

Abstract
Background/Objective:
Proteus syndrome, caused by a mosaic activating AKT1 variant, typically presents in toddlers with progressive, asymmetric overgrowth of the skin and bones. We aimed to define the spectrum of dermatologic disease in individuals with genetically confirmed Proteus syndrome.
Methods:
We conducted a retrospective review of records from dermatologic examinations of individuals evaluated at the NIH with a molecular diagnosis of Proteus syndrome. The types, prevalence, and localization of dermatologic findings were assessed.
Results:
Fifty-one individuals (29 males, 22 females, mean age: 9 years) with clinical features of Proteus syndrome had the mosaic c.49G>A, p.Glu17Lys AKT1 variant. Fifty (98%) had at least one cutaneous feature constituting current clinical diagnostic criteria, including vascular malformations in 42 (82%), epidermal nevus in 41 (80%), volar cerebriform connective tissue nevi in 34 (67%), and adipose dysregulation in 30 (59%). Forty-nine (96%) had at least one dermatologic finding not included within the diagnostic criteria, including confluent volar skin-colored to hypopigmented papules or nodules (n=33, 65%), papules or nodules on the digits or face (n=27, 53%), and nonlinear epidermal nevi (n=15, 29%). Other frequently observed features include nail changes (n=28, 55%), hyperpigmented macules (n=27, 53%), patchy dermal hypoplasia (n=18, 35%), gingival/oral mucosal overgrowth (n=17, 33%), hypopigmented macules (n=16, 31%), dental enamel changes (n=9, 18%), acrochordons (n=6, 12%), and lingual overgrowth (n=4, 8%).
Conclusions:
The range of mucocutaneous features occurring in Proteus syndrome is broader than previously considered. These observations may assist in earlier diagnosis and management and provide novel insights regarding the pathogenesis of the condition.
Keywords: Genetic disease/mechanisms, genodermatoses, neoplasms – benign, skin signs of systemic disease, vascular malformations
Introduction
Proteus syndrome is a rare condition caused by a somatic activating variant in AKT1, a gene that mediates cell cycle progression, cell survival, and apoptosis. Affected individuals present with progressive and asymmetric overgrowth in the skin, soft tissue, bones, and various organs. The condition may be diagnosed clinically by specific diagnostic criteria,1 which include an array of dermatologic features, such as cerebriform connective tissue nevi (CTN), epidermal nevus, subcutaneous adipose tissue dysregulation, and vascular malformations.2 Some cutaneous findings, most notably epidermal nevus and hypertrichotic patches, may follow the lines of Blaschko, indicating mosaicism.3 In recent years, molecular testing for an AKT1 activating variant has been increasingly used to confirm the diagnosis,4 as other overgrowth disorders, such as PIK3CA-related segmental overgrowth and Klippel-Trénaunay syndrome, may present with similar constellations of clinical findings.
Accurate diagnosis of Proteus syndrome is crucial for appropriate screening and management, particularly given the rise of targeted therapy for the disorder.5 However, this can be challenging in patients who do not present with severe overgrowth or prominent skin findings. Many cutaneous features that are not included in the diagnostic criteria are not well-described in the existing literature. Furthermore, the frequencies of well-known dermatologic features of Proteus syndrome have yet to be defined in a large, genetically verified cohort. We sought to investigate cutaneous features associated with genetically confirmed Proteus syndrome to characterize the spectrum of associated dermatologic findings and facilitate early recognition.
Methods
Individuals were evaluated for overgrowth syndromes at the National Institutes of Health (NIH) Clinical Center in Bethesda, Maryland between November 5, 1996 and June 13, 2019. Each individual, or their parents, provided consent according to protocol 94-HG-0132, which was approved by the National Human Genome Research Institute Institutional Review Board at the NIH. As a component of evaluation, individuals underwent total-body dermatologic examination by a board-certified dermatologist (T.N.D.). Those with clinical features of Proteus syndrome underwent genetic testing using a custom restriction-enzyme assay of affected tissue to determine presence of the mosaic c.49G>A, p.(Glu17Lys) AKT1 variant, which is pathogenic for Proteus syndrome. A retrospective review of written medical records and clinical photographs from total-body dermatologic examinations was conducted on individuals with the confirmed AKT1 variant. The appearance, prevalence, and localization of mucocutaneous findings, including features which have been previously described as well as those which may be unusual or distinctive, were assessed.
