

Abstract
Despite advances in breast cancer treatment, residual disease driven by dormant tumor cells continues to be a significant clinical problem. Leukemia inhibitory factor receptor (LIFR) promotes a dormancy phenotype in breast cancer cells and LIFR loss is correlated with poor patient survival. Herein we demonstrate that histone deacetylase inhibitors (HDACi), which are in phase III clinical trials for breast cancer, epigenetically induced LIFR and activated a pro-dormancy program in breast cancer cells. HDACi slowed breast cancer cell proliferation and reduced primary tumor growth. Primary breast tumors from HDACi-treated patients had increased LIFR levels and reduced proliferation rates compared to pre-treatment levels. Recent Phase II clinical trial data studying entinostat and azacitidine in metastatic breast cancer revealed that induction of several pro-dormancy genes post-treatment was associated with prolonged patient survival. Together, these findings suggest HDACi as a potential therapeutic avenue to promote dormancy, prevent recurrence and improve patient outcomes in breast cancer.
Introduction
Breast cancer remains the most diagnosed cancer and second leading cause of cancer deaths in women. Recurrence arises from residual tumor cells within the primary tumor microenvironment or distant metastatic site that have remained in a prolonged dormant state. Dormant tumor cells may lie latent as single quiescent cells or small cell clusters in which dormancy is maintained through balanced proliferation and apoptosis [1–4]. In response to tumor-intrinsic or microenvironmental cues, residual tumor cells become reactivated and develop into clinically detectable lesions [1, 5]. Further investigation into the molecular mechanisms underlying tumor cell dormancy would significantly advance the current field and provide potential therapeutic avenues to target residual disease.
Histone modifications play a key role in gene expression and are predominantly controlled by the balance of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Aberrant HDAC expression and activity are frequently observed in cancer and result in enhanced proliferation, angiogenesis, and metastasis [6]. HDAC inhibitors (HDACi) have emerged as promising cancer therapeutics by inducing differentiation, cell cycle arrest, and apoptosis [7]. Several HDACi are FDA approved for hematological malignancies [8] and several including entinostat and vorinostat are currently being tested in ~70 clinical trials for early, advanced, and metastatic breast cancer [9] [clinicaltrials.gov]. However, little is known about the molecular mechanisms by which HDACi influence tumor cell growth and progression.
Leukemia inhibitory factor (LIF) receptor (LIFR) was previously identified as a breast tumor suppressor and metastasis suppressor [10, 11]. Recent evidence indicates that LIFR activation and downstream STAT3 signaling maintains breast tumor cells in a dormant state [12]. Loss of this signaling axis results in enhanced tumor cell proliferation and bone destruction in preclinical metastasis models [12]. Currently, the upstream regulators of LIFR expression and activity in breast cancer remain largely unclear. Previous work by our laboratory [12] and Zeng et al. [13] suggests that HDACi stimulate LIFR expression in breast cancer cells in vitro.
Our studies herein aimed to determine the extent and mechanism by which HDACi activate LIFR signaling and whether these inhibitors slow the outgrowth of breast cancer cells in vivo, presenting a viable therapeutic strategy to re-program breast cancer cells for long-term dormancy and prevent breast cancer relapse.
Results
HDAC inhibitors stimulate LIFR expression in breast cancer cells regardless of estrogen receptor status
We sought to test a panel of diverse HDACi with varying structural properties and HDAC isoform selectivity for their ability to stimulate LIFR expression. Based on their pharmacological properties and clinical relevance, this panel consisted of entinostat, panobinostat, romidepsin, and vorinostat. HDACi treatment of human breast cancer cell lines MCF7 (ER+), SUM159 (TNBC), and MDA-MB-231b (bone metastatic clone [14, 15]; TNBC) significantly increased LIFR mRNA expression between 6 and 24 hours in a dose-dependent manner (Fig. 1A–D and Fig. S1A). Similar results were observed in mouse mammary carcinoma cells D2.0R (ER+), D2A1 (TNBC), and 4T1BM2 (bone metastatic clone; (TNBC) (Fig. S2A–C). LIFR protein was greater than mRNA levels in both human and mouse cell lines (Fig. 1E–J, Fig. S1B, and Fig. S2D–F). Analysis of the NCI-60 dataset showed a similar increase in LIFR mRNA across several human breast cancer cell lines following vorinostat treatment (Fig. S1C, D). Notably, each HDACi stimulated LIFR expression in all the cell lines we investigated, regardless of estrogen receptor status.
Figure 1. HDAC inhibitors induce LIFR mRNA and protein expression in breast cancer cells.
