

Abstract
Objective
To identify the value of key differentially expressed genes (DEGs) regulated by differentially methylated regions (DMRs) in predicting the prognosis of human colon cancer.
Materials and Methods
RNA sequencing data and DNA methylation data of 455 colon adenocarcinoma (COAD) cases and 41 normal controls were downloaded from The Cancer Genome Atlas (TCGA). Gene Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed by the DAVID database. To identify the hub genes regulated by methylation, univariate Cox and multivariate Cox regression analyses were carried out. A nomogram based on the risk score was built to identify the power of the hub genes to predict prognosis in patients with colon cancer.
Results
A total of 133 DEGs regulated by DMRs were identified through analyzing RNA sequencing data and DNA methylation data from TCGA. GO functional enrichment and KEGG pathway enrichment analysis showed the genes involved in the initiation and progression of colon cancer. Univariate Cox regression analysis and multivariate Cox regression analysis focused on the seven hub genes (CDH4, CR2, KRT85, LGI4, NPAS4, RUVBL1 and SP140) associated with overall survival, the expression of which negatively correlated with their methylation level. The risk score and nomogram model showed that the hub genes served as potential biomarkers for the prognosis prediction of patients with colon cancer.
Conclusion
Our findings suggest that the DEGs regulated by DMRs are involved in the carcinogenesis and development of colon cancer, and the aberrantly methylated DEGs associated with overall survival of patients may be potential diagnostic and therapeutic targets for colon cancer.
Keywords: colon cancer, DNA methylation, gene expression regulation, prognosis prediction
Introduction
In recent years, the morbidity and mortality of colon cancer have increased rapidly, both being ranked fourth worldwide. Although surgery-based comprehensive treatments improve the prognosis of colon cancer, because of the lack of available means for early diagnosis, the mortality level remains high for patients with advanced-stage cancer. The carcinogenesis and development of colon cancer are very complicated, its origin lies in aberrant gene expression, and various factors that can change gene expression levels are involved in the development of the cancer. Hence, studying the specific biomarkers and therapeutic targets is of great value in improving the prognosis of colon cancer.
Accumulating evidence has validated that epigenetic modification can promote carcinogenesis and the development of colon cancer via regulating the expression of various genes. DNA methylation is an important epigenetic modification, with oncogenes or antioncogenes exhibiting unregulated expression if the gene-regulatory regions exhibit abnormal DNA methylation. For example, the oncogene CCAAT/enhancer-binding protein-beta (C/EBP-β), long interspersed nuclear element-1 (LINE-1), F2RL3 and AHRR undergo overexpression owing to hypomethylation in the promoter sites.1–4 Therefore, studying the methylation-regulated differentially expressed genes (DEGs) is necessary for understanding the mechanisms of cancer initiation and development. However, studies on the correlations between DNA methylation and the regulation of gene expression remain inadequate, and the potential value of correlations between differentially methylated regions (DMRs) and DEGs in predicting prognosis in human colon cancer still needs to be studied in depth.
In the past few decades, with the development of next generation sequencing technology and microarray platforms, accumulating DEGs and epigenetic alterations such as DMRs have been revealed by bioinformatics analysis. For instance, Liu et al validated that the DNA methyltransferase inhibitor guadecitabine (SGI-110) altered the expression of oncogenes or antioncogenes by regulating DNA methylation.5 Qu et al analyzed 57 patients with acute myeloid leukemia (AML) with normal karyotype using Illumina’s HumanMethylation 450K BeadChip platform, and showed that abnormal DNA methylation was altered significantly at enhancer regions and that the methylation levels at specific enhancers predicted overall survival of AML patients.6 However, there is still a lack of integrated analysis of the gene expression regulated by DNA methylation in human colon cancer. Similarly, studies on DNA methylation in predicting the prognosis of patients in large cohorts are deficient.