Results
Fifty-one individuals with clinical features of Proteus syndrome were found to have the mosaic c.49G>A, p.(Glu17Lys) variant of AKT1. This cohort included 29 males and 22 females with a mean age of 9 years at first visit. In the majority of individuals, the pathogenic AKT1 variant was identified by testing biopsies of affected skin.
Table 1 summarizes the mucocutaneous features of the 51 individuals. At least one cutaneous feature constituting the current clinical diagnostic criteria for Proteus syndrome1 was present in 50 (98%) individuals, including vascular malformations in 42 (82%), epidermal nevus in 41 (80%), cerebriform CTN in 34 (67%), and adipose tissue dysregulation in 30 (59%). All cerebriform CTN (in contrast to some of the non-cerebriform CTN described below) were on the soles or, less commonly, the palms. Twenty-eight individuals had lipomas or other manifestations of increased subcutaneous fatty tissue, while nine had partial lipohypoplasia.
Table 1:
Dermatologic findings in 51 individuals with genetically confirmed Proteus syndrome
| Mucocutaneous feature | Number of individuals (out of 51) | % of total |
|---|---|---|
| Connective tissue nevus | 50 | 98 |
| Cerebriform connective tissue nevus | 34 | 67 |
| Confluent papules or nodules (on palms or soles) | 33 | 65 |
| Discrete papules or nodules (on digits, eyelids, perinasal skin, lip) | 27 | 53 |
| Vascular malformation | 42 | 82 |
| Capillary malformation | 23 | 45 |
| Venous malformation | 24 | 47 |
| Capillary-venous malformation | 15 | 29 |
| Epidermal nevus | 41 | 80 |
| Linear/Blaschkoid | 30 | 59 |
| Non-linear | 15 | 29 |
| Subcutaneous adipose dysregulation | 30 | 59 |
| Increased subcutaneous fatty tissue (including lipoma) | 28 | 55 |
| Partial lipohypoplasia | 9 | 18 |
| Patchy dermal hypoplasia | 18 | 35 |
| Hyperpigmented macules | 27 | 53 |
| Café au lait macules | 9 | 18 |
| Hypopigmented macule | 16 | 31 |
| Nail changes | 28 | 55 |
| Brachyonychia | 9 | 18 |
| Nail ridging | 5 | 10 |
| Onychoschizia | 4 | 8 |
| Leukonychia | 4 | 8 |
| Single nail onycholysis | 3 | 6 |
| Tooth aberrancies | 12 | 24 |
| Enamel hypoplasia or discoloration | 9 | 18 |
| Asymmetric tooth size | 7 | 14 |
| Dental pitting | 2 | 4 |
| Large teeth | 1 | 2 |
| Conical teeth | 1 | 2 |
| Papules, nodules, or overgrowth of gingiva/oral mucosa | 17 | 33 |
| Papules, nodules, or overgrowth of tongue | 4 | 8 |
| Acrochordons | 6 | 12 |
Forty-nine (96%) individuals had at least one dermatologic finding not included within the current diagnostic criteria. Thirty-three (65%) individuals had plaques on the palms or soles comprised of confluent hypopigmented papules or nodules; of these individuals, 12 did not have cerebriform CTN elsewhere. These non-cerebriform CTN progressed to cerebriform CTN over time (Figure 1A, B). Isolated skin-colored to hypopigmented, firm papules and nodules on the digits, eyelids, perinasal skin, or lip (Figure 2A, B) were seen in 27 individuals (53%), including 15 who did not have cerebriform CTN on palms or soles. Fifteen (29%) individuals had a nonlinear epidermal nevus with morphologies including ovoid, phylloid, and amorphous (Figure 3A). Two individuals had a thick epidermal nevus with a papillomatous appearance with accompanying terminal hair growth (Figure 3B), and one individual had a nevus sebaceus on the scalp.
