

Abstract
Obesity elevates the plasma level of leptin, which has been associated with hypertension. Our recent studies in mice demonstrated that leptin increases blood pressure by activating the carotid sinus nerve, which transmits the chemosensory input from carotid bodies (CBs) to the medullary centers, and that the effect of leptin is mediated via Trpm7 (TRP [transient receptor potential] melastatin 7) channels in CB glomus cells. We also found that Trpm7 overexpression and Trpm7 promoter demethylation in CBs correlate positively with the hyperleptinemia and leptin receptor overexpression in CBs. Hence, we postulated that leptin epigenetically regulates Trpm7 expression in CBs. We addressed our hypothesis by using rat adrenal pheochromocytoma (PC12) cells as a model of CB glomus cells. PC12 cells expressing LEPRb (long, active form of leptin receptor) showed dramatic induction of the promoter activity and expression of Trpm7 upon leptin treatment. The increased Trpm7 expression coincided with the reduction of CpG site–specific methylation and trimethylation of H3K27 (H3 [histone 3] K27 [lysine 27]) and the increase of acetylation of H3K27 and trimethylation of H3K4 (H3 lysine 4) at the Trpm7 promoter. The inhibitor of STAT3 (signal transducer and activator of transcription 3) signaling, SD1008, reversed the leptin-induced Trpm7 promoter activity via modulations of the binding of pSTAT3 (phosphorylated STAT3) and DNMT3B (DNA methyltransferase 3B) and modifications of H3K27 and H3K4 at the Trpm7 promoter. Our results suggest that leptin-activated pSTAT3 epigenetically regulates the transcription of Trpm7 through DNA methylation and histone modifications. Because epigenetic changes are reversible, targeting epigenetic modifications of Trpm7 may serve as a new therapeutic approach for the treatment of hypertension in obesity.
Keywords: leptin, TRPM7, DNA methylation, histone modifications, LEPRb
Clinical Relevance
Because epigenetic changes are reversible, targeting epigenetic modifications of Trpm7 (TRP [transient receptor potential] melastatin 7) in carotid bodies may serve as a new therapeutic approach for the treatment of hypertension in obesity.
Hypertension in obesity is linked to high levels of circulating leptin (1–3). We have shown that leptin acts in the carotid bodies (CBs) to regulate blood pressure (4). CBs are chemoreceptors located at the bifurcation of the common carotid artery, which function as the homeostatic, oxygen-sensing system regulating the hypoxic ventilatory response and carotid sinus nerve activity (5). We and others demonstrated that leptin acts on the hypoxia-sensing glomus cells of CBs via the LEPRb (long, active form of leptin receptor) and its downstream intracellular signaling pathways (6, 7). Shirahata and colleagues provided the first evidence that the leptin-augmented carotid sinus nerve response to hypoxia is mediated via the nonspecific cation TRP (transient receptor potential) channels (8). The TRP superfamily consists of six subfamilies in mammals: TRPC, TRPV, TRPM, TRPA, TRPP, and TRPML (9). Among these Trp cation channels, Trpm7 (TRP melastatin 7) is the most abundant transcript in CBs and is differentially expressed in mouse CBs and the petrosal ganglion compared with the superior cervical ganglion and the brain (8). TRPM7 is an ion channel for divalent cations with an intrinsic serine and threonine protein kinase activity that allows for transporting ions across the cell membrane and transducing signals to the cytosol (10). We have reported that leptin-induced hypertension is dependent on Trpm7 activity (4). More importantly, impairment of leptin signaling in the CBs of LEPRb-deficient db/db mice and leptin-deficient ob/ob mice is accompanied by hypermethylation of the Trpm7 promoter and decreased Trpm7 expression (4). Reintroduction of LEPRb to the CBs of db/db mice using a Leprb-carrying adenovirus restored Trpm7 expression and demethylated the Trpm7 promoter (4). These novel findings suggest that Trpm7 expression in CBs could be regulated epigenetically by a leptin-dependent mechanism. In other cell types, binding of leptin to LEPRb causes JAK2 (Janus kinase 2)-dependent phosphorylation of STAT3 (signal transducer and activator of transcription 3) (11). The pSTAT3 (phosphorylated STAT3) dimer translocates to the nucleus, where it binds to the gene promoter and activates transcription (12).
The goal of the present study was to identify the molecular mechanisms by which leptin regulates Trpm7 expression by using rat adrenal pheochromocytoma (PC12) cells stably expressing LEPRb (PC12LEPRb cells) (13). This model was chosen because PC12 cells 1) are oxygen sensitive and widely used in chemoreception studies and 2) express endogenous JAK2 (13) and high levels of Trpm7. We determined the effect of leptin on the transcription activity and epigenetic modifications (not limited to promoter methylation and histone methylation/acetylation) of the Trpm7 promoter. In addition, we examined the link between leptin-induced LEPRb/STAT3 signaling and epigenetic regulation of Trpm7 transcription.
Methods
Detailed methods are described in the online supplement.
Cell Culture
PC12LEPRb cells were cultured as described in Reference 13. Cells (∼70% confluency) were transfected with Luc (luciferase) reporters and/or treated with leptin (R&D Systems), SD1008 (Tocris Bioscience), or decitabine (5-Aza-2′-deoxycytidine [5-aza-dC]) (Tocris Bioscience) for 3 days.
