

Abstract
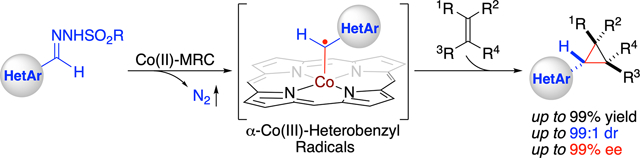
A highly efficient catalytic method has been developed for asymmetric radical cyclopropanation of alkenes with in situ-generated α-heteroaryldiazomethanes via Co(II)-based metalloradical catalysis (MRC). Through fine-tuning the cavity-like environments of newly-synthesized D2-symmetric chiral amidoporphyrins as the supporting ligand, the optimized Co(II)-based metalloradical system is broadly applicable to α-pyridyl and other α-heteroaryldiazomethanes for asymmetric cyclopropanation of wide-ranging alkenes, including several types of challenging substrates. This new catalytic methodology provides a general access to valuable chiral heteroaryl cyclopropanes in high yields with excellent both diastereoselectivities and enantioselectivities. Combined computational and experimental studies further support the underlying stepwise radical mechanism of the Co(II)-based olefin cyclopropanation involving α- and γ-metalloalkyl radicals as the key intermediates.
Graphical Abstract
INTRODUCTION
Radical chemistry has been increasingly explored for the development of new synthetic tools in modern organic synthesis.1 Despite tremendous endeavors, long-standing challenges associated with control of reactivity and enantioselectivity remain largely unresolved for many radical reactions.2 Among recent advances,3 metalloradical catalysis (MRC), which involves the generation and utilization of metal-stabilized organic radicals as catalytic intermediates to harness the potential of radical chemistry, has emerged as a conceptually new approach to guide the discovery of catalytic solutions toward controlling the reactivity and stereoselectivity of radical processes.4–6 As stable 15e-metalloradicals, cobalt-(II) complexes of porphyrins ([Co(Por)]) exhibit the unique capability of homolytically activating diazo compounds to generate α-Co(III)-alkyl radicals as key intermediates for various radical transformations.7 Specifically, with D2-symmetric chiral amidoporphyrins (D2-Por*) as the supporting ligands, these Co-stabilized carbon-centered radicals can engage in asymmetric radical cyclopropanation of alkenes for the preparation of optically active three-membered carbocycles.8 While donor-substituted diazo compounds such as in situ-generated α-aryldiazomethanes have recently been demonstrated as suitable radical precursors for Co(II)-based asymmetric radical cyclopropanation,8k,n the analogous α-heteroaryldiazomethanes have remained underexploited for stereoselective synthesis of valuable chiral heteroaryl cyclopropanes. With this in mind, we sought to explore the feasibility of developing a catalytic process that would employ α-heteroaryldiazomethanes for asymmetric cyclopropanation of alkenes via Co(II)-MRC (Scheme 1). In view of the intrinsic properties of heteroaryl moieties, the proposed Co(II)-based catalytic process presented several fundamental challenges. Besides the concerns with efficiency of metalloradical activation of α-heteroaryldiazomethanes 1′ generated in situ from the corresponding hydrazones 1 in the presence of base, whether the subsequent radical addition of the initially formed α-Co(III)-heterobenzyl radicals I to the alkene substrates 2 could be rendered enantioselective is an unanswered question, primarily owing to the potential competitive coordination of the heteroaryl moieties to the metal center. On this basis, additional uncertainty of controlling reactivity and diastereoselectivity might also arise from the following 3-exo-tet cyclization of the resulting γ-Co(III)-alkyl radicals II while forging the second C–C bond (Scheme 1). Furthermore, the presence of heteroatoms was anticipated to engage in potential H-bonding interactions with the amide units of the amidoporphyrin ligands that could pose potential complication in controlling both reactivity and selectivity in these Co(II)-based radical processes. To address these and related issues, we envisioned the prospect of designing a suitable D2-symmetric chiral amidoporphyrin ligand with proper steric, electronic, and chiral environments that could direct the Co(II)-based catalysis for productive cyclopropanation with effective stereocontrol. If realized, it would enable the development of a new catalytic system for asymmetric olefin cyclopropanation with in situ-generated α-heteroaryldiazomethanes to furnish chiral heteroaryl cyclopropanes 3, which are ubiquitous structural motifs in many pharmaceuticals and biologically important molecules (see Figure S1 in Supporting Information).9
Scheme 1.
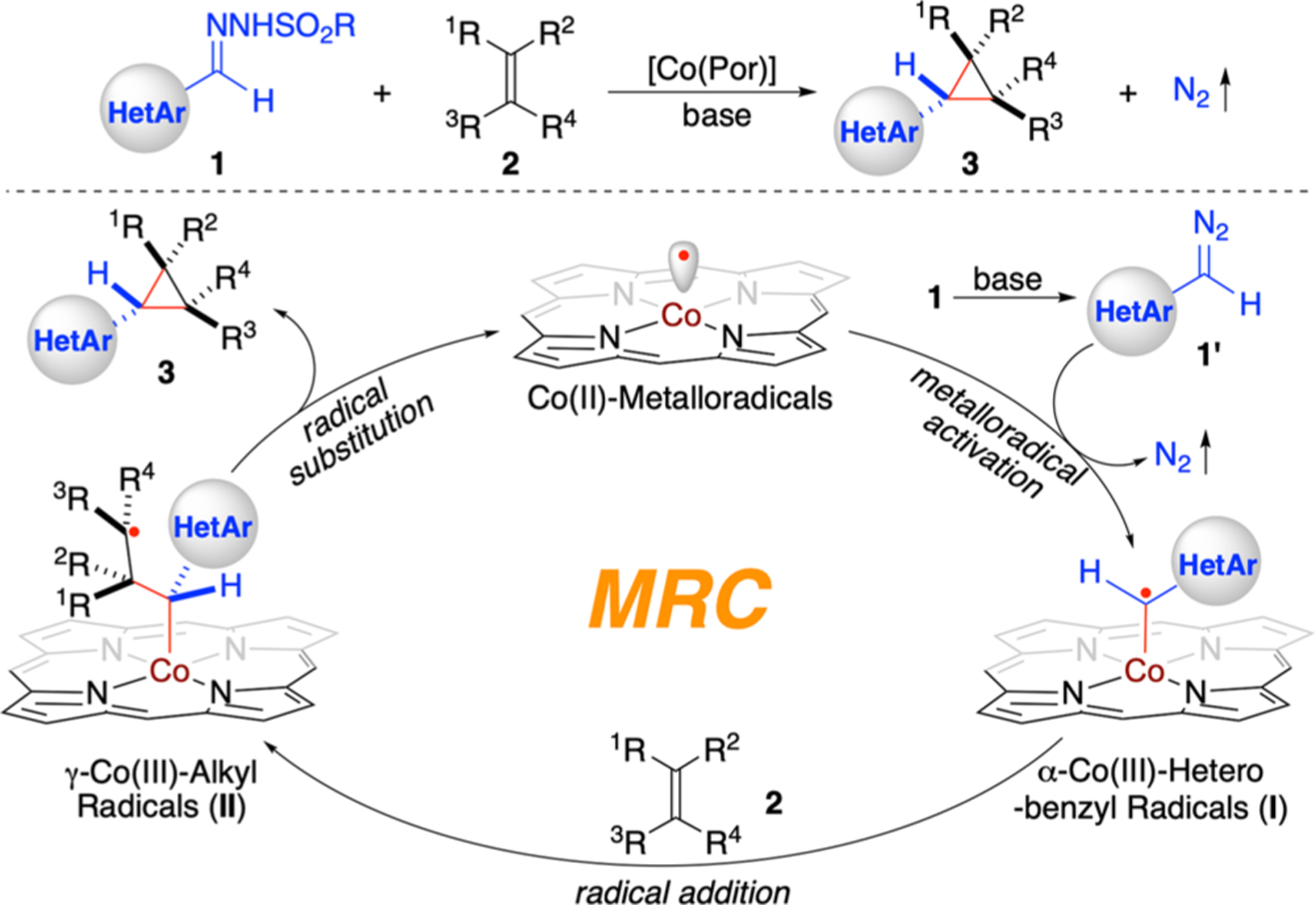
Working Proposal for Synthesis of Heteroaryl Cyclopropanes from Alkenes via Co(II)-Based MRC
