

Abstract
While alkyl radicals have been well demonstrated to undergo both 1,5- and 1,6-hydrogen atom abstraction (HAA) reactions, 1,4-HAA is typically a challenging process both entropically and enthalpically. Consequently, chemical transformations based on 1,4-HAA have been scarcely developed. Guided by the general mechanistic principles of metalloradical catalysis (MRC), 1,4-HAA has been successfully incorporated as a key step, followed by 4-exo-tet radical substitution (RS), for the development of a new catalytic radical process that enables asymmetric 1,4-C–H alkylation of diazoketones for stereoselective construction of cyclobutanone structures. The key to success is the optimization of the Co(II)-based metalloradical catalyst through judicious modulation of D2-symmetric chiral amidoporphyrin ligand to adopt proper steric, electronic, and chiral environments that can utilize a network of noncovalent attractive interactions for effective activation of the substrate and subsequent radical intermediates. Supported by an optimal chiral ligand, the Co(II)-based metalloradical system, which operates under mild conditions, is capable of 1,4-C–H alkylation of α-aryldiazoketones with varied electronic and steric properties to construct chiral α,β-disubstituted cyclobutanones in good to high yields with high diastereoselectivities and enantioselectivities, generating dinitrogen as the only byproduct. Combined computational and experimental studies have shed light on the mechanistic details of the new catalytic radical process, including the revelation of facile 1,4-HAA and 4-exo-tet-RS steps. The resulting enantioenriched α,β-disubstituted cyclobutanones, as showcased with several enantiospecific transformations to other types of cyclic structures, may find useful applications in stereoselective organic synthesis.
Graphical Abstract
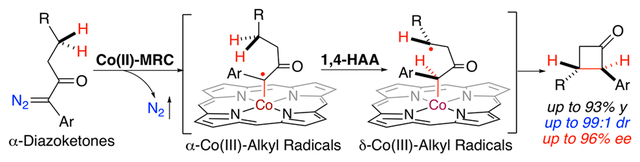
INTRODUCTION
In the past decades, radical reactions have attracted the growing attention of synthetic organic chemists in view of their rich reactivities and attractive characteristics.1 Notably, hydrogen atom abstraction (HAA) by free radicals has long been recognized as one of the most general pathways for C–H activation,2 a fundamental radical reaction that can potentially be utilized for direct functionalization of prevalent C–H bonds in organic molecules. However, the realization of this immense potential faces longstanding challenges that are associated with governing reactivity as well as controlling selectivity of the departing radicals from HAA for ensuing bond formation. To address this and related challenges, metalloradical catalysis (MRC) provides a conceptually new approach for achieving controllable reactivity and selectivity in radical reactions through catalytic generation as well as subsequent regulation of metal-stabilized organic radical.3–5 As stable 15e-metalloradicals, Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] enjoy the unique capability of activating diazo compounds homolytically to generate α-Co(III)-alkyl radicals.6 These Co-stabilized C-centered radical intermediates can undergo common radical reactions, such as radical addition and hydrogen atom abstraction as well as following radical substitution, leading to new catalytic processes for stereoselective radical transformations.7 In particular, Co(II)-based MRC was successfully applied for the development of enantioselective radical C–H alkylation of diazo compounds involving 1,5-HAA as the key step for stereoselective construction of 5-membered ring structures.8 To broaden the synthetic applications of Co(II)-MRC for solving more challenging problems, we were intrigued by the possibility of constructing strained 4-membered ring structures such as cyclobutanones through radical 1,4-C–H alkylation of α-aryldiazoketones (Scheme 1). However, this proposed radical process presented several potential challenges. In view of their unique electronic and steric properties compared with other types of diazo compounds, it was uncertain whether donor/acceptor-substituted diazo compounds 1 could be effectively activated by [Co(D2-Por*)] to generate the corresponding α-Co(III)-alkyl radicals I. Given that 1,4-HAA is known to be an inherently challenging process due to both unfavorable entropic and enthalpic factors,9 how could the resulting tertiary radical intermediate I be promoted for the desired intramolecular hydrogen atom abstraction to form δ-Co(III)-alkyl radicals II? Apart from the reactivity concerns, could the 1,4-HAA of α-Co(III)-alkyl radicals I be rendered enantioselective? Furthermore, the subsequent 4-exo-tet radical cyclization of the alkyl radicals II via intramolecular radical substitution was expected to be equally challenging in light of the high strain associated with four-membered transition state. What factors could be utilized to facilitate the ring closure for product formation? Additionally, how could the C–C bond formation be achieved with effective diastereoselective control during radical cyclization? We reasoned that all these and related questions could be potentially addressed through the optimization of [Co(D2-Por*)] catalyst by the fine-tuning of the D2-symmetric chiral amidoporphyrin ligand to adopt suitable environments that govern the course of the catalytic radical process (Scheme 1). If realized successfully, it would give rise to the first catalytic radical process for asymmetric intramolecular C–H alkylation involving 1,4-HAA as the key step for stereoselective construction of highly strained cyclobutanone structures, the four-membered carbocycles that are important moieties in natural products and pharmaceuticals (Figure S1).10
Scheme 1.
Working Proposal for Construction of Cyclobutanones by Radical 1,4-C–H Alkylation via Co(II)-Based Metalloradical Catalysis
Catalytic asymmetric intramolecular 1,4-C–H alkylation of diazo compounds represents a potentially attractive strategy for the stereoselective construction of four-membered cyclic compounds.11 Due to the high strain associated with four-membered ring structures, the catalytic process for 1,4-C–H alkylation has been largely underdeveloped. While there have been a few reports on the asymmetric synthesis of β-lactams12 and β-lactones13 from α-diazoamides and α-diazoesters, respectively, by Ru, Rh, and Ir-based catalytic systems, asymmetric synthesis of cyclobutanones via 1,4-C–H alkylation from α-diazoketones has not been previously realized.14 The absence of a catalytic system for cyclobutanone synthesis is presumably attributed to the augmented challenge of 1,4-C–H alkylation for α-diazoketones because of their relatively higher conformational flexibility than α-diazoamides and α-diazoesters. As an exciting new application of Co(II)-based MRC for stereoselective organic synthesis, we herein report the development of the first catalytic system that is highly effective for asymmetric 1,4-C–H alkylation of α-diazoketones to construct chiral cyclobutanones. Supported by a new-generation D2-symmetric chiral amidoporphyrin ligand, the Co(II)-based metalloradical system, which enjoys operational simplicity and mild conditions, can activate α-aryldiazoketones with varied electronic and steric properties for 1,4-alkylation of C(sp3)–H bonds, enabling stereoselective construction of chiral α,β-disubstituted cyclobutanones. We show the importance of catalyst development through the fine-tuning of the ligand environments in achieving high reactivity and stereoselectivity in this new radical process. Furthermore, our combined experimental and computational studies on the mechanism of the Co(II)-based metalloradical system have shed light on the underlying stepwise radical pathway, including the key steps of 1,4-HAA and 4-exo-tet-RS. A series of further transformations of the resulting enantioenriched α,β-disubstituted cyclobutanones are provided to showcase their synthetic applications.