Luc Reporter Assay
PC12LEPRb cells were transfected with pEZX-Luc-Trpm7 (a Luc reporter of Trpm7) (NC_005102 chr3−: 125872776–125871275, −1262 to +239 from transcription start site [TSS]) (number RPRM43179-PG02) or pEZX-Luc (an empty promoter Luc reporter) (number NEG-PG02) (Genecopoeia). Five hours after transfection, cells were treated with 1–100 ng/ml recombinant mouse leptin and/or 5 μM SD1008 for 3 days. Luc activity was assayed by using a Gaussia Luc Flash Assay Kit (Invitrogen) according to the manufacturer’s instructions. The relative luminescence units were normalized by using the total protein input.
Measurement of mRNA Level of Genes by Using Quantitative PCR
After treating the cells with leptin or 5-aza-dC for 3 days, RNA was extracted. Five hundred nanograms of total RNA was reverse transcribed by using iScript Reverse Transcriptase (Bio-Rad) before subjecting the RNA to SYBR Green–based quantitative PCR (qPCR). The 2−ΔΔCt method was used to calculate the relative expression ratio of the Trpm7 transcript that was normalized by housekeeping gene Rpl19. The qPCR primers are listed in Table E1 in the online supplement.
Bisulfite Sequencing
After treating the cells with leptin or 5-aza-dC for 3 days, genomic DNA was extracted and subjected to bisulfite treatment by using an EZ DNA Methylation Kit (Zymo Research) before bisulfite-sequencing PCR (BSPCR) amplification of the 5′ Trpm7 promoter. The PCR amplicon was subcloned into the pCR2.1 vector (Thermo Fisher Scientific) and sequenced. The BSPCR primers are listed in Table E1.
Chromatin IP and qPCR
Chromatin IP (ChIP) was performed by using the ChIP-IT Express Kit (Active Motif). The sheared chromatin was immunoprecipitated with antibodies against DNMT3B (DNA methyltransferase 3B) (EpiGentek), PolIIA (RNA polymerase IIA), acetylation of H3K27 (H3 [histone 3] K27 [lysine 27]) (H3K27Ac), trimethylation of H3K4 (H3 lysine 4) (H3K4M3) (Active Motif), trimethylation of H3K27 (H3K27M3) (Abcam), and pSTAT3 (Cell Signaling). DNA was released after reverse cross-linking and purification, which were followed by qPCR. One percent of starting chromatin was used as an input. The primers are listed in Table E1.
Statistical Analysis
Technical duplicates (from three independent sets of experiments) were included in all of the assays. Results were expressed as the mean ± SEM. An ordinary one-way ANOVA with Tukey multiple comparisons was used to determine whether the data between the treatment groups were statistically significant. All the data were analyzed and plotted with Prism 8.4 (GraphPad).
Results
Our previous study in mouse models revealed that the expression of Trpm7 in CBs is dependent on the availability of the LEPRb-dependent signaling pathway (4). We, herein, examined whether leptin regulates the transcriptional activity of Trpm7 in the in vitro model. PC12LEPRb cells were transfected with pEZX-Luc-Trpm7 or a control (CTL) vector (pEZX-Luc), which was followed by leptin treatment for 3 days. In the absence of leptin, PC12LEPRb cells transfected with pEZX-Luc-Trpm7 showed an induction of Trpm7 promoter activity as compared with cells transfected with the CTL vector, indicating that Trpm7 promoter activity was induced endogenously in PC12LEPRb cells. In response to 1 and 10 ng/ml leptin, the Trpm7 promoter activity increased significantly as compared with the untreated CTLs (Figure 1). Leptin at a low physiologic concentration (1 ng/ml) was able to activate Trpm7 transcription activity; therefore, this concentration was chosen for gene expression and epigenetic studies.
Figure 1.

Leptin-induced Trpm7 (TRP [transient receptor potential] melastatin 7) promoter activity in rat adrenal pheochromocytoma (PC12) cells expressing LEPRb (long, active form of leptin receptor) (PC12LEPRb cells). PC12LEPRb cells were transfected with pEZX-Luc-Trpm7 (a Luc [luciferase] reporter of Trpm7) or a control (CTL) vector (pEZX-Luc [an empty promoter Luc reporter]) followed by treatment with leptin at 1, 10, or 100 ng/ml for 3 days. Luc activity was examined. There was no change in luminescence in the PC12LEPRb cells transfected with CTL vector upon leptin treatment (data not shown). All data are represented as a ratio of relative luminescence units (RLU) of the pEZX-Luc-Trpm7 to the RLU of the CTL vector pEZX-Luc and are shown as mean ± SEM values. An ordinary one-way ANOVA with Tukey multiple comparisons was used to determine the significant differences between groups. *P < 0.05.
We have previously demonstrated the decrease in the Trpm7 mRNA level and hypermethylation of the Trpm7 promoter in CBs of db/db and ob/ob mice as compared with wild-type mice, suggesting an important role of leptin and LEPRb signaling in epigenetic regulation of Trpm7 (4). It led us to examine the effect of leptin on DNA methylation at the Trpm7 promoter in PC12LEPRb cells. Figure 2 shows that treatment of PC12LEPRb cells with 5-aza-dC effectively enhanced Trpm7 transcription and caused demethylation of Trpm7 promoter, suggesting that Trpm7 transcription is regulated by DNA methylation. Similar to 5-aza-dC treatment, leptin treatment increased the mRNA level of Trpm7 by 80% (Figure 2A). The increase in Trpm7 mRNA was concordant with a decrease in Trpm7 promoter methylation by 30% (Figure 2B). Bisulfite sequencing showed that leptin or 5-aza-dC caused similar DNA demethylation at CpG sites 1–3, 18–19, 28–29, and 31–32 (Figure 2C). In silico analysis (LASAGNA-Search) (14) revealed that these differentially methylated CpG sites were located at the TSS and at the putative binding sites for transcription factors such as pSTAT3 and the NF-κB p50 and p65 subunits. It is likely that leptin induces DNA demethylation at these CpG sites, allowing the binding of transcriptional activators to facilitate Trpm7 transcription.