Transition-metal catalyzed asymmetric cyclopropanation of alkenes with heteroaryldiazomethanes represents an appealing approach for the synthesis of valuable chiral heteroaryl cyclopropanes with the potential to control both diastereoselectivity and enantioselectivity.9b,10 In contrast to the well-precedented asymmetric cyclopropanation with other types of diazo compounds,11 only a few catalytic systems involving the use of heteroaryldiazomethanes have been reported.12 This underdevelopment is largely attributed to their inherent instability as well as high propensity for unwanted formal dimerization.12,13 Moreover, it is known that rhodium- and other existing metal-based catalytic systems of cyclopropanation could suffer from the notorious catalyst poisoning effect in the presence of nitrogen- and sulfur-containing heterocycles.9b,10g Recently, Chattopadhyay and co-workers14f reported a [Co(TPP)]-catalyzed (TPP = 5,10,15,20-tetraphenylporphyrin) metalloradical cyclopropanation with 2-pyridyldiazomethanes, which could be generated in situ from readily accessible N-tosylhydrazone precursors in the presence of base.14 While this in situ protocol offers a novel alternative for catalytic synthesis of 2-pyridylcyclopropanes in their racemic forms, the enantioselective variant of this transformation is an attractive process that remains elusive. In addition to 2-pyridylcyclopropanes, it would be desirable to develop new catalytic systems that are generally applicable for stereoselective synthesis of diverse types of chiral heteroaryl cyclopropanes. We herein report the development of a new Co(II)-based catalytic system that is highly efficient for asymmetric cyclopropanation of alkenes with in situ-generated α-heteroaryldiazomethanes. Through the support of a new bridged D2-symmetric chiral amidoporphyrin ligand, the Co(II)-catalyzed system allows for efficient activation of 2-pyridyldiazomethanes and other common α-heteroaryldiazomethanes for asymmetric cyclopropanation of a broad range of alkenes, affording the valuable chiral heteroaryl cyclopropanes in high yields with excellent both diastereoselectivities and enantioselectivities. Furthermore, we present detailed computational and experimental studies that shed light on the underlying stepwise radical mechanism.
RESULTS AND DISCUSSION
Catalyst Development.
At the outset of this project, 2-pyridyldiazomethane (1a’), which was in situ generated from the corresponding tosylhydrazone 1a in the presence of Cs2CO3, was investigated as the representative α-heteroaryldiazomethane for asymmetric radical cyclopropanation of styrene (2a) by Co(II)-based metalloradical catalysts [Co-(Por)] (Scheme 2). It was found that the Co(II) complex of D2h-symmetric achiral amidoporphyrin [Co(P1)] (P1 = 3,5-DitBu-IbuPhyrin)15 could effectively catalyze the cyclopropanation reaction to afford the desired 2-pyridylcyclopropane 3a in nearly quantitative yield (99%) with moderate diastereoselectivity (44% de). To evaluate the feasibility of asymmetric induction during the proposed catalytic cycle, Co(II) complexes of a series of D2-symmetric chiral amidoporphyrin ligands [Co(D2-Por*)] were employed as the catalysts. While first-generation chiral metalloradical catalyst [Co(P2)] (P2 = 3,5-DitBu-ChenPhyrin)8a could furnish 3a in a similarly high yield (95%) with higher diastereoselectivity (72% de), it only exhibited insignificant asymmetric induction (5% ee). Switching to second-generation metalloradical catalyst [Co(P3)] (P3 = 3,5-DitBu-Tao(tBu)-Phyrin)16 bearing chiral amide units with ester moieties resulted in the formation of 3a in 86% yield with further improved diastereoselectivity (88% de) and a significant level of enantioselectivity (40% ee). To further enhance the asymmetric induction of this catalytic system, we then turned our attention to new-generation metalloradical catalysts [Co(HuPhyrin)], the Co(II) complexes of bridged D2-symmetric chiral amidoporphyrins featuring more rigid cavity-like environments. When the C6-bridged [Co(P4)] (P4 = 3,5-DitBu-Hu(C6)Phyrin)16 was employed as the catalyst under the same conditions, it indeed enhanced both reactivity and stereoselectivities of the cyclopropanation reaction substantially, generating 3a in high yield (94%) with excellent both diastereoselectivity (98% de) and enantioselectivity (92% ee). Subsequent use of analogous catalyst [Co(P5)] (P5 = 2,6-DiMeO-Hu(C6)Phyrin),17 which bears 2,6-dimethoxyphenyl instead of 3,5-di-tert-butylphenyl groups as the 5,15-diaryl substituents, led to the production of 3a in a comparable yield (91% yield) with the same stereoselectivities (98% de and 92% ee). Aiming at further improving the catalytic system, we synthesized a new C6-bridged catalyst [Co(P6)] (P6 = 2,6-DiPhO-Hu(C6)Phyrin) by replacing the methoxy groups in P5 with phenoxy groups. Gratifyingly, [Co(P6)] could catalyze the cyclopropanation reaction to afford 2-pyridylcyclopropane 3a in almost quantitative yield (99%) with high diastereoselectivity (92% de) and outstanding enantioselectivity (99% ee).
Scheme 2. Ligand Effect on Co(II)-Catalyzed Radical Cyclopropanation of Styrene with 2-Pyridyldiazomethanesa.
