

Abstract
Aminoacyl-tRNA synthetases (ARSs) are widely found in organisms, which can activate amino acids and make them bind to tRNA through ester bond to form the corresponding aminoyl-tRNA. The classic function of ARS is to provide raw materials for protein biosynthesis. Recently, emerging evidence demonstrates that ARSs play critical roles in controlling inflammation, immune responses, and tumorigenesis as well as other important physiological and pathological processes. With the recent development of genome and exon sequencing technology, as well as the discovery of new clinical cases, ARSs have been reported to be closely associated with a variety of cardiovascular diseases (CVDs), particularly angiogenesis and cardiomyopathy. Intriguingly, aminoacylation was newly identified and reported to modify substrate proteins, thereby regulating protein activity and functions. Sensing the availability of intracellular amino acids is closely related to the regulation of a variety of cell physiology. In this review, we summarize the research progress on the mechanism of CVDs caused by abnormal ARS function and introduce the clinical phenotypes and characteristics of CVDs related to ARS dysfunction. We also highlight the potential roles of aminoacylation in CVDs. Finally, we discuss some of the limitations and challenges of present research. The current findings suggest the significant roles of ARSs involved in the progress of CVDs, which present the potential clinical values as novel diagnostic and therapeutic targets in CVD treatment.
Keywords: aminoacyl-tRNA synthetase, aminoacylation, angiogenesis, mitochondrial cardiomyopathy, mutation
Graphical abstract
Zou et al. reviewed the functional roles of aminoacyl-tRNA synthetases (ARSs) in cardiovascular diseases (CVDs) and discussed the involved molecular mechanisms. These reviews provide important insights into the potential value of ARS and provide potential diagnosis biomarker or therapeutic targets for CVDs, especially for angiogenesis and cardiomyopathy.
Introduction
As the leading cause of morbidity and mortality globally, cardiovascular disease (CVD) is normally caused by abnormal cellular biological function and inflammatory processes, which may lead to hardening of the blood vessels and the accumulation of fibrotic scar tissue in the heart. CVD mainly includes hypertension, coronary heart disease, vascular calcification, arrhythmia, hypertrophy of the heart, cardiomyopathy, myocardial infarction, and heart failure (HF). It is associated with a complex series of events, including cell proliferation,1 migration,1 cell pyrodeath,2 autophagy,3 extracellular vesicle signaling,4 and epigenetic modification (e.g., DNA methylation5 and histone acetylation5). In addition, enzymes (arginase,6 angiotensin [ang]-converting enzyme 2 [ACE2],7 and the sirtuin [SIRT] family8) are also involved in the occurrence and development of CVD. More recently, emerging evidence demonstrates that aminoacyl-tRNA synthetase (ARS) is closely related to the occurrence and development of CVD.
ARS is an enzyme necessary for normal metabolism of organisms, in which it is ubiquitously expressed. ARSs catalyze the formation of aminoacyl-tRNA from amino acids (aas) and corresponding tRNA, providing raw materials for protein translation. ARS catalyzes the covalent bonding of each amino acid to its tRNA counterpart in a two-step aminoacylation process. Amino acids and ATP first bind to ARS to form amyladenylate intermediates. ARS then binds to homologous tRNA to promote the transfer of amino acids to form acyl tRNA.9,10 Acyl tRNA subsequently transfers amino acids to the growing polypeptide chain and ensures accurate translation of the genetic information. In all known organisms, translating genetic code into proteins is crucial for cell survival. A typical animal cell has 36 ARSs, 16 of which act only in the cytoplasm and 17 in mitochondria. These genes are encoded on nuclear chromosomes and are involved in protein biosynthesis of cytoplasm and mitochondrial proteins. There are three ARSs with double positioning: lysyl-tRNA synthetase (KARS), glutaminyl-tRNA synthetase (QARS), and glycyl-tRNA synthetase (GARS). Interestingly, mitochondrial synthetases are named in the same way as cytoplasmic synthetases but with the suffix 2 (such as histidyl-tRNA synthetase [HARS] and HARS2); they promote protein synthesis accurately and efficiently in both cell chambers under various conditions.
Although ARS is known for its role in mRNA translation, it is increasingly recognized for its “non-classical” role in transcriptional regulation, intron splicing, immune function, angiogenesis, apoptosis, and cellular stress.11,12 Methylthio-tRNA synthetase (MARS) affects cell-cycle progression by activating the mechanistic target of the rapamycin complex 1 (mTORC1) signaling pathway.13 It also induces cell apoptosis by regulating oxidative stress levels, which ultimately leads to neural tube defects (NTDs) and congenital heart defects (CHDs).14 The MARS inhibitor acetylhomocysteine thioether (AHT) has shown a good therapeutic effect in animal models. Leucyl-tRNA synthetase (LARS) can also regulate the mTORC1 pathway to negatively mediate myoblast differentiation and skeletal muscle regeneration, whereas LARS inhibitor BC-LI-0186 has a good intervention effect. Several other non-classical functions have been reported, such as the promotion of inflammatory responses by KARS15 and the regulation of apoptosis by QARS.16
Increasing evidence has demonstrated that pathological ARS mutations are involved in various human diseases. The first cytoplasmic ARS mutation associated with human disease was Charcot–Marie–Tooth disease (CMT), which was found in GARS.17 Later, it was found that the mutations of five ARS loci were related to CMT and associated neurophenotypes. These include alanyl-tRNA synthetase (AARS), tyrosyl-tRNA synthetase (YARS), HARS, tryptophanyl-tRNA synthetase (WARS), and KARS.18, 19, 20, 21 With the recent development of genetic detection techniques (such as whole genome sequencing and whole exome sequencing) and the discovery of new clinical cases, six ARS gene mutations are reportedly related to angiogenesis. Furthermore, ARS gene mutations have been associated with other CVDs, mainly including hypertrophic cardiomyopathy (HCM) and abdominal aortic aneurysm (AAA). Among these pathogenic mutations, some resulted in impaired aminoacylation activity or redaction activity of ARS or altered gene-expression levels. Some pathogenic mutations do not affect the catalytic activity of ARS but affect the non-classical function of ARS by changing the interaction between ARS and other protein factors. Understanding and defining the underlying mechanisms of phenotypic heterogeneity in ARS-related diseases are considered as potential strategies in developing therapies for these diseases.
In this paper, we introduce the clinical phenotypes and characteristics of CVD, particularly angiogenesis and cardiomyopathy, which are related to reported ARS gene mutations. We summarize the research progress on the pathogenesis of ARS gene mutations and non-classical functions. We also discuss the newly reported aminoacylation and its potential roles in this process. The current findings reveal that ARS is crucially involved in the occurrence and development of CVD through complex mechanisms.
Angiogenesis and ARS
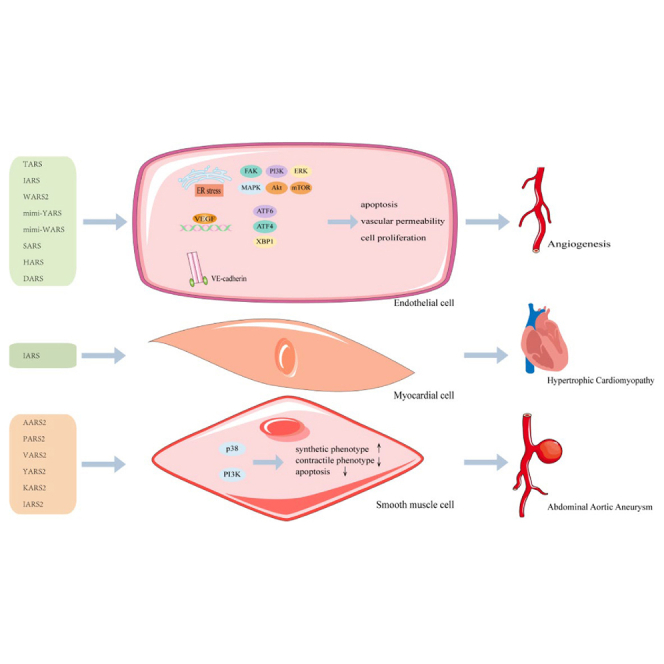
Angiogenesis is the formation of new blood vessels by the existing vascular system; it is an important physiological process that balances the metabolic demand of tissues with oxygen and nutrient supply. In a diseased heart, the metabolic demands of cardiomyocytes may exceed capillary growth, leading to reduced oxygen delivery and cardiac ischemia.22, 23, 24 Some mutations in the ARS gene cause vascular defects, especially in zebrafish. At present, it is found that the abnormal function of ARS regulates angiogenesis mostly through vascular endothelial growth factor (VEGF). Interestingly, the deficiency of several ARS genes (isoleucyl-tRNA synthetase [IARS], seryl-tRNA synthetase [SARS], and threonyl-tRNA synthetase [TARS]) leads to an increase in the branches of the intersegmental vessels (ISVs), indicating that the ARS genes go through both a pro- and antiangiogenic process.25, 26, 27, 28 The angiogenesis process is strictly regulated by the balance between pro- and antiangiogenic factors. VEGF promotes angiogenesis by activating static endothelial cells and provides one of the most important proangiogenic signals.29 Loss of myocardial VEGFA or VEGF receptor 2 (VEGFR2) has been shown to prevent normal coronary artery development in mice.30 VEGF signaling also regulates endothelial cell functions, including proliferation, migration, and survival. A diagram of the involvement of ARS in angiogenesis is presented in Figure 1. Table 1 summarizes the ARS gene variants associated with angiogenesis.
Figure 1.

The regulatory mechanism of ARS in angiogenesis
SARS can directly bind to the VEGFA promoter and inhibit the expression of VEGFA. SARS directly competes with c-myc in the proximal CRE of VEGFA, thereby regulating VEGFA expression. SARS binds to DNA and recruits Sirt2 to regulate the acetylation of c-Myc, thus blocking gene transcription. SARS binds to the transcription factor YY1 to form the SARS/YY1 complex and competitively binds to VEGFA distal CRE between NF-κB1 to regulate the expression of VEGFA. miRNA-1 regulates upstream of VEGFA by inhibiting the expression of SARS, whereas miRNA-206 directly regulates VEGFA mRNA expression. When endothelial cells (ECs) are stimulated by IFN-γ, the expression level of WARS increases, and p53 is partially activated, thereby leading to anti-angiogenesis and anti-proliferation functions. The absence of WARS2 can induce G2/M-phase arrest, inhibit EC proliferation, and promote apoptosis. HARScq34 increased the expression of VEGFA and VE-cadherin.
Table 1.