In the current study, we downloaded RNA sequencing data, DNA methylation data and clinical data on colon cancer from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) project.7 DEGs and DMRs were identified, and DEGs regulated by DMRs were screened by analyzing their correlations. For further study on the function and value of the genes in predicting prognosis, Gene Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed. Moreover, the correlations among methylation status, gene expression level and oversurvival of colon cancer patients were analyzed, and risk analysis of prognosis-related DMRs was performed, to determine potential biomarkers for diagnosis and predicting prognosis in colon cancer patients.
Materials and Methods
Data and Sources
The raw data, expression profiles and clinical information were downloaded from the TCGA-COAD project (https://cancergenome.nih.gov/).7 This dataset includes 455 COAD and 41 normal (non-tumor) samples. The COAD samples were randomly separated into two subsets with equal size: the training dataset (228 tumor/41 normal samples) and testing dataset (227 tumor/0 normal samples).
Identification of DEGs with Altered Methylation Status
The training dataset included colon cancer and normal samples, which were utilized to identify the DEGs with altered methylation status. In brief, DESeq28 was applied to identify DEGs by comparing colon cancer and normal samples.8 The Benjamini–Hochberg method was used to adjust the P-value. We selected DEGs under the condition of FDR <0.05 and |log2FC|>1. Then, differentially methylated sites were identified in the colon cancer group compared with the normal group by the Wilcoxon test. We retained DMRs with a P-value <0.05.9 Furthermore, we identified hypermethylated genes and hypomethylated genes based on the loci relating to the DMRs in the colon cancer and normal samples. The overlapping methylated DEGs were determined via Venn diagrams.
GO Functional Enrichment and KEGG Pathway Enrichment Analysis
To explore the function of DEGs regulated by methylation in the carcinogenesis and development of colon cancer, GO enrichment analysis was performed using the DAVID database (https://david.ncifcrf.gov/) and three categories, namely cellular component (CC), molecular function (MF) and biological process (BP), were analyzed.10 In addition, KEGG pathways were analyzed using KEGG Orthology-Based Annotation System 3.0 (http://kobas.cbi.pku.edu.cn/).11 Differences with P<0.05 were regarded as statistically significant.
Establishment of a Prognostic Model Based on Methylated DEGs
The associations between the expression level of each gene and the overall survival were evaluating by univariate Cox regression analysis in our training dataset. We performed univariate Cox regression using the survival package to obtain 32 genes related to overall survival. Then, a multivariate Cox regression analysis on the seven prognosis-related genes (CDH4, CR2, KRT85, LGI4, NPAS4, RUVBL1 and SP140) was used to construct a prognostic model. Hazard ratios (HRs) and 95% confidence intervals (CIs) were confirmed. Subsequently, survival analysis of the remaining genes was performed by the Kaplan–Meier method with the log-rank test; genes with log-rank P-value <0.05 were retained. The prognostic risk score was defined as follows:12
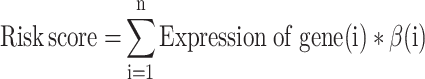 |
whereβ is the regression coefficient of the gene, which represents the contribution of the gene to the prognostic risk score. Based on the risk score, patients can be assigned to a high-risk or low-risk group according to the median cut-off of the prognosis risk scores. Then, Kaplan–Meier survival curves were calculated to compare survival and recurrence risk between the high- and low-risk groups. In addition, to evaluate the predictive accuracy and sensitivity of our prognostic model, time-dependent receiver operating characteristics (ROC) curve analysis within 1 year, 3 years and 5 years were built using the timeROC package.
Association Analysis of Risk Score and Clinical Features
The prognostic effects of various clinicopathological features, including age, gender and tumor stage, were evaluated by univariate Cox regression analysis and multivariate Cox regression analysis. Then, the nomogram was constructed based on the results of the multivariate Cox regression analyses of the risk score and clinicopathological features using the rms package.13
Statistical Analysis
The Kaplan–Meier method was applied to compare the overall survival between the high- and low-risk groups. R software, with the packages rms, survival and timeROC, was used for data analysis. HRs and 95% CIs were computed. Data were presented as mean ± standard deviation (SD). All tests were two sided, and P<0.05 was considered statistically significant.
