

Abstract
Cystic fibrosis (CF) is characterized by chronic airway infection, inflammation, and tissue damage that lead to progressive respiratory failure. NLRP3 and NLRC4 are cytoplasmic pattern recognition receptors that activate the inflammasome, initiating a caspase-1–mediated response. We hypothesized that gain-of-function inflammasome responses are associated with worse outcomes in children with CF. We genotyped nonsynonymous variants in NLRP3 and the NLRC4 pathway from individuals in the EPIC (Early Pseudomonas Infection Control) Observational Study cohort and tested for association with CF outcomes. We generated knockouts of NLRP3 and NLRC4 in human macrophage–like cells and rescued knockouts with wild-type or variant forms of NLRP3 and NLRC4. We identified a SNP in NLRP3, p.(Q705K), that was associated with a higher rate of P. aeruginosa colonization (N = 609; P = 0.01; hazard ratio, 2.3 [Cox model]) and worsened lung function over time as measured by forced expiratory volume in 1 second (N = 445; P = 0.001 [generalized estimating equation]). We identified a SNP in NLRC4, p.(A929S), that was associated with a lower rate of P. aeruginosa colonization as part of a composite of rare variants (N = 405; P = 0.045; hazard ratio, 0.68 [Cox model]) and that was individually associated with protection from lung function decline (P < 0.001 [generalized estimating equation]). Rescue of the NLRP3 knockout with the p.(Q705K) variant produced significantly more IL-1β in response to NLRP3 stimulation than rescue with the wild type (P = 0.020 [Student’s t test]). We identified a subset of children with CF at higher risk of early lung disease progression. Knowledge of these genetic modifiers could guide therapies targeting inflammasome pathways.
Keywords: NLRP3, NLRC4, inflammasome, cystic fibrosis
Clinical Relevance
This research identifies a subset of children with cystic fibrosis who are at higher risk of early lung disease progression. Knowledge of these genetic modifiers could guide therapies targeting inflammasome pathways.
The cystic fibrosis (CF) lung is susceptible to organisms that produce chronic infection, but the ensuing inflammatory response is often unable to eliminate the infection and is responsible for much of the pathologic damage (1–4). To date, there is no widely adopted antiinflammatory therapy for people with CF (5). Host-directed therapies, including corticosteroids (e.g., for maintenance and for pulmonary exacerbations) and nonsteroidal antiinflammatory drugs (e.g., high-dose ibuprofen for maintenance care), have been used to treat CF lung disease with some success, but overall progress in mitigating the inflammatory damage has been modest (6). Understanding which inflammatory processes are beneficial and which are harmful is critical to improving the efficacy of host-directed therapeutics in patients with CF lung disease.
Inflammasomes are cytoplasmic protein complexes that sense microbial patterns and oligomerize to recruit and cleave procaspase 1. Active caspase-1 initiates a downstream inflammatory response that includes the secretion of IL-1β, IL-18, and inflammatory eicosanoids as well as the formation of gasdermin pores in the cell membrane, resulting in a form of programmed cell death called pyroptosis and possibly leading to gasdermin-mediated bacterial killing (7–9). Activation of the canonical inflammasome pathway requires two signals (10). Signal 1 is usually mediated by TLR (Toll-like receptor)/NF-κB signaling and upregulates expression of pro–IL-1β. Signal 2 is inflammasome specific and is produced by a variety of pathogen-associated molecular patterns and damage-associated molecular patterns (11). The NLRC4 inflammasome recognizes intracellular flagellin and components of the bacterial type 3 secretion system (12, 13). The NLRP3 inflammasome is activated by a variety of mechanisms, including potassium efflux and mitochondrial reactive oxygen species generation (11, 14). Pseudomonas species can activate both the NLRC4 and the NLRP3 inflammasome, although the process is complex because bacterial exotoxins can also inhibit inflammasome activation (15, 16). Recent evidence suggests that in neutrophils, inflammasomes are constitutively active and require only activation of the TLR/NF-κB pathway (17). Although inflammasome activation can be protective, the inflammatory response can also be harmful to host tissues. Understanding this balance in CF is crucial to the understanding risks and benefits of potential therapeutic agents.
Inflammasome-mediated inflammation leads to lung injury in several mouse models (8, 18, 19). In mice with CF, inflammasome activity is dysregulated, and IL-1β contributes to lung pathology (20). The use of a small molecule inhibitor of NLRP3 in mice significantly reduced levels of IL-1β in the lung and improved clearance of P. aeruginosa (17). In a mouse model of P. aeruginosa pneumonia, macrophage depletion or deletion of NLRC4 enhanced bacterial clearance and reduced pathologic damage to the lung (21). Aspergillus species are commonly isolated from the lungs of patients with CF and may be associated with worsened outcomes (22). Aspergillus species induce inflammasome activation in mice (20), and the suppression of NLRP3 reduces lung damage in a mouse model of invasive pulmonary aspergillosis (23).
In humans, genetic variants in inflammasomes, particularly in NLRP3, are associated with a range of autoinflammatory conditions collectively referred to as the cryopyrin-associated periodic syndromes (CAPSs) (24). The CAPS disorders are caused by gain-of-function mutations in NLRP3 that display variable penetrance and are characterized primarily by symptoms of an innate immune response, including fever, rash, and systemic inflammation, without evidence of autoantibodies or antigen-specific T cells (25). Less is known about inflammasome activity in humans with CF. BAL samples from people with CF have higher levels of proinflammatory cytokines, including IL-1β, than BAL samples from age-matched healthy control subjects (26), and higher IL-1β levels are associated with worse clinical outcomes, including those for lung function (17). In human monocytes and airway epithelia, CF-associated mutations result in dysregulation of epithelial sodium channels, leading to NLRP3-mediated inflammation (27). In a case series of CF lung explants, P. aeruginosa from segments of the lung with radiographic evidence of increased lung damage were found to hypersecrete the type 3 secretion system components (28). P. aeruginosa colonization is common in children with CF and is associated with poor outcomes (29, 30), although whether chronic P. aeruginosa clinical isolates are able to activate the inflammasome in adults is not clear (31).
In the following study, we examine candidate genetic variants in the NLRP3 and NLRC4 inflammasome pathway for association with chronic P. aeruginosa infection and lung function in a large cohort of children with CF. We identify two variants in NLRP3 and one variant in NLRC4 with significant associations with clinical outcomes in this cohort. We then study the mechanistic impact of these variants in human macrophage–like cells to test our hypothesis that hyperinflammatory inflammasome variants are harmful in CF and that hypoinflammatory variants are protective.
Methods
EPIC Observational Study Cohort
The EPIC (Early Pseudomonas Infection Control) Observational Cohort consists of 1,794 children with CF enrolled at 59 recruitment centers in the United States (32). Two P. aeruginosa entry criteria were used: criterion 1 includes children who had never had a P. aeruginosa–positive respiratory culture result (“never P. aeruginosa” group). Criterion 2 includes children who had had a positive P. aeruginosa respiratory culture result in the past but who had had negative respiratory culture results for the two years preceding enrollment (“past P. aeruginosa” group) (32).