Overview of ARS gene variants related angiogenesis
| Gene | Protein | Unique variants | Cell function/signaling pathway | Diseases | Inhibitor | Ref. | |
|---|---|---|---|---|---|---|---|
| TARS | threonyl-tRNA synthetase | zebrafish tarscq16 | activate VEGFA expression | disorganize vessels abnormal branching of the established ISVs and brain vascular network | VEGF receptor inhibitor; human TARS mRNA | 32 | |
| zebrafish tarsy58 | UPR pathway was upregulated; activate VEGF expression | activate branching of angiogenic vessels in both the trunk and the head | ATF4 inhibitor | 25 | |||
| IARS | isoleucyl-tRNA synthetase | zebrafish tarsy68 | UPR pathway was upregulated; activate VEGF expression | activate branching of angiogenic vessels in both the trunk and the head | N/A | 25 | |
| WARS2 | tryptophanyl-tRNA synthetase 2 | rat WARS2 (L53F) | suppress EC cell cycle in G2/M and proliferation | defects in angiogenic sprouting of ISVs | human WARS mRNA | 35 | |
| Mini-YARS/mini-WARS | mini-tyrosyl-tRNA synthetase/mini-tryptophanyl-tRNA synthetase | N/A | promote EC proliferation and migration via VEGFR2-ERK signal pathway | pro-angiogenic/anti-angiogenic; rats/mice/rhesus monkeys with acute myocardial infarction | N/A | 41,49,109 | |
| SARS | seryl-tRNA synthetase | zebrafish adr/sars | N/A | dilate the aortic arch vasculature and regulate branching of the hindbrain capillary network | N/A | 27 | |
| zebrafish ko095 | activate VEGFA-VEGFR2 signal pathway | disorganize vessels abnormal branching of the established intersegmental vessels | VEGF receptor inhibitor; human SARS mRNA | 51 | |||
| HARS | histidyl-tRNA synthetase | zebrafish harscq34 | activate VE-cadherin expression and altered VE-cadherin localization; activate VEGFA expression | hyperbranching cranial and intersegmental blood vessels | VEGF receptor inhibitor; human HARS mRNA | 59 | |
TARS
Studies have shown that human umbilical vein endothelial cells (HUVECs) secrete TARS upon stimulation by tumor necrosis factor α (TNF-α) and VEGF and can promote HUVEC migration and angiogenesis in vitro.31 In addition, BC194 is a TARS inhibitor, and its inhibitory effect on TARS in zebrafish can inhibit angiogenesis.28 BC194 is related to the competition of threonine substrates, which can stimulate the amino acid starvation response (AAR) and cell apoptosis. On the contrary, other studies have reported vascular disorders, abnormal branches of established ISVs, and abnormal patterns of cerebrovascular networks in zebrafish cq16 mutants, wherein the cq16 mutant gene encodes TARS.32 Further studies revealed that TARS influenced vascular branching by regulating VEGFA expression. VEGFA expression levels were elevated in the TARScq16 mutants, and VEGFA receptor blockers inhibited abnormal vascular branches in the mutants. In addition, part of the TARS protein is reportedly distributed in the nucleus, suggesting that TARS may directly regulate nuclear VEGFA transcription. In another study using zebrafish-recessive mutants y58 and y68, these mutants corresponded to the TARS and IARS genes, respectively.25 In zebrafish mutants TARSy58 and IARSy68, the unfolded protein response (UPR) pathway is activated, and VEGFA expression is upregulated, resulting in an increase in branched blood vessels. Consistent with previous studies, the UPR pathway activates VEGFA expression. Interestingly, injections of human TARS mRNA were effective in treating abnormal blood vessel branching in mutants.32 Therefore, the loss of TARS in humans may be associated with certain CVDs, and zebrafish TARS mutants may be a promising model of human disease. Further investigations into the mechanisms of angiogenesis and remodeling in zebrafish and mammals will provide further insights. However, there is still a lack of relevant studies elucidating the mechanism by which TARS affects angiogenesis in mammals, which requires further in-depth exploration.
WARS2
In human genome-wide association studies, the WARS2 locus is associated with the cardiometabolic phenotype associated with the VEGFA locus. However, genetic studies in mice have shown that WARS2 is associated with skin capillary formation.33 WARS2 variation and decreased enzyme activity can reveal the atypical role of ARS, which may be tissue specific. The effect of WARS2 knockdown in zebrafish embryos is similar to that previously observed in mutants with impaired VEGF function. WARS2 mutation reduces endothelial cell proliferation, activates proapoptotic pathways, and damages brown adipose tissue functions, which may be involved in the balance between VEGF and Notch-dependent signaling.34,35 Interestingly, in the mouse WARS2V117L/V117L mutant model studied by Agnew et al.36, upregulation of murine tissue-specific mitochondrial biogenesis occurred compared with respiratory chain deletion. Increased Peroxisome proliferators-activated receptor γ coactivator-1 a (Pgc1a) expression in WARS2V117L/V117L mutant mouse embryonic fibroblasts prevented impaired respiratory chain function by upregulating mitochondrial mass and function. This observation was supported by other findings that increased Pgc1a expression, and thus upregulated mitochondrial biogenesis can reduce disease characteristics in mouse models and human patient cell lines and increase mitochondrial respiratory capacity.37,38 However, it is unclear how only some tissues in WARS2V117L/V117L mice regulate Pgc1a and are protected. In general, WARS2 is a key factor in angiogenesis in the heart and other tissues, enabling endothelial cell migration and proliferation. In zebrafish, inhibition of WARS2 results in a lack of trunk blood vessels, impaired myocardial–endocardial contact, and impaired cardiac function.35 Inhibition of WARS2 in rats results in defective cardiac angiogenesis and reduced cardiac capillary density. A mouse WARS2 mutant model was observed to have complex tissue-specific pathology, including adipose tissue dysfunction, lipodystrophy, hearing loss, and HCM.36
Mini-YARS/mini-WARS
YARS and WARS have also been shown to regulate angiogenesis in mammals.39 When interferon-γ (IFN-γ)-stimulated endothelial cells, WARS expression was increased, thereby promoting p53 phosphorylation (p-p53) and activation.40 Activation of p53 is associated with strong antiangiogenic and antiproliferative functions. In mammalian cells, activation of YARS/WARS by extracellular leukocyte elastase produces two distinct cytokines that generate pro- and antiangiogenic signals. YARS were hydrolyzed to obtain two polypeptides: the N terminus containing the glutamate-leucine-arginine (ELR) motif (mini-YARS) and the C terminus containing the endothelial monocyte-activating polypeptide II (EMAP II) domain (EMAP II-YARS). The binding of mini-YARS to VEGFR2 promotes endothelial angiogenesis.41 Mini-YARS stimulated neutrophil activation and chemotaxis in vitro and angiogenesis in endothelial cell culture, chorioallantoic membrane, and matrix gel implants in mice.42,43 Full-length YARS showed no angiogenic activity. Similarly, the N-terminal WHEP domain (termed by its presence in WARS, HARS, and glutamyl-prolyl-tRNA synthetases [EPRS]) was removed by hydrolysis of WARS to obtain C-terminal peptide WARS-C (mini-WARS).44 The N-terminal of tryptophanyl-tRNA synthetase (TRPR) contains the WHEP domain whose spatial structure is helix-turn-helix. Mini-WARS binds to the extracellular domain of vascular endothelial cadherin (VE-cadherin), thereby inhibiting the extracellular signal-regulated kinase (ERK) signaling pathway induced by VEGF and ultimately inhibiting angiogenesis.45 Mini-WARS inhibits new blood vessel formation without affecting established blood vessels.39,40 The antiangiogenic activity of mini-WARS was confirmed in vivo.44,46 Similarly, full-length WARS cannot bind to VE-cadherin, so they do not inhibit angiogenesis. Interestingly, mini-WARS inhibited mini-YARS-induced angiogenesis; subsequent studies have shown that this is regulated by the VEGFR-2/VE-cadherin pathway (Figure 2).47
Figure 2.

The involvement of mini-YARS/mini-WARS in angiogenesis
Full-length of YARS is extracellular cleaved by polymorphonuclear neutrophil (PMN) elastase or other protease molecules into C- and N-terminal fragments (mini-YARS), which can regulate angiogenesis or immune response, respectively. Mini-YARS can also transactivate VEGFR. Full length of WARS is further processed outside of cells by PMN elastase or other proteases to form mini-WARS-like fragments. Mini-WARS inhibits angiogenesis by binding to the extracellular domain of VE-cadherin. When mini-WARS is combined with VE-cadherin, it inhibits the VEGF-induced signaling pathway and ultimately suppresses the proliferation, migration, and angiogenesis of ECs. In addition, mini-WARS could inhibit mini-YARS-induced angiogenesis.
In addition, mini-WARS inhibits the activation of ERK, Akt, and endothelial nitric oxide (NO) synthase (eNOS), and shear force signal transduction, thereby regulating cytoskeleton reorganization and gene expression; this inhibition ultimately inhibits new blood vessel formation and stability.48 This also suggests the role of mini-WARS in vascular remodeling, blood pressure regulation, and atherosclerosis. It was later found that the mini-YARS/mini-WARS-mediated effect on the proliferation and migration of rat coronary venular endothelial cells (RCVECs) to ischemic tissues may occur through the activation of phosphatidylinositol 3-kinase (PI3K)/Akt and ERK.49 It was first found that mini-YARS inhibited angiogenesis at low concentrations and stimulated angiogenesis at high concentrations. In vitro studies have shown that hypoxia-induced VEGF-dependent signaling can reduce the level of mini-YARS.50 These results suggest that endogenous mini-YARS may play a vasoinhibitory role in some physiological environments. Low or high levels of mini-YARS may provide treatment options for limiting or increasing angiogenesis in certain diseases. It was found that mini-YARS/mini-WARS affected the myocardial infarction area by affecting angiogenesis and microvessel density in a rat model of myocardial infarction.49 These results suggest the possibility of using mini-YARS/mini-WARS as angiogenic therapy in ischemic disease. Therefore, the natural fragments of YARS and WARS involved in translation have opposing effects on angiogenesis. The balance between the two tRNA synthetases affects the balance between proangiogenesis and antiangiogenesis. More studies must be done in the future to elucidate the mechanisms of these actions and the relationship between YARS and WARS.
SARS
Functional analysis of the mutant zebrafish with SARS demonstrated that SARS could regulate vascular development.27,51 Furthermore, SARS and VEGF signal transduction can regulate angiogenesis.51 Because of the increased VEGFA expression induced by SARS mutants, vascular branching and dilatation phenotypes can be inhibited by blocking VEGF receptor function. Importantly, by knocking down SARS in HUVECs, the VEGF level is increased, resulting in increased vascular branching points.27 Interestingly, an injection of human SARS mRNA was effective in treating the abnormal vascular branches observed in mutant zebrafish.51 Therefore, human SARS damage may be associated with certain CVDs in humans, and zebrafish SARS mutants may be a promising model of human disease. Interestingly, aminoacylated, defective SARS (T429A) mRNA also restored the vascular phenotype, suggesting that the role of SARS in vascular development is independent of aminoacylation.27
A recent study found that the carboxyl terminus of SARS has a unique domain (called UNE-S) found in all vertebrates, from fish to humans.52 As the nuclear localization signal (NLS) was introduced by UNE-S, this new, additional UNE-S domain in HUVECs and HEK293T cells not only mobilizes the transfer of SARS from the cytoplasm to the nucleus but also promotes the interaction of SARS with potential nuclear targets.26 Interestingly, UNE-S mutation or deletion did not affect the aminoacylation reaction. Consistent with this finding, SARS mutants that were previously associated with vascular dysplasia deleted or isolated NLS into other conformations. Furthermore, aminoacylated, defective SARS that rescued vascular dysplasia had the same nuclear localization as wild type (WT), suggesting a high correlation between vascular dysplasia and lack of SARS nuclear localization. Further studies showed that SARS could regulate VEGFA transcription in the nucleus. Therefore, the involvement of SARS in regulating vascular development may be dependent on UNE-S but not on its enzyme activity. These results demonstrate for the first time the important role of other domains related to tRNA synthetase at the biological level. The findings also suggest that UNE-S is involved in the regulation of cardiovascular phylogeny in vertebrates.
