is to perform studies in the context of the in vivo environment.
Acknowledgments
Acknowledgment
Figures were created by using BioRender (https://biorender.com/).
Footnotes
Supported by National Institutes of Health grants 5R35CA197532 and P01AG049665 (N.S.C.) and 5K08HL143138 (S.H.), the American Heart Association Career Development Award 19CDA34630070 (S.H.), and the Parker B. Francis Fellowship (S.H.).
Originally Published in Press as DOI: 10.1165/rcmb.2020-0550TR on April 9, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Chandel NS. Navigating metabolism. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- 2. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, et al. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med. 1986;315:657–663. doi: 10.1056/NEJM198609113151101. [DOI] [PubMed] [Google Scholar]
- 4. Zhao H, Dennery PA, Yao H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;314:L544–L554. doi: 10.1152/ajplung.00521.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu G, Summer R. Cellular metabolism in lung health and disease. Annu Rev Physiol. 2019;81:403–428. doi: 10.1146/annurev-physiol-020518-114640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. 2020;2:127–129. doi: 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 7. Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 2015;13:1895–1908. doi: 10.1016/j.celrep.2015.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 9. Chuang CH, Dorsch M, Dujardin P, Silas S, Ueffing K, Hölken JM, et al. Altered mitochondria functionality defines a metastatic cell state in lung cancer and creates an exploitable vulnerability. Cancer Res. 2020:canres.1865.2020. doi: 10.1158/0008-5472.CAN-20-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473. doi: 10.1126/science.aaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kong H, Chandel NS. Regulation of redox balance in cancer and T cells. J Biol Chem. 2018;293:7499–7507. doi: 10.1074/jbc.TM117.000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Che M, Wang R, Li X, Wang HY, Zheng XFS. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov Today. 2016;21:143–149. doi: 10.1016/j.drudis.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177–178. doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 14. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 15. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016;17:1037–1052. doi: 10.1016/j.celrep.2016.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158:84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- 18. Losman JA, Koivunen P, Kaelin WG., Jr 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer. 2020;20:710–726. doi: 10.1038/s41568-020-00303-3. [DOI] [PubMed] [Google Scholar]
- 19. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 21. Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Jr, et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of ump synthase. Cell. 2017;169:258–272.e17. doi: 10.1016/j.cell.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dey P, Kimmelman AC, DePinho RA. Metabolic codependencies in the tumor microenvironment. Cancer Discov. doi: 10.1158/2159-8290.CD-20-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014;19:558–573. doi: 10.1016/j.cmet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 24. Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013;75:95–126. doi: 10.1146/annurev-physiol-030212-183804. [DOI] [PubMed] [Google Scholar]
- 25. Hagan G, Southwood M, Treacy C, Ross RM, Soon E, Coulson J, et al. 18FDG PET imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: a proof-of-principle study. Pulm Circ. 2011;1:448–455. doi: 10.4103/2045-8932.93543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu J, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci USA. 2019;116:13394–13403. doi: 10.1073/pnas.1821401116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl) 2011;89:771–783. doi: 10.1007/s00109-011-0762-2. [DOI] [PubMed] [Google Scholar]
- 29. McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 30. Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 31. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9:eaao4583. doi: 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- 32. Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2:44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 33. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016;126:3313–3335. doi: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS One. 2014;9:e88727. doi: 10.1371/journal.pone.0088727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nigdelioglu R, Hamanaka RB, Meliton AY, O’Leary E, Witt LJ, Cho T, et al. Transforming growth factor (TGF)-β promotes de novo serine synthesis for collagen production. J Biol Chem. 2016;291:27239–27251. doi: 10.1074/jbc.M116.756247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hamanaka RB, Nigdelioglu R, Meliton AY, Tian Y, Witt LJ, O’Leary E, et al. Inhibition of phosphoglycerate dehydrogenase attenuates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2018;58:585–593. doi: 10.1165/rcmb.2017-0186OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho SJ, Moon JS, Lee CM, Choi AM, Stout-Delgado HW. Glucose transporter 1–dependent glycolysis is increased during aging-related lung fibrosis, and phloretin inhibits lung fibrosis. Am J Respir Cell Mol Biol. 2017;56:521–531. doi: 10.1165/rcmb.2016-0225OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. 