Statistical Analysis
Outcomes included age of onset of chronic P. aeruginosa (33, 34) and longitudinal forced expiratory volume in 1 second (FEV1) based on Global Health Initiative prediction equations. We performed association tests for those with the wild type (WT) versus those who were variant carriers by using the Cox model for the age of chronic P. aeruginosa onset and by using generalized estimating equations for differences in longitudinal FEV1. Analyses were performed separately by using enrollment criteria because of the high potential for criteria to differentially select for underlying genetic factors and were performed by using both groups combined when no difference between groups was found.
Genotyping and SNP Selection
Genotyping was performed for EPIC individuals in two phases; whole exome sequencing (n = 189) and exome chip sequencing (N = 989) were performed during the Exome Sequencing Project (34), which were followed by a second phase of targeted sequencing using single-molecule molecular inversion probes for candidate genes (n = 1,201 with complete data). The NLRP3 variants p.(V198M) and p.(Q705K) were selected on the basis of existing data suggesting their association with the autoinflammatory condition CAPS (24, 35). For NLRC4, pathway analysis using phase 1 data was performed with a variable comprising the presence or absence of the uncommon allele of any of 14 variants in the NLRC4 pathway (NAIP, PYCARD, NLRC4, and CASP1 [13]; see Table E1 in the data supplement).
Cell Lines, Genetic Manipulation, and Inflammasome Assays
We used CRISPR/Cas9 to generate NLRP3 and NLRC4 knockouts in the human monocyte–like cell lines THP-1 and U937. Gene rescue (“knockin”) was performed by reintroducing either the WT or variant version of the knocked-out gene. Western blotting was performed to quantify protein expression (Figure E1). For inflammasome assays, cell lines were plated in PMA (50 ng/ul) for 48 hours, washed with Hanks’ balanced salt solution, and rested overnight in media (RPMI with 10% FBS). For NLRP3-specific stimulation, cells were incubated with nigericin (10 μm) for 4 hours after 2-hour prestimulation with LPS (0.1 ng/ml). For NLRC4-specific stimulation, a fusion of Burkholderia thailandensis needle protein with the Bacillus anthracis lethal factor was cytoplasmically delivered via the B. anthracis protective antigen (gifts of Russel Vance, University of California Berkeley) for 4 hours. Supernatant cytokines were measured by using a sandwich ELISA (R&D Systems).
P.aeruginosa macrophage assays
The P. aeruginosa laboratory strain O1 with a multiplicity of infection of 5 (PAO1) (gift of Matthew Parsek, University of Washington) or a clinical isolate (gift of Pradeep Singh, University of Washington) was picked from a single colony on a freshly streaked Luria broth plate and grown overnight in Luria broth media. The overnight culture was back-diluted to an optical density of 0.01 and grown to log-phase, at which point cell lines were exposed to bacteria at the indicated multiplicity of infection.
For further details, see the data supplement.
Results
Association of NLRP3 Variants with CF Outcomes in the EPIC Cohort
To examine whether inflammasome variants are associated with clinical outcomes in CF, we studied genetic variants in NLRP3 and the NLRC4 pathway for association with clinical outcomes in the EPIC cohort. Enrolled children either had never had a an airway (e.g., oropharyngeal or sputum) culture positive for P. aeruginosa (never P. aeruginosa group) or had had an airway culture positive for P. aeruginosa in the past but had cleared their cultures for the two years preceding enrollment (past P. aeruginosa group). We used a hypothesis-driven approach to determine whether SNPs with suspected hyperinflammatory phenotypes influenced clinical outcomes in CF. Of the CAPS-associated rare variants we included in our analysis (Table E2), two were present in the EPIC cohort: p.(Q705K) and p.(V198M). The p.