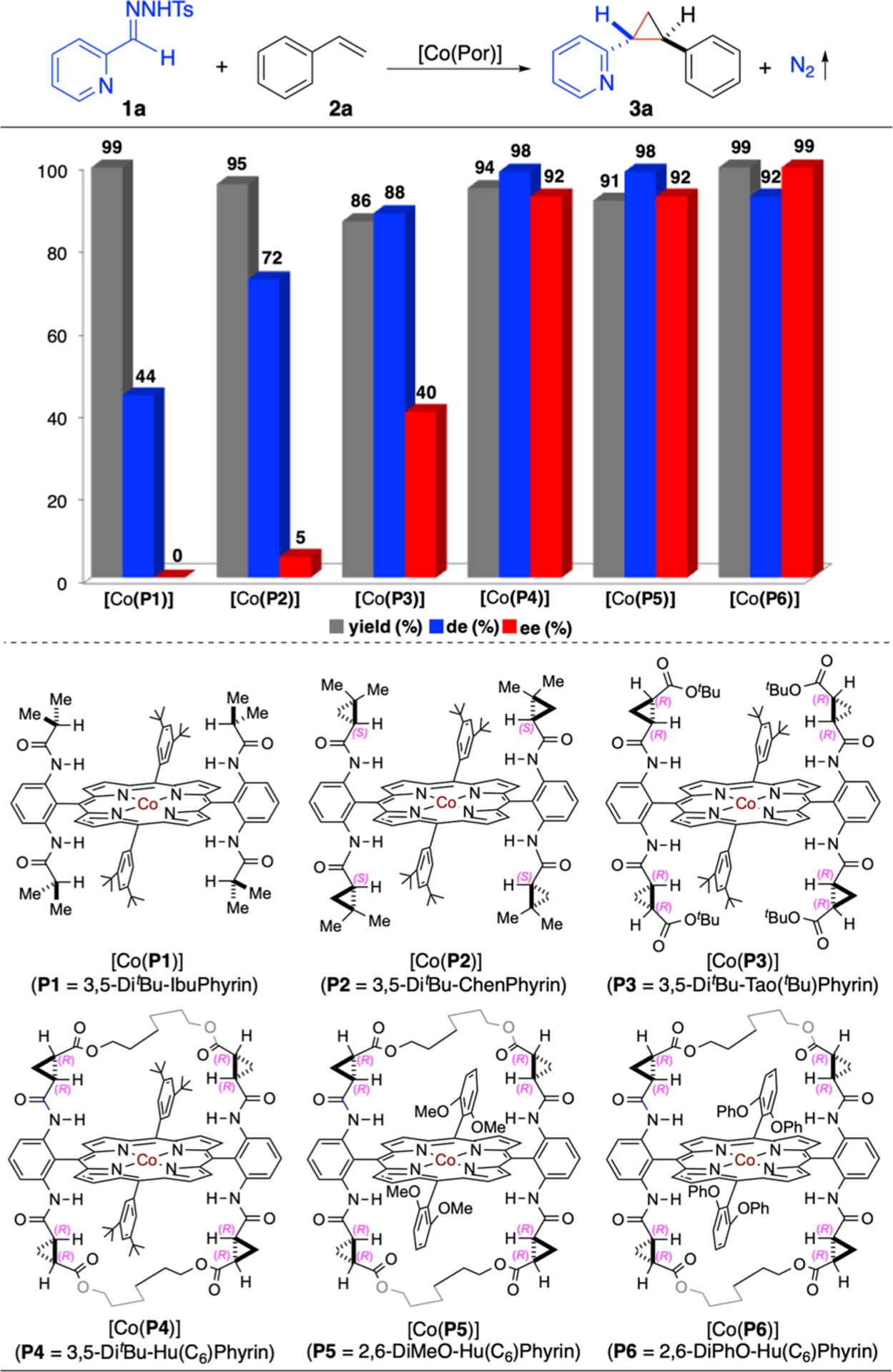
aCarried out with 1a (0.10 mmol), 2a (0.15 mmol), and Cs2CO3 (0.20 mmol) using [Co(Por)] (2 mol %) in toluene (1.0 mL) at 80 °C for 16 h; Isolated yields; Diastereomeric excess (de) determined by 1H NMR of crude reaction mixture; Enantiomeric excess (ee) of the major (E)-isomer determined by chiral HPLC; Ts = 4-toluenesulfonyl.
Substrate Scope.
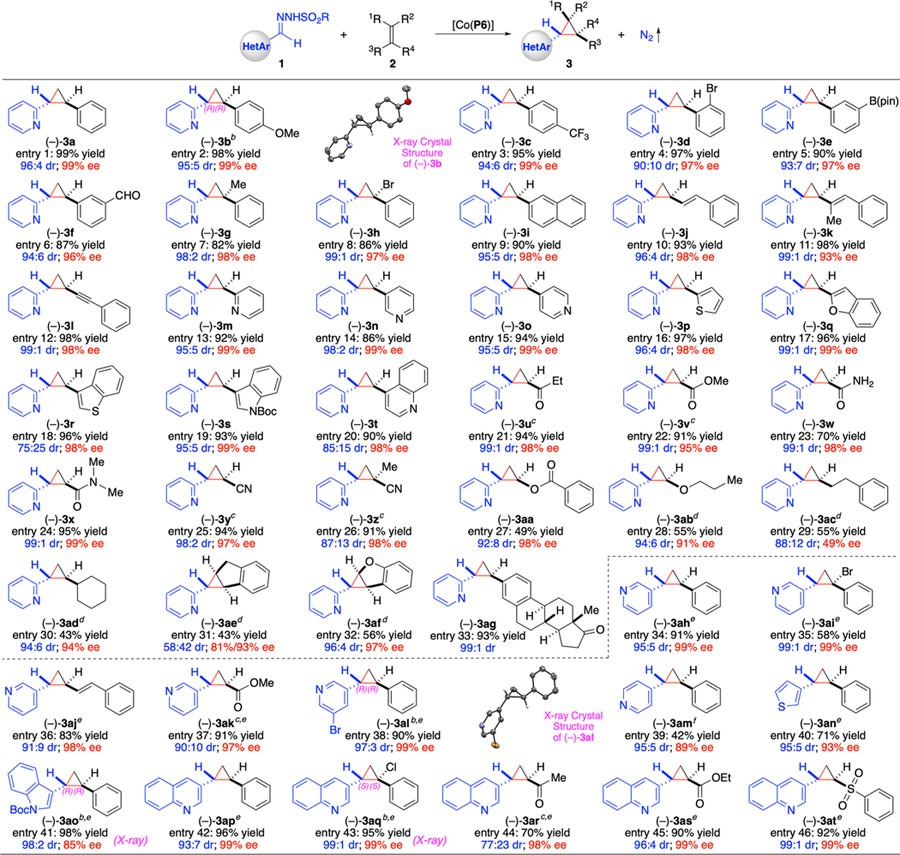
Under the optimized conditions, the scope and versatility of [Co(P6)]-catalyzed asymmetric cyclopropanation with in situ-generated 2-pyridyldiazomethane (1a’) were explored by employing different types of alkenes as the substrates (Table 1). Like formation of 3a from styrene (entry 1), its derivatives bearing electron-donating and electron-withdrawing aryl substituents could also be effectively cyclopropanated by [Co(P6)] with 1a’, producing the desired cyclopropanes 3b and 3c in similarly high yields and stereoselectivities (entries 2 and 3). The absolute configuration of the major enantiomer of 3b was established as (R,R). Additionally, this [Co(P6)]-based metalloradical system was shown to tolerate various functional groups as exemplified by the stereoselective formation of 3d–3f containing aryl substituents of halogen, pinacolborane, and formyl functionalities at different positions (entries 4–6). Besides monosubstituted olefins, 1,1-disubstituted olefins like α-substituted styrenes could serve as suitable substrates as well, affording the trisubstituted cyclopropanes 3g and 3h with excellent control of the newly generated quaternary stereogenic centers (entries 7 and 8). In addition to the extended aromatic olefins such as formation of 3i from 2-vinylnaphthalene (entry 9), both conjugated dienes and enynes could be regio- and chemo-selectively cyclopropanated to form cyclopropanes 3j–3l in high yields with excellent stereoselectivities (entries 10–12). The Co(II)-based cyclopropanation was further highlighted by its unique reactivity toward various heteroaromatic olefins as shown with the highly stereoselective synthesis of 1,2-bisheteroaryl cyclopropanes 3m–3t containing pyridine, thiophene, benzofuran, benzothiophene, indole, and quinoline (entries 13–20). Given that both heteroarene and cyclopropane are prevalent structural motifs in bioactive compounds, the access of bisheteroaryl cyclopropanes in high enantiopurity may find potential applications in drug research and development. Moreover, electron-deficient olefins such as acrylketones, acrylates, acrylamides, and acrylonitriles, which are known to be challenging substrates, could all be utilized for asymmetric cyclopropanation by [Co(P6)], furnishing the functionalized electrophilic cyclopropanes 3u–3z in high yields with excellent control of stereoselectivities (entries 21–26). Similar to electron-deficient olefins, the catalytic cyclopropanation could also be applied to electron-rich olefins such as vinyl benzoate and vinyl propyl ether for highly stereoselective formation of cyclopropyl ester 3aa and cyclopropyl ether 3ab albeit in relatively lower yields (entries 27 and 28). Notably, the [Co(P6)]-based system proved to be similarly effective for the asymmetric cyclopropanation of aliphatic olefins, affording the alkyl-substituted pyridylcyclopropanes 3ac and 3ad in moderate yields with moderate to excellent stereoselectivities (entries 29 and 30). Gratifyingly, internal olefins such as indene and benzofuran, which are typically challenging substrates for asymmetric cyclopropanation due to steric factors, could also be cyclopropanated to form the fused cyclopropanes 3ae and 3af in moderate yields with high enantioselectivities despite varied diastereoselectivities (entries 31 and 32). It is worth mentioning that the Co(II)-based system was amenable to late-stage derivatization of biologically complex molecules as exemplified by the high-yielding formation of cyclopropane derivative of estrone 3ag with excellent diastereoselectivity (entry 33).