Figure 1:
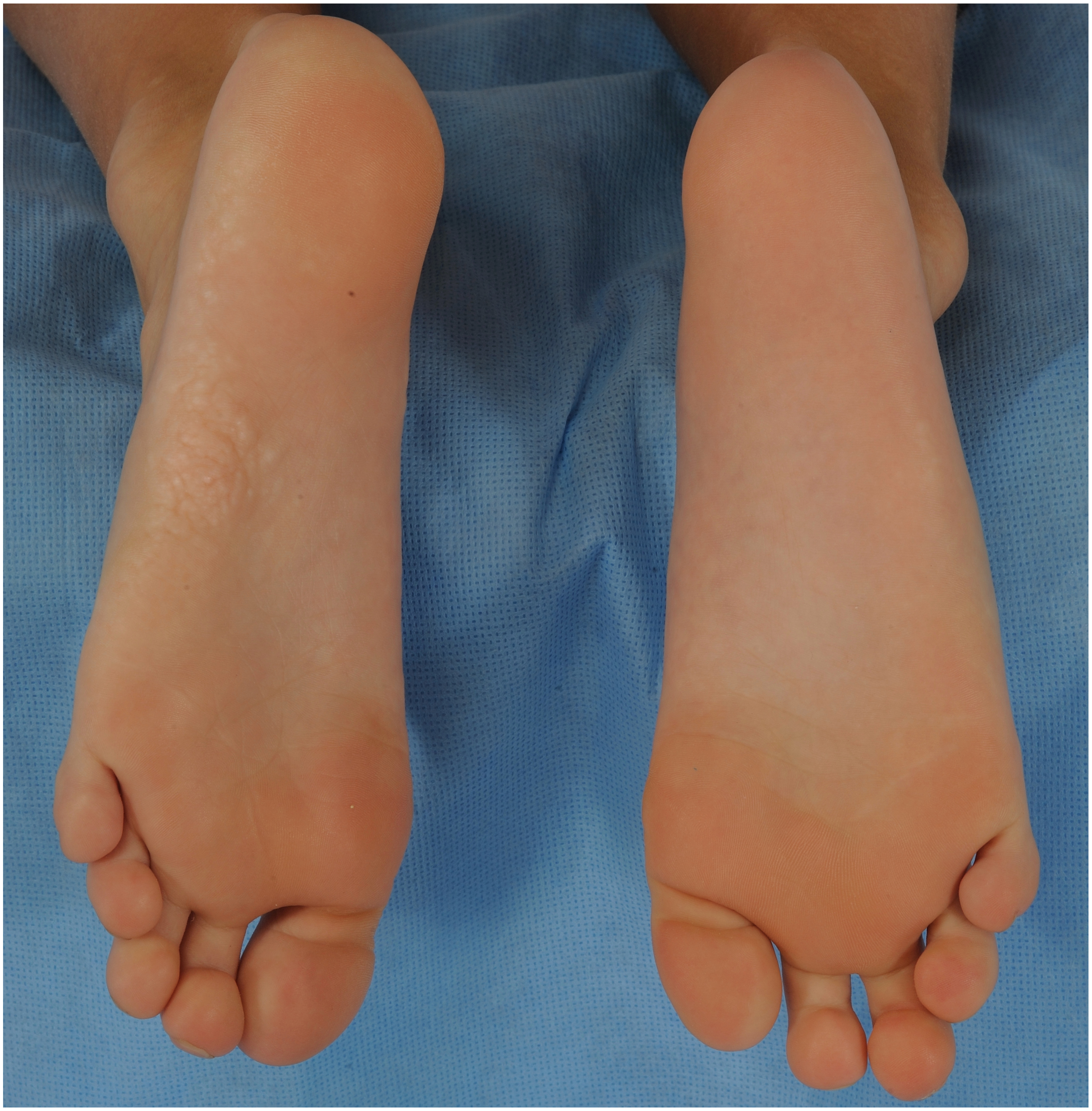

Development of the cerebriform connective tissue nevus in Proteus syndrome. A. A 5-year-old boy has a plaque on his left sole consisting of confluent, slightly hypopigmented papules. B. At the age of 13 years, the plaque on the left foot has expanded and developed into a characteristic cerebriform connective tissue nevus, while the sole of the right foot has developed a plaque with a few areas showing the beginnings of cerebriform morphology.