RESULTS AND DISCUSSION
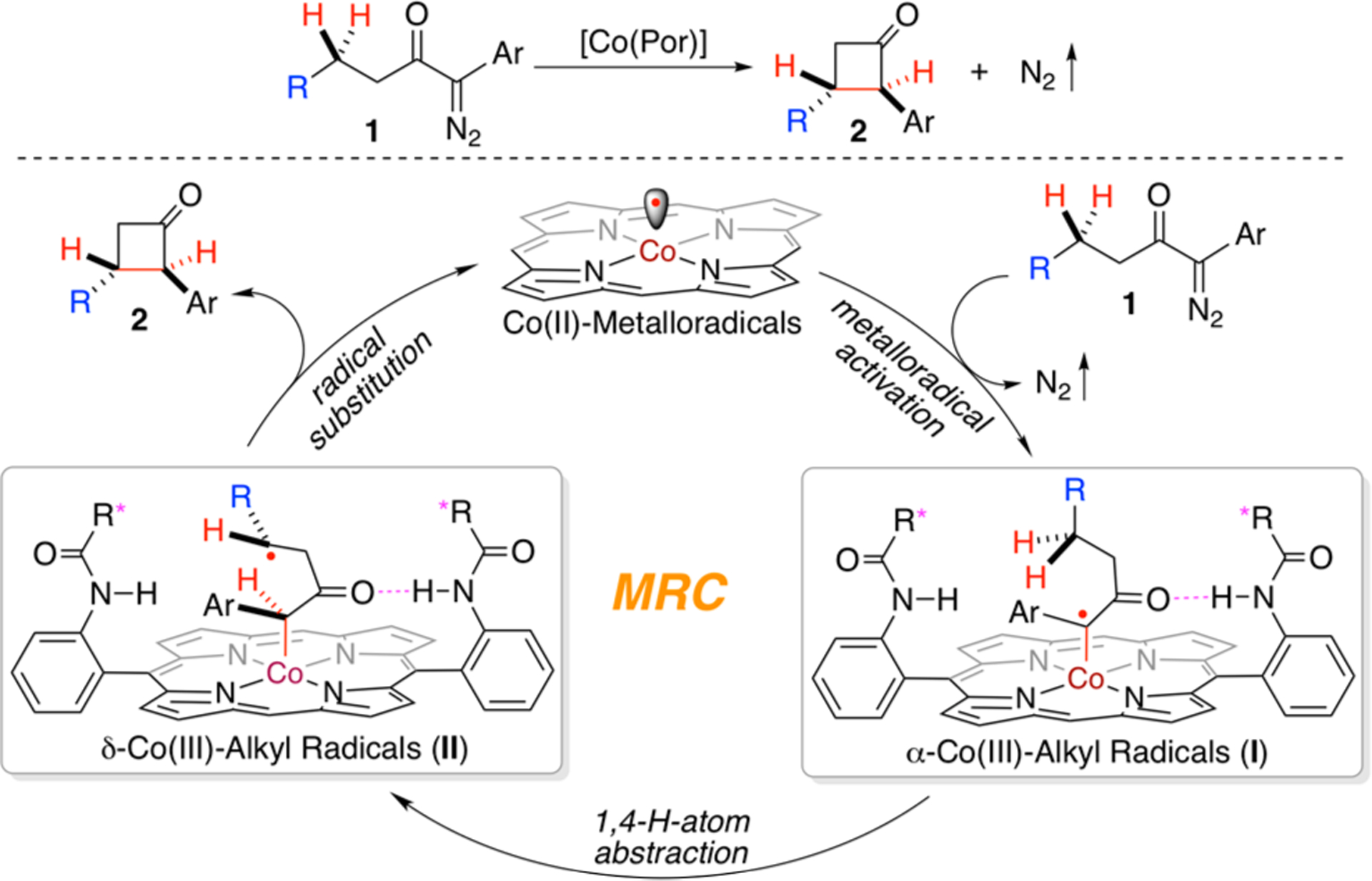
Catalyst Development.
At the outset of this research project, 1-diazo-1,4-diphenylbutan-2-one (1a) was used as the model substrate to examine the feasibility of the proposed catalytic process for 1,4-C–H alkylation (Scheme 2). Simple achiral metalloradical catalyst [Co(TPP)] (TPP = 5,10,15,20-tetraphenylporphyrin) was shown to be incapable of activating 1a for the intramolecular C–H alkylation reaction, failing to generate any cyclobutanone product. Instead, diazoketone 1a was thermally decomposed to the carboxylic acid derivative via Wolff rearrangement, followed by nucleophilic reaction of the corresponding ketene intermediate with water. Excitingly, when the achiral amidoporphyrin catalyst [Co(P1)] (P1 = 3,5-DitBu-IbuPhyrin)15 was used, it could productively catalyze the C–H alkylation reaction to deliver the desired product 2,3-diphenylcyclobutan-1-one (2a) in 54% yield with 40% de. The dramatic difference in reactivity between [Co(P1)] and [Co(TPP)] might be attributed to rate acceleration through the potential hydrogen bonding interaction between amide units of the porphyrin and the carbonyl group of the α-diazoketone. Employment of first-generation chiral metalloradical catalyst [Co(P2)] (P2 = 3,5-DitBu-ChenPhyrin)7a enabled asymmetric induction in the 1,4-C–H alkylation reaction, affording cyclobutanone 2a with moderate enantioselectivity (46% ee) without significantly affecting the product yield (50%) and diastereoselectivity (52% de). When second-generation chiral metalloradical catalyst [Co(P3)] (P3 = 3,5-DitBu-TaoPhyrin) bearing chiral amide units with ester moieties was used for the catalytic reaction,16 it led to the increase in both product yield (71%) and diastereoselectivity (86% de) but the decrease in enantioselectivity (15% ee). When switching to new-generation C6-bridged metalloradical catalyst [Co(P4)] (P4 = 3,5-DitBu-Hu(C6)Phyrin) featuring more rigid cavity-like environments,16 dramatic improvements in both diastereoselectivity (94% de) and enantioselectivity (78% ee) were observed even if in relatively lower product yield (61%). Subsequent use of analogous catalyst [Co(P5)] (P5 = 2,6-DiMeO-Hu(C6)Phyrin),17 which bears 2,6-dime-thoxyphenyl instead of 3,5-di-tert-butylphenyl groups as the 5,15-diaryl substituents, further improvements in enantioselectivity (96% ee) as well as yield (80%) were achieved without significantly affecting the diastereoselectivity (82% de). These results demonstrate that the rigidification of ligand environment of chiral metalloradical catalyst [Co(D2-Por*)] plays an important role in achieving both high reactivity and stereoselectivity for the 1,4-C–H alkylation process. It should be emphasized that the diastereomeric mixtures of the resulting cyclobutanone 2a from all the catalytic reactions were isomerized to trans-enriched 2a with 96% de after purification by column chromatography on silica gel as a result of the relative acidity of the tertiary α-C–H bond, regardless the original diastereoselectivities before purification.
Scheme 2. Ligand Effect on Co(II)-Based Catalytic System for Asymmetric 1,4-C–H Alkylation of α-Aryldiazoketonea.