It is well known that c-Myc is a major transcription factor that promotes VEGFA gene expression in the nucleus and thus plays a key role in vascular development. Shi et al.54 found that SARS is an antagonist of c-Myc. SARS control vascular development in zebrafish embryos by inhibiting the activity of c-Myc. This process is implemented in two different ways: direct competition between SARS and c-Myc in the proximal cyclic AMP (cAMP) response element (CRE) of VEGFA to regulate VEGFA expression. In addition, SARS directly recruit histone deacetylase (SIRT2) to remove the chemical tags that c-Myc proteins add to the histones. Interestingly, when SIRT2 was knocked out by RNAi or inhibited by inhibitors, SARS inhibition of VEGFA expression was observed to be completely reversed. Recent studies have found that SARS and transcription factor Yin Yang 1 (YY1) interact to form the SARS/YY1 complex.55 Thus, the SARS/YY1 complex and nuclear factor κB1 (NF-κB1) regulate angiogenesis by controlling VEGFA production. Since SARS affects the VEGF signaling pathway during the vascular development of zebrafish, SARS may become an effective target for treating CVD and cancers, among which anti-VEGF therapy has achieved certain efficacy.56 Further studies in zebrafish and mammalian SARS will contribute to a comprehensive understanding of the precise molecules involved in vascular network formation and remodeling.
Another study showed that noncoding genes can affect the process of vascular development during zebrafish angiogenesis by regulating the level of SARS protein.57 MicroRNA (miRNA)-1 directly inhibited the SARS protein level and indirectly upregulated the VEGFA protein level, whereas miRNA-206 directly inhibited the VEGFA protein level and had an antiangiogenic effect. Interestingly, miRNA-1 and miRNA-206 are highly similar and evolutionally conserved miRNAs. Their seed sequences were identical, with differences of only 4 nt in the sequence outside the seed.58 Future studies will focus on whether other ncRNAs are involved in the regulation of ARS and thus have a role in the regulation of angiogenesis, as well as the specificity of their target genes in targeting mechanisms and their physiological correlation. For example, the mechanisms by which miRNA, long noncoding RNA (lncRNA), circular RNA (circRNA), and other noncoding RNAs (ncRNAs) regulate ARS remains to be further studied.
HARS
Ni and Luo59 showed that abnormal blood vessel branching is increased in zebrafish HARS mutants. Moreover, functional defects in HARS lead to increased VE-cadherin and VEGFA expression and altered VE-cadherin localization. VE-cadherin is located at the intercellular contact point and is the main component of endothelial adhesion, which is necessary for tissue-stable vascular endothelium. Correct localization of VE-cadherin plays a key role in establishing the vascular network. VE-cadherin knockout reduced abnormal branching, but the vasculature thinned in mutants. This suggests that VE-cadherin is involved in regulating intercellular connectivity, leading to disordered connectivity in mutants. Interestingly, the use of VEGF receptor inhibitors blocked the vascular defects observed in the mutants, suggesting that HARS is dependent on the regulation of VEGFA signaling to participate in vascular development. More importantly, injection of human HARS mRNA effectively treated vascular defects in the mutant, suggesting that the role of HARS in angiogenesis is conserved between zebrafish and humans. Since this mutation does not affect the aminoacylation function of HARS, it reveals a new atypical function of HARS, broadening our knowledge of the regulatory network of angiogenesis. Therefore, HARS may be a potential therapeutic target in diseases related to vascular development.
Mitochondrial cardiomyopathy and ARS2
Mitochondrial cardiomyopathy refers to myocardial damage caused by mitochondrial dysfunction. Mutations in a variety of ARS genes are associated with mitochondrial cardiomyopathy (Table 2). As a hypermetabolic organ, the heart relies on the mitochondrial ATP of cardiomyocytes for normal production and synchronous contraction of the myocardium; therefore, the heart is particularly sensitive to abnormal ARS2 function. The typical clinical phenotypes of linear cardiomyopathy are HCM and dilated cardiomyopathy, arrhythmia, and the left ventricular densification insufficiency. The clinical manifestations can range from asymptomatic to fatal multiorgan diseases. Severe cardiac symptoms include HF and ventricular arrhythmias, which can worsen dramatically with sudden cardiac death when metabolic disorders occur. Mitochondrial diseases are often caused by genetic defects in pyruvate oxidation pathways. At the heart of this pathway is the oxidative phosphorylation (OXPHOS) system, the components of which are encoded by the nuclear and mitochondrial genomes. In addition to tRNAs and rRNAs encoded by mitochondrial DNA, mitochondrial translators include a variety of nuclear-encoded proteins, which are synthesized in the cytoplasm and then introduced into the mitochondrial matrix. These include mitochondrial ARS2, which catalyzes the connection of amino acids to specific tRNA.
Table 2.
Overview of nuclear-encoded ARS2 gene variants related with cardiomyopathy
| Gene | Locus | Protein | Disease phenotype(s) | Unique variants | Reference | |
|---|---|---|---|---|---|---|
| AARS2 | 6p21.1 | alanyl-tRNA synthetase 2 (mtAlaRS) | hypertrophic cardiomyopathy; left ventricular function reducing; cardiac failure; lactic acidosis; respiratory failure leukoencephalopathy with ovarian failure; multiple respiratory chain complex defects | c.1774C > T (p.Arg592Trp) | 60,62,64 | |
| c.1738C > T (p.Arg580Trp) | ||||||
| c.464T > G (p.Leu155Arg) | ||||||
| c.2872C > T (p.(Arg958∗)) | ||||||
| c.647dup (p.Cys218Leufs∗6) | ||||||
| c.1616A > G (p.Tyr539Cys) | ||||||
| c.2882C > T (p.Ala961Val) | ||||||
| c.209T > C (p.Phe70Ser) | ||||||
| PARS2 | 3p21.31 | prolyl-tRNA synthetase 2 (mtProRS) | heart failure; dilated cardiomyopathy; left ventricular hypertrophy, multiorgan failure; infantile-onset developmental delay; epilepsy | c.239T > C/p.(Ile80Thr) | 65 | |
| c.1091C > G/p.(Pro364Arg) | ||||||
| c.1130dupC/p.(Lys378∗) | ||||||
| c.836C > T/p.(Ser279Leu) | ||||||
| c.283G > A (p.Val95Ile) | ||||||
| c.1091C > G (p.Pro364Arg) | ||||||
| VARS2 | 6p21.3 | valyl-tRNA synthetase 2 (mtValRS) | hypertrophic cardiomyopathy; left ventricular function reducing; biventricular dilation; pericardial effusion; pulmonary hypertension; lactic acidosis; multiple respiratory chain complex defects; epileptic; cerebral atrophy | c.1100C > T (p.Thr367lle) | 67,110 | |
| c.1490G > A (p.Arg497His) | ||||||
| c.1258G > A (p.Ala420Thr) | ||||||
| c.2557-2A > G (p.Ala747Thr) | ||||||
| c.1150G > A (p.Asp384Asn) | ||||||
| c.1546G > T (p.Glu516∗) | ||||||
| c.2239G > A (p.Ala747Thr) | ||||||
| c.1135G > A (p.Ala397Thr) | ||||||
| c.1877C > A (p.Ala626Asp) | ||||||
| c.601C > T (p.Arg201Trp) | ||||||
| c.643C > T (p.His215Tyr) | ||||||
| c.1354A > G (p.Met452Val) | ||||||
| YARS2 | 12p11.21 | tyrosyl-tRNA synthetase 2 (mtTyrRS) | concentric hypertrophic cardiomyopathy; myopathy/exercise intolerance; respiratory insufficiency; multiple respiratory chain complex defects; lactic acidosis; sideroblastic anemia; MLASA syndrome | c.1175T > C (p.Leu392Ser) | 71 | |
| c.1106G > A (p.Cys369Tyr) | ||||||
| c.1147_1164dup (p.Val383_Glu388dup) | ||||||
| c.137G > A (p.Gly46Asp) | ||||||
| KARS2 | 16q23.1 | lysyl-tRNA synthetase 2 (mtLysRS) | cardiomyopathy; Charcot–Marie–Tooth polyneuropathy; autosomal recessive non-syndromic hearing loss; congenital visual impairment; mild psychomotor delay and mild myopathy | c.1049T > A (p.L350 > H) | 73 | |
| c.1169C > G (p.P390 > R) | ||||||
| IARS2 | 1q41 | isoleucyl-tRNA synthetase 2 (mtIleRS) | hypertrophic cardiomyopathy; left ventricular hypertrophy; progressive hearing impairment; multiple respiratory chain complex defects | m.4277T.C | 76 | |
| DARS2 | 1q25.1 | aspartyl-tRNA synthetase 2 (mtAspRS) | CHD; neurodevelopmental disabilities (NDDs); and other congenital anomalies (CAs) | c.228-16C > A (p.Arg76-SerfsX5) | 80 | |
| c.716T > C (p.Leu239Pro) | ||||||
AARS2
A mutation of the AARS2 gene is clinically manifested as fatal early-onset cardiomyopathy. Patients died of severe cardiomyopathy within 10 months of birth, and OXPHOS complexes in the heart and parts of the skeletal muscle and brain were significantly reduced. To date, only 8 cases of perinatal or infantile fatal cardiomyopathy have been reported in which mitochondrial alanine tRNA synthetase deficiency has led to incorrect tRNA aminoacylation.60,61 The AARS2 gene defect was identified by exon sequencing in two patients with hypertrophic mitochondrial cardiomyopathy with defects in complex I and IV of the cardiac respiratory chain.60 Interestingly, modeling analysis of the protein structure of AARS2 showed that one patient had a mutation that resulted in incorrect tRNA aminoacylation by affecting the editing domain, whereas the other patient had a mutation that severely interfered with catalytic function and prevented tRNA aminoacylation. Another study showed that uneven red fibular images, respiratory chain complex I and IV deficiency, and histological evaluation showed large mitochondrial accumulation and cytochrome C oxidase (COX)-negative fibers in patients with AARS2 deficiency.62
Aminoacylation domains exist in all ARS2. Only AARS2 and TARS2 have editing activity that redacts erroneous products. A slight decrease in this proofreading activity resulted in increased mouse embryonic mortality.63 At present, AARS2 mainly has a p.Arg592Trp mutation and p.Arg580Trp leading to HCM. Previous studies have shown that cardiomyopathy phenotypes are caused by a single allele, leading to amino acid changes in the editing domain of AARS2 (the p.Arg592Trp mutation), whereas leukocyte dystrophic mutations are located in other domains of the synthetase.64 AARS2 mutations severely impair amination in patients with cardiomyopathy, whereas mutations found in patients with leucocyte dystrophy partially retain amination. All mutations were predicted by structural analysis to decrease the aminoacylation activity of the synthetase because all AARS2 domains contribute to tRNA binding for aminoacylation. Computer simulation of p.Arg580Trp showed that Arg580 was exposed to the surface, but in vitro studies showed that the variant had little effect on the aminoacylation function and had an effect on the stability and folding of AARS2 proteins.64 In contrast, Arg592 forms a variety of non-covalent bonds with its neighbor Arg. These residues indicate the importance of arginine at this location for protein stability. It is not located near the active site of aminoacylation, so there is no accumulation of mischarged tRNA[Alanine (Ala)] during aminoacylation. Patients with p.Arg580Trp had significantly lower levels of AARS2 in their fibroblasts than those with at least one p.Arg592Trp mutation. In the analysis of the protein lysates of skeletal muscle cells, cardiomyocytes, and fibroblasts in the patients, the reduced AARS2 protein level was associated with multiple severe OXPHOS defects in skeletal muscle and myocardium, but this phenomenon was not observed in the fibroblasts of the patients. It reveals that skeletal muscle cells and cardiomyocytes are more prone to deficiency in mitochondrial protein synthesis function, and AARS2 activity is crucial for early development of cardiac muscle cells.
These findings support the hypothesis that other AARS2-associated variants may only cause a decrease in AARS2 protein levels or loss of aminoacylation activity and are not present in the heart. In other words, the same ARS2 mutation leads to two very different tissues involved in different diseases. Phenotypic differences in AARS2-related diseases are due to differences in aminoacylation. However, stable AARS2 protein levels in vivo have not been evaluated in tissues extracted from patients with other AARS2 dysfunction-related diseases. In addition, there has been a lack of in vitro studies confirming the effects of the p.Arg592Trp mutation on amino acid activation, aminoacylation, and misaminoacylation activities. This will help to analyze whether the two mutations share a common biological mechanism or which may be two mechanisms of clinically fatal cardiomyopathy.