Results
Selection of DEGs and DMRs in Colon Cancer Samples
The study procedures are shown in Figure 1A. We downloaded RNA-seq data from 455 colon cancer and 41 normal tissues, and screened 4180 up-regulated and 7821 down-regulated DEGs. The expression of DEGs is shown as a heatmap in Figure 1B. We also analyzed DMR data from TCGA database, and identified 373 hypermethylated genes and 571 hypomethylated genes. The expression of DMRs is shown as a heatmap in Figure 1C. Next, we analyzed the intersection of down-regulated genes and hypermethylated genes and found 95 genes that met the condition (Figure 1D); similarly, there were 38 genes in the intersection of up-regulated genes and hypomethylated genes (Figure 1E). As shown in Figure 1F, the expression levels of DEGs negatively correlated with the methylation levels. Therefore, a total of 133 genes met our requirements and were the candidate genes for further analysis.
Figure 1.

The landscape of gene expression and methylation in colon cancer. (A) Flowchart of the data analysis pipeline in this study. (B) Heatmap showing the differentially expressed genes in the colon cancer and normal groups. (C) Heatmap showing the differentially methylated regions associated with genes in the colon cancer and normal groups. (D) Venn diagram showing the down-regulated genes associated with hypermethylated sites in colon cancer. (E) Venn diagram showing the up-regulated genes associated with hypomethylated sites in colon cancer. (F) Scatter plot showing the correlation between differentially expressed genes and differentially methylated regions in colon cancer.
Functional and Pathway Enrichment Analysis for Candidate Methylated Genes
To understand the potential biological function of the 133 candidate methylated genes, GO functional and KEGG pathway enrichment analysis was performed. In total, 17 enriched GO terms in biological process (BP) and six terms in molecular function (MF) were identified (Figure 2A). From the results of GO analysis, we found that the candidate genes were mainly enriched in cancer-associated functions, such as xenobiotic glucuronidation, cellular hormone metabolic and second-messenger-mediated signaling. In addition, 10 pathways were found to be significantly enriched from the results of KEGG pathway enrichment analysis (Figure 2B). “Drug metabolism–cytochrome P450”, “Complement and coagulation cascades” and “Chemical carcinogenesis” are involved in the carcinogenesis and development of colon cancer based on previous reports.14–16 Therefore, the candidate methylated genes participate in colon cancer.
Figure 2.

GO and KEGG pathways enrichment based on candidate methylated genes. (A) GO analysis of the differentially methylated regions (DMRs) associated with differentially expressed genes (DEGs) in colon cancer. (B) KEGG analysis of the differentially methylated regions associated with DEGs in colon cancer. The size of the solid circle indicates the number of genes.
Identification of Key Methylated DEGs Associated with Poor Prognosis
The univariate Cox regression analysis confirmed 32 genes that were significantly correlated with prognosis. Subsequently, the multivariate Cox regression analysis focused on seven genes, which were CDH4, CR2, KRT85, LGI4, NPAS4, RUVBL1 and SP140 (Table 1). Next, we selected the seven genes for further study. The expression levels of the seven genes in cancer and normal tissues is shown in Figure 3A. The expression of CDH14, KRT85 and RUVBL1 was significantly up-regulated, while that of CR2, LGI4, NPAS4 and SP140 was remarkably down-regulated in the cancer tissues (n=455) compared with the normal tissues (n=41). The methylation levels of these seven genes in cancer and normal tissues are shown in Figure 3B. The methylation levels of CDH14, CR2, LGI4, NPAS4 and SP140 were markedly up-regulated, while KRT85 and RUVBL1 were remarkably down-regulated in the cancer tissues (n=455) compared with the normal tissues (n=41). Kaplan–Meier survival analysis showed that the expression level of RUVBL1 significantly correlated with the overall survival rate of colon cancer patients (Figure 3C). Collectively, methylated RUVBL1 was related to prognosis in colon cancer.