Figure 2:


Papular and nodular connective tissue nevi in Proteus syndrome. A. The only lesions of a connective tissue nevus in this 14-year-old girl were a papule on the left fourth toe and a nodule on the left fifth toe. Biopsy of the nodule confirmed connective tissue nevus and the presence of the AKT1 variant. B. Firm, skin-colored papules and nodules on the lower eyelid, perinasal region, and the lower vermilion border in an 8-year-old boy.
Figure 3:


Variations in appearance of epidermal nevus in Proteus syndrome. A. Verrucous nonlinear epidermal nevus on the back of the right hand, with a surgical scar. B. Epidermal nevus on the lateral trunk with a thick, papillomatous or acrochordon-like appearance and terminal hair growth in a 17-year-old boy.
Twenty-seven (53%) individuals had hyperpigmented macules and 16 (31%) had hypopigmented macules; seven individuals had both cutaneous hyperpigmentation and hypopigmentation (Figure 4A, B). The morphologies of these lesions included round, ovoid, linear, and mottled, and their borders ranged from ill-defined to sharply demarcated. Twenty-three individuals had capillary malformations; two individuals had a nevus anemicus, both of which were adjacent to a telangiectatic nevus. Patchy dermal hypoplasia was present in 35% of individuals (n=18). Each individual with dermal hypoplasia had some variation of CTN or dermal growth elsewhere on the body.
Figure 4:


Hyper- and hypopimented macules in a 12-year-old boy with Proteus syndrome. A. On the right lower back, there is a light brown macule with irregular borders. B. On the right upper back of the same child, there is a faint hypopigmented macule with a mottled pattern.
Nail changes were observed in 55% of individuals (n=28). Nine individuals had brachyonychia, including seven with this manifestation on a single digit, one with involvement of two separate digits on opposite feet, and one with involvement of all toenails of both feet. Longitudinal or horizontal ridging of nails was present in five individuals; three had involvement of multiple nails and two had this finding localized to a single nail. Four individuals had onychoschizia, including two with involvement of a single nail and two with involvement of multiple nails. Single nail onycholysis was observed in three individuals, one of whom also had associated brachyonychia in the same digit which likely contributed to upward displacement of nail. Four individuals had fingernail leukonychia, including two with Terry’s nails.
Dental changes were observed in 12 individuals (24%), including nine with enamel hypoplasia or discoloration (Figure 5), seven with asymmetry of tooth size, two with dental pitting, one with generalized large teeth causing dental crowding, and one with conical teeth. Papules, nodules, and asymmetric overgrowth of oral mucosa were present in 17 individuals (33%). Two individuals had asymmetric overgrowth of the tongue, one had a firm white lingual papule, and one had lingual papillary hypertrophy. Numerous acrochordons were observed in 12% (n=6) of the cohort, including three individuals with asymmetric axillary acrochordons which were more numerous on the overgrown side of the body and two with acrochordon-like structures within an epidermal nevus (Figure 3B). The majority (5/6, 83%) were under 18 years of age when this finding was documented. One to two individuals each were noted to have other cutaneous hamartomas, including nevus lipomatosus superficialis, dermoid cyst, and neurofibroma.