Table 1.
Scope of Co(II)-Catalyzed Asymmetric Radical Cyclopropanation of Alkenes with Heteroaryldiazomethanesa
|
Carried out with 1 (0.10 mmol), 2 (0.15 mmol), and Cs2CO3 (0.20 mmol) at 80 °C for 16 h using [Co(P6)] (2 mol %) in toluene (1.0 mL); R = 4-methylphenyl; Isolated yields; Diastereomeric ratio (dr) determined by 1H NMR of crude reaction mixture; Enantiomeric excess (ee) of the major isomer determined by chiral HPLC.
Absolute configuration determined by X-ray crystallography.
With 2 (0.30 mmol).
With 2 (1.0 mmol).
At 22 °C; R = 2,4,6-triisopropylphenyl.
At 60 °C.
In addition to the representative 2-pyridyldiazomethane (1a’), it was demonstrated that metalloradical catalyst [Co(P6)] could effectively activate different types of α-heteroaryldiazomethanes for asymmetric cyclopropanation of alkenes (Table 1). For instance, 3-pyridyldiazomethane (1b’) generated from the corresponding trishydrazone (2,4,6-triisopropylbenzenesulfonyl hydrazone) was found to be a competent radical precursor even at room temperature for the Co(II)-based asymmetric cyclopropanation. As shown with styrene (monosubstituted olefin), α-bromostyrene (1,1-disubstituted olefin), 1-phenyl-1,3-butadiene (conjugated diene), and methyl acrylate (electron-deficient olefin) as representative substrates, [Co(P6)] could effectively activate in situ-generated 1b’ at room temperature for highly asymmetric cyclopropanation reactions, leading to productive formation of the corresponding 3-pyridylcyclopropanes 3ah, 3ai, 3aj, and 3ak with exceptional control of stereoselectivities (entries 34–37). Likewise, other heteroaryldiazomethanes, including those generated in situ from the trishydrazones derived from 5-bromo-3-pyridyl, 4-pyridyl, 3-thienyl, 3-indolyl, and 3-quinolinyl carboxaldehydes, were all shown to be effective radical precursors for [Co(P6)]-catalyzed asymmetric olefin cyclopropanation as exemplified with the room temperature reactions of styrene as the model substrate, affording the corresponding heteroaryl cyclopropanes 3al–3ap in moderate to high yields with excellent stereoselectivities (entries 38–42). The absolute configurations of the newly generated stereogenic centers in 3al and 3ao were both established as (R,R) by X-ray crystallography. In addition, 3-quinolinyldiazomethane (1g’) was also investigated for Co(II)-based cyclopropanation reactions of selected alkenes ranging from α-chlorostyrene to electron-deficient olefins. Gratifyingly, almost all of these alkene substrates could be effectively cyclopropanated, allowing for the high-yielding formation of 3-quinolinylcyclopropanes 3aq–3at with excellent stereoselectivities (entries 43–46). The only exception was observed for the reaction of methyl vinyl ketone, which afforded the corresponding cyclopropane 3ar with excellent enantioselectivity but in lower yield with diminished diastereoselectivity (entry 44). The absolute configuration of the major enantiomer of 3aq was determined to be (S,S) by X-ray crystallography.
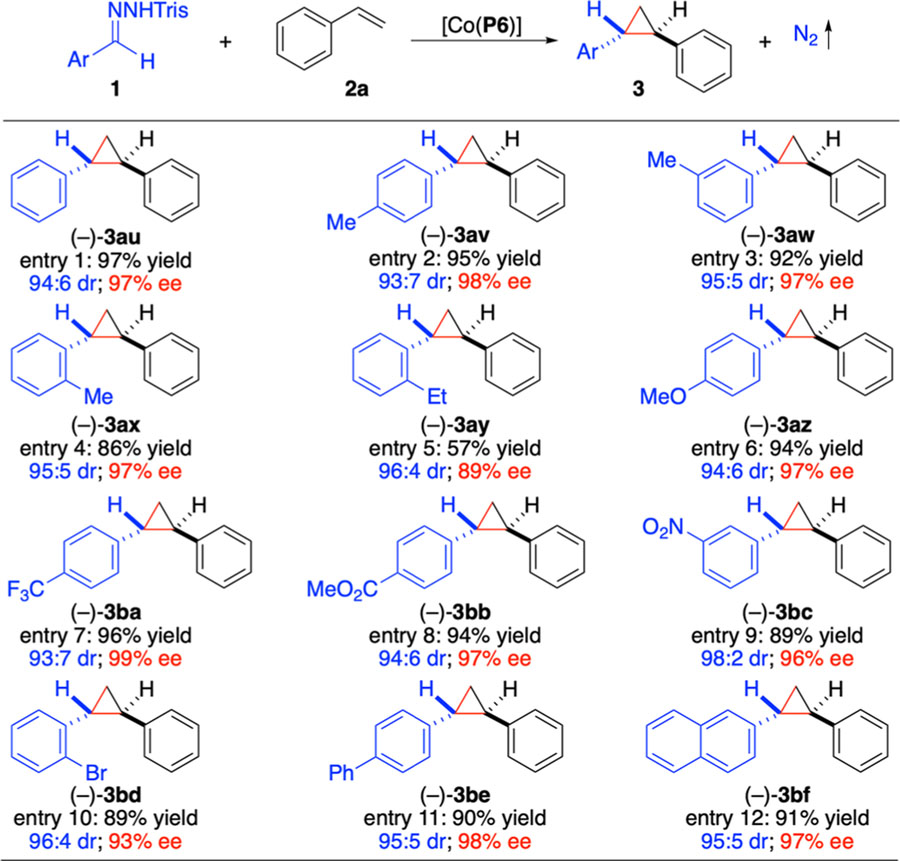
Considering that the [Co(P6)]-based catalytic system could productively utilize various α-heteroaryldiazomethanes containing heteroatom at different positions, we sought to explore the possibility of employing α-aryldiazomethanes for asymmetric cyclopropanation (Table 2), which has been largely limited to those with α-aryl groups containing H-bonding acceptors at the ortho-position.8k,n To our delight, it was found that [Co(P6)] could effectively activate α-phenyldiazomethane (1h’) derived from benzaldehyde trishydrazone (1h) as the radical precursor for asymmetric cyclopropanation of styrene (2a) under the standard conditions, affording the desired 1,2-diphenylcyclopropane (3au) in high yield with high diastereoselectivity and excellent enantioselectivity (entry 1). Encouraged by this positive outcome, we then evaluated a wide array of α-aryldiazomethanes without H-bonding acceptors for asymmetric cyclopropanation by [Co(P6)]. In addition to nonsubstituted α-phenyldiazomethane, α-aryldiazomethanes bearing methyl substituent at different aryl positions, including p-Me (1i’), m-Me (1j’), and o-Me (1k’), could all be efficiently activated by [Co(P6)] for cyclopropanation of 2a, furnishing the corresponding arylcyclopropanes 3av–3ax in similarly high yields with the same high level of stereoselectivities (entries 2–4). Notably, the sterically encumbered o-ethylphenyldiazomethane (1l’) was found to be also suitable for the catalytic reaction, forming cyclopropane 3ay with high stereoselectivities albeit in lower yield (entry 5). It was further shown that the [Co(P6)]-based system could use α-aryldiazomethanes containing substituents with varied electronic properties at different aryl positions, such as p-OMe (1m’), p-CF3 (1n’), p-CO2Me (1o’), and m-NO2(1p’), for the reaction, enabling high-yielding formation of the desired cyclopropanes 3az–3bc with excellent stereoselectivities (entries 6–9). Additionally, halogenated aryldiazomethanes were also suitable for the catalytic process as exemplified by the stereoselective synthesis of cyclopropane 3bd with o-bromophenyldiazomethane (1q’) (entry 10). Furthermore, the Co(II)-based catalytic system could be applicable to α-aryldiazomethanes bearing extended aromatic systems, including p-biphenyldiazomethane (1r’) and 2-naphthyldiazomethane (1s’), delivering the corresponding cyclopropanes 3be and 3bf in high yields with excellent control of stereoselectivities. Evidently, [Co(P6)] represents a powerful new catalyst that is generally applicable for asymmetric olefin cyclopropanation with both α-heteroaryldiazomethanes and α-aryldiazomethanes.