Table 1.
Multivariate Cox Regression Analysis of Clinical Factors and Hub Genes
| Univariate Cox | Multivariate Cox | |||||
|---|---|---|---|---|---|---|
| Log (HR) | 95% CI | P | Log (HR) | 95% CI | P | |
| Age | 0.1012 | −0.5091; 0.7115 | n.s. | 0.2830 | −0.3472; 0.9133 | n.s. |
| Gender | 0.0518 | −0.4836;, 0.5873 | n.s. | 0.1129 | −0.4392; 0.6649 | n.s. |
| Tumor stage | 0.9370 | 0.0885;, 1.7854 | 0.03 | 1.2712 | 0.3904; 2.1520 | 0.004 |
| Risk score | 0.5552 | 0.3628, 0.7477 | <0.001 | 0.6304 | 0.4274; 0.8335 | <0.001 |
| CR2 | 0.0589 | 0.0275, 0.0903 | <0.001 | 0.2046 | 0.0236; 0.3856 | 0.03 |
| KRT85 | 2.0704 | 0.9047, 3.2361 | <0.001 | 2.5114 | 1.0993; 3.9234 | <0.001 |
| LGI4 | 0.1448 | 0.0054;, 0.2842 | 0.04 | 0.1983 | 0.0131; 0.3835 | 0.04 |
| NPAS4 | 3.8793 | 0.4403, 7.3182 | 0.03 | −19.9721 | −38.0522, −1.8921 | 0.03 |
| SP140 | 0.4395 | 0.2049; 0.6742 | <0.01 | 0.7001 | 0.1804; 1.2197 | 0.008 |
| RUVBL1 | 0.0830 | 0.0113; 0.1547 | 0.02 | 0.1327 | 0.0267; 0.2388 | 0.01 |
| CDH4 | 2.4616 | 1.1367; 3.7865 | <0.001 | 1.9639 | 0.1010; 3.8268 | 0.04 |
Figure 3.

The expression and methylation profiles of the selected genes differ between the colon cancer and normal groups. The boxplots show the expression level (A) and methylation level (B) of the selected genes (CDH4, CR2, KRT85, LG14, NPAS4, RUVBL1 and SP140) in the colon cancer and normal groups. (C) Kaplan–Meier survival curve for the expression of RUVBL1. Data are presented as the mean ± SD. *P<0.05. **P<0.01.
Construction of the Prognostic Methylation Model for Patients with Colon Cancer
To estimate the prognosis of the key methylated DEGs in colon cancer, a risk score was built. We divided 228 colon cancer samples into high-risk (n=114) and low-risk groups (n=114). In the training cohort, patients in the high-risk group had a shorter overall survival than patients in the low-risk group (Figure 4A and B). The heatmap showed the expression levels of the seven DEGs. With an increased risk score in patients with colon cancer, the levels of SP140, CR2, KRT85, LGI4, RUVBL1 and CDH4 were obviously increased, whereas the level of NPAS4 was decreased. In addition, an ROC model was built, and we found that the AUC values for 1-, 3- and 5-year overall survival were greater than 0.5 (Figure 4C). Similarly, in validating cohort, patients in the high-risk group also had a shorter overall survival than patients in the low-risk group (Figure 4D and E). The heatmap showed that the levels of SP140 and NPAS4 were obviously increased, while CR2, KRT85 and LGI4 were decreased as the risk score of patients with colon cancer increased. The risk score model also achieved prognostic prediction power, with an AUC of 0.57 for 5-year overall survival (Figure 4F). Therefore, these results reveal that this prognostic prediction model has good potential for application in clinical practice.
Figure 4.