Figure 2.

Leptin increased the Trpm7 mRNA level and demethylated the Trpm7 promoter. PC12LEPRb cells were treated with 1 ng/ml leptin or 0.5 μM 5-aza-dC for 3 days and were subjected to gene expression and DNA methylation analysis. (A) mRNA levels of Trpm7 assayed by using quantitative PCR (qPCR). (B) Average values of Met% were determined by using bisulfite sequencing. (C) Schematic diagram of the 5′ promoter region of Trpm7. In silico analysis identified putative transcription factor binding sites at the Trpm7 promoter. The methylation status of individual CpG sites at the Trpm7 promoter was analyzed by using bisulfite sequencing. Unmethylated (open circles) or methylated (solid circles) CpG sites are indicated. Each row of circles represents an individual clone sequenced. The boxed area illustrates the specific CpG sites showing differential methylation as compared with vehicle CTLs. Results shown in A and B are expressed as the mean ± SEM. An ordinary one-way ANOVA with Tukey multiple comparisons was used to determine the significant differences between groups. **P < 0.01 and ****P < 0.0001 compared with CTL. The qPCR-amplified regions for the chromatin IP assay (ChIP) for the Region 1 (R1) through R3 regions (data shown in Figures 3–4) are indicated by lines. 5-aza-dC = 5-Aza-2′-deoxycytidine; ATF6 = activating transcription factor 6; GATA3 = GATA binding protein 3; Met% = Trpm7 promoter methylation; p50 = NF-κB p50 subunit; p65 = NF-κB p65 subunit; pSTAT3 = phosphorylated STAT3; RER = relative expression ratio; STAT3 = signal transducer and activator of transcription 3; TSS = transcription start site; USF = upstream stimulatory factor.
Next, we explored the signaling mechanisms underlying the effect of leptin on Trpm7 transcription. Application of SD1008, a specific JAK2/STAT3 inhibitor (15), significantly reduced the leptin-induced increase in the Trpm7 mRNA level and the Trpm7 promoter activity to the level of vehicle CTLs (Figure 3A), suggesting that the transcription effect was indeed mediated by this signaling pathway. SD1008 diminished the change in leptin-induced Trpm7 promoter demethylation by ∼60%. SD1008 alone did not alter Trpm7 gene transcription in the absence of leptin. We further examined the binding of pSTAT3 and PolIIA at the differentially demethylated clusters of Trpm7 promoter regions (Region 1 [R1]: CpG sites 1–3, R2: CpG sites 18–19, and R3: CpG sites 28–29 and 31–32). Leptin induced a fivefold increase in pSTAT3 binding in the R1 region and a modest increase in the R2 region but had no effect on pSTAT3 binding in the R3 region (Figure 3C). In contrast, leptin caused a significant increase in PolIIA binding at the R3 region and caused a marginal increase at the R2 region but failed to induce PolIIA binding in the R1 region (Figure 3B). The leptin-induced pSTAT3 and PolIIA binding at the Trpm7 promoter were completely reversed by SD1008. These results clearly validated the specific binding sites of pSTAT3 and PolIIA at the Trpm7 promoter and indicated that the binding of both transcriptional activators is mediated by leptin and STAT3 activation.
Figure 3.
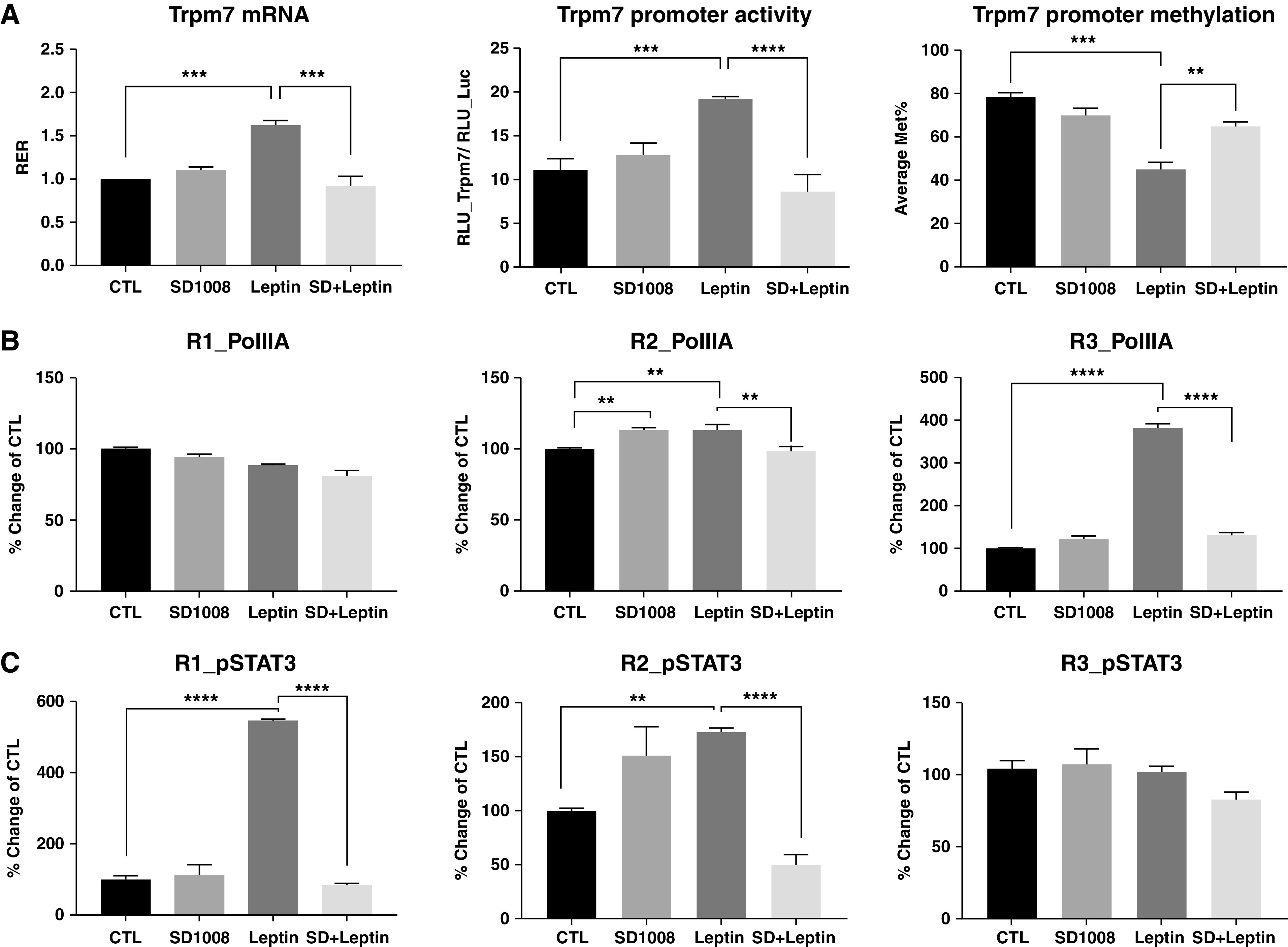