Mitochondrial prolyl-tRNA synthetase (PARS2)
PARS2 made prolyl-tRNA of mitochondria charge homologous proline (P). Clinical features observed in patients with PARS2 dysfunction included abnormalities of the brain and muscles and other systemic abnormalities, including dilated cardiomyopathy. Patients with PARS2 dysfunction usually have similar features and symptoms, and most die between 0 and 10 years old. PARS2 mutations are rare, and only 10 patients have previously been reported. To date, patients with the longest lifespan of lesions caused by the biallelic PARS2 have been reported.65 Unlike most previous cases, the patient did not have chronic lactic acidemia, and cardiomyopathy did not occur until the age of 19. He died of HF at 21. This may be because he has been receiving supportive antioxidant supplements since infancy to treat mitochondrial dysfunction. In addition, his older brother, who had the same phenotype, received no treatment and died of HF at the age of 5. Normally, treatment strategies for some mitochondrial diseases include direct involvement of natural substances in ATP production, such as creatine, carnitine, and coenzyme Q10, through mitochondrial function.66 Mitochondrial cytopathic disease is associated with decreased aerobic energy transduction, increased occurrence of oxidative stress response, apoptosis, and necrosis. Theoretically, the combination of specific nutritional drugs may provide an alternative source of energy by bypassing defects in mitochondrial respiration. Although many factors may have influenced the difference in disease progression between the longest-lived patient and his older brother, antioxidant supplements may have reduced lactate levels, thereby delaying the onset of cardiomyopathy. Currently, there is a lack of clinical and laboratory validation of the therapeutic effects of antioxidants and other vitamins in patients with PARS2 dysfunction and even in ARS-related diseases, especially in cardiomyopathy,
Mitochondrial valyl-tRNA synthetase (VARS2)VARS2 is a protein of 1,093 amino acids located on chromosome 6p21.3 of the mitochondrial matrix. It is associated with encephalopathy or hypertrophic non-obstructive cardiomyopathy and often leads to chronic disabilities and poor prognosis such as short stature, growth hormone deficiency, and hypogonadism.67, 68, 69 VARS2 mutations are rare, and only 14 patients have been reported so far. Ten of the fourteen patients had HCM. This suggests that HCM may be a common feature of this mutation. Echocardiography showed different degrees of double ventricular hypertrophy, which can be accompanied by asymmetric ventricular septal hypertrophy and biventricular dilation. The left ventricular function may be reduced, with pericardial effusion, hepatosplenomegaly, and pulmonary hypertension. Postmortem examination of the hearts of some patients revealed double ventricular hypertrophy, asymmetrical ventricular septal hypertrophy, and immature cardiomyocytes. Microscopic sections showed diffuse vacuolar degeneration of muscle cells. Dysfunction of VARS2 should be included in the differential diagnosis of the etiology of neonatal mitochondrial brain cardiomyopathy.
However, the same genetic defects affect specific tissues or organs and thus exhibit very different clinical phenotypes. Little is known about how the same genetic defects lead to these heterogeneous phenotypes. The authors suggest that it may be related to the concentration of tissue-related amino acids and the different sensitivity of cells to impaired mitochondrial protein synthesis.67 This hypothesis may explain the heterotypic phenotype of the same gene defect. Interestingly, the clinical phenotype of multiple patients with a variant of c.1100C > T (p.Thr367Ile) suggests that the mutation may have a minor effect on the heart.67,70 In addition, the authors found that VARS2 had two mutations near the binding pocket of homologous valine (p.Arg497His and p.Ala626Asp).67 By analyzing the changes of VARS2 structure and physicochemical properties of VARS2, it was found that some VARS2 mutations may modify the conformation of the valine-binding region, thus destroying the function of the enzyme.
YARS2
The YARS2 gene encodes a mitochondrial protein that catalyzes the binding of tyrosine to tRNA. YARS2 mutations mainly occur in the autosomal recessive MLASA syndrome (myopathy, lactic acidosis, and sideroblastic anemia), including myopathy, lactic acidosis, and sideroblastic anemia, and are associated with complex defects in the mitochondrial respiratory chain from infants to children. Only 17 patients with mitochondrial myopathy associated with YARS2 mutation have been reported.71 Among the 17 patients with YARS2-related mitochondrial myopathy, 88% had elevated blood lactic acid levels, accompanied by systemic myopathy. Sideroblastic anemia was present in 71% of patients. HCM (53%) and respiratory insufficiency (47%) were also prominent clinical features. Unlike other ARS2 mutations, the central nervous system is rarely affected. These findings underscore the importance of regular monitoring of respiratory and cardiac function. In addition, early encouragement of supportive therapies, such as non-invasive ventilation and drug-induced heart remodeling drugs, can delay disease progression. Muscle histochemical studies showed that all patients tested had severe COX deficiency. The muscle biopsy results of severe COX deficiency in muscle fibers were consistent with the diagnosis of mitochondrial myopathy. Interestingly, western blotting of fibroblasts and myoblasts in some patients showed no decrease in YARS2 protein levels. This may reflect the tissue specificity of YARS2 and other ARS mutations, as well as the changes in the expression threshold between different individuals and tissues. Sommerville et al.71 hypothesized that one factor contributing to this change might be the rate of protein replacement in individual tissues. These data showed that the decreased levels of mitochondrial cytochrome oxidase subunit I and II (mt-COI and mt-COII) in fibroblasts and myoblasts of the patients were slight, although this was supported by a significant deficiency of COX in muscles. However, there is still a lack of evaluation of YARS2 protein levels in cardiomyocytes. Besides, other in vitro studies on the YARS2 mutation were lacking to confirm the effects of amino acid activation, aminoacylation, and misaminoacylation activities on myocardial cells, to further study the mechanism of cardiomyopathy.
KARS
Mutations in KARS encode mitochondrial and cytoplasmic KARS. The clinical phenotypes of KARS mutations include the autosomal-recessive CMT multiple neuropathies; autosomal-recessive non-syndromic hearing loss; and congenital visual impairment, progressive microcephaly, and cognitive deficits. To date, eight mutations have been identified in KARS, but only two of them have recently been reported to affect mitochondrial function and to be associated with neurophenotypes, lactic acidosis, HCM, and mitochondria respiratory complexes I (CI) and IV (CIV) binding defects.72 Consistently, muscle biopsies in patients showed a significant reduction in COX in the muscle.
By targeting sequencing, Verrigni et al.73 identified two new variants (L350H mutation, P390R mutation) in KARS. The two mutations occurred in the enzyme’s catalytic domain and damaged two amino acid residues, which are highly conserved from advanced organisms to single-celled organisms. The L350H mutation is expected to destroy the enzyme activity, whereas the P390R mutation will impair the enzyme activity by eliminating the proline-specific skeleton angle restriction and modifying the salt-bridge pattern, resulting in incorrect protein-interface folding related to the KARS intermolecular interaction. All of these will affect the interaction between KARS and P38, thus affecting the formation of the multisynthetase complex (MSC).74 This is important because in advanced eukaryotes, many ARS bind together to form high molecular weight MSC, in which at least 9 ARS and 3 helper proteins (p18, p38, p43) are linked to perform different functions other than aminoacylation. Verrigni and co-workers’73 findings support the possibility that KARS mutations may cause myopathy and HCM due to altered mitochondrial function. Therefore, mitochondrial dysfunction caused by KARS mutation may be the cause of childhood myopathy and HCM. However, the level of KARS2 protein in cardiomyocytes, skeletal muscle cells, and fibroblasts has not been evaluated.
IARS2
IARS2 is a 1,012-amino acid protein located at chromosome 1q41 in the mitochondrial matrix. IARS2 mutations have been reported in patients with cataracts, partial sensorineural deafness, short stature growth hormone deficiency, peripheral neuropathy, or Leigh syndrome.75 In a recent study, skeletal muscle biopsies of patients with IARS2 gene deficiency revealed many COX composite defects in fibrous and respiratory chain complex I, III, and IV and reduced homeostasis of IARS2 mutations in skeletal muscle.76 In addition, mitochondrial genome sequencing revealed the presence of homotypic m.4277T > C mutations in IARS2. Forcing cells to utilize OXPHOS in galactose medium resulted in increased (reactive oxygen species) ROS production in mutant transmitochondrial cybrids and decreased cell viability due to widespread cell death. This suggests that homoplasmic mitochondrial tRNA (mt-tRNA) mutations with a cardiac limited clinical phenotype can themselves induce mitochondrial dysfunction. The misfolding of mt-tRNA molecules was proved by electrophoretic migration of mutated mt-tRNA molecules. Surprisingly, this mutation did not affect the normal aminoacylation function of IARS2, but structural changes can destroy its stability, and the change of structure will damage its stability. Interestingly, all homogeneous mtDNA mutations are characterized by very different penetrance even among members of the same family. Pathogenic mtDNA mutations can be heterogenous or homogeneous. For the first time, high levels of constitutive ARS were observed in human tissues to prevent the phenotypic expression of homoplasmic mt-tRNA point mutations. One hypothesis is that differences in the efficiency of compensation mechanisms (e.g., mitochondrial biological compensation) are associated with the production of ROS resulting from impaired OXPHOS.77 In addition, it is speculated that nuclear modifiers, mtDNA haplotypes, and epigenetic or environmental factors may influence the tissue specificity and severity of homoplasmic mt-tRNA mutations. It is necessary to further study the pathogenic biochemical pathway of the IARS2 mutation in order to understand its related diseases.
Aspartyl-tRNA synthetase (DARS)
In a case-control study, genetic variants of the three SNPs of the DARS gene were observed to influence susceptibility to isolated ventricular septal defect (VSD), and the risk increased significantly with increased alleles.78 Transcription factors are known to play a fundamental role in all stages of cardiac development, including compartment formation, valvular and septal generation, and cardiac pedigree determination.79 DARS2 knockout mouse model, the DARS2 deficient type is damaged the stability of mitochondrial protein in mouse heart tissue. This results in increased expression of stress-related transcription activators to C/EBP homology proteins (CHOP) and activated transcription factors 4 and 5(Atf4 and Atf5), as well as increased mitochondrial biology and autophagy of mitochondrial non-folded proteins, resulting in metabolic changes.80 This tissue-specific adaptive stress response was not related to respiratory chain defects. The heart is the body’s most energy-hungry organ, whether these adaptive stress responses promote rather than delay the progression of cardiomyopathy. Previous studies have shown that increased mitochondrial biogenesis and metabolic changes in tissue-specific mitochondrial transcription factor A (TFAM)-deficient hearts at the late stage of disease do not delay the progression of HF but rather promote it.81 In addition, overexpression of PGC-1α reduces autophagy. Interestingly, elevated PGC-1α expression levels were beneficial for the heart of neonates, whereas in adult mice, they resulted in mitochondrial structural abnormalities, mitochondrial biogenesis, and a slight increase in cardiomyopathy.82 In contrast, skeletal muscle has intrinsic mechanisms that depend on the slow transformation of mitochondrial transcripts and high protein static buffering capacity. Increased understanding of tissue-specific pathophysiology may lead to the discovery of targets that may be easier to treat than respiratory chain dysfunction itself, to alleviate the development and progression of ARS2-related mitochondrial cardiomyopathy.
HF
HF is usually accompanied by cardiac fibrosis due to increased fibrin synthesis following activation of cardiac fibroblasts (CFs). Since many fibrins are rich in proline, such as collagen, the translation function involved in EPRS may play a key role in fibrin synthesis during the development of cardiac fibrosis. EPRS catalyzes the binding of the two amino acids glutamic acid (E) and proline (P) to their homologous tRNA for protein synthesis.