2015;192:1462–1474. doi: 10.1164/rccm.201504-0780OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Win T, Thomas BA, Lambrou T, Hutton BF, Screaton NJ, Porter JC, et al. Areas of normal pulmonary parenchyma on HRCT exhibit increased FDG PET signal in IPF patients. Eur J Nucl Med Mol Imaging. 2014;41:337–342. doi: 10.1007/s00259-013-2514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang YP, Lee SB, Lee JM, Kim HM, Hong JY, Lee WJ, et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res. 2016;15:1717–1724. doi: 10.1021/acs.jproteome.6b00156. [DOI] [PubMed] [Google Scholar]
- 41. Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 2012;186:740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao YD, Yin L, Archer S, Lu C, Zhao G, Yao Y, et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: a metabolomic study. BMJ Open Respir Res. 2017;4:e000183. doi: 10.1136/bmjresp-2017-000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hamanaka RB, O’Leary EM, Witt LJ, Tian Y, Gökalp GA, Meliton AY, et al. Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am J Respir Cell Mol Biol. 2019;61:597–606. doi: 10.1165/rcmb.2019-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schwörer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, et al. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J. 2020;39:e103334. doi: 10.15252/embj.2019103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley-Usmar VM, et al. Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J Biol Chem. 2018;293:1218–1228. doi: 10.1074/jbc.RA117.000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ge J, Cui H, Xie N, Banerjee S, Guo S, Dubey S, et al. Glutaminolysis promotes collagen translation and stability via α-ketoglutarate-mediated mTOR activation and proline hydroxylation. Am J Respir Cell Mol Biol. 2018;58:378–390. doi: 10.1165/rcmb.2017-0238OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem. 2013;288:770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beeh KM, Beier J, Haas IC, Kornmann O, Micke P, Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:1119–1123. doi: 10.1183/09031936.02.00262402. [DOI] [PubMed] [Google Scholar]
- 51. Tsubouchi K, Araya J, Yoshida M, Sakamoto T, Koumura T, Minagawa S, et al. Involvement of GPx4-regulated lipid peroxidation in idiopathic pulmonary fibrosis pathogenesis. J Immunol. 2019;203:2076–2087. doi: 10.4049/jimmunol.1801232. [DOI] [PubMed] [Google Scholar]
- 52. Skoulidis F, Neal JW, Akerley WL, Paik PK, Papagiannakopoulos T, Reckamp KL, et al. A phase II randomized study of telaglenastat, a glutaminase (GLS) inhibitor, versus placebo, in combination with pembrolizumab (Pembro) and chemotherapy as first-line treatment for KEAP1/NRF2-mutated non-squamous metastatic non-small cell lung cancer (mNSCLC) [abstract] J Clin Oncol. 2020;38:TPS9627. [Google Scholar]
- 53. Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32:341–352. doi: 10.1016/j.cmet.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arrieta O, Barron F, Padilla MS, Aviles-Salas A, Ramirez-Tirado LA, Arguelles Jimenez MJ, et al. Effect of metformin plus tyrosine kinase inhibitors compared with tyrosine kinase inhibitors alone in patients with epidermal growth factor receptor-mutated lung adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:e192553. doi: 10.1001/jamaoncol.2019.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP, et al. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol. 2009;158:1285–1294. doi: 10.1111/j.1476-5381.2009.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li S, Han D, Zhang Y, Xie X, Ke R, Zhu Y, et al. Activation of AMPK prevents monocrotaline-induced extracellular matrix remodeling of pulmonary artery. Med Sci Monit Basic Res. 2016;22:27–33. doi: 10.12659/MSMBR.897505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhai C, Shi W, Feng W, Zhu Y, Wang J, Li S, et al. Activation of AMPK prevents monocrotaline-induced pulmonary arterial hypertension by suppression of NF-κB-mediated autophagy activation. Life Sci. 2018;208:87–95. doi: 10.1016/j.lfs.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 58. Liu Y, Xu Y, Zhu J, Li H, Zhang J, Yang G, et al. Metformin prevents progression of experimental pulmonary hypertension via inhibition of autophagy and activation of adenosine monophosphate-activated protein kinase. J Vasc Res. 2019;56:117–128. doi: 10.1159/000498894. [DOI] [PubMed] [Google Scholar]
- 59. Yoshida T, Matsuura K, Goya S, Ma D, Shimada K, Kitpipatkun P, et al. Metformin prevents the development of monocrotaline-induced pulmonary hypertension by decreasing serum levels of big endothelin-1. Exp Ther Med. 2020;20:149. doi: 10.3892/etm.2020.9278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Omura J, Satoh K, Kikuchi N, Satoh T, Kurosawa R, Nogi M, et al. Protective roles of endothelial amp-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ Res. 2016;119:197–209. doi: 10.1161/CIRCRESAHA.115.308178. [DOI] [PubMed] [Google Scholar]