aCarried out with 1a (0.10 mmol) using [Co(Por)] (2 mol %) in tert-butyl methyl ether (TBME) (0.5 mL) at 40 °C for 12 h. bIsolated yield. cDiastereomeric excess (de) determined by 1H NMR analysis of crude reaction mixture before purification. dIsomerized to trans-enriched products with 96% de after purification by silica gel column chromatography for all catalytic reactions. eEnantiomeric excess (ee) of trans-diastereomer determined by chiral HPLC after purification
Substrate Scope.
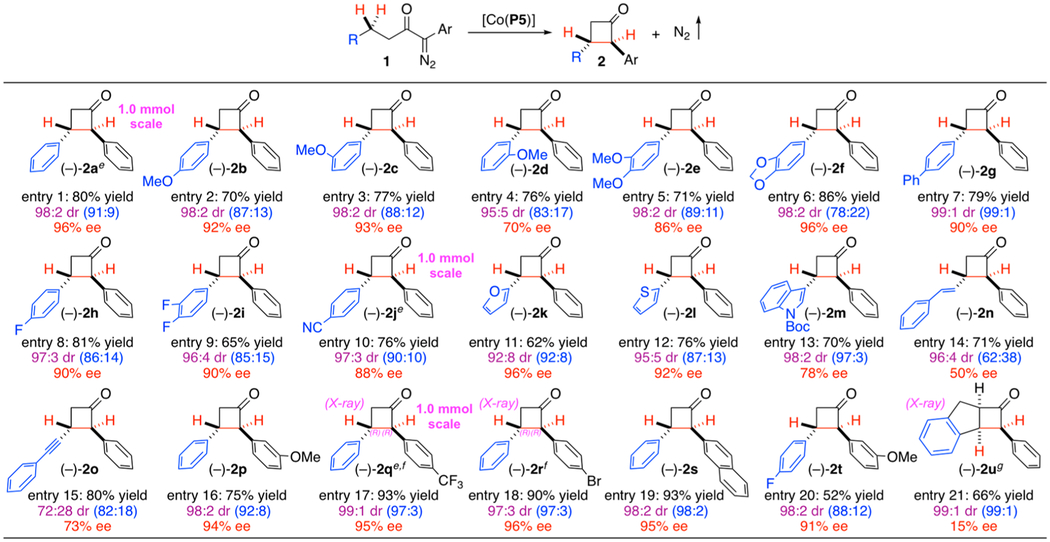
Under the optimized conditions, the substrate scope of the [Co(P5)]-catalyzed intramolecular 1,4-C–H alkylation was evaluated with α-aryldiazoketones 1 containing different types of C–H bonds (Table 1). Like the parent 1a, 1,4-diaryl-α-diazoketone derivatives containing 4-aryl substituents with various steric and electronic properties at different positions, including 4-OMe (1b), 3-OMe (1c), 2-OMe (1d), 3,4-di-OMe (1e), 1’,3’-dioxolane-3,4-fused (1f), 4-Ph (1g), 4-F (1h), 3,4-di-F (1i), and 4-CN (1j), could be efficiently alkylated by [Co(P5)] at the benzylic C–H bonds, delivering the corresponding cyclobutanones 2b–2j in good yields with high enantioselectivities (Table 1; entries 2–10). The Co(II)-catalyzed 1,4-C–H alkylation was shown to be compatible with substrates containing heteroarenes, such as furan (1k), thiophene (1l) and indole (1m), allowing for stereoselective construction of β-heteroarylcyclobutanones 2k–2m in similarly good yields with the same high enantioselectivities (Table 1; entries 11–13). Furthermore, the Co(II)-based catalytic system could chemoselectively alkylate allylic C–H bonds without affecting the typically more reactive C═C π bonds as demonstrated by the productive formation of β-alkenylcyclobutanone 2n from the reaction of 4-alkenyl-substituted diazoketone (Z)-1n in good yield albeit with lower enantioselectivity (Table 1; entry 14). It was noted that the olefin configuration was completely isomerized from (Z) to (E) during the catalytic process (see Scheme 3D for detailed discussion). Likewise, propargylic C–H bonds could also be chemoselectively alkylated by [Co(P5)] without reacting with the C≡C π bonds, as exemplified by the reaction of 4-alkynyl-substituted diazoketone 1o to generate β-alkynylcyclobutanone 2o in good yield with better enantioselectivity (Table 1; entry 15). In addition to the substrates with different 4-aryl substituents, the Co(II)-based metalloradical system was shown to be applicable to α-diazoketones containing 1-aryl substituents with various steric and electronic properties at different positions. For example, catalytic 1,4-C–H alkylation reactions of 1-aryl-4-phenyl-α-diazoketones, such as those bearing 3-OMe (1p), 4-CF3 (1q), and 4-Br (1r) phenyl groups as well as 2-naphthyl group (1s), proceeded smoothly to afford the corresponding cyclobutanones 2p–2s in good to high yields with excellent enantioselectivities (Table 1; entries 16–19). 1,4-Diaryl-α-diazoketones containing both 1- and 4-aryl substituents were found to work equally well as shown with the successful reaction of α-diazoketone 1t for formation of the desired α,β-bisarylcyclobutanone 2t with excellent level of enantioselectivity despite in moderate yield (Table 1; entry 20). Furthermore, the Co(II)-based catalytic system could be also applicable to cyclic substrates such as 2-indane-derived α-diazoketone 1u, resulting in asymmetric desymmetrization of the two benzylic C–H sites in the indane ring to deliver cyclobutanone 2u (Table 1; entry 21). It is remarkable that the strained tricyclic structure with fused 4-/5-membered rings could be constructed through catalytic 1,4-C–H alkylation in good yield despite with low enantioselectivity. A new Co(II)-metalloradical catalyst supported by a different type of D2-symmetric chiral amidoporphyrin ligand would likely be needed in order to achieve high enantioselectivity for the asymmetric desymmetrization 1,4-C–H alkylation process. Finally, [Co(P5)] was found to be ineffective for 1,4-alkylation of nonbenzylic C–H bonds due to the competition from predominant 1,5-C–H alkylation. However, preliminary results from the catalytic reaction of 1-diazo-1,7-diphenylheptane-2-one with C–H bonds at different positions indicated that site-selective 1,4- over 1,5-C–H alkylation could be potentially achieved through fine-tuning of the D2-symmetric chiral amidoporphyrin as the supporting ligand for Co(II)-metalloradical catalyst (see Scheme S1 in Supporting Information for the details). As aforementioned, the diastereomeric mixtures of the resulting cyclobutanones 2, regardless the original diastereoselectivities from the catalytic reactions, were all further enriched to give trans-dominant products with excellent diastereoselectivities after purification by column chromatography on silica gel (Table 1), which was realized by isomerization as a result of the relative acidity of the tertiary α-C–H bonds. The only exception was observed for product 2o, the diastereoselectivity of which was decreased after the purification (Table 1; entry 15), which is presumably a result of the less steric hindrance of the alkyne group. It is worth mentioning that the Co(II)-based catalytic process for the synthesis of cyclobutanone derivatives could be readily scaled up under the same condition as exemplified by the stereoselective syntheses of optically active cyclobutanones 2a, 2j and 2q on 1.0 mmol scale in similarly good yields with the same level of high enantioselectivities (Table 1; entries 1, 10 and 17).