Wu et al.83 found that mRNA and protein levels of EPRS were elevated in failing hearts. This study further demonstrated that gene knockout of an allele of EPRS significantly reduced the degree of heart fibrosis by approximately 50% in a mouse model of HF. The EPRS-specific inhibitor Halofuginone (Halo) blocks the binding of EPRS to proline and tRNA[Proline (Pro)] and blocks their connection.84 Halo activates key signaling elements in the AAR genes (asna and chop) and AAR pathways (general control nonderepressible 2 [GCN2] and eukaryotic initiation factor 2a [eIF2a]) and extensively regulates the transcriptome and proteome of CFs. By activating the AAR pathway of cardiomyocytes, pulmonary congestion can be reduced, left ventricular function and left ventricular remodeling/fibrosis can be improved, ischemic myocardium can be reduced, and survival rate can be improved. However, the therapeutic mechanisms of Halo inhibit atypical transforming growth factor (TGF) signaling and the activated amino acid hunger response (AASR) only at the transcriptional level.84, 85, 86 Studies have demonstrated the protective and antifibrosis effects of Halo in a variety of mouse HF models.85 In addition, some newly identified proline-rich genes (e.g., Ltbp2 and Sulf1) can also be regulated by EPRS translation. Sulf1 is abundantly expressed in human and mouse myofibroblasts. In the CF culture system, the level of SULF1 expression influences collagen deposition and CF activation. SULF1 overexpression promotes collagen deposition and myocardial fibroblast activation and partially antagonizes the antifibrosis effect of Halo therapy. This finding indicates that SULF1 is a new biomarker of cardiac fibrosis and suggests that ERAS may also affect collagen deposition and myocardial fibroblast activation by regulating SULF1 expression.
AAA
Recent studies have found that ARS is also associated with the development and progression of AAA.87 AAA is a chronic degenerative condition that causes permanent focal dilation of the walls of the abdominal aorta. Several factors are at risk of developing AAA, including age, high blood pressure, smoking, dyslipidemia, and a family history of AAA and other CVDs. Li et al.87 searched and analyzed the Gene Expression Omnibus (GEO) database and found that IARS mRNA expression was markedly higher in the aortas of AAA patients than in those of patients without AAA. The imbalance of medial aortic injury and repair is an important feature of aortic disease. Inhibiting IARS expression levels in mice significantly reduced AAA formation and reduced phenotypic transformation and apoptosis in vascular smooth muscle cells (VSMCs). The expression of p-p38, Bcl-2-associated X protein (Bax), and osteopontin (OPN) was significantly increased in AAA tissues, whereas the levels of Smooth muscle protein 22-α (SM22α) and p-PI3K were significantly decreased. In addition, the activation state of the PI3K and p38 mitogen-activated protein kinase (MAPK) pathways also changed correspondingly. Consistently, angII-induced phenotypic transformation and apoptosis of VSMCs were reduced after IARS inhibition in vitro. IARS promotes the phenotypic transformation and apoptosis of VSMCs through the p38 MAPK and PI3K signaling pathways, respectively, thus promoting the occurrence and development of AAA (Figure 3). This study may contribute to further understanding of the mechanism of medial aortic degeneration, and IARS may be a potential target for preventing the development of AAA.
Figure 3.

ARS promotes angiogenesis through an unfolded protein response pathway
Defects in TARS and IARS lead to endoplasmic reticulum stress and activate the unfolded protein response pathway. The unfolded protein response genes ATF4, ATF6, X-box protein 1 (XBP1), and VEGFA were upregulated in TARSy58 and IARSy68 mutants.
Application of ARS drug research and development
Protein translation is the classical function of ARS; this function has been widely validated and used as a target for the development of antimicrobial and antimalarial drugs.88 ARS inhibitors have several targets: amino acid binding sites, ATP binding sites, editing decision regions, and tRNA recognition folding regions. In addition, there are amino acid-AMP analog inhibitors, amino acid-tRNA analog inhibitors, and inhibitors that inhibit the binding of ARS to related proteins. Most inhibitors are amino acid binding site inhibitors. An increasing number of innovative drugs targeting ARS have been discovered and modified, and those that have been marketed have demonstrated that targeting ARS is an important direction for antimicrobial drug development. However, the lack of bioavailability and selectivity of ARS inhibitors are common problems at present; thus, few drugs have entered clinical studies.
As mentioned earlier, there have been few studies on ARS inhibitors in the treatment of CVD. Currently, Halo is the most widely studied mechanism of ARS for treating CVD. Halo is an EPRS inhibitor with excellent antimalarial activity, and its mechanism of action is the consumption of ATP and EPRS binding to the active site. Most proline-rich proteins are secreted or transmembrane proteins, and many of them are ligands/receptors for extracellular matrix or cellular signaling that promote cardiac fibrosis. In vitro and in vivo experiments on human CFs, cardiomyocytes, and mouse models of HF confirmed the antifibrosis effect of Halo.85 In addition, Borrelidin (BN) is a TARS inhibitor and an effective antimicrobial and antimalarial drug, and it has been shown to inhibit angiogenesis in a rat aortic angiogenesis model.89 Notably, BN has cytotoxic effects, possibly because it induces AAR, which leads to cell-cycle arrest and apoptosis.28 The toxicity of the BN derivative BC194 to endothelial cells is greatly reduced, but it can still inhibit angiogenesis. Treatment of endothelial cells with BC194 inhibited endothelial cell migration and increased cell-to-cell contact. At the same time, in vivo studies of BC194 application in zebrafish resulted in incomplete vascular development, increased ectopic branches, and reduced vascular lumen. There is a lack of research on the efficacy and side effects of CVD in mammals. These studies have revealed the potential of ARS as a treatment for CVD; however, whether ARS can be used as a key target for the treatment of CVD needs further study.
Aminoacylation
The classic function of ARS is to precisely attach each type of amino acid to a specific tRNA in a process known as aminoacylation. Recently, significant studies have found that both cytoplasmic ARS and mitochondrial ARS2 can undergo aminoacylation in their corresponding substrate proteins.12 Each ARS/ARS2 transfers the homologous amino acid to the ε-amine of lysine in the modified protein. ARS can also be used as an amino acid sensor to sense the sufficiency of its homologous amino acids. The process by which amino acids are covalently linked to proteins is called protein aminoacylation. Protein aminoacylation has three characteristics: (1) dependence on ARS as an aminoacyl transferase and tRNA as its metabolic carrier; (2) specificity, which depends on the corresponding ARS; and (3) variability, which differs depending on substrate specificity and covalent bond formation. More importantly, protein aminoacylation can affect its activity and thus its function.
He et al.12 have shown that LARS can induce leucinization of Rag heterodimer (RagA/B); this leads to an increase in guanosine triphosphate (GTP) load of RagA/B, followed by mTORC1 activation when amino acid levels reach their physiological levels. The extracellular apoptotic signal sensor (apoptosis signal-regulating kinase 1 [ASK1]) was glutamated to negatively regulate the kinase activity of ASK1 and inhibit cell apoptosis. Lysine aminoacylation markers can be removed by deacetylases such as SIRT1 and SIRT3, dynamically regulating the level of aminoacylation. Therefore, ARS can also be used as a direct intracellular amino acid sensor to modify the ε-amine of lysine in proteins, thus maintaining intracellular homeostasis through aminoacylation. This is another important non-classical feature of ARS. Because amino acids have multiple sensors, and the affinity and subcellular localization of these sensors are different, different pathways can coordinate and accurately detect amino acid signals. Amino acid modification of proteins enriches the mechanism of amino acid signaling to biological networks. Notably, He at al.12 found that the lysine-aminoacylated protein is widely present in human cells, and its functions cover almost all aspects of cell physiology, including cardioventricular myogenesis, myocardial fiber development, cardiac rhythm, and blood pressure regulation. The proteins related to cardiac rhythm regulation include desmoplakin, dystrophin, and desmin. Developmental regulation-related proteins (e.g., GTP-binding protein 1 and caspase-9), angiogenesis-related proteins (e.g., cyclin-dependent kinase-like 5), and key molecules in the signaling pathway (e.g., MAPK-activated protein kinase 2) can all be modified by lysine aminoacylation. This indicates that metabolite-derived modification, especially aminoacylation modification proteins, may have great research prospects in the pathogenesis of CVD and may therefore shed light on a new dimension of this process. These findings show the potential role of various ARSs in the development, diagnosis, and treatment of CVD, as well as present considerable prospects for clinical application.
Discussion
With the consideration that various ARS mutants play a key role in the development of CVDs, different ARS mutants have different clinical manifestations and individual growth prognoses. These include angiogenesis, HCM, abnormal early development of the heart (especially in early cardiomyocytes development), dysfunction of the mitochondria of the heart leading to HF, myocardial infarction, atherosclerosis, and AAA, among other CVDs.
Lack of ARS can lead to cardiac angiogenesis defects and lower capillary density. Endothelial cell migration and proliferation play an important role in angiogenesis. The absence of WARS2 can affect endothelial cell migration and proliferation, thereby affecting cardiac angiogenesis and reducing capillary density. ARS cannot only affect angiogenesis by regulating endothelial cell function but can also regulate VEGF expression by regulating signal transduction pathways, thus regulating angiogenesis. Interestingly, the natural fragments of YARS and WARS have no effect on angiogenesis, whereas the small fragments produced by hydrolysis can affect angiogenesis. Mini-YARS/mini-WARS affect angiogenesis by regulating endothelial cell proliferation and migration and the expression level of the vascular chemokine VEGF. Furthermore, the effects of mini-YARS on angiogenesis are different at high and low levels. Mini-WARS also reportedly inhibits mini-YARS-induced angiogenesis. The balance between the two types of ARS affects the balance between angiogenesis and antiangiogenesis, providing new possibilities for angiogenesis therapy in ischemic diseases. SARS also has the nuclear localization domain UNE-S and can form a complex with the transcription factor YY1, which enriches the mechanism by which ARS regulates VEGF.
In recent years, an increasing number of studies have reported the close association between ncRNAs and the development of CVD.90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105 Meanwhile, ncRNAs can be used as an ARS regulator. For example, miRNA-1 regulates the protein level of SARS and indirectly upregulates the protein level of VEGF to promote angiogenesis. miRNA-206 can directly inhibit the VEGF protein level and play an antagonistic role in angiogenesis. However, it is still necessary to study the regulatory effect of ncRNA on ARS and its targeting mechanism under different physiological and pathological conditions, which may provide insight into a new dimension of the ARS regulatory mechanism. IARS promotes the regulation of phenotypic transformation and VSMC apoptosis, which promotes the occurrence and development of AAA. This suggests that IARS may contribute to the AAA diagnosis as new diagnostic molecules and even as a potential therapeutic target. At present, only some studies have shown that mRNA injection of human-related ARS can effectively treat abnormal blood vessel branching in zebrafish mutants; these studies have included the injection of TARS, SARS, and HARS. This suggests that the role of ARS in regulating zebrafish and human angiogenesis may be conserved. This finding contributes to the possibility of using zebrafish ARS mutants to model human disease in the future and presents possibilities for further study on the mechanisms of angiogenesis and related angiogenic molecules.