Risk analysis of the selected genes in colon cancer. (A) Distribution of risk score, survival status and expression level in the training dataset. (B) Kaplan–Meier survival curve for risk score in the training dataset. (C) Time-dependent ROC analysis of 1-year (orange), 3-year (blue) and 5-year (red) overall survival in the training dataset using the selected genes. (D) Distribution of risk score, survival status and expression level in the testing dataset. (E) Kaplan–Meier survival curve for risk score in the testing dataset. (F) Time-dependent ROC analysis of 1-year (orange), 3-year (blue) and 5-year (red) overall survival in the testing dataset using the selected genes.
Nomogram Analysis for Prognosis Prediction
We investigated whether the prognostic model was independent of other clinical properties, using multivariate Cox regression analysis. We found that the tumor stage and risk score correlated with poor survival (Figure 5A). Then, we constructed a simple-to-use nomogram based on risk score and clinical characteristics, such as gender, age at diagnosis and pathological stage of patients with colon cancer (Figure 5B). The nomogram demonstrated that the risk score for the prognostic model showed good performance in predicting the 1-, 3- and 5-year survival rates of patients with colon cancer.
Figure 5.

The risk score is a good predictor of 3- and 5-year overall survival in patients with colon cancer. (A) Multivariate Cox regression analysis comparing the independent prognostic factors for overall survival of patients with colon cancer. (B) Nomogram for predicting clinical outcomes with risk score.
Discussion
Many reports have validated that epigenetic regulation is involved in the carcinogenesis and development of cancers, and DNA methylation, which is the most common form of epigenetic modification, plays an important role in the regulation of gene expression. The alteration of DNA methylation in the gene promoter region changes the expression level of the gene. In general, hypermethylation inhibits gene expression, whereas hypomethylation promotes gene expression.17–19 DNA methylation alterations are found in many patients with colon cancer, for example, SST1 pericentromeric repeats exhibited hypomethylation, which resulted in the mutation of TP53, and the mutated TP53 was associated with genome damage, which was related to the tumorigenesis and development of colon cancer.20 Similarly, hypermethylation appeared in the promoter of antioncogenes SFPR1, SFPR2 and WIF1, leading to down-regulated expression of genes, inhibition of gene function, activation of the Wnt/β-catenin signal pathway and promotion of colon cancer.21 ADHFEI is also an antioncogene; the hypermethylation of ADHFEI promotes the proliferation of colon cancer cells via regulating cell-cycle progression.22 The abnormal methylation of the above genes predicted poor prognosis of colon cancer. However, the systematic analysis of the correlation between DEGs and DMRs was still deficient. Therefore, it is necessary to further study the gene expression regulated by DNA methylation to provide potential therapeutic targets.
Our study also validated that a lot of DEGs and DMRs existed in colon cancer tissues. Based on the methylation pattern of regulation, we focused on 133 genes, 95 of which were down-regulated but hypermethylated and 38 of which were up-regulated but hypomethylated. Similarly, a negative regulated correction was found between DEGs and DMRs. DAVID gene enrichment analysis and KEGG pathway enrichment analysis are useful for predicting the function and pathway of a gene set. From the results of GO functional analysis, we found that the 133 DEGs regulated by DMRs played an important role in carcinogenesis and the development of colon cancer. In the biological process (BP) module of GO enrichment analysis, as many as nine terms were related to metabiotic regulation. Many reports show that the retinoic acid metabolic process is associated with colonic tumorigenesis and metastasis; for example, the retinoic acid metabolizing enzymes CYP26B1, LRAT and CYP26A1 are overexpressed in colorectal cancer tissues, and LRAT and CYP26B1 are significantly associated with the prognosis of colorectal cancer.23 Xenobiotic metabolic processes are also involved in the carcinogenesis and development of cancer, because the colonic epithelium is exposed to various compounds from the diet; these compounds can be metabolized to procarcinogens, which can lead to colon cancer.24,25 Similarly, in the cellular companion (CC) module, there were four terms relating to molecular binding. Some reports found that retinoid acid binding receptor inhibited cancer cell apoptosis by regulating the miR-22/NUR77 axis.25,26 From the results of KEGG pathway enrichment analysis, we found that the genes were mainly enriched in molecular metabolism, and drug, retinoid and porphyrin metabolism has been shown to be involved in unregulated tumor cell metabolism, and resulted in chemotherapy resistance.27