Table 1.
Asymmetric Synthesis of Cyclobutanones by Co(II)-Catalyzed 1,4-C–H Alkylation of α-Diazoketonesa,b,c,d
|
Carried out with 1 (0.10 mmol) and [Co(P5)] (2 mol %) in TBME (0.5 mL) at 40 °C for 12 h.
Isolated yield.
Diastereomeric ratio (dr) determined by 1H NMR analysis: trans-enriched products after purification by silica gel column chromatography due to isomerization; value in parathesis determined from reaction mixture before purification.
Enantiomeric excess (ee) of trans-diastereomer determined by chiral HPLC after purification.
Reaction performed in 1.0 mmol scale.
Absolute configuration determined by X-ray crystallography.
Relative configuration determined by X-ray crystallography.
Scheme 3. Mechanistic Studies on Co(II)-Catalyzed 1,4-C–H Alkylation of α-Aryldiazoketones.
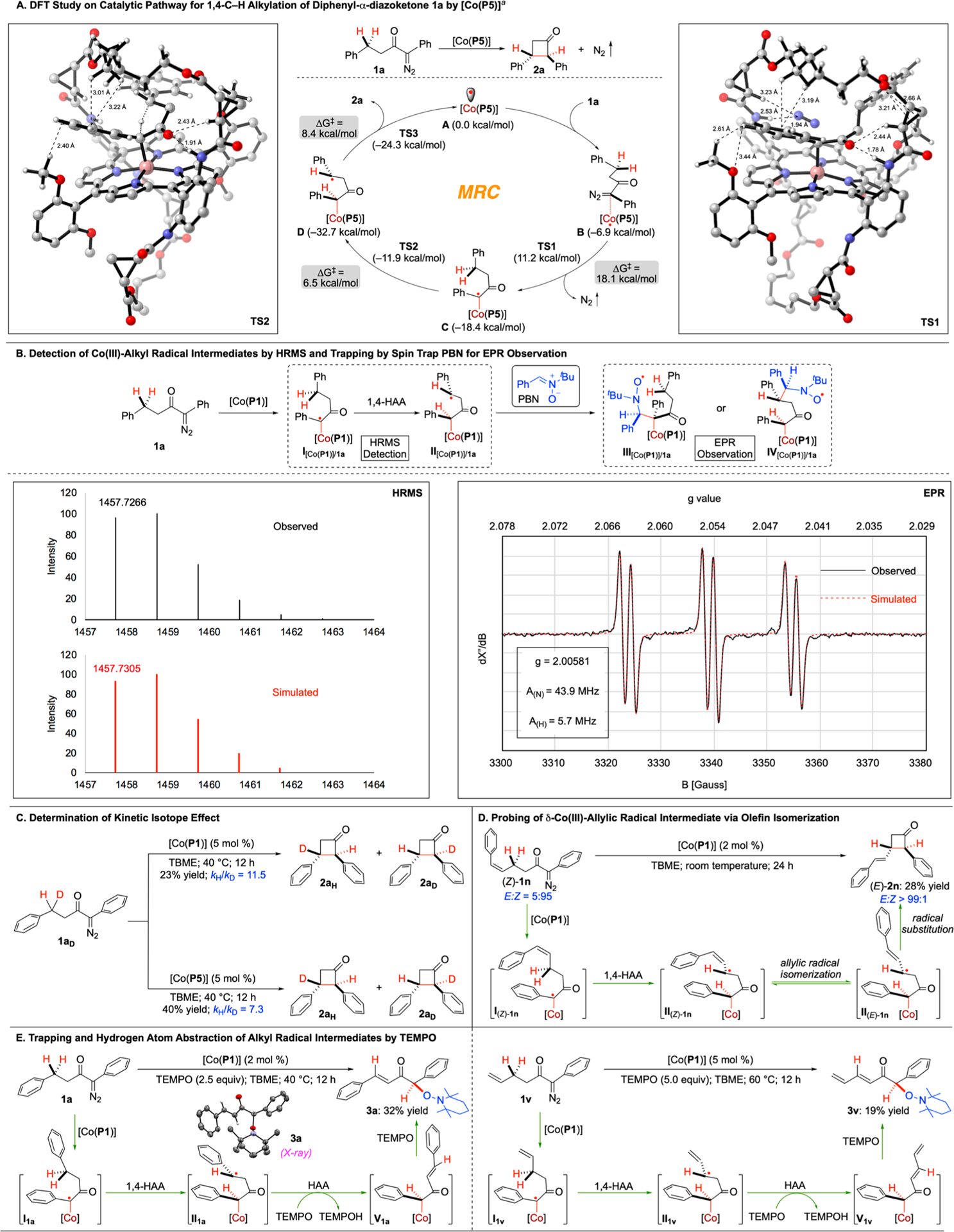
aFree energy profile of the [Co(P5)]-catalyzed 1,4-C–H alkylation. Density functional theory calculations were performed at SMD(diisopropylether)-BP86-D3(BJ)/def2TZVPP//BP86-D3(BJ)/def2SVP level of theory.
Mechanistic Studies.
To gain insights into this metalloradical process, combined computational and experimental studies were conducted to explore the proposed stepwise radical mechanism for the Co(II)-catalyzed 1,4-C–H alkylation (Scheme 1). First, density functional theory (DFT) calculations were performed to elucidate the catalytic pathway for 1,4-C–H alkylation reaction of α-aryldiazoketone 1a with the use of the actual catalyst [Co(P5)] (Scheme 3A; see Supporting Information for details). The computational study reveals the initial formation of intermediate B between diazo 1a and catalyst [Co(P5)] through a network of noncovalent attractions, including multiple H-bonds and π-interactions. This complexation process, which is exergonic by 6.9 kcal/mol, places the substrate underneath the bridge of the catalyst and positions the α-carbon atom of diazo 1a in a close proximity to the Co center of [Co(P5)] (C---Co: ~ 2.70 Å) for further interaction. Upon metalloradical activation (MRA) by [Co(P5)], the bound 1a undergoes the extrusion of dinitrogen to generate α-Co(III)-alkyl radical C. The metalloradical activation step, which is exergonic by 11.5 kcal/mol, is found to be associated with a relatively high but accessible activation barrier (TS1: ΔG‡ = 18.1 kcal/mol). Subsequent 1,4-HAA of intermediate C, which is exergonic by 14.3 kcal/mol, gives rise to the corresponding δ-Co(III)-alkyl radical intermediate D with a relatively low activation barrier (TS2: ΔG‡ = 6.5 kcal/mol). Such a low barrier for 1,4-HAA revealed by the DFT computation, which is uncommon for free radical processes,18 may be attributed to the presence of the multiple noncovalent interactions that stabilize transition state TS2. As illustrated by the computed model of TS2 (Scheme 3A), these cooperative noncovalent attractive interactions orient the reacting substrate within the catalyst cavity in proximity with proper conformation to facilitate the 1,4-HAA. According to the DFT calculations, the final step of 4-exo-tet cyclization of alkyl radical D via intramolecular radical substitution also has a relatively low activation barrier (TS3: ΔG‡ = 8.4 kcal/mol), leading to the formation of cyclobutanone 2a while regenerating catalyst [Co(P5)].