Table 2.
Scope of Co(II)-Catalyzed Asymmetric Radical Cyclopropanation of Styrene with Aryldiazomethanesa
|
Carried out with 1 (0.10 mmol), 2a (0.15 mmol), and Cs2CO3 (0.20 mmol) using [Co(P6)] (2 mol %) in toluene (1.0 mL) at 22 °C for 16 h; Isolated yields; Diastereomeric ratio (dr) determined by 1H NMR of crude reaction mixture; Enantiomeric excess (ee) of the major (E)-isomer determined by chiral HPLC; Tris = 2,4,6-triisopropylbenzene sulfonyl.
Mechanistic Studies.
To gain insight into the proposed stepwise radical mechanism (Scheme 1), combined computational and experimental studies were conducted (Scheme 3). First, density functional theory (DFT) calculations were performed to elucidate the details of the catalytic pathway and associated energetics for the cyclopropanation reaction of styrene (2a) with 2-pyridyldiazomethane (1a’) by [Co(P6)] (Scheme 3A; see Supporting Information for details). The DFT calculations reveal the initial formation of intermediate B between the catalyst and 2-pyridyldiazomethane through multiple noncovalent attractive interactions, including H-bonding and π-stacking interactions. The noncovalent complexation, which is exergonic by 8.7 kcal/mol, positions the α-carbon atom of diazo 1a’ in close proximity to the Co(II)-metalloradical center of [Co(P6)] (C–Co: ~2.91 Å) for further interactions (Scheme 3A; see Scheme S7 in Supporting Information). The ensuing metalloradical activation, which is slightly exergonic by 1.2 kcal/mol, is found to be associated with a relatively high but accessible activation barrier (TS1: ΔG‡ = 19.7 kcal/mol), affording α-Co(III)-pyridyl radical intermediate C with the release of dinitrogen as the byproduct (see Scheme S6 in Supporting Information). The subsequent radical addition of the resulting radical intermediate C to alkene 2a, which is highly exergonic by 22.4 kcal/mol, proceeds through an exceedingly low activation barrier (TS2: ΔG‡ = 2.3 kcal/mol), delivering γ-Co(III)-alkyl radical intermediate D (see Scheme S6 in Supporting Information). As illustrated in the DFT-optimized structure of TS2 (Scheme 3A; see Scheme S7 in Supporting Information), there exists a network of noncovalent attractive interactions, such as multiple H-bonding and π-stacking interactions, between the substrates and the catalyst that synergistically lower the activation barrier of the transition state. According to the DFT calculations, the final step of 3-exo-tet cyclization of γ-Co(III)-alkyl radical intermediate D, which is exergonic by 12.6 kcal/mol (see Scheme S6 in Supporting Information), is a nearly barrierless process, leading to the formation of cyclopropane product 3a while regenerating the metalloradical catalyst [Co(P6)].
Scheme 3. Mechanistic Studies on Co(II)-Catalyzed Radical Olefin Cyclopropanation with Heteroaryldiazomethanesa,b,c.
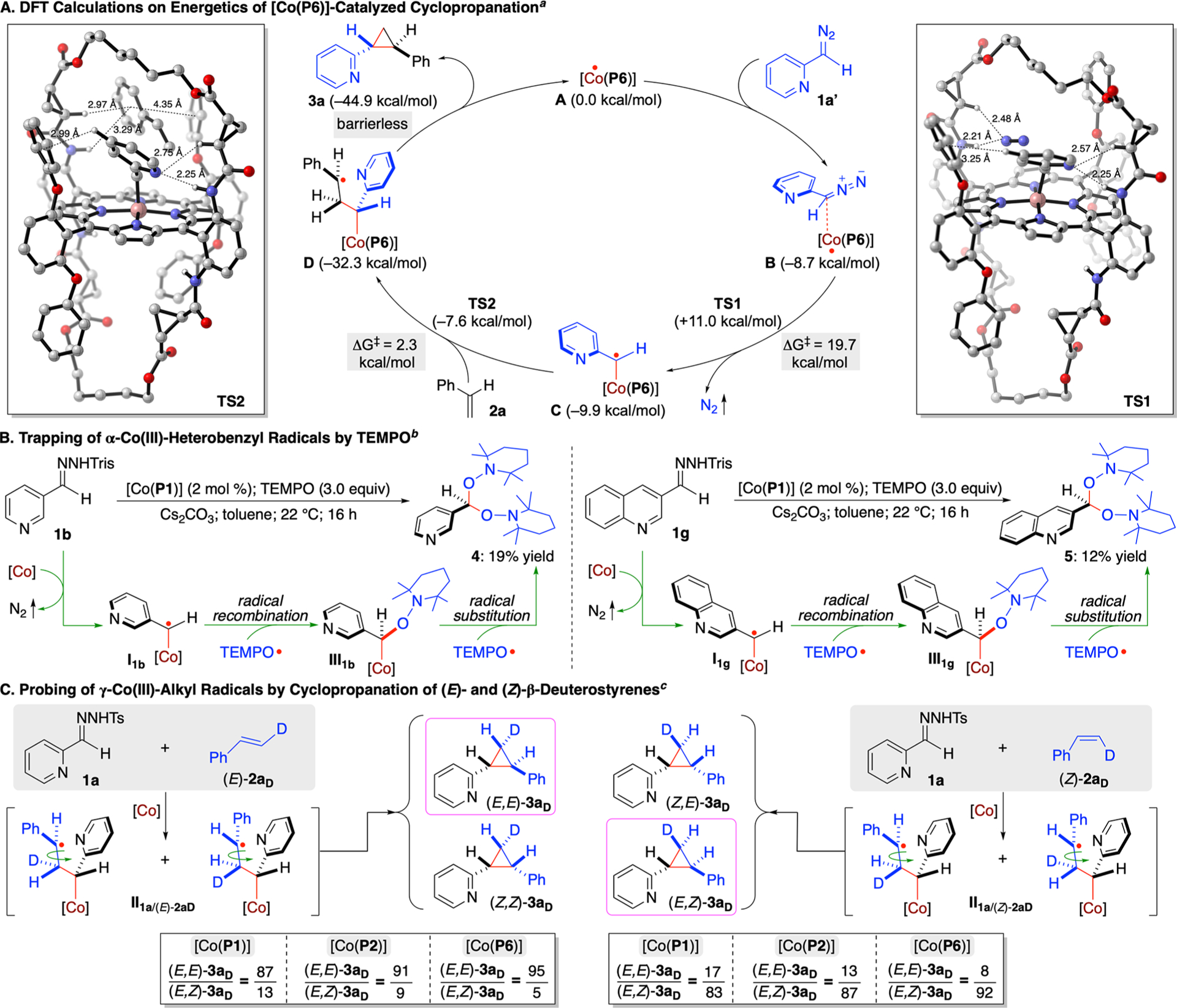
aDFT calculations on energetics for catalytic cyclopropanation of styrene (2a) with 2-pyridyldiazomethane (1a’) by [Co(P6)]. bTEMPO-trapping experiments for metalloradical activation of 3-pyridyl trishydrazone (1b) and 3-quinolinyl trishydrazone (1g) by [Co(P1)]. cCatalytic cyclopropanation reactions of (E)- and (Z)-β-deuterostyrenes with 2-pyridyl tosylhydrazone (1a) by [Co(P1)], [Co(P2)], and [Co(P6)].