To screen the genes that are associated with prognosis in colon cancer patients, univariate and multivariate Cox regression analysis was performed to test the independent significance of different factors. We found that seven genes from the candidate genes were significantly related to the overall survival of colon cancer patients. Meanwhile, the expression levels of the seven genes were negatively related to their methylation levels, which proved that the expression of the genes was regulated by methylation. From previous reports, we discovered that the seven genes are involved in the carcinogenesis and development of various cancers. For example, in glioblastoma, CDH4 played the role of an oncogene by proliferating and infiltrating the brain parenchyma.28 Similarly, CDH4 acted as an oncogene by initiating and maintaining epigenetic suppression of multiple tumor suppressor genes in colorectal cancer.29 In human basal cell carcinoma, the abnormal expression of KRT85 resulted in cancer stem cell exhaustion.30 LGI4 interacts with ADAM to promote the proliferation and differentiation of neuronal precursors and adipocytes. In breast cancer, the interactions between LGI4 and ADAM are also related to carcinogenesis.31 In the present study, although the abnormal expression of the seven genes could be seen as independent risk factors for overall survival in colon cancer patients, the expression levels of the genes were not related to the overall survival rate of patients, except for RUVBL1. RUVBL1 is an oncogene that is related to the prognosis of patients with various cancers, and it promotes carcinogenesis through regulating the Wnt/β-catenin signal pathway.32–34 Therefore, RUVBL1 has a potential value in prognosis and as a therapeutic target for cancer patients.
Unregulated DNA methylation is an important factor in carcinogenesis, and it can be used for diagnosis and for predicting prognosis in cancer patients. In our study, we built a risk score to estimate the power of the seven hub genes in predicting the prognosis of colon patients. We found that the seven hub genes had a good performance in prognosis prediction, which illustrated that aberrant methylation in cancer tissues could be used as a diagnostic marker and therapeutic target. Next, our nomogram showed that the risk score was a good predictor for 1-, 3- or 5-year overall survival in colon cancer patients; it demonstrated the correlation between the risk score and clinical features, including age and tumor stage. However, these results should be validated by further studies.
Finally, some limitations in this study should be noted. First, our RNA sequencing, DNA methylation and clinical data were obtained from TCGA database, but no clinical samples were used for validating the results. Therefore, it is necessary to select large clinical samples for testing the effectiveness of the biomarkers associated with the hub genes in this study. Second, we screened the biomarkers using the methods of statistical and bioinformatics analysis, but not biological experiments. Therefore, the mechanisms of the biomarkers are still unknown, and biological experiments are needed to understand the roles of the candidate markers in the carcinogenesis and development of colon cancer. Third, there is potential bias in this study, because we did not analyze some important clinical information, especially regarding treatment factors (such as operation, chemotherapy and radiotherapy). Therefore, prospective studies and multicenter clinical trials are necessary for further validation.
In conclusion, we constructed a novel prognostic model consisting of seven DEGs regulated by methylation (CDH4, CR2, KRT85, LGI4, NPAS4, RUVBL1 and SP140). This model can be used for the prognostic prediction of patients with colon cancer. The DEGs regulated by methylation could serve as independent risk factors in the clinical treatment of colon cancer.
Acknowledgment
This study is supported by grants from Guangzhou Medical and Health Research Project (no. 2018A010007, 20201A010005).
Highlights
RNA sequencing data and DNA methylation data of COAD containing clinical information from TCGA were analyzed.