To experimentally detect α-Co(III)-alkyl radical I and δ-Co(III)-alkyl radical II, the reaction mixture of α-aryldiazoketone 1a with catalyst [Co(P1)] was analyzed by high-resolution mass spectrometry (HRMS) with electrospray ionization (ESI) in the absence of any additives as electron carriers (Scheme 3B). The obtained spectrum clearly reveals a signal corresponding to [(P1)Co–C(C6H5)(C(O)-CH2CH2C6H5)]+ (m/z = 1457.7266), which resulted from neutral α-Co(III)-alkyl radical intermediate I[Co(P1)]/1a or δ-Co(III)-alkyl radical intermediate II[Co(P1)]/1a by the loss of one electron. Both the exact mass and the pattern of isotope distribution determined by ESI-HRMS matches almost perfectly with those calculated from the formula [C92H102CoN8O5]+ (see Supporting Information for details). In addition to the HRMS detection, we made multiple attempts to observe I[Co(P1)]/1a and II[Co(P1)]/1a by electron paramagnetic resonance (EPR) without success. This negative outcome of EPR observation was attributed to the fleeting nature of the alkyl radical intermediates as a result of facile 1,4-HAA and RS steps, as revealed from DFT calculations. Accordingly, the common spin trap phenyl N-tert-butyl-α-phenylnitrone (PBN) was employed to trap I[Co(P1)]/1a or II[Co(P1)]/1a, which resulted in the generation of radicals III[Co(P1)]/1a or IV[Co(P1)]/1a that could be successfully observed by EPR at room temperature (Scheme 3B; see Supporting Information for details). The observed triplet of doublet signals in the isotropic EPR spectrum could be fittingly simulated on the basis of hyperfine couplings by 14N (I = 1) and 1H (I = 1/2): g = 2.00581; A(N) = 43.9 MHz; A(H) = 5.7 MHz.
To further study the 1,4-HAA step in the catalytic process, monodeuterated diazo 1aD was prepared as the substrate for the study of kinetic isotopic effect (KIE) using both achiral catalyst [Co(P1)] and chiral catalyst [Co(P5)] (Scheme 3C). The KIE values for the catalytic reaction of diazo 1aD by [Co(P1)] and [Co(P5)] were determined to be 11.5 and 7.3, respectively. These large values of primary KIE are in good agreement with the proposed step of homolytic C–H bond cleavage via intramolecular H-atom abstraction by α-Co(III)-alkyl radical intermediate I. To further probe the existence of δ-Co(III)-alkyl radical intermediate II, (Z)-1n was employed as the substrate for 1,4-C–H alkylation reaction using [Co(P1)] as the catalyst (Scheme 3D). Like the catalytic reaction by [Co(P5)] (Table 1; entry 14), it was found that (E)-2n was generated as the only product, indicating isomerization of the olefin configuration during the catalytic process. The formation of (E)-2n from (Z)-1n implies the involvement of δ-Co(III)-allylic radical II(E)-1n, which could be generated through isomerization of δ-Co(III)-allylic radical II(Z)-1n. To directly trap the alkyl radical intermediate, the catalytic reaction of 1a using [Co(P1)] as the catalyst was conducted in the presence of TEMPO (Scheme 3E). Interestingly, TEMPO-trapping product 3a was isolated in 32% yield, the structure of which was confirmed by X-ray crystallography. Formation of 3a further implies the initial generation of α-Co(III)-alkyl radical I1a by MRA, followed by 1,4-HAA to deliver δ-Co(III)-alkyl radical II1a in the catalytic reaction. Instead of radical recombination with TEMPO, it is presumed to be more favorable for intermediate II1a to undergo HAA by TEMPO, furnishing Co(III)-alkyl intermediate V1a with the extended conjugation. Subsequent radical substitution with another molecule of TEMPO delivered the TEMPO-trapping product 3a. In a similar pathway, the catalytic reaction of allyl diazoketone 1v by [Co(P1)] in the presence of TEMPO could give TEMPO trapping product 3v, indicating the involvement of the corresponding radical intermediates I1v, II1v and V1v. Furthermore, catalytic reactions of allyl diazoketone bearing a cyclopropyl ring as radical-clock substrate were performed with [Co(P5)] in both absence and presence of TEMPO to probe the lifetime of the corresponding δ-Co(III)-alkyl radical intermediate. Since there was no evidence for formation of ring-opening product in both reactions, it was concluded that the C–C and related bond formation were faster than the ring-opening of the cyclopropylcarbinyl radical within the cavity-like ligand environment of the catalyst (see Supporting Information for details).
Synthetic Applications.
Considering that the resulting enantioenriched cyclobutanones contain both versatile carbonyl group and strained four-membered structure, they may serve as useful intermediates for stereoselective organic synthesis (Scheme 4). To demonstrate the synthetic utility of this methodology, we carried out a series of synthetic transformations using enantioenriched cyclobutanone 2a (96% ee) as the model reactant. For example, cyclobutanone 2a could undergo efficient Horner-Wadsworth-Emmons reaction to provide the corresponding chiral methylenecyclobutane 4a in high yield without the loss of either diastereopurity or enantiopurity. Cyclobutanone 2a could also be stereospecifically converted to the corresponding O-benzoyl oxime 5a, which is known to undergo various ring-opening transformations.19 In addition, 2a could undergo both efficient reduction and reductive amination, allowing for the production of chiral cyclobutanol 6a and cyclobutanamine 7a, respectively, with excellent diastereoselectivities. Moreover, the ketone functionality in cyclobutanone 2a could undergo nucleophilic addition with Grignard reagents such as benzylmagnesium chloride, vinylmagnesium bromide, and ethynylmagnesium bromide to provide corresponding cyclobutanols 8a, 9a, and 10a bearing a newly-formed quaternary stereogenic center in excellent yields with effective control of diastereoselectivities. It is worth mentioning that tertiary cyclobutanols such as 8a, 9a, and 10a can be potentially employed for downstream functionalization via C–C bond cleavage.20 In view of the wide applications of the chiral γ-lactone scaffold,21 it was shown that γ-lactone 11a could be efficiently generated from cyclobutanone 2a through Baeyer-Villiger oxidation with the retention of the original stereochemical purity. Gratifyingly, chiral dihydroquinazoline 12a, as a family of heterocycles with interesting biological activities,22 could be synthesized from the reaction of 2a with o-aminobenzylamine through simple two-step transformation.23
Scheme 4. Synthetic Transformations of Resulting Chiral Cyclobutanones from Co(II)-Catalyzed 1,4-C–H Alkylationa.