Blockade of STAT3 signaling decreased leptin induction of Trpm7 gene transcription, promoter activity, and promoter demethylation through alteration of the pSTAT3 binding in PC12LEPRb cells. (A) PC12LEPRb cells were treated with 1 ng/ml leptin and/or 5 μM SD1008 for 3 days. The Trpm7 mRNA level and promoter methylation were assessed by using qPCR and bisulfite sequencing, respectively. To measure promoter activity, PC12LEPRb cells were transfected with pEZX-Luc-Trpm7 or pEZX-Luc CTL vector, which was followed by treatment with 5 μM SD1008, 1 ng/ml leptin, or combined treatment with SD + Leptin for 3 days. Luc activity was examined. Data are represented as a ratio of the RLU of the pEZX-Luc-Trpm7 to the RLU of the CTL vector pEZX-Luc. Binding of (B) PolIIA (RNA polymerase IIA) and (C) pSTAT3 at R1, R2, and R3 clusters in the Trpm7 promoter were assayed by using ChIP-qPCR. PC12LEPRb cells were treated with 1 ng/ml leptin and/or 5 μM SD1008 for 3 days before the ChIP assay. Results are shown as the percent change in binding as compared with the PC12LEPRb CTLs. All data are present as mean ± SEM values. A one-way ANOVA with Tukey multiple comparisons was used to determine the significant differences between groups. **P < 0.01, ***P < 0.001, and ****P < 0.0001. SD + Leptin = combined treatment with SD1008 and leptin.
It is well recognized that histone modifications can act conjointly with DNA methylation and transcription activators/repressors to modulate gene transcription. We further examined the roles of histone modifications in the regulation of Trpm7 transcription. Because there are large numbers of histone modifications, we first determined whether leptin modulates the activity of histone modification enzymes, HAT (histone acetyltransferase) and HMT (histone methyltransferase), resulting in the covalent modification at specific histone variants (Table E2). Leptin significantly increased the activity of HAT and HMT at H3K4, whereas it decreased activity of HMT at H3K27 (by more than 25%). In particular, the induction of H3K4M3 and H3K27Ac by leptin could be reversed by SD1008. In contrast, leptin decreased H3K27M3, which was reversed by SD1008. In addition, leptin treatment caused a reduction in the activity of DNMT3B by 50%. On the basis of these results, we focused on the enrichment of H3K27Ac, H3K4M3, H3K27M3, and DNMT3B in the Trpm7 promoter in response to leptin. Figure 4 shows the effects of leptin and SD1008 on histone modifications and DNMT3B binding at the regions of CpG site–specific demethylation as determined by using ChIP-qPCR. At R1, the region where pSTAT3 binds, leptin caused significant induction of H3K27Ac and reduction of H3K4M3 and DNMT3B binding but had no effect on H3K27M3 (Figure 4A). These responses were reversed by SD1008. Leptin also caused significant demethylation of H3K27 and disrupted DNMT3B binding at the R2 region (Figure 4B) but had no effect on H3K4M3 and H3K27Ac. Application of SD1008 to the leptin-treated cells caused a significant reduction in H3K4M3 and H3K27Ac at R2. These results suggest that leptin enhances the binding of pSTAT3 and PolIIA (Figures 3B–3C) while decreasing binding of DNMT3B and H3K27M3, resulting in an active chromatin at this R2 region. Furthermore, leptin also caused significant epigenetic modifications at the R3 region (CpG sites 28–32) where the TSS is encompassed (Figure 4C). Leptin treatment of PC12LEPRb cells caused a dramatic increase in H3K4M3 at the R3 region, and the response was completely reversed by SD1008. However, there was no change in the methylation of H3K27 or in H3K27Ac in response to leptin and/or SD1008. There was a significant loss of DNMT3B binding by leptin at this region. SD1008 was not able to reverse this loss but caused a further decrease in DNMT3B binding at this region. Although we did not fully understand the mechanisms underlying the reduction of DNMT3B binding by the combination of SD1008 and leptin, this change did not lead to a further reduction in overall DNA methylation of Trpm7 (Figure 3A). These results suggest that leptin may cause posttranslational modification of H3K4 (H3K4M3) together with the recruitment of PolIIA at this region to facilitate Trpm7 transcription.