In an effort to directly trap α-Co(III)-heterobenzyl radical intermediate I, the metalloradical activation of 3-pyridyldiazo-methane (1b’) by [Co(P1)] was carried out in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) without alkene substrates, resulting in the isolation of bis-TEMPO-trapped product 4 in 19% yield (Scheme 3B). The observation of compound 4 evidently implies the initial formation of α-Co(III)-pyridyl radical I1b, which was presumably captured by TEMPO through radical recombination to generate Co(III)-alkyl intermediate III1b. Subsequent radical substitution reaction of intermediate III1b with a second molecule of TEMPO was likely responsible for the final formation of 4. Similarly, bis-TEMPO-trapped product 5 was isolated in 12% yield from the metalloradical activation of 3-quinolinyldiazo-methane (1g’) by [Co(P1)] in the presence of TEMPO, indicating the existence of α-Co(III)-heterobenzyl radical intermediate I1g and the following Co(III)-alkyl intermediate III1g (Scheme 3B).
To probe the involvement of the γ-Co(III)-alkyl radical intermediate II in the proposed mechanism (Scheme 1), both isotopomers of β-deuterostyrene (E)-2aD and (Z)-2aD were employed as substrates for Co(II)-catalyzed cyclopropanation with 2-pyridyl tosylhydrazone (1a). Unlike a concerted mechanism that results in stereospecific cyclopropane products, a stepwise radical mechanism may give rise to the formation of four possible diastereomers of cyclopropanes due to the potential rotation of the β-C–C bond in γ-Co(III)-alkyl radicalintermediate II before cyclization. As expected, both reactions of (E)-2aD and (Z)-2aD with 1a afforded the cyclopropane products as a mixture of four different diastereomers: (E,E)-3aD, (Z,Z)-3aD, (Z,E)-3aD, and (E,Z)-3aD (Scheme 3C; see Supporting Information for details). Among them, the ratio between isotopomers (E,E)-3aD and (E,Z)-3aD could be accurately determined by the combination of 1H and 2H NMR analysis. When the bridged [Co(P6)] was used as the catalyst, the isotopomeric ratio of (E,E)-3aD to (E,Z)-3aD was determined to be 95:5 and 8:92 for the cyclopropanation reactions of (E)-2aD and (Z)-2aD, respectively. This observation of both (E)- and (Z)-isotopomers of (E)-3a in both reactions evidently suggested the rotation of β-C–C bond in the corresponding γ-Co(III)-alkyl radical intermediates II1a/(E)-2aD and II1a/(Z)-2aD. When the nonbridged [Co(P2)] was employed as the catalyst, the isotopomeric ratio of (E,E)-3aD to (E,Z)-3aD changed for both reactions of (E)-2aD (from 95:5 to 91:9) and (Z)-2aD (from 8:92 to 13:87), indicating a higher degree of the β-C–C bond rotation in the less-hindered catalyst environment. Accordingly, the use of even less-hindered catalyst [Co(P1)] allowed a further increase in the degree of the β-C–C bond rotation, changing the isotopomeric ratio of (E,E)-3aD to (E,Z)-3aD to 87:13 and 17:83, respectively, for the two reactions. Collectively, these experimental results, together with the DFT calculations, provided corroborating evidence for the proposed stepwise radical mechanism of the Co(II)-catalyzed asymmetric cyclopropanation with heteroaryldiazomethanes.
CONCLUSIONS
In summary, we have applied Co(II)-based metalloradical catalysis (MRC) for the successful development of asymmetric radical cyclopropanation of alkenes with heteroaryldiazomethanes. With the newly synthesized bridged D2-symmetric chiral amidoporphyrin 2,6-DiPhO-Hu(C6)Phyrin as the optimal supporting ligand, the Co(II)-based metalloradical system can effectively activate different types of heteroaryldiazomethanes even at room temperature for olefin cyclopropanation, offering a general approach for stereoselective synthesis of chiral heteroaryl cyclopropanes. In addition to styrene derivatives, the Co(II)-catalyzed cyclopropanation is highlighted by an extraordinarily broad scope of alkenes, including several types of challenging substrates, affording a diverse range of heteroaryl cyclopropanes in high yields with excellent both diastereoselectivities and enantioselectivities. Furthermore, our combined computational and experimental studies have provided several lines of evidence in elucidating the underlying stepwise radical mechanism of the Co(II)-based olefin cyclopropanation involving α- and γ-metalloalkyl radicals as the key intermediates. In view of the ubiquity of the resulting enantioenriched heteroaryl cyclopropanes in biologically important compounds, we hope this Co(II)-catalyzed asymmetric radical cyclopropanation process will find wide applications in organic synthesis related to drug discovery.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (R01-GM102554) and in part by NSF (CHE 1900375).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04655.
Experimental details and analytical data for all new compounds (PDF)
Accession Codes
CCDC 2083336–2083339 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c04655
The authors declare no competing financial interest.
Contributor Information
Xiaoxu Wang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States;.
Jing Ke, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States;.
Yiling Zhu, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Arghya Deb, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Yijie Xu, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States;.