(Q705K) (rs35829419) variant was associated with a higher rate of chronic P. aeruginosa infection (N = 609; P = 0.01; hazard ratio [HR], 2.3 [at age 10, Cox model]) in children from the past P. aeruginosa enrollment group (Figure 1A). The p.(Q705K) variant was also associated with worsened lung function over time as measured by the FEV1 (N = 445; P = 0.001 [generalized estimating equation]) in enrollees from the past P. aeruginosa group (Figure 1B). A second nonsynonymous rare variant in NLRP3, p.(V198M) (rs121908147), was not associated with rate of P. aeruginosa colonization but showed a trend toward a protective effect (N = 1,348; P = 0.07; HR, 0.50 [Cox model]) in the combined enrollment group averaged over all ages (Figure 1C). The p.(V198M) SNP was not associated with lung function over time in the EPIC cohort (data not shown).
Figure 1.
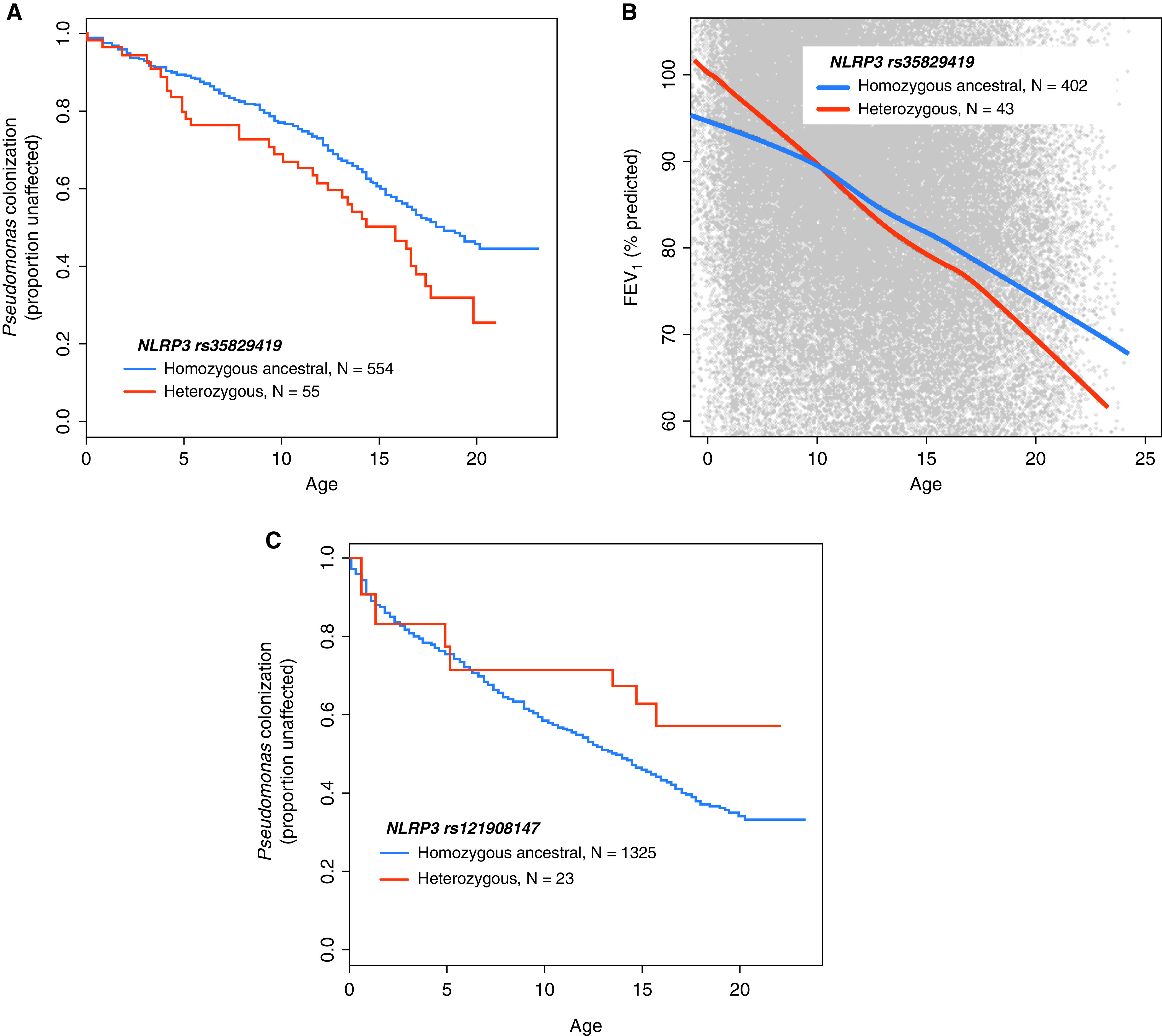
The p.(Q705K) and p.(V198M) NLRP3 variants and clinical outcomes in the EPIC (Early Pseudomonas Infection Control) cohort. (A) Proportion of individuals in the EPIC cohort without P. aeruginosa in the sputum by age based on the rs35829419 [p.(Q705K)] genotype (N = 609; P = 0.01; hazard ratio [HR], 2.3 [at age 10, Cox model, “past P. aeruginosa” enrollees only]). (B) The forced expiratory volume in 1 second (FEV1) percent predicted relative to the Global Lung Initiative standard by age based on the rs35829419 genotype (N = 445; P = 0.001 [generalized estimating equation, past P. aeruginosa enrollees only]). (C) Proportion of individuals in the EPIC cohort without P. aeruginosa in the sputum by age based on the rs121908147 [p.(V198M)] genotype (N = 1,348; P = 0.07; HR, 0.50 [Cox model]). The model was adjusted for age at enrollment, CFTR residual function, and CAV2 rs8940 status. CAV2 = caveolin 2; CFTR = cystic fibrosis transmembrane conductance regulator; NLRP3 = NLR family pyrin domain containing 3; P. aeruginosa = Pseudomonas aeruginosa.
Association of an NLRC4 Rare Variant with CF Outcomes in the EPIC Cohort
To further understand the relationship between the inflammasome and CF outcomes, we investigated a group of variants in the NLRC4 pathway. Most variants were synonymous and had a minor allele frequency less than 5% in the EPIC cohort (Table E1); they included polymorphisms in CASP1, NAIP, NLRC4, and ASC. To increase our statistical power for the initial analysis, we collapsed all variants into a single variable on the basis of the presence of any single variant. The presence of any of 14 rare variants in the NLRC4 pathway was associated with a delayed time to chronic P. aeruginosa infection (P = 0.045; HR, 0.68 [Cox model]; N = 405; adjusted for age at enrollment and CFTR residual function; Figure 2A) among individuals with a high-risk CAV2 genotype (WT at locus rs8940). The sample size was large enough for individual variant testing of the FEV1 outcome for the A929S NLRC4 variant (rs61754192). We examined the NLRC4 A929S variant for association with lung function among EPIC enrollees from the never P. aeruginosa group and found that the presence of this variant was associated with higher FEV1 percentiles (difference = +8.2%; P < 0.001 [generalize estimating equation]) than those of CF children without a pathway variant (Figure 2B).
Figure 2.
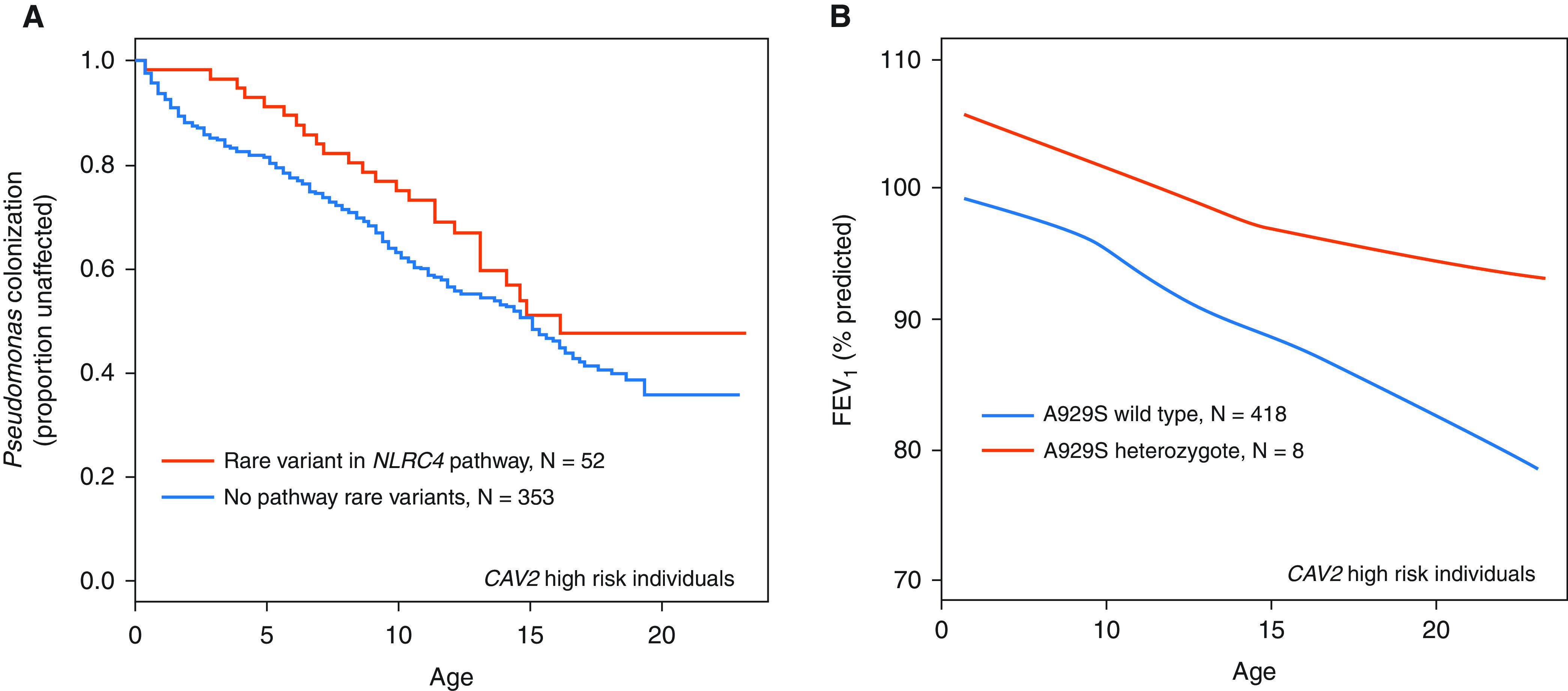
The p.(A929S) NLRC4 variant and clinical outcomes in the EPIC cohort. (A) Proportion of individuals without P. aeruginosa in the sputum by age based on a yes/no composite for the presence of any one of 14 rare nonsynonymous variants in NLRC4, NAIP, CASP1, and ASC (N = 405; P = 0.045; HR, 0.68 [Cox model]). Individuals were from the “never P. aeruginosa” enrollment group and had the high-risk CAV2 genotype (rs8940, wild type [WT]). (B) The FEV1 percent predicted relative to the Global Lung Initiative standard by age based on the rs61754192 [p.(A929S)] genotype (difference = +8.2%; P < 0.001 [generalized estimating equation]). ASC = apoptosis-associated speck-like protein containing a CARD; NAIP = NLR family apoptosis inhibitory protein; NLRC4 = NLR family CARD domain containing 4.