Figure 4.
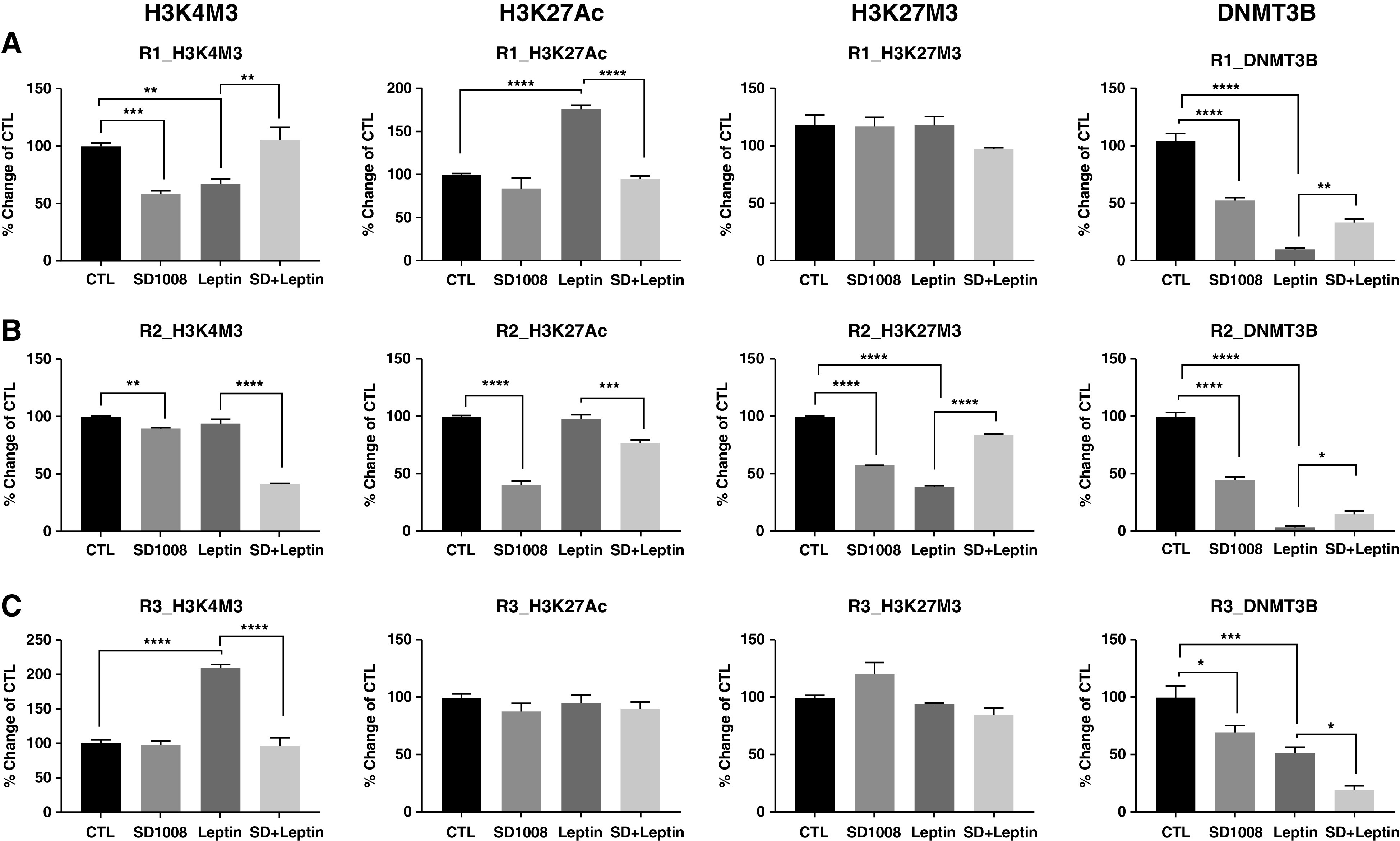
Leptin modulated H3K4M3, H3K27Ac, H3K27M3, and binding of DNMT3B (DNA methyltransferase 3B) at the Trpm7 promoter. PC12LEPRb cells were treated with 1 ng/ml leptin and/or 5 μM SD1008 for 3 days. Epigenetic modifications at (A) R1, (B) R2, and (C) R3 of the Trpm7 promoter were examined by using ChIP-qPCR. Results are shown as the percent change in enrichment as compared with the PC12LEPRb CTLs and are presented as mean ± SEM values. A one-way ANOVA with Tukey multiple comparisons was used to determine the significant differences between groups. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. H3 = histone 3; H3K4M3 = trimethylation of H3K4; H3K27Ac = acetylation of H3K27; H3K27M3 = trimethylation of H3K27; K4 = lysine 4; K27 = lysine 27.