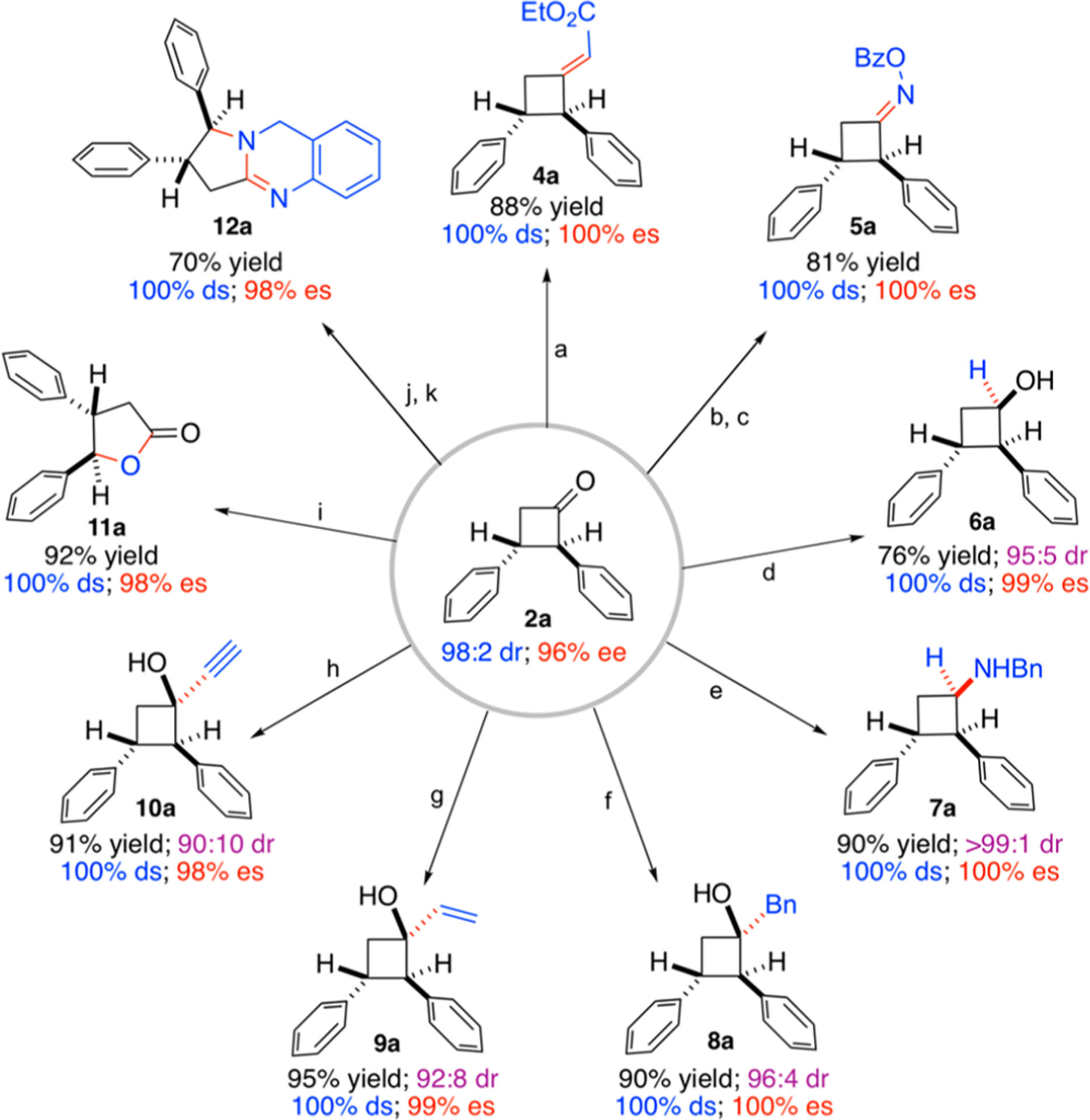
a(a) Triethyl Phosphonoacetate (1.5 equiv); Sodium Hydride (1.2 equiv); THF; RT; 12 h. (b) Hydroxylamine Hydrochloride (2.0 equiv); Pyridine; RT; 2 h. (c) Benzoyl Chloride (1.5 equiv); Triethylamine (2.0 equiv); DCM; 0 °C; 6 h. (d) Sodium Borohydride (1.0 equiv); MeOH; −78 °C; 10 h. (e) Benzylamine (1.1 equiv); Sodium Triacetoxyborohydride (2.0 equiv); DCM; RT; 12 h. (f) Benzylmagnesium Chloride (1.5 equiv); THF; 0 °C; 1 h. (g) Vinylmagnesium Bromide (1.5 equiv); THF; 0 °C; 1 h. (h) Ethynylmagnesium Bromide (1.5 equiv); THF; 0 °C; 1 h. (i) m-CPBA; DCM; 0 °C; 2 h. (j) o-Aminobenzylamine (1.1 equiv); CHCl3; 60 °C; 9 h. (k) NCS (1.5 equiv); DCM; 0 °C; 2 h.
CONCLUSIONS
In summary, we have demonstrated the first catalytic system for asymmetric radical 1,4-C–H alkylation via Co(II)-based MRC that involves the typically challenging 1,4-hydrogen atom abstraction. The key to the successful development is the judicious modulation of D2-symmetric chiral amidoporphyrin ligand to adopt desired steric, electronic and chiral environments around the Co(II)-metalloradical center that maximize a network of noncovalent attractive interactions in catalytic intermediates. With the bridged D2-symmetric chiral amidoporphyrin 2,6-DiMeO-Hu(C6)Phyrin as the optimal supporting ligand, the Co(II)-based metalloradical system, which operates under mild conditions, can catalyze asymmetric 1,4-C–H alkylation of α-aryldiazoketones with varied electronic and steric properties to construct chiral α,β-disubstituted cyclobutanones in good yields with high diastereoselectivities and enantioselectivities. The combined computational and experimental studies have shed light on the working details of this new catalytic process that proceeds through a stepwise radical mechanism, which is fundamentally different from the concerted C–H insertion by the existing catalytic systems involving metallocarbenes. As showcased with several enantiospecific transformations to other types of cyclic structures from the resulting enantioenriched α,β-disubstituted cyclobutanones, this Co(II)-based metalloradical system for asymmetric 1,4-C–H alkylation should find broad applications in organic synthesis. We envision that catalytic radical processes incorporating 1,4-H-atom abstraction as the key step may offer a general strategy for asymmetric construction of highly strained four-membered cyclic structures.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (R01-GM132471) and in part by NSF (CHE-1900375).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04968.
Experimental details and analytical data for all new compounds (PDF)
Accession Codes
CCDC 2083314–2083317 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c04968
The authors declare no competing financial interest.