Generation of Inflammasome Knockouts Using CRISPR/Cas9
To study the impact of inflammasome genetic variants on macrophage inflammatory responses, we first generated NLRP3 and NLRC4 knockouts in human macrophage–like cells (36, 37) (U937 for the NLRP3 knockout; U937 and THP-1 for the NLRC4 knockout). A restriction fragment-length polymorphism was used to select for successful targeting of NLRP3 (Figure 3A) and NLRC4 (Figure 3B) (38). After stimulation with nigericin, an NLRP3-specific potassium ionophore (39), the NLRP3-knockout cell line showed a dramatic reduction in IL-1β production relative to the WT cell line or to the NLRC4-knockout cell line (Figure 3C). Similarly, NLRC4-knockout U937 cells (data shown) and THP-1 cells (data not shown) showed a dramatic reduction in the IL-1β response to needle protein (Figure 3C). IL-1β was also reduced in the NLRP3-knockout cells relative to the WT cells after stimulation with the NLRC4-specific B. thailandensis needle protein, but this was not shown to the same degree as with the NLRP3-specific stimulus. Levels of IL-6 production in response to LPS were not significantly different in the knockout lines relative to the WT U937 line (Figure 3D).
Figure 3.
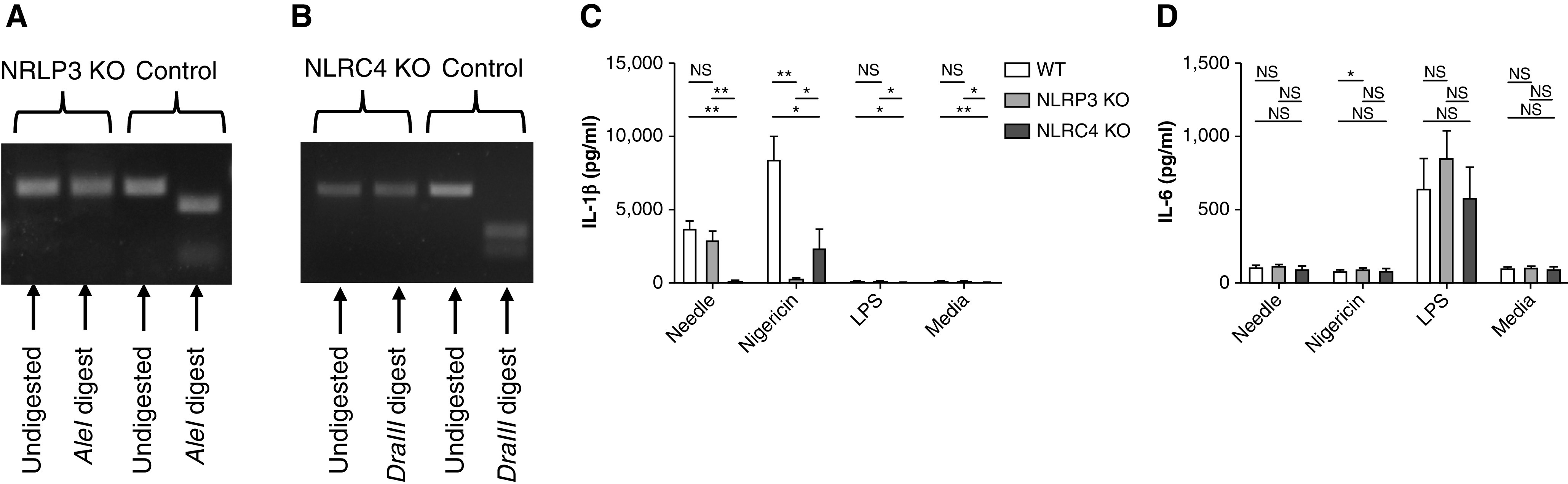
Inflammasome KOs in human monocyte-like cells. The restriction fragment-length polymorphism of CRISPR/Cas9 targets in (A) NLRP3 and (B) NLRC4 is shown. Control lines were generated by using a nontargeting guide RNA but otherwise were exposed to the same CRISPR/Cas9 delivery system. A short region of DNA containing the target site was PCR amplified and exposed to the indicated restriction endonuclease. The (C) IL-1β and (D) IL-6 responses to Burkholderia thailandensis needle protein (Needle), nigericin (10 μM), LPS (50 ng/ml), or Media. The WT and KO variants were generated in U937 cells. Needle was administered at 8 ng/ml in conjunction with 16 ng/ml Bacillus anthracis protective antigen. *P < 0.05 and **P < 0.005 (Student’s t test). KO = knockout; Media = media control (RPMI + 10% FBS); NS = not significant.
NLRP3 Rare Variants and Inflammatory Responses in Human Macrophage–like Cell Lines
To understand the mechanism by which rare NLRP3 variants influence CF outcomes, we generated a gene rescue of either WT NLRP3 or the p.(V198M) or p.(Q705K) NLRP3 variants in the U937 NLRP3-knockout cells. IL-1β and IL-6 production increased relative to that in the knockout cells after rescue with WT NLRP3 (Figure 4A, 4B). Relative to WT rescue, rescue with the p.(Q705K) variant resulted in a mildly greater (but significant) level of IL-1β production after stimulation with NLRP3-specific nigericin (P = 0.004), but this was not shown after NLRC4-specific stimulation or infection with PAO1 (Figure 4C). IL-6 levels were similarly increased after nigericin stimulation (P = 0.04) but were not increased in other conditions (Figure 4D). The association with nigericin-induced IL-1β was reproducible across three experiments, each with six replicates, as was the lack of association in needle protein– or PAO1-induced IL-1β. In addition to evaluating responses to PAO1, we also evaluated cytokine responses to a P. aeruginosa clinical isolate (28). The response elicited by the clinical isolate was similar to that elicited by PAO1 (Figures E2A and E2B). In the experiment shown, the response elicited by both strains of P. aeruginosa was significantly greater in the p.(Q705K) variant–rescue cell line than in the WT-rescue cell line (Figure E2A). However, this difference between the WT and p.(Q705K) cell lines was not consistently observed with repeat experiments.
Figure 4.