Discussion
We have previously reported that TRPM7 protein expression was colocalized with LEPRb in glomus cells of CBs for mediating leptin-induced hypertensive effects (4). The expression and promoter methylation status of Trpm7 in CBs is dependent on the availability of leptin and LEPRb. In the present study, we use the unique PC12LEPRb cell model to delineate the signaling pathway and transcription factors, the CpG site–specific methylation, and the types of histone modification that are responsible for leptin-induced Trpm7 transcription. Our key findings are 1) that leptin induced Trpm7 expression through the STAT3 signaling pathway; 2) that it promoted demethylation of the Trpm7 promoter at specific CpG sites putative for pSTAT3, p50, and p65 binding and increased binding of pSTAT3 and PolIIA to specific Trpm7 promoter regions; 3) that leptin facilitated H3K27Ac and suppressed DNMT3B binding at the pSTAT3 binding regions; and 4) that leptin induced H3K4M3 and H3K27 demethylation and reduced DNMT3B binding at the PolIIA binding regions. Taken together, these findings have demonstrated that leptin induces epigenetic modifications through promoter demethylation and posttranslational modifications of histone proteins, resulting in the increase of Trpm7 transcription. These effects of leptin on the epigenetic regulation of Trpm7 transcription are mediated by LEPRb-dependent STAT3 activation.
There is mounting evidence suggesting that leptin-induced transcription regulation of a wide spectrum of genes occurs through epigenetic mechanisms, including posttranslational modifications of histone proteins, DNA methylation, and microRNAs, which play a critical role in the pathogenesis of metabolic disorders (16). However, the mechanism of leptin-induced Trpm7 transcription has not been examined. Our results demonstrated for the first time that leptin-induced Trpm7 expression is mediated by activation of STAT3 and that its efficacy of binding to the Trpm7 promoter is tightly regulated by DNA methylation and histone modification. This notion is supported by the evidence that the pSTAT3 inhibitor SD1008 completely abolished leptin-induced Trpm7 gene transcription and binding of pSTAT3 to the promoter (Figure 3A, 3C). The pSTAT3-dependent Trpm7 transcription is facilitated by CpG site–specific DNA demethylation. Leptin or 5-aza-dC treatment selectively demethylated multiple specific CpG sites in the Trpm7 promoter, including a specific CpG site cluster (in the R1 region) with the putative pSTAT3 binding motif (Figure 2C). The increase in binding was completely reversed in the presence of SD1008 (Figure 3C). Demethylation of these CpG sites could be explained by the reduced binding of the de novo DNA methyltransferase DNMT3B (Figure 4). This would allow the binding of transcription activators and chromatin remodeling proteins to initiate Trpm7 transcription. It is noted that SD1008 has no effect on Trpm7 transcription or pSTAT3 binding without leptin treatment. This is presumably due to the hypermethylation status in the R1 region of the untreated CTL PC12LEPRb cells, suggesting the presence of an inactive chromatin for transcription (Figure 2C). When the cells are exposed to leptin, CpG site–specific demethylation occurs and allows pSTAT3 binding to facilitate Trpm7 transcription.
pSTAT3 binding at the promoter may also recruit other transcription activators and epigenetically modified proteins to further facilitate gene transcription (17). Our results show that leptin-induced pSTAT3 binding was accompanied by the binding of PolIIA, which is required for the transcription of all protein coding genes. The binding of PolIIA at the R2 region in close proximity to the enhancer is associated with the formation of a preinitiation complex for gene transcription composed of transcriptional factors and cofactors at the core promoter (18). Moreover, leptin can effectively induce posttranslational modifications of histone proteins in these binding regions (16). Histone modifications could lead to chromatin conformational changes that affect the accessibility of the chromatin (19). H3K27Ac and H3K4M3 are active enhancer marks, commonly associated with the activation of nearby genes (20). H3K27M3 at the promoter region is associated with downregulation of gene expression, and it is recognized as another major epigenetic silencing mechanism in addition to DNA methylation (21). We showed that leptin induced H3K4M3, demethylation of H3K27, and H3K27Ac at the Trpm7 promoter, which could result in the relaxation of chromatin to enhance Trpm7 transcription. It is noteworthy that SD1008 completely reversed the leptin-induced H3K27Ac at the R1 region and H3K27M3 at the R2 region and partially reversed the disruption of DNMT3B binding in the R1 and R2 regions. The H3K4 demethylation at the R1 region by leptin is unexpected, as we observed the increase in Trpm7 promoter activity (Figure 4A). However, we could argue that the significant change of H3K4 methylation at this region (R1) may not be critical for Trpm7 transcription. The binding of other transcription activators such as pSTAT3 may interfere the modification of the H3K4 residue. At the R3 region where the TSS is located, we observed that SD1008 could reverse the leptin-induced H3K4M3 and PolIIA binding. These findings suggest that the above processes are dependent on the activation of STAT3 and that the epigenetic mechanisms (DNA methylation and histone modifications) work conjointly with transcription activators (pSTAT3 and PolIIA) to activate Trpm7 transcription.