113 DEGs regulated by DMRs participated in tumorigenesis and the development of COAD.
The hub genes we screened had potential value as prognostic and therapeutic targets for patients with colon cancer.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
- 1.Xiong L, Wu F, Wu Q, et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat Commun. 2019;10(1):335. doi: 10.1038/s41467-018-08245-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hur K, Cejas P, Feliu J, et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut. 2014;63(4):635–646. doi: 10.1136/gutjnl-2012-304219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Touzart A, Boissel N, Belhocine M, et al. Low level CpG island promoter methylation predicts a poor outcome in adult T-cell acute lymphoblastic leukemia. Haematologica. 2020;105:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alhamdow A, Lindh C, Hagberg J, et al. DNA methylation of the cancer-related genes F2RL3 and AHRR is associated with occupational exposure to polycyclic aromatic hydrocarbons. Carcinogenesis. 2018;39(7):869–878. doi: 10.1093/carcin/bgy059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M, Zhang L, Li H, et al. Integrative epigenetic analysis reveals therapeutic targets to the DNA methyltransferase inhibitor guadecitabine (SGI-110) in hepatocellular carcinoma. Hepatology. 2018;68(4):1412–1428. doi: 10.1002/hep.30091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qu Y, Siggens L, Cordeddu L, et al. Cancer-specific changes in DNA methylation reveal aberrant silencing and activation of enhancers in leukemia. Blood. 2017;129(7):e13–e25. doi: 10.1182/blood-2016-07-726877 [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Jensen MA, Zenklusen JC. A practical guide to the cancer genome atlas (TCGA). Methods Mol Biol. 2016;1418:111–141. [DOI] [PubMed] [Google Scholar]
- 8.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi A, Park IH, Wen B, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41(12):1350–1353. doi: 10.1038/ng.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gene Ontology Consortium. The gene ontology project in 2008. Nucleic Acids Res. 2008;36:D440–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Draghici S, Khatri P, Tarca AL, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17(10):1537–1545. doi: 10.1101/gr.6202607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SK, Kim SY, Kim JH, et al. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol Oncol. 2014;8(8):1653–1666. doi: 10.1016/j.molonc.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S, Tong YX, Zhang XH, et al. A novel and validated nomogram to predict overall survival for gastric neuroendocrine neoplasms. J Cancer. 2019;10(24):5944–5954. doi: 10.7150/jca.35785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Yang J, Edin ML, et al. Targeted metabolomics identifies the cytochrome P450 monooxygenase eicosanoid pathway as a novel therapeutic target of colon tumorigenesis. Cancer Res. 2019;79(8):1822–1830. doi: 10.1158/0008-5472.CAN-18-3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing S, Wang Y, Hu K, Wang F, Sun T, Li Q. WGCNA reveals key gene modules regulated by the combined treatment of colon cancer with PHY906 and CPT11. Biosci Rep. 2020;40(9):BSR20200935. doi: 10.1042/BSR20200935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roscilli G, Marra E, Mori F, et al. Carnitines slow down tumor development of colon cancer in the DMH-chemical carcinogenesis mouse model. J Cell Biochem. 2013;114(7):1665–1673. doi: 10.1002/jcb.24508 [DOI] [PubMed] [Google Scholar]