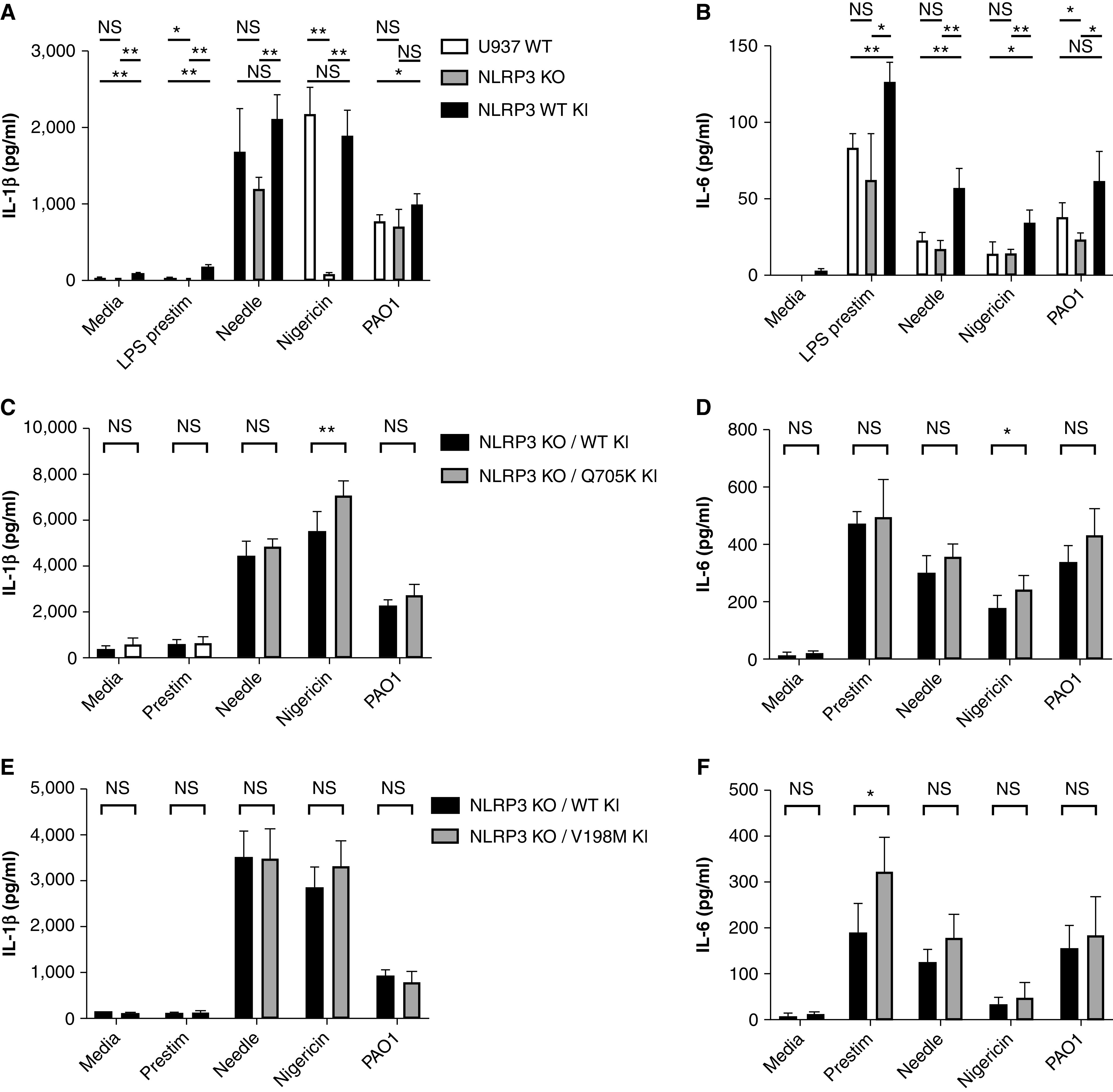
NLRP3 gene rescue and rare-variant inflammatory responses to inflammasome stimuli. The (A) IL-1β and (B) IL-6 responses to stimuli in U937 WT cells, NLRP3-KO cells, and NLRP3-KO cells after stable replacement of the knocked-out gene with WT human NLRP3 (NLRP3 WT knockin [KI]). (B) IL-6 concentrations were below detectable limits for the WT and NLRP3-KO lines. The (C) IL-1β and (D) IL-6 responses to stimuli in NLRP3-KO cells rescued with either the WT or the p.(Q705K) variant of human NLRP3 (NLRP3 KO/Q705K KI). The (E) IL-1β and (F) IL-6 responses to stimuli in NLRP3-KO cells rescued with either WT or the p.(V198M) variant of human NLRP3 (NLRP3 KO/V198M KI). All stimulation conditions other than the media control received LPS prestim. Nigericin at a 10uM concentration was used. Needle was administered at 8 ng/ml in conjunction with 16 ng/ml of B. anthracis protective antigen. *P < 0.05 and **P < 0.005 (Student’s t test). LPS prestim = prestimulation with LPS at 0.1 ng/ml; PAO1 = P. aeruginosa laboratory strain O1 with a multiplicity of infection of 5.
To determine whether the observed effect on IL-1β between the WT cell line and the p.(Q705K)-variant cell line was due to a difference in protein expression or protein function, we compared the relative expression of NLRP3 in the knockin lines by using Western blotting (Figures E1A and E1B). The WT-knockin cell line had relatively more expression than either variant-knockin cell line, suggesting that the increase in IL-1β in the p.(Q705K) variant was due to the change in protein function rather than degree of expression. The normalization of IL-1β concentration to protein expression increased the difference between nigericin-mediated IL-1β in the WT-rescue cell line compared with the p.(Q705K)-variant cell line (P = 0.00003) and also showed increased IL-1β in the variant relative to the WT cell line after stimulation with media (P = 0.04), needle protein (P = 0.0002), and PAO1 (P = 0.002) for the experiment shown in Figure 4C (normalized data not shown). IL-6 was also significantly greater in the p.(Q705K)-variant cell line than in the WT cell line after stimulation with needle protein (P = 0.002), nigericin (P = 0.003), and PAO1 (P = 0.003) for the experiment shown in Figure 4D after normalization (normalized data not shown).
Gene rescue with the p.(V198M) variant did not yield a consistent association with the various stimulation conditions and IL-1β production relative to the WT (Figure 4E). Although IL-6 production was significantly greater with low-dose LPS in the experiment shown (Figure 4F), this association was not reproducible across multiple experiments. We also compared cytokine responses between the WT- and p.(V198M)-rescue lines by using a P. aeruginosa clinical isolate (Figures E2C and E2D). In these experiments, neither the clinical isolate nor PAO1 elicited a different IL-1β response between the WT- and p.(V198M)-rescue lines.
When normalized for protein expression based on Western blotting (Figures E1A and E1B), IL-1β production was greater with the p.(V198M) variant than with the WT rescue for media alone (P = 0.0003), LPS prestimulation (P = 0.4), needle protein (P = 0.009), and nigericin (P = 0.0005) but was not greater with PAO1 in the experiment shown in Figure 4E (normalized data not shown). IL-6 production remained significantly greater with low-dose LPS and became significant for needle protein stimulation (P = 0.002) for the experiment shown in Figure 4F (normalized data not shown).