In addition to pSTAT3, activation of LEPRb signaling may facilitate other kinase signaling pathways (e.g., PI3K, ERK, and AMPK) and transcriptional factors (e.g., NFκB and SOCS3) (22), which may contribute to the active transcription of Trpm7. SD1008 did not fully reverse leptin-induced Trpm7 promoter demethylation and DNMT3B binding (Figure 3A-C), indicating the contribution of alternative transcription factors to promoter demethylation. For example, our in silico analysis and bisulfite sequencing revealed a putative binding site of NFκB subunits in the R2 region of the Trpm7 promoter (CpG sites 18–19), where leptin-induced demethylation occurs. Nevertheless, inhibition of STAT3 by SD1008 could completely block leptin-induced Trpm7 transcription (Figure 3A) and histone modifications (Figure 4), suggesting that pSTAT3 is the key player for the epigenetic regulation of Trpm7 transcription. SD1008 is known to inhibit SRC (nonreceptor tyrosine kinase), in addition to JAK2 and STAT3. It has been suggested that Src family members are candidates for the non-JAK2 kinases that also mediate leptin signaling by activating STAT3 (13). Therefore, it is possible that SD1008 inhibits pSTAT3 via JAK2 and/or non-JAK2 kinases. Other transcription factors such as NFκB may act synergistically, with STAT3 signaling, to further facilitate Trpm7 transcription. The role of LEPRb-mediated signaling pathways in Trpm7 transcription and the cross-talk between these signaling pathways warrant further future investigations.
In summary, our results show that the Trpm7 promoter is hypermethylated in the absence of leptin and that DNMT3B binding and H3K27M3 contribute to the promoter methylation and gene silencing. When PC12LEPRb cells are exposed to leptin, leptin binds to LEPRb and activates STAT3 signaling. The binding of pSTAT3 cooperates with epigenetic mechanisms, CpG site–specific DNA demethylation, H3K27Ac, and H3K4M3, which ultimately activate Trpm7 transcription (Figure 5). We were the first group to report leptin-induced hypertension through the TRPM7 channel (4). In this study, we further demonstrated the link between leptin-mediated LEPRb/STAT3 signaling and epigenetic regulation of Trpm7 transcription. In the future, we may use the PC12 cell model to determine whether epigenetic regulation of Trpm7 (by using modifiers for DNMT, HMT, HAT, and HDAC or methylated oligonucleotides targeting CpG site–specific methylation of the Trpm7 promoter) could modify the CB activity. Moreover, we may apply the pharmaceutical interventions of epigenetic or STAT3 signaling modulators on the in vivo model as described by Shin and colleagues (4) for modulating the CB activity and leptin-mediated hypoxic ventilatory response. Epigenetic modifiers may serve as a new therapeutic approach for suppression of TRPM7, which in turn modulates leptin-mediated hypertension.
Figure 5.
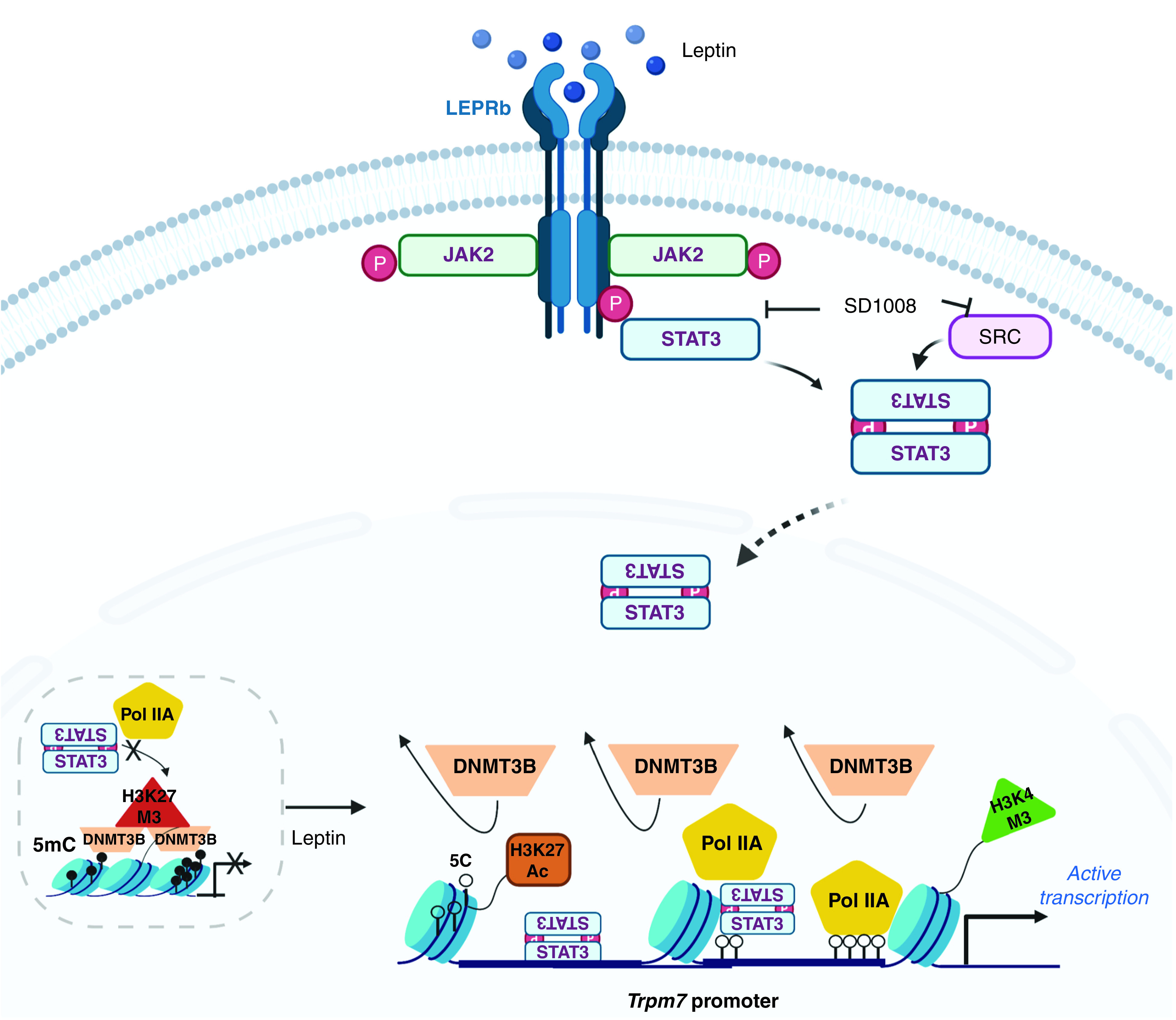
A schematic of the epigenetic mechanisms underlying Trpm7 transcription in response to leptin. 5C = 5-cytosine; 5mC = 5-methylcytosine; JAK2 = Janus kinase 2; P = phosphorylation; SRC = nonreceptor tyrosine kinase.