REFERENCES
- (1).For selected books and reviews, see:; (a) Zard SZ Radical Reactions in Organic Synthesis; Oxford University Press, 2003. [Google Scholar]; (b) Chatgilialoglu C; Studer A Encyclopedia of Radicals in Chemistry, Biology, and Materials; John Wiley & Sons: 2012. [Google Scholar]; (c) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3296. [DOI] [PubMed] [Google Scholar]; (d) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (e) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (f) Quiclet-Sire B; Zard SZ Fun with Radicals: Some New Perspectives for Organic Synthesis. Pure Appl. Chem 2011, 83, 519–551. [Google Scholar]; (g) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (2).For selected reviews, see:; (a) Lu Q; Glorius F Radical Enantioselective C(sp3)–H Functionalization. Angew. Chem., Int. Ed 2017, 56, 49–51. [DOI] [PubMed] [Google Scholar]; (b) Stateman LM; Nakafuku KM; Nagib DA Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sarkar S; Cheung KPS; Gevorgyan V C–H Functionalization Reactions Enabled by Hydrogen Atom Transfer to Carbon-Centered Radicals. Chem. Sci 2020, 11, 12974–12993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang C; Li ZL; Gu QS; Liu XY Catalytic enantioselective C(sp3)–H functionalization involving radical intermediates. Nat. Commun 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected examples for radical C–H functionalization, see:; (e) Choi GJ; Zhu QL; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C–H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chu JCK; Rovis T Amide-Directed Photoredox-Catalysed C–C Bond Formation at Unactivated sp3 C–H Bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang W; Wang F; McCann SD; Wang DH; Chen PH; Stahl SS; Liu GS Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hu XQ; Chen JR; Xiao WJ Controllable Remote C–H Bond Functionalization by Visible-Light Photocatalysis. Angew. Chem., Int. Ed 2017, 56, 1960–1962. [DOI] [PubMed] [Google Scholar]; (i) Burg F; Gicquel M; Breitenlechner S; Pothig A; Bach T Site- and Enantioselective C–H Oxygenation Catalyzed by a Chiral Manganese Porphyrin Complex with a Remote Binding Site. Angew. Chem., Int. Ed 2018, 57, 2953–2957. [DOI] [PubMed] [Google Scholar]; (j) Nakafuku KM; Zhang ZX; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C–H Amination for the Synthesis of β-Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Yang CJ; Zhang C; Gu QS; Fang JH; Su XL; Ye L; Sun Y; Tian Y; Li ZL; Liu XY Cu-Catalysed Intramolecular Radical Enantioconvergent Tertiary β-C(sp3)–H Amination of Racemic Ketones. Nat. Catal 2020, 3, 539–546. [Google Scholar]
- (3).For selected reviews and highlights on Co(II)-based MRC, see:; (a) Lu H; Zhang XP Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (c) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (d) Huang H-M; Garduno-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (e) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal 2019, 9, 3604–3617. [Google Scholar]
- (4).For selected examples of Ti(III)-based radical processes, see:; (a) Nugent WA; RajanBabu TV Transition-Metal-Centered Radicals in Organic Synthesis. Titanium(III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]; (b) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Cationic Titanocene(III) Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers Ii RA Tuning the Redox Properties of the Titanocene(III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (e) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (f) Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (g) Ye K-Y; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- (5).For selected examples of metalloradical-mediated radical processes, see:; (a) Wayland BB; Poszmik G; Mukerjee SL; Fryd M Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes. J. Am. Chem. Soc 1994, 116, 7943–7944. [Google Scholar]; (b) Zhang X-X; Wayland BB Rhodium(II) Porphyrin Bimetalloradical Complexes: Preparation and Enhanced Reactivity with CH4 and H2. J. Am. Chem. Soc 1994, 116, 7897–7898. [Google Scholar]; (c) Chan KS; Li XZ; Dzik WI; de Bruin B Carbon–Carbon Bond Activation of 2,2,6,6-Tetramethyl-piperidine-1-oxyl by a RhII Metalloradical: A Combined Experimental and Theoretical Study. J. Am. Chem. Soc 2008, 130, 2051–2061. [DOI] [PubMed] [Google Scholar]; (d) Chan YW; Chan KS Metalloradical-Catalyzed Aliphatic Carbon–Carbon Activation of Cyclooctane. J. Am. Chem. Soc 2010, 132, 6920–6922. [DOI] [PubMed] [Google Scholar]; (e) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (f) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H• Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]; (g) Roy S; Khatua H; Das SK; Chattopadhyay B Iron(II)-Based Metalloradical Activation: Switch from Traditional Click Chemistry to Denitrogenative Annulation. Angew. Chem., Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]
- (6).(a) Dzik WI; Xu X; Zhang XP; Reek JNH; de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]; (b) Belof JL; Cioce CR; Xu X; Zhang XP; Space B; Woodcock HL Characterization of Tunable Radical Metal-Carbenes: Key Intermediates in Catalytic Cyclopropanation. Organometallics 2011, 30, 2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu HJ; Dzik WI; Xu X; Wojtas L; de Bruin B; Zhang XP Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (7).(a) Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]; (b) Zhu SF; Ruppel JV; Lu HJ; Wojtas L; Zhang XP Cobalt-Catalyzed, Asymmetric Cyclopropanation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc 2008, 130, 5042–5043. [DOI] [PubMed] [Google Scholar]; (c) Fantauzzi S; Gallo E; Rose E; Raoul N; Caselli A; Issa S; Ragaini F; Cenini S Asymmetric Cyclopropanation of Olefins Catalyzed by Chiral Cobalt(II)-Binaphthyl Porphyrins. Organometallics 2008, 27, 6143–6151. [Google Scholar]; (d) Xu X; Lu HJ; Ruppel JV; Cui X; de Mesa SL; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]; (e) Cui X; Xu X; Lu HJ; Zhu SF; Wojtas L; Zhang XP Enantioselective Cyclopropenation of Alkynes with Acceptor/Acceptor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2011, 133, 3304–3307. [DOI] [PubMed] [Google Scholar]; (f) Zhang J; Jiang JW; Xu DM; Luo Q; Wang HX; Chen JJ; Li HH; Wang YX; Wan XB Interception of Cobalt-Based Carbene Radicals with α-Aminoalkyl Radicals: A Tandem Reaction for the Construction of β-Ester-γ-Amino Ketones. Angew. Chem., Int. Ed 2015, 54, 1231–1235. [DOI] [PubMed] [Google Scholar]; (g) Reddy AR; Hao F; Wu K; Zhou CY; Che CM Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem., Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]; (h) Wang Y; Wen X; Cui X; Wojtas L; Zhang XP Asymmetric Radical Cyclopropanation of Alkenes with In Situ Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) te Grotenhuis C; van den Heuvel N; van der Vlugt JI; de Bruin B Catalytic Dibenzocyclooctene