The Rare NLRC4 A929S Variant Was Not Associated with Cytokine Responses in Human Macrophage–like Cell Lines
We took a similar approach to study the mechanism by which the NLRC4 p.(A929S) variant influences the time to Pseudomonas infection acquisition and lung function in children with CF. We introduced either the WT or the p.(A929S) variant into NLRC4-knockout macrophage-like cell lines. Replacement of the NLRC4 knockout with either WT or p.(A929S) NRLC4 increased IL-1β production and, to a lesser extent, IL-6, in response to the intracellular needle protein (Figure 5A, 5B). However, we did not observe a significant difference in the cytokine response between the WT and the p.(A929S) variant across a range of needle protein doses, nor did we observe significant differences in response to PAO1 (Figure 5C) or a P. aeruginosa clinical isolate (Figures E2E and E2F). Although the media control showed greater IL-1β in the p.(A929S)-variant cell line (Figure E2E), this was not consistent across repeat experiments. Western blotting of NLRC4 expression between the two knockin lines showed a minimal difference in protein expression between the WT and p.(A929S)-variant cell lines (Figures E1C and E1D).
Figure 5.
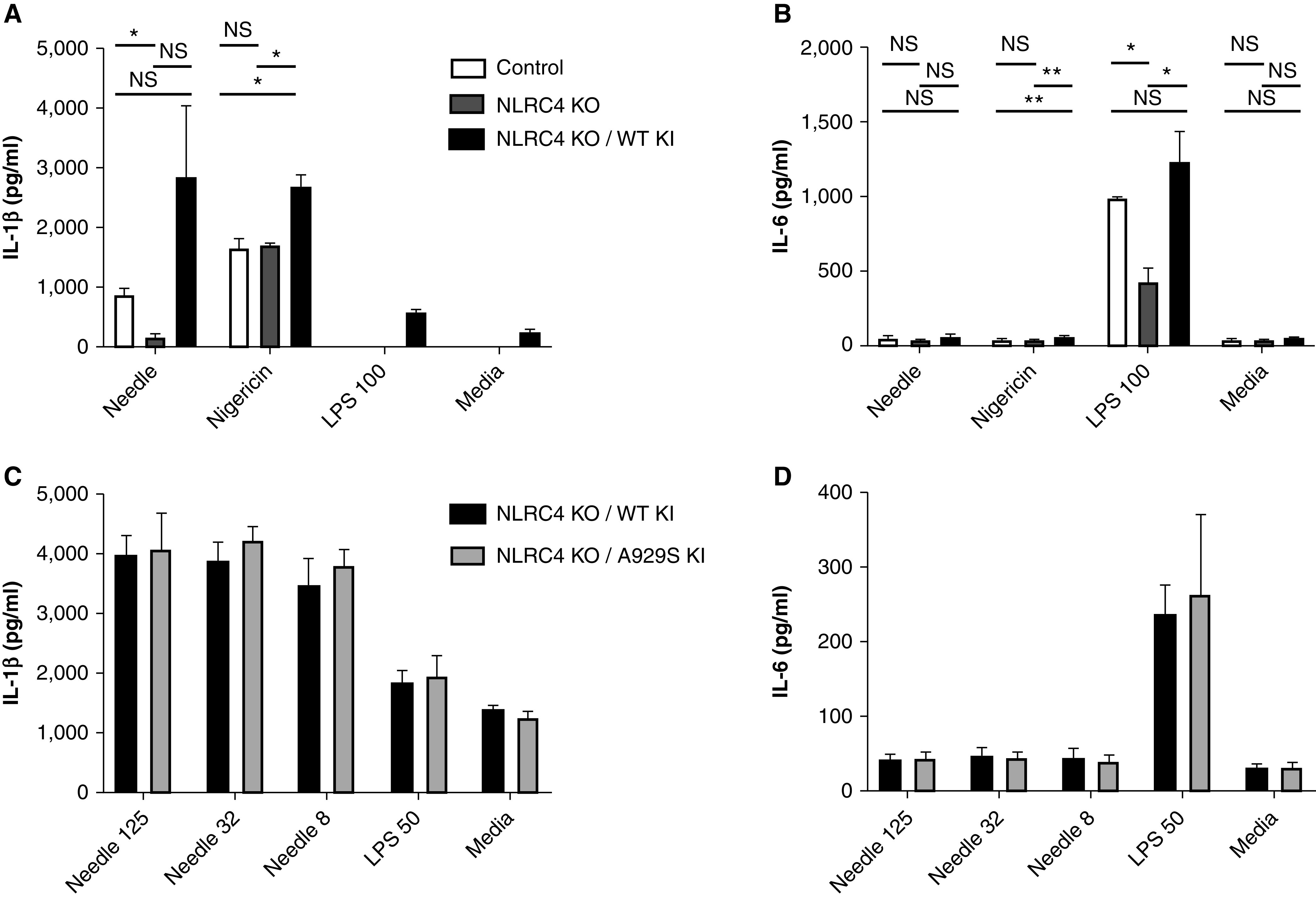
NLRC4 gene rescue and rare-variant inflammatory responses to inflammasome stimuli. The (A) IL-1β and (B) IL-6 responses to stimuli in U937 CRISPR/Cas9 control cells, NLRC4-KO cells, and NLRC4-KO cells after stable replacement of the knocked-out gene with WT human NLRC4 (NLRC4 WT KI). (A) The difference in IL-1β between the NLRC4-KO and NLRC4 WT–KI cells after Needle stimulation was borderline significant, with a P value of 0.052. The control and NLRC4-KO lines did not produce measurable IL-1β after stimulation with 100 ng/ml of LPS or Media. The (C) IL-1β and (D) IL-6 responses to stimuli in NLRC4-KO cells rescued with either WT or the p.(A929S) variant of human NLRC4 (NLRC4 KO/A929S KI). No differences between the WT- and p.(A929S)-rescue cell lines were significant in C or D. LPS was provided at 50 ng/ml, Needle was administered in doses of 125, 32, or 8 ng/ml in conjunction with B. anthracis protective antigen administered at 250, 64, or 16 ng/ml, respectively. *P < 0.05 and **P < 0.005 (Student’s t test). B. anthracis = Bacillus anthracis.
In summary, we identified three inflammasome nonsynonymous variants with relevance to the clinical outcome in the EPIC cohort; of these, the p.(Q705K) NLRP3 variant showed a consistent hyperinflammatory phenotype relative to WT NLRP3 in human macrophage–like cells.
Discussion
We identified a gain-of-function variant, p.(Q705K), in NLRP3 that was associated both with earlier onset of chronic P. aeruginosa in the airway secretions of children with CF and with an increased rate of lung function decline. The p.(Q705K) variant was also associated with increased levels of IL-1β production in macrophage-like cell lines after stimulation with the NLRP3-specific nigericin, but not after stimulation with NLRC4-specific needle protein, and has previously been reported to underlie the autoinflammatory NLRP3-mediated condition CAPS. The p.(V198M) NLRP3 variant, although also associated with CAPSs, showed a trend toward protection from P. aeruginosa acquisition in children with CF and was not associated with an inflammatory phenotype in our macrophage-like cells lines. Lastly, the p.(A929S) NLRC4 variant, which to our knowledge does not have a previously described functional consequence, was protective against chronic P. aeruginosa infection with a composite of other variants in our cohort and was protective against lung function decline as an individual variant in the EPIC cohort but was not associated with inflammatory phenotype in our macrophage-like cell lines.