Footnotes
Supported by National Heart, Lung, and Blood Institute grant R01 HL133100 (V.Y.P. and J.S.K.S.).
Author Contributions: Participated in research design: B.H.-y.Y., J.S.K.S., V.Y.P., and W.-y.T. Conducted experiments: B.H.-y.Y., K.G., L.B., O.P., and W.-y.T. Performed data analysis: B.H.-y.Y. and W.-y.T. Contributed reagents: L.R., J.S.K.S., V.Y.P., and W.-y.T. Wrote or contributed to the manuscript: B.H.-y.Y., M.-K.S., J.S.K.S., V.Y.P., and W.-y.T.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0374OC on April 23, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Ma D, Feitosa MF, Wilk JB, Laramie JM, Yu K, Leiendecker-Foster C, et al. Leptin is associated with blood pressure and hypertension in women from the National Heart, Lung, and Blood Institute Family Heart Study. Hypertension. 2009;53:473–479. doi: 10.1161/HYPERTENSIONAHA.108.118133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 3. Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 4. Shin MK, Eraso CC, Mu YP, Gu C, Yeung BHY, Kim LJ, et al. Leptin induces hypertension acting on transient receptor potential melastatin 7 channel in the carotid body. Circ Res. 2019;125:989–1002. doi: 10.1161/CIRCRESAHA.119.315338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weir EK, López-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porzionato A, Rucinski M, Macchi V, Stecco C, Castagliuolo I, Malendowicz LK, et al. Expression of leptin and leptin receptor isoforms in the rat and human carotid body. Brain Res. 2011;1385:56–67. doi: 10.1016/j.brainres.2011.02.028. [DOI] [PubMed] [Google Scholar]
- 7. Caballero-Eraso C, Shin M-K, Pho H, Kim LJ, Pichard LE, Wu Z-J, et al. Leptin acts in the carotid bodies to increase minute ventilation during wakefulness and sleep and augment the hypoxic ventilatory response. J Physiol. 2019;597:151–172. doi: 10.1113/JP276900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shirahata M, Tang WY, Shin MK, Polotsky VY. Is the carotid body a metabolic monitor? Adv Exp Med Biol. 2015;860:153–159. doi: 10.1007/978-3-319-18440-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12:218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yee NS, Kazi AA, Yee RK. Cellular and developmental biology of TRPM7 channel-kinase: implicated roles in cancer. Cells. 2014;3:751–777. doi: 10.3390/cells3030751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bjørbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 12. Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 13. Jiang L, Li Z, Rui L. Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J Biol Chem. 2008;283:28066–28073. doi: 10.1074/jbc.M805545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee C, Huang CH. LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques. 2013;54:141–153. doi: 10.2144/000113999. [DOI] [PubMed] [Google Scholar]
- 15. Duan Z, Bradner J, Greenberg E, Mazitschek R, Foster R, Mahoney J, et al. 8-Benzyl-4-oxo-8-azabicyclo[3.2.1]oct-2-ene-6,7-dicarboxylic acid (SD-1008), a novel Janus kinase 2 inhibitor, increases chemotherapy sensitivity in human ovarian cancer cells. Mol Pharmacol. 2007;72:1137–1145. doi: 10.1124/mol.107.038117. [DOI] [PubMed] [Google Scholar]
- 16. Wróblewski A, Strycharz J, Świderska E, Drewniak K, Drzewoski J, Szemraj J. et al. Molecular insight into the interaction between epigenetics and leptin in metabolic disorders. Nutrients. 2019;11:1–23. doi: 10.3390/nu11081872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kruczyk M, Przanowski P, Dabrowski M, Swiatek-Machado K, Mieczkowski J, Wallerman O, et al. Integration of genome-wide of Stat3 binding and epigenetic modification mapping with transcriptome reveals novel Stat3 target genes in glioma cells. Biochim Biophys Acta. 2014;1839:1341–1350. doi: 10.1016/j.bbagrm.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 18. Wang J, Zhao S, He W, Wei Y, Zhang Y, Pegg H, et al. A transcription factor IIA-binding site differentially regulates RNA polymerase II-mediated transcription in a promoter context-dependent manner. J Biol Chem. 2017;292:11873–11885. doi: 10.1074/jbc.M116.770412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. 2013;58:439–445. doi: 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- 21. Igolkina AA, Zinkevich A, Karandasheva KO, Popov AA, Selifanova MV, Nikolaeva D, et al. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 histone tags suggest distinct regulatory evolution of open and condensed chromatin landmarks. Cells. 2019;8:1034. doi: 10.3390/cells8091034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci. 2011;16:2771–2793. doi: 10.2741/3885. [DOI] [PubMed] [Google Scholar]