- 61. Dean A, Nilsen M, Loughlin L, Salt IP, MacLean MR. Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension. 2016;68:446–454. doi: 10.1161/HYPERTENSIONAHA.116.07353. [DOI] [PubMed] [Google Scholar]
- 62. Goncharov DA, Goncharova EA, Tofovic SP, Hu J, Baust JJ, Pena AZ, et al. Metformin therapy for pulmonary hypertension associated with heart failure with preserved ejection fraction versus pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;198:681–684. doi: 10.1164/rccm.201801-0022LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lai YC, Tabima DM, Dube JJ, Hughan KS, Vanderpool RR, Goncharov DA, et al. SIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation. 2016;133:717–731. doi: 10.1161/CIRCULATIONAHA.115.018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang L, Halliday G, Huot JR, Satoh T, Baust JJ, Fisher A, et al. Treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in pulmonary hypertension associated with heart failure with preserved ejection fraction. Arterioscler Thromb Vasc Biol. 2020;40:1543–1558. doi: 10.1161/ATVBAHA.119.313883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ranchoux B, Nadeau V, Bourgeois A, Provencher S, Tremblay É, Omura J, et al. Metabolic syndrome exacerbates pulmonary hypertension due to left heart disease. Circ Res. 2019;125:449–466. doi: 10.1161/CIRCRESAHA.118.314555. [DOI] [PubMed] [Google Scholar]
- 66. Song Y, Wu Y, Su X, Zhu Y, Liu L, Pan Y, et al. Activation of AMPK inhibits PDGF-induced pulmonary arterial smooth muscle cells proliferation and its potential mechanisms. Pharmacol Res. 2016;107:117–124. doi: 10.1016/j.phrs.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 67. Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, et al. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011;25:1922–1933. doi: 10.1096/fj.10-175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liao S, Li D, Hui Z, McLachlan CS, Zhang Y. Metformin added to bosentan therapy in patients with pulmonary arterial hypertension associated with congenital heart defects: a pilot study. ERJ Open Res. 2018;4:00060-2018. doi: 10.1183/23120541.00060-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xiao H, Zhang J, Xu Z, Feng Y, Zhang M, Liu J, et al. Metformin is a novel suppressor for transforming growth factor (TGF)-β1. Sci Rep. 2016;6:28597. doi: 10.1038/srep28597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res. 2016;17:107. doi: 10.1186/s12931-016-0420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. 2018;24:1121–1127. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Choi SM, Jang AH, Kim H, Lee KH, Kim YW. Metformin reduces bleomycin-induced pulmonary fibrosis in mice. J Korean Med Sci. 2016;31:1419–1425. doi: 10.3346/jkms.2016.31.9.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat Commun. 2019;10:2987. doi: 10.1038/s41467-019-10839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li L, Huang W, Li K, Zhang K, Lin C, Han R, et al. Metformin attenuates gefitinib-induced exacerbation of pulmonary fibrosis by inhibition of TGF-β signaling pathway. Oncotarget. 2015;6:43605–43619. doi: 10.18632/oncotarget.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xiao H, Huang X, Wang S, Liu Z, Dong R, Song D, et al. Metformin ameliorates bleomycin-induced pulmonary fibrosis in mice by suppressing IGF-1. Am J Transl Res. 2020;12:940–949. [PMC free article] [PubMed] [Google Scholar]
- 76. Gamad N, Malik S, Suchal K, Vasisht S, Tomar A, Arava S, et al. Metformin alleviates bleomycin-induced pulmonary fibrosis in rats: pharmacological effects and molecular mechanisms. Biomed Pharmacother. 2018;97:1544–1553. doi: 10.1016/j.biopha.2017.11.101. [DOI] [PubMed] [Google Scholar]
- 77. Salomon JJ, Ehrhardt C. Organic cation transporters in the blood-air barrier: expression and implications for pulmonary drug delivery. Ther Deliv. 2012;3:735–747. doi: 10.4155/tde.12.51. [DOI] [PubMed] [Google Scholar]
- 78. Ota S, Horigome K, Ishii T, Nakai M, Hayashi K, Kawamura T, et al. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem Biophys Res Commun. 2009;388:311–316. doi: 10.1016/j.bbrc.2009.07.164. [DOI] [PubMed] [Google Scholar]
- 79. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Spagnolo P, Kreuter M, Maher TM, Wuyts W, Bonella F, Corte TJ, et al. Metformin does not affect clinically relevant outcomes in patients with idiopathic pulmonary fibrosis. Respiration. 2018;96:314–322. doi: 10.1159/000489668. [DOI] [PubMed] [Google Scholar]
- 81. Kreuter M, Lederer DJ, Cottin V, Kahn N, Ley B, Vancheri C, et al. Concomitant medications and clinical outcomes in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54:1901188. doi: 10.1183/13993003.01188-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021:S1550-4131(21)00057-7. doi: 10.1016/j.cmet.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]