X. Peter Zhang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States;.
REFERENCES
- (1).(a) For selected books, see:Zard SZ Radical Reactions in Organic Synthesis; Oxford University Press: 2003. [Google Scholar]; (b) Chatgilialoglu C; Studer A Encyclopedia of Radicals in Chemistry, Biology, and Materials; John Wiley & Sons: 2012. [Google Scholar]; (c) For selected reviews, see:Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (d) Narayanam JM; Stephenson CR Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (e) Quiclet-Sire B; Zard SZ Fun with Radicals: Some New Perspectives for Organic Synthesis. Pure Appl. Chem 2010, 83, 519–551. [Google Scholar]; (f) Prier CK; Rankic DA; MacMillan DW Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected reviews, see:Bar G; Parsons AF Stereoselective Radical Reactions. Chem. Soc. Rev 2003, 32, 251–263. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]; (c) Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]
- (3).(a) For selected examples on approaches to controlling radical reactivity and stereoselectivity, see:Du JN; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (c) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang W; Wang F; McCann SD; Wang DH; Chen PH; Stahl SS; Liu GS Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kern N; Plesniak MP; McDouall JJW; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (f) Morrill C; Jensen C; Just-Baringo X; Grogan G; Turner NJ; Procter DJ Biocatalytic Conversion of Cyclic Ketones Bearing -Quaternary Stereocenters into Lactones in an Enantioselective Radical Approach to Medium-Sized Carbocycles. Angew. Chem., Int. Ed 2018, 57, 3692–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]; (h) Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Favoenzymes Enables a Stereoselective Radical Cyclization. Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Huang H-M; McDouall JJW; Procter DJ SmI2-Catalysed Cyclization Cascades by Radical Relay. Nat. Catal 2019, 2, 211–218. [Google Scholar]; (j) Nakafuku KM; Zhang Z; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C–H Amination for the Synthesis of β-Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) For selected reviews and highlights on Co(II)-based MRC, see:Lu HJ; Zhang XP Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (c) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (d) Huang H-M; Garduño-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (e) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar]
- (5).(a) For selected examples of Ti(III)-based radical processes, see:Nugent WA; RajanBabu TV Transition-Metal-Centered Radicals in Organic Synthesis. Titanium(III)-Induced Cyclization of Epoxyolefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]; (b) Rajanbabu TV; Nugent WA Selective Generation of Free-Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic-Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Cationic Titanocene(III) Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (d) Funken N; Mühlhaus F; Gansäuer A General, Highly Selective Synthesis of 1,3-and 1,4-Difunctionalized Building Blocks by Regiodivergent Epoxide Opening. Angew. Chem., Int. Ed 2016, 55, 12030–12034. [DOI] [PubMed] [Google Scholar]; (e) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (f) Yao CB; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (g) Ye KY; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- (6).(a) For selected examples of metalloradical-mediated radical processes, see:Wayland BB; Poszmik G; Mukerjee SL; Fryd M Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes. J. Am. Chem. Soc 1994, 116, 7943–7944. [Google Scholar]; (b) Zhang X-X; Wayland BB Rhodium(II) Porphyrin Bimetalloradical Complexes: Preparation and Enhanced Reactivity with CH4 and H2. J. Am. Chem. Soc 1994, 116, 7897–7898. [Google Scholar]; (c) Chan KS; Li XZ; Dzik WI; de Bruin B Carbon-Carbon Bond Activation of 2,2,6,6-Tetramethyl-piperidine-1-oxyl by a RhII Metalloradical: A Combined Experimental and Theoretical Study. J. Am. Chem. Soc 2008, 130, 2051–2061. [DOI] [PubMed] [Google Scholar]; (d) Chan YW; Chan KS Metalloradical-Catalyzed Aliphatic Carbon-Carbon Activation of Cyclooctane. J. Am. Chem. Soc 2010, 132, 6920–6922. [DOI] [PubMed] [Google Scholar]; (e) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co-H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (f) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H· Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]; (g) Roy S; Khatua H; Das SK; Chattopadhyay B Iron(II)-Based Metalloradical Activation: Switch from Traditional Click Chemistry to Denitrogenative Annulation. Angew. Chem., Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]; (h) Das SK; Roy S; Khatua H; Chattopadhyay B Iron-Catalyzed Amination of Strong Aliphatic C(sp3)–H Bonds. J. Am. Chem. Soc 2020, 142, 16211–16217. [DOI] [PubMed] [Google Scholar]; (i) Zhang Z; Gevorgyan V Co-Catalyzed Transannulation of Pyridotriazoles with Isothiocyanates and Xanthate Esters. Org. Lett 2020, 22, 8500–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Dzik WI; Xu X; Zhang XP; Reek JNH; de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]; (b) Belof JL; Cioce CR; Xu X; Zhang XP; Space B; Woodcock HL Characterization of Tunable Radical Metal-Carbenes: Key Intermediates in Catalytic Cyclopropanation. Organometallics 2011, 30, 2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu H; Dzik WI; Xu X; Wojtas L; de Bruin B; Zhang XP Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (8).(a) Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]; (b) Caselli A; Gallo E; Ragaini F; Ricatto F; Abbiati G; Cenini S Chiral Porphyrin Complexes of Cobalt(II) and Ruthenium(II) in Catalytic Cyclopropanation and Amination Reactions. Inorg. Chim. Acta 2006, 359, 2924–2932. [Google Scholar]; (c) Chen Y; Ruppel JV; Zhang XP Cobalt-Catalyzed Asymmetric Cyclopropanation of Electron-Deficient Olefins. J. Am. Chem. Soc 2007, 129, 12074–12075. [DOI] [PubMed] [Google Scholar]; (d) Fantauzzi S; Gallo E; Rose E; Raoul N; Caselli A; Issa S; Ragaini F; Cenini S Asymmetric Cyclopropanation of Olefins Catalyzed by Chiral Cobalt(II)-Binaphthyl Porphyrins. Organometallics 2008, 27, 6143–6151. [Google Scholar]; (e) Zhu S; Ruppel JV; Lu H; Wojtas L; Zhang XP Cobalt-Catalyzed Asymmetric Cyclopropanation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc 2008, 130, 5042–5043. [DOI] [PubMed] [Google Scholar]; (f) Zhu S; Xu X; Perman JA; Zhang XP A General and Efficient Cobalt(II)-Based Catalytic System for Highly Stereoselective Cyclopropanation of Alkenes with α-Cyanodiazoacetates. J. Am. Chem. Soc 2010, 132, 12796–12799. [DOI] [PubMed] [Google Scholar]; (g) Xu X; Lu HJ; Ruppel JV; Cui X; de Mesa SL; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]; (h) Paul ND; Mandal S; Otte M; Cui X; Zhang XP; de Bruin B Metalloradical Approach to 2H-Chromenes. J. Am. Chem. Soc 2014, 136, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Reddy AR; Hao F; Wu K; Zhou CY; Che CM Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem., Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]; (j) Chirila A; Gopal Das B; Paul ND; de Bruin B Diastereoselective Radical-Type Cyclopropanation of Electron-Deficient Alkenes Mediated by the Highly Active Cobalt(II) Tetramethyltetraaza[14]annulene Catalyst. ChemCatChem 2017, 9, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang Y; Wen X; Cui X; Wojtas L; Zhang XP Asymmetric Radical Cyclopropanation of Alkenes with In Situ-Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wang Y; Wen X; Cui X; Zhang XP Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C–H Alkylation. J. Am. Chem. Soc 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Wen X; Wang Y; Zhang XP Enantioselective Radical Process for Synthesis of Chiral Indolines by Metalloradical Alkylation of Diverse C(sp3)–H Bonds. Chem. Sci 2018, 9, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Lee W-CC; Wang