ARS affects not only the coronary arteries and aorta, which provide blood oxygen, but also cardiomyocytes and fibroblasts. HCM patients, whose ARS gene defects lead to mitochondrial respiratory defects, have poor prognoses. Some ARS patients are reported to be newborns to 10 years old, whereas others may even experience perinatal death. However, relatively normal lactic acid levels and delayed disease progression can reportedly be promoted in individual patients through regular respiratory and cardiac function tests, in addition to early support via therapies such as specific nutritional and vitamin combinations, noninvasive ventilation, and cardiac remodeling drugs. These patients often have other systemic diseases, and the same mtARS mutation can lead to different diseases involving different tissues. One hypothesis suggests that phenotypic differences in the same ARS-related diseases are due to mutations exerting different effects on aminoacylation. That is, cardiac abnormalities may be present when aminoacylation is severely impaired, and other systemic diseases may be present when aminoacylation is partially reduced. At the same time, different organs have different sensitivities to mitochondrial functional changes at different developmental stages. This suggests that ARS activity is critical to the heart during early development, which somewhat explains the younger age of onset of HCM caused by defects in the ARS gene. Research on ARS dysfunction has great prospects for elucidating the mechanism by which the same ARS gene defects cause heterogeneous phenotypes. This research may also elucidate the signaling pathways, cytokines, and abnormal proteins involved, thus contributing to future clinical diagnosis and disease prognosis.
As previously mentioned, the specific inhibitor of EPRS, Halo, can reportedly regulate the transcriptome and proteome of CFs by activating signal transduction elements. In addition, its antifibrosis effect has been verified in various mouse models of HF, with its application improving left ventricular function and remodeling and improving survival rate. At the same time, the protein of the proline-rich gene Sulf1 is enriched in human and mouse muscle fibroblasts after translation by EPRS. High levels of Sulf1 protein expression promote collagen deposition and CF activation and partially antagonize the antifibrosis effect of Halo. This suggests that there are a number of new proteins that are rich in certain amino acids that need to be studied in the context of CVD. The effects of ARS on organelles include not only mitochondria but also organelles associated with autophagy. Studies conducted in the DARS2-deficient mouse model have found that the impaired protein stability of the mitochondria in the heart tissue leads to abnormal increases in CHOP, Atf4, and Atf5 expression, which not only causes changes in mitochondrial function but also damages the autophagy pathway. This suggests that ARS gene defects may affect changes in organelle function through a variety of pathways, leading to cell-function changes and disease. This suggests that the ARS-translated proteins and activated cytokines may be novel markers of cardiac dysfunction, and these may play a therapeutic role in the future by regulating the expression of related proteins. Furthermore, the signal transduction, cytokine, and protein expression of various ARSs can affect organelle function and lead to HCM caused by cardiomyocytes and fibroblasts. This knowledge will contribute to the development of clinically targeted therapies and protocols and the discovery of new markers of cardiac dysfunction.
Although many clinical cases of pathogenic mutations in cytoplasmic and mitochondrial ARS genes have been reported, there are still many problems to be solved in the study of pathogenic mechanism, especially regarding the relationship between nonclassical dysfunction and disease phenotype. Mitochondrial ARS gene mutations lead to impaired mitochondrial protein synthesis, resulting in mitochondrial productivity defects; however, the relationship between the occurrence of such defects and mitochondria–nucleus communication has not been extensively studied. Despite this, existing studies have also shown the complexity of the pathogenic mechanism of ARS. The mechanisms by which several mutated ARSs cause CVD differ, and these mechanisms involve new and unknown cellular functions of ARS. The questions of why the pathogenic effects of ARS target only certain types of cells, why symptoms appear at a certain age, and what other aspects of the cellular functional network are involved in the pathogenic process must be studied further. At present, some articles point out that ARS is a biomarker for diagnosis and monitoring in tumors. For example, plasma KARS1 is a new biomarker for diagnosis of colorectal cancer.106 MARS is a useful diagnostic marker for lymph node metastasis of non-small cell lung cancer.107 However, there is no clear article pointing out that ARS has been used as a biomarker of CVDs. At present, it is found that the abnormal function of ARS affects the proliferation and migration of endothelial cells and regulates the expression of VEGF and affects angiogenesis. ARS also affects its phenotype transformation and apoptosis in smooth muscle cells. Therefore, we speculate that ARS may be a potential biomarker for detection and/or treatment in CVDs, such as myocardial infarction, HCM, and early abnormal development of the heart, needing further deep investigation.
Conclusions
The widespread involvement of ARS in CVD has aroused a high level of research interest. In particular, recent studies have shown that ARS modifies the function of regulatory proteins through aminoacylation; this regulation may be widely present in various cell components, molecular functions, and biological processes. Further studies are required to determine whether ARS can be used as a potential therapeutic target for treating CVD.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number [no.] 81870331), Natural Science Foundation of Shandong Province (grant no. ZR201910310323), and Qingdao Municipal Science and Technology Bureau Project (grant no. 21-1-4-rkjk-12-nsh).
Author contributions
Y.Z., Y.Y., T.Z., X.F., X.H., X.L., L.H.H.A., and Z.W. wrote and revised the manuscript.
Declaration of interests
The authors have no competing interests.
Contributor Information
Zhibin Wang, Email: m17853291291@163.com.
Tao Yu, Email: yutao0112@qdu.edu.cn.
References
- 1.Dubois-Deruy E., Peugnet V., Turkieh A., Pinet F. Oxidative Stress in Cardiovascular Diseases. Antioxidants. 2020;9:864. doi: 10.3390/antiox9090864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel P., Karch J. Regulation of cell death in the cardiovascular system. Int. Rev. Cell Mol. Biol. 2020;353:153–209. doi: 10.1016/bs.ircmb.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Gao J., Chen X., Shan C., Wang Y., Li P., Shao K. Autophagy in cardiovascular diseases: role of noncoding RNAs. Mol. Ther. Nucleic Acids. 2020;23:101–118. doi: 10.1016/j.omtn.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao X.F., Wang Z.M., Wang F., Gu Y., Zhang J.J., Chen S.L. Exosomes in Coronary Artery Disease. Int. J. Biol. Sci. 2019;15:2461–2470. doi: 10.7150/ijbs.36427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X., Yang Y., Guo J., Meng Y., Li M., Yang P., Liu X., Aung L.H.H., Yu T., Li Y. Targeting the epigenome in in-stent restenosis: from mechanisms to therapy. Mol. Ther. Nucleic Acids. 2021;23:1136–1160. doi: 10.1016/j.omtn.2021.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wernly B., Pernow J., Kelm M., Jung C. The role of arginase in the microcirculation in cardiovascular disease. Clin. Hemorheol. Microcirc. 2020;74:79–92. doi: 10.3233/CH-199237. [DOI] [PubMed] [Google Scholar]
- 7.Guo L., Yin A., Zhang Q., Zhong T., O’Rourke S.T., Sun C. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac hypertrophy via a Sirt3-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2017;312:H980–H991. doi: 10.1152/ajpheart.00768.2016. [DOI] [PubMed] [Google Scholar]
- 8.Wang W.R., Li T.T., Jing T., Li Y.X., Yang X.F., He Y.H., Zhang W., Lin R., Zhang J.Y. SIRT1 Regulates the Inflammatory Response of Vascular Adventitial Fibroblasts through Autophagy and Related Signaling Pathway. Cell. Physiol. Biochem. 2017;41:569–582. doi: 10.1159/000457878. [DOI] [PubMed] [Google Scholar]
- 9.Delarue M. Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol. 1995;5:48–55. doi: 10.1016/0959-440x(95)80008-o. [DOI] [PubMed] [Google Scholar]
- 10.Oprescu S.N., Griffin L.B., Beg A.A., Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113:139–151. doi: 10.1016/j.ymeth.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 12.He X.D., Gong W., Zhang J.N., Nie J., Yao C.F., Guo F.S., Lin Y., Wu X.H., Li F., Li J. Sensing and Transmitting Intracellular Amino Acid Signals through Reversible Lysine Aminoacylations. Cell Metab. 2018;27:151–166.e6. doi: 10.1016/j.cmet.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Kim E.Y., Jung J.Y., Kim A., Kim K., Chang Y.S. Methionyl-tRNA synthetase overexpression is associated with poor clinical outcomes in non-small cell lung cancer. BMC Cancer. 2017;17:467. doi: 10.1186/s12885-017-3452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mei X., Qi D., Zhang T., Zhao Y., Jin L., Hou J., Wang J., Lin Y., Xue Y., Zhu P. Inhibiting MARSs reduces hyperhomocysteinemia-associated neural tube and congenital heart defects. EMBO Mol. Med. 2020;12:e9469. doi: 10.15252/emmm.201809469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park S.G., Kim H.J., Min Y.H., Choi E.C., Shin Y.K., Park B.J., Lee S.W., Kim S. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. USA. 2005;102:6356–6361. doi: 10.1073/pnas.0500226102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko Y.G., Kim E.Y., Kim T., Park H., Park H.S., Choi E.J., Kim S. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001;276:6030–6036. doi: 10.1074/jbc.M006189200. [DOI] [PubMed] [Google Scholar]
- 17.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayençon M., Sarcey A., Broussolle E. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 20.Tsai P.C., Soong B.W., Mademan I., Huang Y.H., Liu C.R., Hsiao C.T., Wu H.T., Liu T.T., Liu Y.T., Tseng Y.T. A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain. 2017;140:1252–1266. doi: 10.1093/brain/awx058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vester A., Velez-Ruiz G., McLaughlin H.M., Lupski J.R., Talbot K., Vance J.M., Züchner S., Roda R.H., Fischbeck K.H., Biesecker L.G., NISC Comparative Sequencing Program A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat. 2013;34:191–199. doi: 10.1002/humu.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sano M., Minamino T., Toko H., Miyauchi H., Orimo M., Qin Y., Akazawa H., Tateno K., Kayama Y., Harada M. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 23.Oka T., Akazawa H., Naito A.T., Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014;114:565–571. doi: 10.1161/CIRCRESAHA.114.300507. [DOI] [PubMed] [Google Scholar]