Figure 5:

Asymmetric gingival overgrowth, aberrancies in tooth shape, and abnormal dental enamel layer with exposed yellowish dentin in a 5-year-old girl with Proteus syndrome.
Discussion
Almost all individuals with genetically confirmed Proteus syndrome in this cohort had cutaneous findings included in the current diagnostic criteria, but importantly, also had dermatologic findings not included in these criteria. We observed both cerebriform and non-cerebriform morphologies of CTN. The palms and soles commonly had confluent, hypopigmented papules and nodules, which represent an early stage of cerebriform CTN development.6 Hence, in young children, it may be possible to make an earlier diagnosis by biopsy and genetic testing of this tissue before its transition to a cerebriform appearance. Some non-cerebriform CTN, appearing as isolated, firm, skin-colored to hypopigmented papules and nodules, occurred on the face and digits, and we have used these as a tissue source for molecular genetic analyses as well.
Some epidermal nevi observed in this cohort also had distinctive morphologies which have not been previously described. Epidermal nevi in Proteus syndrome are classically described as thin, velvety plaques with linear morphology.1 In this cohort, we also observed ovoid, phylloid, and amorphous morphologies, and sometimes thick, papillomatous plaques. The accompanying hypertrichosis within some epidermal nevi and our recent finding of hypertrichotic patches in Proteus syndrome may represent the effects of AKT1 activation on hair follicles.3
As anticipated, overgrowth of mucocutaneous tissues was common in this cohort, including overgrowth of epidermal (in epidermal nevi), dermal (in CTN), adipose (in lipomas), and vascular (in capillary and venous malformations) tissues.7–9 We also observed hyperpigmented macules, and it remains to be determined whether this may represent increased numbers of melanocytes or increased production of pigment in Proteus syndrome. Interestingly, findings consistent with undergrowth of mucocutaneous tissues were also observed, including patchy dermal hypoplasia, lipohypoplasia, nevus anemicus, hypopigmented macules, and enamel hypoplasia. Co-existence of hyperplastic and hypoplastic cutaneous and extracutaneous features in individuals with Proteus syndrome have been reported by Happle.10–12 Undergrowth may hence be a common but under-recognized characteristic of Proteus syndrome.
Furthermore, many mucocutaneous features reported here have been previously unreported or underreported in Proteus syndrome, including changes in the nails, dental enamel, gingiva, and skin pigmentation. These characteristics may be attributed to the AKT1 variant in affected cells, since such findings are frequently observed in other conditions involving the PI3K-Akt-mTOR signaling pathway.13,14 A remarkable proportion of individuals within this cohort had numerous acrochordons, a finding that is classically associated with insulin resistance and also observed in other genodermatoses that involve activation of the PI3K-Akt-mTOR pathway. Cutaneous tags are present in up to 28% of individuals with tuberous sclerosis complex15 and are also a feature of Birt-Hogg-Dubé syndrome, an entity discovered by Hornstein and Knickenberg.16 This may be a consequence of the regulation of the PI3K-Akt-mTOR signaling pathway by the insulin signaling pathway.17 The presence of numerous acrochordons in 12% of individuals with Proteus syndrome in this study, of which half had more numerous acrochordons on the overgrown side of the body, further supports involvement of mTOR in generating this phenotype.
Limitations of this study include the retrospective, observational design and lack of longitudinal follow-up for evolution of many of the mucocutaneous features. Future histologic and molecular analyses are warranted to more thoroughly investigate the pathogenesis of less frequently described dermatologic findings in Proteus syndrome.
Conclusions
The findings in this genetically confirmed cohort show that the range of mucocutaneous features occurring in Proteus syndrome is broader than previously considered. Knowledge of these findings may aid in earlier and more accurate diagnosis and foster appropriate management. These results not only broaden the phenotypic spectrum of cutaneous features beyond those included in the current diagnostic criteria for Proteus syndrome, but also provide novel insights regarding the pathogenesis of the condition.