Previous studies have implied that increased lung damage and worse outcomes are due to higher IL-1β–mediated responses (17, 19, 20). We used genetic models with well-defined and clinically important criteria to study the associations between inflammasome genetics and outcomes in children. To our knowledge, this is the largest analysis of inflammasome genetics and clinical outcomes in CF to date. The use of a pediatric cohort was particularly relevant because an earlier age of P. aeruginosa onset has been shown to be strongly associated with the likelihood of severe lung disease later in life (30). The p.(Q705K) variant has been identified as relevant in other diseases aside from autoinflammatory conditions and has now been identified in CF. One recent study found an association between the NLRP3 p.(Q705K) variant and poor outcomes in Ethiopian patients treated for tuberculosis (40). Another study evaluated the functional consequences of the p.(Q705K) NLRP3 variant in macrophage-like cells (35). The authors transiently transfected THP-1 WT cells with either WT NLRP3 or the p.(Q705K) variant and measured IL-1β output; they found a small but significant increase in IL-1β concentration in the p.(Q705K)-variant cells at baseline. Our molecular data support this result yet show that the phenotypic difference occurred after exposure to an NLRP3-specific stimulus rather than at baseline, which is an important and potentially clinically relevant distinction when considering the use of host-directed therapeutics targeting inflammatory pathways.
Our results with the p.(Q705K) variant support the hypothesis that inflammasome-mediated inflammation is harmful in CF and suggest that macrophages are an important mediator of this inflammation. The results of both our genetic and molecular studies with the p.(V198M) NLRP3 variant, which is also associated with autoinflammatory CAPSs, are not consistent with the p.(Q705K) result. The genetic analysis shows a trend toward a protective effect, whereas the molecular analysis does not show a consistent hyper- or hypoinflammatory response in macrophages with the p.(V198M) variant. There are a few potential explanations for this inconsistency. Multiple SNPs in NLRP3 are associated with CAPSs, many of which display variable penetrance (24). It is possible that the p.(V198M) mutation does not produce the same degree of inflammatory response in this population. Given the protective effect of the NLRC4 p.(A929S) variant in the EPIC cohort, we predicted a hypoinflammatory response in macrophages, which we did not observe in our macrophage-like cells. It is possible that such a phenotype does exist in macrophages but that it exists under different conditions; we tested a range of NLRC4-specific conditions as well as broader inflammasome stimulation conditions but still did not observe an effect. It may also be the case that such a phenotype only becomes evident in the absence of a functioning CFTR. Exposure to neither a laboratory strain nor a clinical isolate of P. aeruginosa consistently produced a significant difference in IL-1β production between the WT and NLRP3-variant cell lines. In this context, the clinical significance of our significant finding with nigericin is less clear. It is similarly possible that such a phenotype requires the absence of a functioning CFTR or that overexpression of the inflammasome variants in our rescue lines obscured a difference that might be seen with endogenous levels of protein expression.
We focused primarily on the role of inflammasome genetic variants in macrophage-like cells. IL-1β secretion has been best characterized in monocytes and macrophages (41–44), and blood monocytes may be the primary producers of IL-1β (45). In CF, BAL samples show the expansion of a small macrophage population with surface markers that are more consistent with newly migrated monocytes than with alveolar macrophages (46). However, it is possible that macrophages are not the most important cell type responsible for our observations in the EPIC cohort. CF is primarily an epithelial-cell disorder, and the airway epithelia themselves could be an important mediator of inflammasome responses; NLRP3-specific inflammation in both monocytes and epithelial cells was shown to be enhanced by CF-mediated alterations in epithelial sodium channels (27). In a murine CF model, neutrophils can produce IL-1β in the absence of signal 2, and NLRP3 inhibition reduces airway inflammation (although the NLRP3 inhibition was not specific to only neutrophils) (17). Taken together, our results suggest an important role for inflammasomes in the CF lung acting via mechanisms that we are only starting to understand.
Our study has several limitations. Sequencing was limited to exons and then to a subset of SNPs within inflammasome genes of interest. Some of our variants of interest are rare, which limited our statistical power to identify associations with clinical outcomes. For this reason, we used a composite of NLRC4 nonsynonymous variants. However, it is possible that variants could have opposing hyper- or hypoinflammatory phenotypes that we would miss by grouping them together. The use of two enrollment criteria, never P. aeruginosa and past P. aeruginosa, in the EPIC cohort has subtle but important implications for the generalizability of our results. When evaluating for genetic variants that influence the early acquisition of P. aeruginosa, enrolling older children who have never had P. aeruginosa by definition biases the cohort away from containing these variants. Such a bias could explain why we only saw the effect of the p.(Q705K) NLRP3 variant in children from the past P. aeruginosa enrollment group. For NLRC4, our genetic analysis found an association only in the subgroup of children without a protective variant in CAV2, rs8940. The variant rs8940 is common in European Americans (minor allele frequency = 0.19) and was found by our group to be a disease modifier in children with CF (34). CAV2 is clustered with CAV1 on a 300-kb region on chromosome 7, with the function of the latter being far better characterized. In a rat bleomycin-induced idiopathic pulmonary fibrosis model, caveolin-1 overexpression was protective against IL-1β–mediated fibrogenesis of the lungs, suggesting a role for caveolin-1 in inflammasome regulation (47). Both caveolin-1 and caveolin-2 seem to be structural components of lipid rafts that are important for bacterial endocytosis, and caveolin-2 may act as a caveolin-1 antagonist (48); in mouse lung epithelial cells, caveolin-2 was found to be necessary for invasion by P. aeruginosa (49). The importance of the CAV2 genotype for NLRC4-variant outcomes suggests the possibility of a mechanistic relationship between the two molecules that will be important for further study.
Both Food and Drug Administration– approved and experimental medications are currently available that alter inflammasome pathways and could provide benefit in the CF lung under specific circumstances of chronic airway infection and inflammation. These therapies include IL-1β antagonism (anakinra) (20), IL-18 neutralization (50), and eicosanoid pathway inhibition (ibuprofen) (6). Our work provides evidence that inflammasome genetics influences inflammatory responses in the CF lung in humans and specifically supports the hypothesis that hyperinflammatory variants are harmful to the CF lung. We also characterized a genetic variant whose carriers are likely at a higher risk of lung damage and who may benefit the most from host-directed therapies. The recognition of host genetic variants outside of CFTR that alter inflammatory responses allows for the tailoring of host-directed therapies for subpopulations of individuals with CF who will receive the most benefit from the treatment.
Acknowledgments
Acknowledgment
The authors thank Nihar Mahajan for assistance with development of the NLRP3-knockout cell line. They thank Bruce Marshall, M.D., and the CF Foundation for providing National Patient Registry data. We also thank all the site investigators and research coordinators, as well as all the participants in the EPIC Observational Study and their families.