- 24.Taqueti V.R., Hachamovitch R., Murthy V.L., Naya M., Foster C.R., Hainer J., Dorbala S., Blankstein R., Di Carli M.F. Global coronary flow reserve is associated with adverse cardiovascular events independently of luminal angiographic severity and modifies the effect of early revascularization. Circulation. 2015;131:19–27. doi: 10.1161/CIRCULATIONAHA.114.011939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castranova D., Davis A.E., Lo B.D., Miller M.F., Paukstelis P.J., Swift M.R., Pham V.N., Torres-Vázquez J., Bell K., Shaw K.M. Aminoacyl-Transfer RNA Synthetase Deficiency Promotes Angiogenesis via the Unfolded Protein Response Pathway. Arterioscler. Thromb. Vasc. Biol. 2016;36:655–662. doi: 10.1161/ATVBAHA.115.307087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X., Shi Y., Zhang H.M., Swindell E.C., Marshall A.G., Guo M., Kishi S., Yang X.L. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat. Commun. 2012;3:681. doi: 10.1038/ncomms1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzog W., Müller K., Huisken J., Stainier D.Y. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circ. Res. 2009;104:1260–1266. doi: 10.1161/CIRCRESAHA.108.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirando A.C., Fang P., Williams T.F., Baldor L.C., Howe A.K., Ebert A.M., Wilkinson B., Lounsbury K.M., Guo M., Francklyn C.S. Aminoacyl-tRNA synthetase dependent angiogenesis revealed by a bioengineered macrolide inhibitor. Sci. Rep. 2015;5:13160. doi: 10.1038/srep13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu X., Zong T., Yang P., Li L., Wang S., Wang Z., Li M., Li X., Zou Y., Zhang Y. Nicotine: Regulatory roles and mechanisms in atherosclerosis progression. Food Chem. Toxicol. 2021;151:112154. doi: 10.1016/j.fct.2021.112154. [DOI] [PubMed] [Google Scholar]
- 30.Wu B., Zhang Z., Lui W., Chen X., Wang Y., Chamberlain A.A., Moreno-Rodriguez R.A., Markwald R.R., O’Rourke B.P., Sharp D.J. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams T.F., Mirando A.C., Wilkinson B., Francklyn C.S., Lounsbury K.M. Secreted Threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci. Rep. 2013;3:1317. doi: 10.1038/srep01317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao Z., Wang H., Mao X., Luo L. Noncanonical function of threonyl-tRNA synthetase regulates vascular development in zebrafish. Biochem. Biophys. Res. Commun. 2016;473:67–72. doi: 10.1016/j.bbrc.2016.03.051. [DOI] [PubMed] [Google Scholar]
- 33.Liu F., Smith J., Zhang Z., Cole R., Herron B.J. Genetic heterogeneity of skin microvasculature. Dev. Biol. 2010;340:480–489. doi: 10.1016/j.ydbio.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pravenec M., Zídek V., Landa V., Mlejnek P., Šilhavý J., Šimáková M., Trnovská J., Škop V., Marková I., Malínská H. Mutant Wars2 gene in spontaneously hypertensive rats impairs brown adipose tissue function and predisposes to visceral obesity. Physiol. Res. 2017;66:917–924. doi: 10.33549/physiolres.933811. [DOI] [PubMed] [Google Scholar]
- 35.Wang M., Sips P., Khin E., Rotival M., Sun X., Ahmed R., Widjaja A.A., Schafer S., Yusoff P., Choksi P.K. Wars2 is a determinant of angiogenesis. Nat. Commun. 2016;7:12061. doi: 10.1038/ncomms12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agnew T., Goldsworthy M., Aguilar C., Morgan A., Simon M., Hilton H., Esapa C., Wu Y., Cater H., Bentley L. A Wars2 Mutant Mouse Model Displays OXPHOS Deficiencies and Activation of Tissue-Specific Stress Response Pathways. Cell Rep. 2018;25:3315–3328.e6. doi: 10.1016/j.celrep.2018.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastin J., Aubey F., Rötig A., Munnich A., Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J. Clin. Endocrinol. Metab. 2008;93:1433–1441. doi: 10.1210/jc.2007-1701. [DOI] [PubMed] [Google Scholar]
- 38.Khan N.A., Auranen M., Paetau I., Pirinen E., Euro L., Forsström S., Pasila L., Velagapudi V., Carroll C.J., Auwerx J., Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ewalt K.L., Schimmel P. Activation of angiogenic signaling pathways by two human tRNA synthetases. Biochemistry. 2002;41:13344–13349. doi: 10.1021/bi020537k. [DOI] [PubMed] [Google Scholar]
- 40.Sajish M., Zhou Q., Kishi S., Valdez D.M., Jr., Kapoor M., Guo M., Lee S., Kim S., Yang X.L., Schimmel P. Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-γ and p53 signaling. Nat. Chem. Biol. 2012;8:547–554. doi: 10.1038/nchembio.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg Y., King M., Kiosses W.B., Ewalt K., Yang X., Schimmel P., Reader J.S., Tzima E. The novel fragment of tyrosyl tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 2008;22:1597–1605. doi: 10.1096/fj.07-9973com. [DOI] [PubMed] [Google Scholar]
- 42.Park S.G., Kim S. Do aminoacyl-tRNA synthetases have biological functions other than in protein biosynthesis? IUBMB Life. 2006;58:556–558. doi: 10.1080/15216540600735974. [DOI] [PubMed] [Google Scholar]
- 43.Yang X.L., Schimmel P., Ewalt K.L. Relationship of two human tRNA synthetases used in cell signaling. Trends Biochem. Sci. 2004;29:250–256. doi: 10.1016/j.tibs.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Otani A., Slike B.M., Dorrell M.I., Hood J., Kinder K., Ewalt K.L., Cheresh D., Schimmel P., Friedlander M. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. USA. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tzima E., Reader J.S., Irani-Tehrani M., Ewalt K.L., Schwartz M.A., Schimmel P. VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J. Biol. Chem. 2005;280:2405–2408. doi: 10.1074/jbc.C400431200. [DOI] [PubMed] [Google Scholar]
- 46.Kise Y., Lee S.W., Park S.G., Fukai S., Sengoku T., Ishii R., Yokoyama S., Kim S., Nureki O. A short peptide insertion crucial for angiostatic activity of human tryptophanyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2004;11:149–156. doi: 10.1038/nsmb722. [DOI] [PubMed] [Google Scholar]
- 47.Zeng R., Chen Y.C., Zeng Z., Liu X.X., Liu R., Qiang O., Li X. Inhibition of mini-TyrRS-induced angiogenesis response in endothelial cells by VE-cadherin-dependent mini-TrpRS. Heart Vessels. 2012;27:193–201. doi: 10.1007/s00380-011-0137-1. [DOI] [PubMed] [Google Scholar]
- 48.Wakasugi K., Slike B.M., Hood J., Ewalt K.L., Cheresh D.A., Schimmel P. Induction of angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J. Biol. Chem. 2002;277:20124–20126. doi: 10.1074/jbc.C200126200. [DOI] [PubMed] [Google Scholar]
- 49.Zeng R., Chen Y.C., Zeng Z., Liu W.Q., Jiang X.F., Liu R., Qiang O., Li X. Effect of mini-tyrosyl-tRNA synthetase/mini-tryptophanyl-tRNA synthetase on ischemic angiogenesis in rats: proliferation and migration of endothelial cells. Heart Vessels. 2011;26:69–80. doi: 10.1007/s00380-010-0032-1. [DOI] [PubMed] [Google Scholar]
- 50.Cheng G., Zhang H., Yang X., Tzima E., Ewalt K.L., Schimmel P., Faber J.E. Effect of mini-tyrosyl-tRNA synthetase on ischemic angiogenesis, leukocyte recruitment, and vascular permeability. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;295:R1138–R1146. doi: 10.1152/ajpregu.90519.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukui H., Hanaoka R., Kawahara A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ. Res. 2009;104:1253–1259. doi: 10.1161/CIRCRESAHA.108.191189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo M., Yang X.L., Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010;11:668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Y., Xu X., Zhang Q., Fu G., Mo Z., Wang G.S., Kishi S., Yang X.L. tRNA synthetase counteracts c-Myc to develop functional vasculature. eLife. 2014;3:e02349. doi: 10.7554/eLife.02349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu C.Y., Wang P.C., Tsai H.J. Competitive binding between Seryl-tRNA synthetase/YY1 complex and NFKB1 at the distal segment results in differential regulation of human vegfa promoter activity during angiogenesis. Nucleic Acids Res. 2017;45:2423–2437. doi: 10.1093/nar/gkw1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olsson A.K., Dimberg A., Kreuger J., Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 57.Lin C.Y., Lee H.C., Fu C.Y., Ding Y.Y., Chen J.S., Lee M.H., Huang W.J., Tsai H.J. MiR-1 and miR-206 target different genes to have opposing roles during angiogenesis in zebrafish embryos. Nat. Commun. 2013;4:2829. doi: 10.1038/ncomms3829. [DOI] [PubMed] [Google Scholar]
- 58.Chen J.F., Mandel E.M., Thomson J.M., Wu Q., Callis T.E., Hammond S.M., Conlon F.L., Wang D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ni R., Luo L. A noncanonical function of histidyl-tRNA synthetase: inhibition of vascular hyperbranching during zebrafish development. FEBS Open Bio. 2018;8:722–731. doi: 10.1002/2211-5463.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Götz A., Tyynismaa H., Euro L., Ellonen P., Hyötyläinen T., Ojala T., Hämäläinen R.H., Tommiska J., Raivio T., Oresic M. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011;88:635–642. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor R.W., Pyle A., Griffin H., Blakely E.L., Duff J., He L., Smertenko T., Alston C.L., Neeve V.C., Best A. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312:68–77. doi: 10.1001/jama.2014.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mazurova S., Magner M., Kucerova-Vidrova V., Vondrackova A., Stranecky V., Pristoupilova A., Zamecnik J., Hansikova H., Zeman J., Tesarova M., Honzik T. Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: case reports and review of the literature. Cardiol. Young. 2017;27:936–944. doi: 10.1017/S1047951116001876. [DOI] [PubMed] [Google Scholar]
- 63.Hilander T., Zhou X.L., Konovalova S., Zhang F.P., Euro L., Chilov D., Poutanen M., Chihade J., Wang E.D., Tyynismaa H. Editing activity for eliminating mischarged tRNAs is essential in mammalian mitochondria. Nucleic Acids Res. 2018;46:849–860. doi: 10.1093/nar/gkx1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sommerville E.W., Zhou X.L., Oláhová M., Jenkins J., Euro L., Konovalova S., Hilander T., Pyle A., He L., Habeebu S. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Hum. Mol. Genet. 2019;28:258–268. doi: 10.1093/hmg/ddy294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Almuqbil M.A., Vernon H.J., Ferguson M., Kline A.D. PARS2-associated mitochondrial disease: A case report of a patient with prolonged survival and literature review. Mol. Genet. Metab. Rep. 2020;24:100613. doi: 10.1016/j.ymgmr.2020.100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodriguez M.C., MacDonald J.R., Mahoney D.J., Parise G., Beal M.F., Tarnopolsky M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35:235–242. doi: 10.1002/mus.20688. [DOI] [PubMed] [Google Scholar]
- 67.Bruni F., Di Meo I., Bellacchio E., Webb B.D., McFarland R., Chrzanowska-Lightowlers Z.M.A., He L., Skorupa E., Moroni I., Ardissone A. Clinical, biochemical, and genetic features associated with VARS2-related mitochondrial disease. Hum. Mutat. 2018;39:563–578. doi: 10.1002/humu.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chae Y.S., Lee S.J., Moon J.H., Kang B.W., Kim J.G., Sohn S.K., Jung J.H., Park H.Y., Park J.Y., Kim H.J., Lee S.W. VARS2 V552V variant as prognostic marker in patients with early breast cancer. Med. Oncol. 2011;28:1273–1280. doi: 10.1007/s12032-010-9574-4. [DOI] [PubMed] [Google Scholar]