D-S; Zhang C; Xie J; Li B; Zhang XP Asymmetric Radical Cyclopropanation of Dehydroaminocarboxylates: Stereoselective Synthesis of Cyclopropyl α-Amino Acids. Chem. 2021, 7, 1588–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Liu H; Kerdesky FA; Black LA; Fitzgerald M; Henry R; Esbenshade TA; Hancock AA; Bennani YL An Efficient Multigram Synthesis of the Potent Histamine H3 Antagonist GT-2331 and the Reassessment of the Absolute Configuration. J. Org. Chem 2004, 69, 192–194. [DOI] [PubMed] [Google Scholar]; (b) Butcher KJ; Denton SM; Field SE; Gillmore AT; Harbottle GW; Howard RM; Laity DA; Ngono CJ; Pibworth BA Convergent Asymmetric Synthesis of Two Complex TRPV1 Antagonists. Org. Process Res. Dev 2011, 15, 1192–1200. [Google Scholar]; (c) MacKinnon CH; Lau K; Burch JD; Chen Y; Dines J; Ding X; Eigenbrot C; Heifetz A; Jaochico A; Johnson A; Kraemer J; Kruger S; Krülle TM; Liimatta M; Ly J; Maghames R; Montalbetti CA; Ortwine DF; Pérez-Fuertes Y; Shia S; Stein DB; Trani G; Vaidya DG; Wang X; Bromidge SM; Wu LC; Pei Z Structure-based Design and Synthesis of Potent Benzothiazole Inhibitors of Interleukin-2 Inducible T Cell Kinase (ITK). Bioorg. Med. Chem. Lett 2013, 23, 6331–6335. [DOI] [PubMed] [Google Scholar]; (d) Kohn TJ; Du X; Lai S; Xiong Y; Komorowski R; Veniant M; Fu Z; Jiao X; Pattaropong V; Chow D; Cardozo M; Jin L; Conn M; DeWolf WE; Kraser CF; Hinklin RJ; Boys ML; Medina JC; Houze J; Dransfield P; Coward P 5-Alkyl-2-urea-Substituted Pyridines: Identification of Efficacious Glucokinase Activators with Improved Properties. ACS Med. Chem. Lett 2016, 7, 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Talele TT The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- (10).(a) Charette AB; Molinaro C; Brochu C Catalytic Asymmetric Cyclopropanation of Allylic Alcohols with Titanium-TADDOLate: Scope of the Cyclopropanation Reaction. J. Am. Chem. Soc 2001, 123, 12168–12175. [DOI] [PubMed] [Google Scholar]; (b) Davies HML; Townsend RJ Catalytic Asymmetric Cyclopropanation of Heteroaryldiazoacetates. J. Org. Chem 2001, 66, 6595–6603. [DOI] [PubMed] [Google Scholar]; (c) Marcin LR; Denhart DJ; Mattson RJ Catalytic Asymmetric Diazoacetate Cyclopropanation of 1-Tosyl-3-vinylindoles. A Route to Conformationally Restricted Homotryptamines. Org. Lett 2005, 7, 2651–2654. [DOI] [PubMed] [Google Scholar]; (d) Chuprakov S; Kwok SW; Zhang L; Lercher L; Fokin VV Rhodium-Catalyzed Enantioselective Cyclopropanation of Olefins with N-Sulfonyl 1,2,3-Triazoles. J. Am. Chem. Soc 2009, 131, 18034–18035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Grimster N; Zhang L; Fokin VV Synthesis and Reactivity of Rhodium(II) N-Triflyl Azavinyl Carbenes. J. Am. Chem. Soc 2010, 132, 2510–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jin C; Decker AM; Huang X-P; Gilmour BP; Blough BE; Roth BL; Hu Y; Gill JB; Zhang XP Synthesis, Pharmacological Characterization, and Structure-Activity Relationship Studies of Small Molecular Agonists for the Orphan GPR88 Receptor. ACS Chem. Neurosci 2014, 5, 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Bajaj P; Sreenilayam G; Tyagi V; Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem., Int. Ed 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Fu L; Mighion JD; Voight EA; Davies HML Synthesis of 2,2,2,-Trichloroethyl Aryl- and Vinyldiazoacetates by Palladium-Catalyzed Cross-Coupling. Chem. - Eur. J 2017, 23, 3272–3275. [DOI] [PubMed] [Google Scholar]; (i) Moore EJ; Steck V; Bajaj P; Fasan R Chemoselective Cyclopropanation over Carbene Y-H Insertion Catalyzed by an Engineered Carbene Transferase. J. Org. Chem 2018, 83, 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Vargas DA; Khade RL; Zhang Y; Fasan R Biocatalytic Strategy for Highly Diastereo- and Enantioselective Synthesis of 2,3-Dihydrobenzofuran-Based Tricyclic Scaffolds. Angew. Chem., Int. Ed 2019, 58, 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wei B; Sharland JC; Lin P; Wilkerson-Hill SM; Fullilove FA; McKinnon S; Blackmond DG; Davies HML In Situ Kinetic Studies of Rh(II)-Catalyzed Asymmetric Cyclopropanation with Low Catalyst Loadings. ACS Catal. 2020, 10, 1161–1170. [Google Scholar]; (l) Nam D; Steck V; Potenzino RJ; Fasan R A Diverse Library of Chiral Cyclopropane Scaffolds via Chemoenzymatic Assembly and Diversification of Cyclopropyl Ketones. J. Am. Chem. Soc 2021, 143, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Doyle MP Catalytic Methods for Metal Carbene Transformations. Chem. Rev 1986, 86, 919–939 [DOI] [PubMed] [Google Scholar]; (b) Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev 1998, 98, 911–935. [DOI] [PubMed] [Google Scholar]; (c) Intrieri D; Carminati DM; Gallo E The Ligand Influence in Stereoselective Carbene Transfer Reactions Promoted by Chiral Metal Porphyrin Catalysts. Dalton Trans. 2016, 45, 15746–16048. [DOI] [PubMed] [Google Scholar]
- (12).Allouche EMD; Charette AB Cyclopropanation Reactions of Semi-stabilized and Non-stabilized Diazo Compounds. Synthesis 2019, 51, 3947–3963. [Google Scholar]
- (13).(a) Doyle MP; High KG; Oon S-M; Osborn AK Diazirines in Carbenoid Reactions Catalyzed by Rhodium(II) Carboxylates. Tetrahedron Lett. 1989, 30, 3049–3052. [Google Scholar]; (b) Regitz M; Maas G Diazo Compounds: Properties and Synthesis; Academic Press: London, 1996. [Google Scholar]; (c) Aggarwal VK; de Vicente J; Bonnert RV Catalytic Cyclopropanation of Alkenes Using Diazo Compounds Generated in Situ. A Novel Route to 2-Arylcyclopropylamines. Org. Lett 2001, 3, 2785–2788. [DOI] [PubMed] [Google Scholar]; (d) Fulton JR; Aggarwal VK; de Vicente J The Use of Tosylhydrazone Salts as a Safe Alternative for Handling Diazo Compounds and Their Applications in Organic Synthesis. Eur. J. Org. Chem 2005, 2005, 1479–1492. [Google Scholar]
- (14).(a) Zimmerman HE; Ignatchenko A Control of the Stereochemistry of Kinetic Protonation: Intramolecular Proton Delivery. J. Org. Chem 1999, 64, 6635–6645. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J; Quiñones N; Tomás-Gamasa M; Cabal M-P Intermolecular Metal-Free Cyclopropanation of Alkenes Using Tosylhydrazones. Eur. J. Org. Chem 2012, 2012, 2312–2317. [Google Scholar]; (c) Roda NM; Tran DN; Battilocchio C; Labes R; Ingham RJ; Hawkins JM; Ley SV Cyclopropanation Using Flow-Generated Diazo Compounds. Org. Biomol. Chem 2015, 13, 2550–2554. [DOI] [PubMed] [Google Scholar]; (d) Liu Z; Zhang X; Zanoni G; Bi X Silver-Catalyzed Cyclopropanation of Alkenes Using N-Nosylhydrazones as Diazo Surrogates. Org. Lett 2017, 19, 6646–6649. [DOI] [PubMed] [Google Scholar]; (e) Allouche EMD; Al-Saleh A; Charette AB Iron-Catalyzed Synthesis of Cyclopropanes by In Situ Generation and Decomposition of Electronically Diversified Diazo Compounds. Chem. Commun 2018, 54, 13256–13259. [DOI] [PubMed] [Google Scholar]; (f) Roy S; Das SK; Chattopadhyay B Cobalt(II)-based Metalloradical Activation of 2-(Diazomethyl)-pyridines for Radical Transannulation and Cyclopropanation. Angew. Chem., Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]; (g) Zhang Z; Yadagiri D; Gevorgyan V Light-Induced Metal-Free Transformations of Unactivated Pyridotriazoles. Chem. Sci 2019, 10, 8399–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shang Z-H; Zhang Z-X; Weng W-Z; Wang Y-F; Cheng T-W; Zhang Q-Y; Song L-Q; Shao T-Q; Liu K-X; Zhu Y-P A Metal- and Azide-free Oxidative Coupling Reaction for the Synthesis of [1,2,3]Triazolo[1,5-a]quinolines and their Application to Construct C-C and C-P Bonds, 2-Cyclopropylquinolines and Imidazo[1,5-a]quinolines. Adv. Synth. Catal 2021, 363, 490–496. [Google Scholar]
- (15).Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]
- (16).Hu Y; Lang K; Tao J; Marshall MK; Cheng Q; Cui X; Wojtas L; Zhang XP Next-Generation D2-Symmetric Chiral Porphyrins for Cobalt(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging. Angew. Chem., Int. Ed 2019, 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereo-retentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.