Acknowledgements
The authors acknowledge Emily Modlin (NIH/NHGRI) for assistance with data collection.
Funding statement:
This study was funded by the Intramural Research Program of the National Human Genome Research Institute (NHGRI) at the National Institutes of Health (NIH), grant HG200388 04. This research was also made possible by the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, and other private donors. For a complete list, visit the foundation website at http://www.fnih.org.
Conflict of interest statement:
Dr. Biesecker has received in-kind research support from ArQule Inc. (now wholly owned by Merck and Co.) and is a member of the Illumina Corp. Medical Ethics Advisory Board. The remaining authors have no conflicts of interest to report.
Footnotes
Ethics approval statement: Each study participant (or their parent, if a minor) provided written informed consent under the protocol 94-HG-0132, which was approved by the NHGRI Institutional Review Board at the NIH.
Statement of prior presentation: Portions of this work were presented at the Society for Pediatric Dermatology Annual Meeting in Austin, TX (July 11–14, 2019).
Data availability statement: The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The opinions and assertions expressed herein are those of the authors and do not necessarily reflect the official policy or position of the Uniformed Services University, the Department of Defense, or the National Institutes of Health.
References
- 1.Biesecker LG, Sapp JC. Proteus Syndrome. 2012August9 [Updated 2019 Jan 10]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK99495/. [PubMed] [Google Scholar]
- 2.Nguyen D, Turner JT, Olsen C, Biesecker LG, Darling TN. Cutaneous manifestations of proteus syndrome: correlations with general clinical severity. Arch Dermatol. 2004;140(8):947–953. [DOI] [PubMed] [Google Scholar]
- 3.Pithadia DJ RJ, Sapp JC, Biesecker LG, Darling TN. Hypertrichotic patches as a mosaic manifestation of Proteus syndrome. J Am Acad Dermatol. 2021;84(2):415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, et al. Pharmacodynamic Study of Miransertib in Individuals with Proteus Syndrome. Am J Hum Genet. 2019;104(3):484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan NR, Patel R, Crenshaw MM, et al. Pathogenetic insights from quantification of the cerebriform connective tissue nevus in Proteus syndrome. J Am Acad Dermatol. 2018;78(4):725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholson CL, Daveluy S. Epidermal Nevus Syndromes. PubMed StatPearls. 2020. [PubMed] [Google Scholar]
- 8.Winik BC, Boente MC, Asial RA. Cerebriform plantar hyperplasia: ultrastructural study of two cases. Eur J Dermatol. 2000;10(7):551–554. [PubMed] [Google Scholar]
- 9.Cohen MM Jr. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014;85(2):111–119. [DOI] [PubMed] [Google Scholar]
- 10.Happle R. Elatoproteus Syndrome: Delination of an Inverse Form of Proteus Syndrome. Am J Med Genet. 1999;84:25–28. [DOI] [PubMed] [Google Scholar]
- 11.Happle R. Lipomatosis and partial lipohypoplasia in Proteus syndrome: a clinical clue for twin spotting? Am J Med Genet. 1995;56(3):332–333. [DOI] [PubMed] [Google Scholar]
- 12.Happle R, Steijlen PM, Theile U, et al. Patchy dermal hypoplasia as a characteristic feature of Proteus syndrome. Arch Dermatol. 1997;133(1):77–80. [PubMed] [Google Scholar]
- 13.Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol Clin. 2017;35(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150(10):1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baykal C Acrochordons on the neck; a remarkable clinical feature of tuberous sclerosis showing different patterns. J Eur Acad Dermatol Venereol. 2018;32(4):e146–e147. [DOI] [PubMed] [Google Scholar]
- 16.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113(12):1674–1677. [PubMed] [Google Scholar]
- 17.Shah OJ, Hunter T. Tuberous sclerosis and insulin resistance. Unlikely bedfellows reveal a TORrid affair. Cell Cycle. 2005;4(1):46–51. [DOI] [PubMed] [Google Scholar]