- 69.Alsemari A., Al-Younes B., Goljan E., Jaroudi D., BinHumaid F., Meyer B.F., Arold S.T., Monies D. Recessive VARS2 mutation underlies a novel syndrome with epilepsy, mental retardation, short stature, growth hormone deficiency, and hypogonadism. Hum. Genomics. 2017;11:28. doi: 10.1186/s40246-017-0124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diodato D., Melchionda L., Haack T.B., Dallabona C., Baruffini E., Donnini C., Granata T., Ragona F., Balestri P., Margollicci M. VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 2014;35:983–989. doi: 10.1002/humu.22590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sommerville E.W., Ng Y.S., Alston C.L., Dallabona C., Gilberti M., He L., Knowles C., Chin S.L., Schaefer A.M., Falkous G. Clinical Features, Molecular Heterogeneity, and Prognostic Implications in YARS2-Related Mitochondrial Myopathy. JAMA Neurol. 2017;74:686–694. doi: 10.1001/jamaneurol.2016.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kohda M., Tokuzawa Y., Kishita Y., Nyuzuki H., Moriyama Y., Mizuno Y., Hirata T., Yatsuka Y., Yamashita-Sugahara Y., Nakachi Y. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016;12:e1005679. doi: 10.1371/journal.pgen.1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verrigni D., Diodato D., Di Nottia M., Torraco A., Bellacchio E., Rizza T., Tozzi G., Verardo M., Piemonte F., Tasca G. Novel mutations in KARS cause hypertrophic cardiomyopathy and combined mitochondrial respiratory chain defect. Clin. Genet. 2017;91:918–923. doi: 10.1111/cge.12931. [DOI] [PubMed] [Google Scholar]
- 74.Guo M., Ignatov M., Musier-Forsyth K., Schimmel P., Yang X.L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. USA. 2008;105:2331–2336. doi: 10.1073/pnas.0712072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwartzentruber J., Buhas D., Majewski J., Sasarman F., Papillon-Cavanagh S., Thiffault I., Sheldon K.M., Massicotte C., Patry L., Simon M., FORGE Canada Consortium Mutation in the nuclear-encoded mitochondrial isoleucyl-tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with Leigh syndrome. Hum. Mutat. 2014;35:1285–1289. doi: 10.1002/humu.22629. [DOI] [PubMed] [Google Scholar]
- 76.Perli E., Giordano C., Tuppen H.A., Montopoli M., Montanari A., Orlandi M., Pisano A., Catanzaro D., Caparrotta L., Musumeci B. Isoleucyl-tRNA synthetase levels modulate the penetrance of a homoplasmic m.4277T>C mitochondrial tRNA(Ile) mutation causing hypertrophic cardiomyopathy. Hum. Mol. Genet. 2012;21:85–100. doi: 10.1093/hmg/ddr440. [DOI] [PubMed] [Google Scholar]
- 77.Moreno-Loshuertos R., Ferrín G., Acín-Pérez R., Gallardo M.E., Viscomi C., Pérez-Martos A., Zeviani M., Fernández-Silva P., Enríquez J.A. Evolution meets disease: penetrance and functional epistasis of mitochondrial tRNA mutations. PLoS Genet. 2011;7:e1001379. doi: 10.1371/journal.pgen.1001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng Y., Chen R., Mo X. Association of DARS gene polymorphisms with the risk of isolated ventricular septal defects in the Chinese Han population. Ital. J. Pediatr. 2016;42:102. doi: 10.1186/s13052-016-0311-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y., Yang Y., He X., Yang P., Zong T., Sun P., Sun R.C., Yu T., Jiang Z. The cellular function and molecular mechanism of formaldehyde in cardiovascular disease and heart development. J. Cell. Mol. Med. 2021;25:5358–5371. doi: 10.1111/jcmm.16602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dogan S.A., Pujol C., Maiti P., Kukat A., Wang S., Hermans S., Senft K., Wibom R., Rugarli E.I., Trifunovic A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19:458–469. doi: 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 81.Hansson A., Hance N., Dufour E., Rantanen A., Hultenby K., Clayton D.A., Wibom R., Larsson N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M., Saffitz J.E., Medeiros D.M., Valencik M.L., McDonald J.A., Kelly D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 83.Wu J., Subbaiah K.C.V., Xie L.H.T., Jiang F., Khor E.S., Mickelsen D., Myers J.R., Tang W.H.W., Yao P. Glutamyl-Prolyl-tRNA Synthetase Regulates Proline-Rich Pro-Fibrotic Protein Synthesis During Cardiac Fibrosis. Circ. Res. 2020;127:827–846. doi: 10.1161/CIRCRESAHA.119.315999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keller T.L., Zocco D., Sundrud M.S., Hendrick M., Edenius M., Yum J., Kim Y.J., Lee H.K., Cortese J.F., Wirth D.F. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012;8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qin P., Arabacilar P., Bernard R.E., Bao W., Olzinski A.R., Guo Y., Lal H., Eisennagel S.H., Platchek M.C., Xie W. Activation of the Amino Acid Response Pathway Blunts the Effects of Cardiac Stress. J. Am. Heart Assoc. 2017;6:e004453. doi: 10.1161/JAHA.116.004453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Song D.G., Kim D., Jung J.W., Nam S.H., Kim J.E., Kim H.J., Kim J.H., Lee S.J., Pan C.H., Kim S., Lee J.W. Glutamyl-prolyl-tRNA synthetase induces fibrotic extracellular matrix via both transcriptional and translational mechanisms. FASEB J. 2019;33:4341–4354. doi: 10.1096/fj.201801344RR. [DOI] [PubMed] [Google Scholar]
- 87.Li B., Wang Z., Chen R., Hong J., Wu Q., Hu J., Hu Z., Zhang M. Up regulation of isoleucyl-tRNA synthetase promotes vascular smooth muscle cells dysfunction via p38 MAPK/PI3K signaling pathways. Life Sci. 2019;224:51–57. doi: 10.1016/j.lfs.2019.03.052. [DOI] [PubMed] [Google Scholar]
- 88.Kwon N.H., Fox P.L., Kim S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov. 2019;18:629–650. doi: 10.1038/s41573-019-0026-3. [DOI] [PubMed] [Google Scholar]
- 89.Funahashi Y., Wakabayashi T., Semba T., Sonoda J., Kitoh K., Yoshimatsu K. Establishment of a quantitative mouse dorsal air sac model and its application to evaluate a new angiogenesis inhibitor. Oncol. Res. 1999;11:319–329. [PubMed] [Google Scholar]
- 90.Yang P., Yang Y., Sun P., Tian Y., Gao F., Wang C., Zong T., Li M., Zhang Y., Yu T., Jiang Z. βII spectrin (SPTBN1): biological function and clinical potential in cancer and other diseases. Int. J. Biol. Sci. 2021;17:32–49. doi: 10.7150/ijbs.52375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li M., Yang Y., Wang Z., Zong T., Fu X., Aung L.H.H., Wang K., Wang J.X., Yu T. Piwi-interacting RNAs (piRNAs) as potential biomarkers and therapeutic targets for cardiovascular diseases. Angiogenesis. 2021;24:19–34. doi: 10.1007/s10456-020-09750-w. [DOI] [PubMed] [Google Scholar]
- 92.Cheng M., Yang Y., Xin H., Li M., Zong T., He X., Yu T., Xin H. Non-coding RNAs in aortic dissection: From biomarkers to therapeutic targets. J. Cell. Mol. Med. 2020;24:11622–11637. doi: 10.1111/jcmm.15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Q., Liu B., Wang Y., Bai B., Yu T., Chu X.M. The biomarkers of key miRNAs and target genes associated with acute myocardial infarction. PeerJ. 2020;8:e9129. doi: 10.7717/peerj.9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Q., Yang Y., Fu X., Wang Z., Liu Y., Li M., Zhang Y., Li Y., Li P.F., Yu T., Chu X.M. Long noncoding RNA XXYLT1-AS2 regulates proliferation and adhesion by targeting the RNA binding protein FUS in HUVEC. Atherosclerosis. 2020;298:58–69. doi: 10.1016/j.atherosclerosis.2020.02.018. [DOI] [PubMed] [Google Scholar]
- 95.Liu Y., Yang Y., Wang Z., Fu X., Chu X.M., Li Y., Wang Q., He X., Li M., Wang K. Insights into the regulatory role of circRNA in angiogenesis and clinical implications. Atherosclerosis. 2020;298:14–26. doi: 10.1016/j.atherosclerosis.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 96.Yu T., Wang Z., Jie W., Fu X., Li B., Xu H., Liu Y., Li M., Kim E., Yang Y., Cho J.Y. The kinase inhibitor BX795 suppresses the inflammatory response via multiple kinases. Biochem. Pharmacol. 2020;174:113797. doi: 10.1016/j.bcp.2020.113797. [DOI] [PubMed] [Google Scholar]
- 97.Liu S., Yang Y., Jiang S., Xu H., Tang N., Lobo A., Zhang R., Liu S., Yu T., Xin H. MiR-378a-5p Regulates Proliferation and Migration in Vascular Smooth Muscle Cell by Targeting CDK1. Front. Genet. 2019;10:22. doi: 10.3389/fgene.2019.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang N., Jiang S., Yang Y., Liu S., Ponnusamy M., Xin H., Yu T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018;36:e12436. doi: 10.1111/1755-5922.12436. [DOI] [PubMed] [Google Scholar]
- 99.Liu S., Yang Y., Jiang S., Tang N., Tian J., Ponnusamy M., Tariq M.A., Lian Z., Xin H., Yu T. Understanding the role of non-coding RNA (ncRNA) in stent restenosis. Atherosclerosis. 2018;272:153–161. doi: 10.1016/j.atherosclerosis.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 100.Xue Q., He N., Wang Z., Fu X., Aung L.H.H., Liu Y., Li M., Cho J.Y., Yang Y., Yu T. Functional roles and mechanisms of ginsenosides from Panax ginseng in atherosclerosis. J. Ginseng Res. 2021;45:22–31. doi: 10.1016/j.jgr.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang Y., Yu T., Jiang S., Zhang Y., Li M., Tang N., Ponnusamy M., Wang J.X., Li P.F. miRNAs as potential therapeutic targets and diagnostic biomarkers for cardiovascular disease with a particular focus on WO2010091204. Expert Opin. Ther. Pat. 2017;27:1021–1029. doi: 10.1080/13543776.2017.1344217. [DOI] [PubMed] [Google Scholar]
- 102.Tian C., Yang Y., Bai B., Wang S., Liu M., Sun R.-C., Yu T., Chu X.M. Potential of exosomes as diagnostic biomarkers and therapeutic carriers for doxorubicin-induced cardiotoxicity. Int. J. Biol. Sci. 2021;17:1328–1338. doi: 10.7150/ijbs.58786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bai B., Yang Y., Ji S., Wang S., Peng X., Tian C., Sun R.C., Yu T., Chu X.M. MicroRNA-302c-3p inhibits endothelial cell pyroptosis via directly targeting NOD-, LRR- and pyrin domain-containing protein 3 in atherosclerosis. J. Cell. Mol. Med. 2021;25:4373–4386. doi: 10.1111/jcmm.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.He X., Lian Z., Yang Y., Wang Z., Fu X., Liu Y., Li M., Tian J., Yu T., Xin H. Long Non-coding RNA PEBP1P2 Suppresses Proliferative VSMCs Phenotypic Switching and Proliferation in Atherosclerosis. Mol. Ther. Nucleic Acids. 2020;22:84–98. doi: 10.1016/j.omtn.2020.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu H., Shi C., Wang S., Yang C., Wan X., Luo Y., Tian L., Li L. Identification of NCAPH as a biomarker for prognosis of breast cancer. Mol. Biol. Rep. 2020;47:7831–7842. doi: 10.1007/s11033-020-05859-9. [DOI] [PubMed] [Google Scholar]
- 106.Suh J.H., Park M.C., Goughnour P.C., Min B.S., Kim S.B., Lee W.Y., Cho Y.B., Cheon J.H., Lee K.Y., Nam D.H., Kim S. Plasma Lysyl-tRNA Synthetase 1 (KARS1) as a Novel Diagnostic and Monitoring Biomarker for Colorectal Cancer. J. Clin. Med. 2020;9:533. doi: 10.3390/jcm9020533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee J.M., Kim T., Kim E.Y., Kim A., Lee D.K., Kwon N.H., Kim S., Chang Y.S. Methionyl-tRNA Synthetase is a Useful Diagnostic Marker for Lymph Node Metastasis in Non-Small Cell Lung Cancer. Yonsei Med. J. 2019;60:1005–1012. doi: 10.3349/ymj.2019.60.11.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeng R., Wang M., You G.Y., Yue R.Z., Chen Y.C., Zeng Z., Liu R., Qiang O., Zhang L. Effect of Mini-Tyrosyl-tRNA Synthetase/Mini-Tryptophanyl-tRNA Synthetase on Angiogenesis in Rhesus Monkeys after Acute Myocardial Infarction. Cardiovasc. Ther. 2016;34:4–12. doi: 10.1111/1755-5922.12161. [DOI] [PubMed] [Google Scholar]
- 110.Ma K., Xie M., He X., Liu G., Lu X., Peng Q., Zhong B., Li N. A novel compound heterozygous mutation in VARS2 in a newborn with mitochondrial cardiomyopathy: a case report of a Chinese family. BMC Med. Genet. 2018;19:202. doi: 10.1186/s12881-018-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]