Footnotes
Supported by two Cystic Fibrosis Foundation Research Development pilot grants (A.D.G. and T.R.H.) under SINGH15R0 as well as the Cystic Fibrosis Foundation grants GIBSON07K0, OBSERV04K0, and OBSERV12K0 (M.R. and R.L.G.); EPIC0K0 (M.J.B. and R.L.G.); BAMSHA14P (M.J.B. and M.J.E.); and BAMSHA 18XX0 (M.J.B., R.L.G., and M.J.E.). This research was further supported by the National Heart, Lung, and Blood institute Grand Opportunity (GO) Exome Sequencing Project grants RC2 HL-102923 ([LungGO] [M.J.B., R.L.G., and M.J.E.]), RC2 HL-102925 (BroadGO), and RC2 HL-102926 ([SeattleGO] [M.J.B.]). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions: Concept and/or design of the study: A.D.G., W.R.B., M.R., M.J.B., R.L.G., T.R.H., and M.J.E. Data acquisition: A.D.G., W.R.B., K.J.B, F.K.N., L.L.J., and M.J.E. Data analysis: A.D.G., K.J.B., F.K.N., L.L.J., M.J.B., R.L.G., and M.J.E. Interpretation of data: A.D.G., W.R.B., K.J.B., F.K.N., L.L.J., M.R., M.J.B., R.L.G., T.R.H., and M.J.E. All authors contributed to manuscript drafting or revision, provided final approval of the version to be published, and agree to be accountable for the work regarding the investigation of any questions related to accuracy or integrity.
This article has a data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0257OC on April 12, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Bruscia EM, Bonfield TL. Innate and adaptive immunity in cystic fibrosis. Clin Chest Med. 2016;37:17–29. doi: 10.1016/j.ccm.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 2. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am J Respir Cell Mol Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, et al. AREST CF. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros. 2018;17:715–722. doi: 10.1016/j.jcf.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 4. Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 5. Roesch EA, Nichols DP, Chmiel JF. Inflammation in cystic fibrosis: an update. Pediatr Pulmonol. 2018;53:S30–S50. doi: 10.1002/ppul.24129. [DOI] [PubMed] [Google Scholar]
- 6. Lands LC, Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst Rev. 2016;4:CD001505. doi: 10.1002/14651858.CD001505.pub4. [DOI] [PubMed] [Google Scholar]
- 7. Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 13. Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev. 2018;281:115–123. doi: 10.1111/imr.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. 2015;6:6201. doi: 10.1038/ncomms7201. [DOI] [PubMed] [Google Scholar]
- 15. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ince D, Sutterwala FS, Yahr TL. Secretion of flagellar proteins by the Pseudomonas aeruginosa type III secretion-injectisome system. J Bacteriol. 2015;197:2003–2011. doi: 10.1128/JB.00030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McElvaney OJ, Zaslona Z, Becker-Flegler K, Palsson-McDermott EM, Boland F, Gunaratnam C, et al. Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med. 2019;200:1381–1391. doi: 10.1164/rccm.201905-1013OC. [DOI] [PubMed] [Google Scholar]
- 18. Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, et al. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol. 2012;189:2006–2016. doi: 10.4049/jimmunol.1201065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ravi Kumar S, Paudel S, Ghimire L, Bergeron S, Cai S, Zemans RL, et al. Emerging roles of inflammasomes in acute pneumonia. Am J Respir Crit Care Med. 2018;197:160–171. doi: 10.1164/rccm.201707-1391PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun. 2016;7:10791. doi: 10.1038/ncomms10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Breuer O, Schultz A, Garratt LW, Turkovic L, Rosenow T, Murray CP, et al. Aspergillus infections and progression of structural lung disease in children with cystic fibrosis. Am J Respir Crit Care Med. 2020;201:688–696. doi: 10.1164/rccm.201908-1585OC. [DOI] [PubMed] [Google Scholar]
- 23. Moretti S, Bozza S, Oikonomou V, Renga G, Casagrande A, Iannitti RG, et al. IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog. 2014;10:e1004462. doi: 10.1371/journal.ppat.1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. 2015;8:15–27. doi: 10.2147/JIR.S51250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 26. Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 27. Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. eLife. 2019;8:e49248. doi: 10.7554/eLife.49248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe. 2015;18:307–319. doi: 10.1016/j.chom.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 30. Pittman JE, Calloway EH, Kiser M, Yeatts J, Davis SD, Drumm ML, et al. Age of Pseudomonas aeruginosa acquisition and subsequent severity of cystic fibrosis lung disease. Pediatr Pulmonol. 2011;46:497–504. doi: 10.1002/ppul.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huus KE, Joseph J, Zhang L, Wong A, Aaron SD, Mah TF, et al. Clinical isolates of Pseudomonas aeruginosa from chronically infected cystic fibrosis patients fail to activate the inflammasome during both stable infection and pulmonary exacerbation. J Immunol. 2016;196:3097–3108. doi: 10.4049/jimmunol.1501642. [DOI] [PubMed] [Google Scholar]
- 32. Rosenfeld M, Emerson J, McNamara S, Joubran K, Retsch-Bogart G, Graff GR, et al. EPIC Study Group Participating Clinical Sites. Baseline characteristics and factors associated with nutritional and pulmonary status at enrollment in the cystic fibrosis EPIC observational cohort. Pediatr Pulmonol. 2010;45:934–944. doi: 10.1002/ppul.21279. [DOI] [PubMed] [Google Scholar]
- 33. Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, et al. National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project; Lung GO. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44:886–889. doi: 10.1038/ng.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emond MJ, Louie T, Emerson J, Chong JX, Mathias RA, Knowles MR, et al. NHLBI GO Exome Sequencing Project. Exome sequencing of phenotypic extremes identifies CAV2 and TMC6 as interacting modifiers of chronic Pseudomonas aeruginosa infection in cystic fibrosis. PLoS Genet. 2015;11:e1005273. doi: 10.1371/journal.pgen.1005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verma D, Särndahl E, Andersson H, Eriksson P, Fredrikson M, Jönsson JI, et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1β and IL-18 production. PLoS One. 2012;7:e34977. doi: 10.1371/journal.pone.0034977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 40. Abate E, Blomgran R, Verma D, Lerm M, Fredrikson M, Belayneh M, et al. Polymorphisms in CARD8 and NLRP3 are associated with extrapulmonary TB and poor clinical outcome in active TB in Ethiopia. Sci Rep. 2019;9:3126. doi: 10.1038/s41598-019-40121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 42. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. CANTOS Trial Group. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 43. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 44. Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One. 2017;12:e0175336. doi: 10.1371/journal.pone.0175336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Piccioli P, Rubartelli A. The secretion of IL-1β and options for release. Semin Immunol. 2013;25:425–429. doi: 10.1016/j.smim.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 46. Wright AK, Rao S, Range S, Eder C, Hofer TP, Frankenberger M, et al. Pivotal advance. Expansion of small sputum macrophages in CF: failure to express MARCO and mannose receptors. J Leukoc Biol. 2009;86:479–489. doi: 10.1189/jlb.1108699. [DOI] [PubMed] [Google Scholar]
- 47. Lin X, Barravecchia M, Matthew Kottmann R, Sime P, Dean DA. Caveolin-1 gene therapy inhibits inflammasome activation to protect from bleomycin-induced pulmonary fibrosis. Sci Rep. 2019;9:19643. doi: 10.1038/s41598-019-55819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Almeida CJG. Caveolin-1 and caveolin-2 can be antagonistic partners in inflammation and beyond. Front Immunol. 2017;8:1530. doi: 10.3389/fimmu.2017.01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zaas DW, Swan ZD, Brown BJ, Li G, Randell SH, Degan S, et al. Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa . J Biol Chem. 2009;284:9955–9964. doi: 10.1074/jbc.M808629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139:1698–1701. doi: 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]