Synthesis via Cobalt(III)-Carbene Radical and ortho-Quinodimethane Intermediates. Angew. Chem., Int. Ed 2018, 57, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Roy S; Das SK; Chattopadhyay B Cobalt(II)-based Metalloradical Activation of 2-(Diazomethyl)-pyridines for Radical Transannulation and Cyclopropanation. Angew. Chem., Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]; (k) Lee W-CC; Wang D-S; Zhang C; Xie J; Li B; Zhang XP Asymmetric Radical Cyclopropanation of Dehydroaminocarboxylates: Stereoselective Synthesis of Cyclopropyl α-Amino Acids. Chem 2021, 7, 1588–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Cui X; Xu X; Jin LM; Wojtas L; Zhang XP Stereoselective Radical C–H Alkylation with Acceptor/Acceptor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. Chem. Sci 2015, 6, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y; Wen X; Cui X; Zhang XP Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C–H Alkylation. J. Am. Chem. Soc 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wen X; Wang Y; Zhang XP Enantioselective Radical Process for Synthesis of Chiral Indolines by Metalloradical Alkylation of Diverse C(sp3)–H Bonds. Chem. Sci 2018, 9, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For selected reviews, see:; (a) Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nechab M; Mondal S; Bertrand MP 1,n-Hydrogen-Atom Transfer (HAT) Reactions in Which n ≠ 5:: An Updated Inventory. Chem. - Eur. J 2014, 20, 16034–16059. [DOI] [PubMed] [Google Scholar]; (c) Matsubara H; Kawamoto T; Fukuyama T; Ryu I Applications of Radical Carbonylation and Amine Addition Chemistry: 1,4-Hydrogen Transfer of 1-Hydroxylallyl Radicals. Acc. Chem. Res 2018, 51, 2023–2035. [DOI] [PubMed] [Google Scholar]
- (10).(a) Bellus D; Ernst B Cyclobutanones and Cyclobutenones in Nature and in Synthesis. Angew. Chem., Int. Ed 1988, 27, 797–827. [Google Scholar]; (b) Lee-Ruff E; Mladenova G Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev 2003, 103, 1449–1483. [DOI] [PubMed] [Google Scholar]; (c) Namyslo JC; Kaufmann DE The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev 2003, 103, 1485–1537. [DOI] [PubMed] [Google Scholar]; (d) Dembitsky VM Bioactive Cyclobutane-Containing Alkaloids. J. Nat. Med 2008, 62, 1–33. [DOI] [PubMed] [Google Scholar]; (e) Seiser T; Saget T; Tran DN; Cramer N Cyclobutanes in Catalysis. Angew. Chem., Int. Ed 2011, 50, 7740–7752. [DOI] [PubMed] [Google Scholar]
- (11).(a) Davies HML; Dai X, 10.04 - Synthetic Reactions via C–H Bond Activation: Carbene and Nitrene C–H Insertion. In Comprehensive Organometallic Chemistry III, Mingos DMP; Crabtree RH, Eds. Elsevier: Oxford, 2007; pp 167–212. [Google Scholar]; (b) Doyle MP; Duffy R; Ratnikov M; Zhou L Catalytic Carbene Insertion into C–H Bonds. Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- (12).(a) Wang JC; Zhang Y; Xu ZJ; Lo VKY; Che CM Enantioselective Intramolecular Carbene C–H Insertion Catalyzed by a Chiral Iridium(III) Complex of D4-Symmetric Porphyrin Ligand. ACS Catal 2013, 3, 1144–1148. [Google Scholar]; (b) Fu LB; Wang HB; Davies HML Role of Ortho-Substituents on Rhodium-Catalyzed Asymmetric Synthesis of β-Lactones by Intramolecular C–H Insertions of Aryldiazoacetates. Org. Lett 2014, 16, 3036–3039. [DOI] [PubMed] [Google Scholar]
- (13).Huang LZ; Xuan Z; Jeon HJ; Du ZT; Kim JH; Lee SG Asymmetric Rh(II)/Pd(0) Relay Catalysis: Synthesis of α-Quaternary Chiral β-Lactams through Enantioselective C–H Insertion/Diastereoselective Allylation of Diazoamides. ACS Catal 2018, 8, 7340–7345. [Google Scholar]
- (14).For asymmetric synthesis of cyclobutanones through other methods other than 1,4-C–H alkylation, please see:; (a) Kleinbeck F; Toste FD Gold(I)-Catalyzed Enantioselective Ring Expansion of Allenylcyclopropanols. J. Am. Chem. Soc 2009, 131, 9178–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reeves CM; Eidamshaus C; Kim J; Stoltz BM Enantioselective Construction of α-Quaternary Cyclobutanones by Catalytic Asymmetric Allylic Alkylation. Angew. Chem., Int. Ed 2013, 52, 6718–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim DK; Riedel J; Kim RS; Dong VM Cobalt Catalysis for Enantioselective Cyclobutanone Construction. J. Am. Chem. Soc 2017, 139, 10208–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shim SY; Choi Y; Ryu DH Asymmetric Synthesis of Cyclobutanone via Lewis Acid Catalyzed Tandem Cyclopropanation/Semipinacol Rearrangement. J. Am. Chem. Soc 2018, 140, 11184–11188. [DOI] [PubMed] [Google Scholar]
- (15).Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]
- (16).Hu Y; Lang K; Tao JR; Marshall MK; Cheng QG; Cui X; Wojtas L; Zhang XP Next-Generation D2-Symmetric Chiral Porphyrins for Cobalt(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging. Angew. Chem., Int. Ed 2019, 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereo-retentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang XL; Dannenberg JJ Molecular-Orbital Estimation of the Activation Enthalpies for Intramolecular Hydrogen Transfer as Functions of Size of the Cyclic Transition-State and C–H–C Angle. J. Org. Chem 1991, 56, 5421–5424. [Google Scholar]
- (19).Xiao TB; Huang HT; Anand D; Zhou L Iminyl-Radical-Triggered C–C Bond Cleavage of Cycloketone Oxime Derivatives: Generation of Distal Cyano-Substituted Alkyl Radicals and Their Functionalization. Synthesis 2020, 52, 1585–1601. [Google Scholar]
- (20).Murakami M; Ishida N Cleavage of Carbon–Carbon σ-Bonds of Four-Membered Rings. Chem. Rev 2021, 121, 264–299. [DOI] [PubMed] [Google Scholar]
- (21).(a) Pettersson T; Eklund AM; Wahlberg I New Lactones from Tobacco. J. Agric. Food Chem 1993, 41, 2097–2103. [Google Scholar]; (b) Mutou T; Kondo T; Ojika M; Yamada K Isolation and Stereostructures of Dolastatin G and Nordolastatin G, Cytotoxic 35-Membered Cyclodepsipeptides from the Japanese Sea Hare Dolabella auricularia. J. Org. Chem 1996, 61, 6340–6345. [DOI] [PubMed] [Google Scholar]; (c) Song QL; Pascouau C; Zhao JP; Zhang GZ; Peruch F; Carlotti S Ring-opening Polymerization of γ-Lactones and Copolymerization with Other Cyclic Monomers. Prog. Polym. Sci 2020, 110, 101309. [Google Scholar]
- (22).(a) Shang XF; Morris-Natschke SL; Liu YQ; Guo X; Xu XS; Goto M; Li JC; Yang GZ; Lee KH Biologically Active Quinoline and Quinazoline Alkaloids Part I. Med. Res. Rev 2018, 38, 775–828. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shang XF; Morris-Natschke SL; Yang GZ; Liu YQ; Guo X; Xu XS; Goto M; Li JC; Zhang JY; Lee KH Biologically Active Quinoline and Quinazoline Alkaloids Part II. Med. Res. Rev 2018, 38, 1614–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Murai K; Komatsu H; Nagao R; Fujioka H Oxidative Rearrangement of Spiro Cyclobutane Cyclic Aminals: Efficient Construction of Bicyclic Amidines. Org. Lett 2012, 14, 772–775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.