- 17.Parrella P. the value of epigenetic biomarkers in breast cancer. Biomark Med. 2018;12(9):937–940. doi: 10.2217/bmm-2018-0187 [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Bettington ML, Walker NI, et al. CpG Island methylation in sessile serrated adenomas increases with age, indicating lower risk of malignancy in young patients. Gastroenterology. 2018;155(5):1362–1365. doi: 10.1053/j.gastro.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 19.Hao X, Luo H, Krawczyk M, et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A. 2017;114(28):7414–7419. doi: 10.1073/pnas.1703577114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuelsson JK, Dumbovic G, Polo C, et al. Helicase lymphoid-specific enzyme contributes to the maintenance of methylation of SST1 pericentromeric repeats that are frequently demethylated in colon cancer and associate with genomic damage. Epigenomes. 2017;1(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Fu J, Bi H, et al. DNA methylation of SFRP1, SFRP2, and WIF1 and prognosis of postoperative colorectal cancer patients. BMC Cancer. 2019;19(1):1212. doi: 10.1186/s12885-019-6436-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu YH, Ma S, Zhang XN, et al. Hypermethylation of ADHFE1 promotes the proliferation of colorectal cancer cell via modulating cell cycle progression. Onco Targets Ther. 2019;12:8105–8115. doi: 10.2147/OTT.S223423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown GT, Cash BG, Blihoghe D, Johansson P, Alnabulsi A, Murray GI. the expression and prognostic significance of retinoic acid metabolising enzymes in colorectal cancer. PLoS One. 2014;9(3):e90776. doi: 10.1371/journal.pone.0090776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nogacka AM, Gomez-Martin M, Suarez A, Gonzalez-Bernardo O, de Los Reyes-gavilan CG, Gonzalez S. Xenobiotics formed during food processing: their relation with the intestinal microbiota and colorectal cancer. Int J Mol Sci. 2019;20(8):2051. doi: 10.3390/ijms20082051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu JC, Tsai ML, Lai CS, et al. Polymethoxyflavones prevent benzo[a]pyrene/dextran sodium sulfate-induced colorectal carcinogenesis through modulating xenobiotic metabolism and ameliorate autophagic defect in ICR mice. Int J Cancer. 2018;142(8):1689–1701. doi: 10.1002/ijc.31190 [DOI] [PubMed] [Google Scholar]
- 26.Hu Y, French SW, Chau T, et al. RARbeta acts as both an upstream regulator and downstream effector of miR-22, which epigenetically regulates NUR77 to induce apoptosis of colon cancer cells. FASEB J. 2019;33(2):2314–2326. doi: 10.1096/fj.201801390R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kralova J, Kolar M, Kahle M, et al. Glycol porphyrin derivatives and temoporfin elicit resistance to photodynamic therapy by different mechanisms. Sci Rep. 2017;7:44497. doi: 10.1038/srep44497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceresa D, Alessandrini F, Bosio L, et al. Cdh4 down-regulation impairs in vivo infiltration and malignancy in patients derived glioblastoma cells. Int J Mol Sci. 2019;20(16):4028. doi: 10.3390/ijms20164028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia L, Huang W, Bellani M, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell. 2017;31(5):653–668. doi: 10.1016/j.ccell.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan HJ, Benketah A, Olivero C, et al. Hair follicle differentiation-specific keratin expression in human basal cell carcinoma. Clin Exp Dermatol. 2020:45:417–425. [DOI] [PubMed] [Google Scholar]
- 31.Caselli N, Intonti F, Riboli F, Gurioli M. Engineering the mode parity of the ground state in photonic crystal molecules. Opt Express. 2014;22(5):4953–4959. doi: 10.1364/OE.22.004953 [DOI] [PubMed] [Google Scholar]
- 32.Mello T, Materozzi M, Zanieri F, et al. Liver haploinsufficiency of RuvBL1 causes hepatic insulin resistance and enhances hepatocellular carcinoma progression. Int J Cancer. 2020;146:3410–3422. [DOI] [PubMed] [Google Scholar]
- 33.Chen J, Liu G, Wu Y, et al. CircMYO10 promotes osteosarcoma progression by regulating miR-370-3p/RUVBL1 axis to enhance the transcriptional activity of beta-catenin/LEF1 complex via effects on chromatin remodeling. Mol Cancer. 2019;18(1):150. doi: 10.1186/s12943-019-1076-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Tong X, Xu Y, et al. Functional genetic variants of RUVBL1 predict overall survival of Chinese patients with epithelial ovarian cancer. Carcinogenesis. 2019;40(10):1209–1219. doi: 10.1093/carcin/bgz092 [DOI] [PubMed] [Google Scholar]