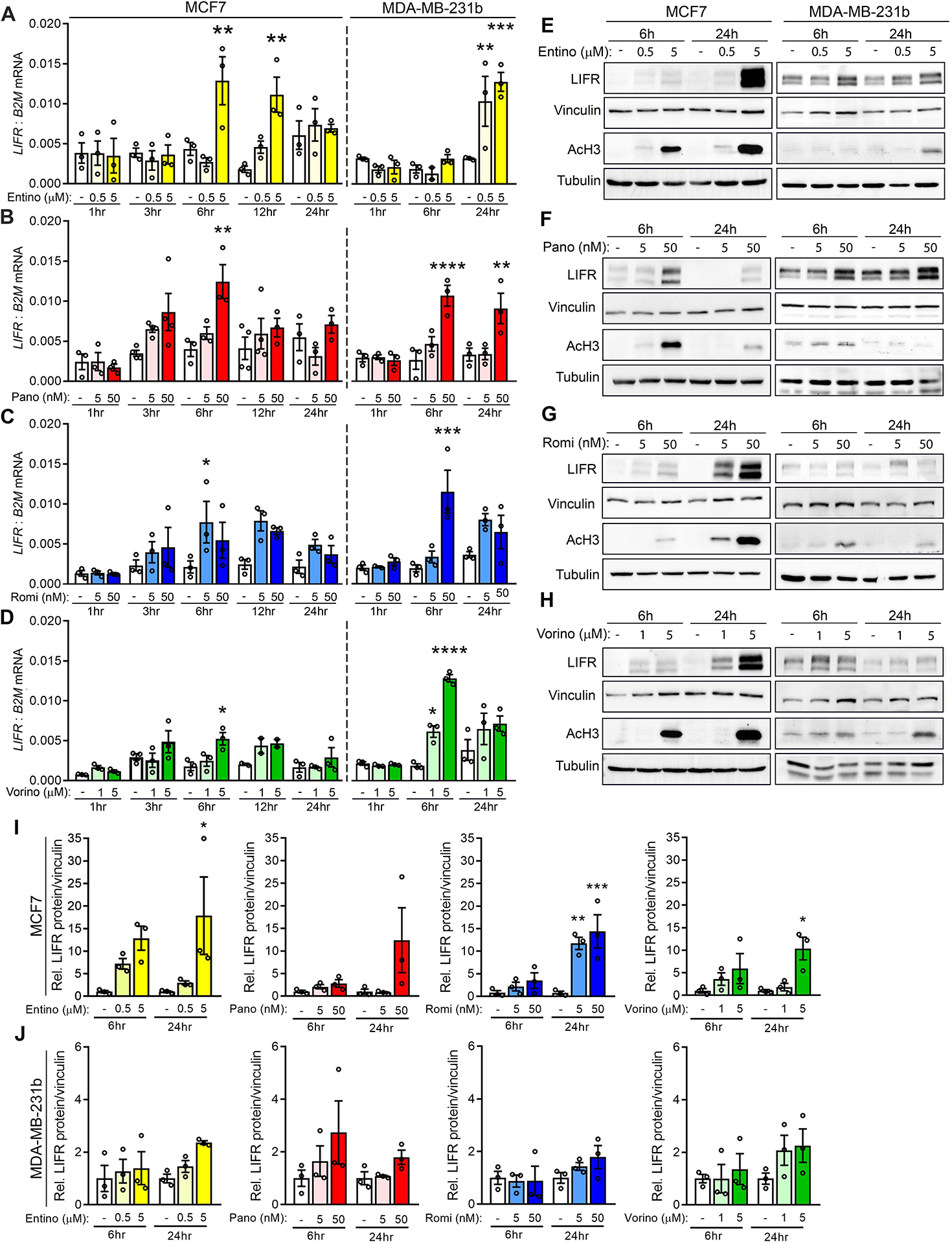
(A-D) LIFR mRNA levels in MCF7 and MDA-MB-231b cells treated with (A) 0.5μM or 5μM entinostat, (B) 5nM or 50nM panobinostat, (C) 5nM or 50nM romidepsin, (D) 1μM or 5μM vorinostat or DMSO (vehicle control) for 1, 3, 6, 12, or 24 hours. (E-H) Representative western blots for LIFR, acetylated histone H3 (AcH3), vinculin (loading control), and tubulin (loading control) protein levels in MCF7 and MDA-MB-231b cells treated with (E) 0.5μM or 5μM entinostat, (F) 5nM or 50nM panobinostat, (G) 5nM or 50nM romidepsin, (H) 1μM or 5μM vorinostat or DMSO (vehicle control) for 6 or 24 hours. (I, J) Quantitation of LIFR protein levels from western blots described in E-H for (I) MCF7 and (J) MDA-MB-231b cells. A-J: n=three independent biological replicates. Bar graphs = mean +/− SEM. A-D, I, J: One-way ANOVA with Sidak’s multiple comparisons tests. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
To determine whether HDACi-stimulated LIFR was functional and enhanced downstream signaling, we explored STAT3 activation following stimulation with recombinant LIF in combination with HDACi. MCF7 cells co-stimulated with LIF and HDACi showed increased phosphorylation of STAT3 Y705, which is essential for STAT3 transcriptional activity [16, 17], compared to LIF treatment alone (Fig. 2A, B). We [12] and others [13] have reported that MDA-MB-231 cells do not express functional LIFR, since LIF treatment does not induce STAT3 signaling. Interestingly, HDACi treatment re-sensitized MDA-MB-231b cells to LIF stimulation resulting in dramatic STAT3 phosphorylation (Fig. 2C, D). A similar response has been reported for MDA-MB-231 cells treated with vorinostat [13] suggesting that multiple HDACi can restore functional LIFR and re-sensitize cells to the ligand. Enhanced STAT3 signaling was not due to any significant HDACi-mediated changes in STAT3 promoter acetylation, total mRNA or protein levels, or basal phospho-STAT3 levels in the absence of ligand (Fig. 2A–D, Fig. S3A, B and Fig. S4L), suggesting that STAT3 signaling originated from upstream HDACi-induced signals.
Figure 2. Epigenetic regulation of LIFR by HDAC inhibitors and activation of downstream STAT3 signaling.
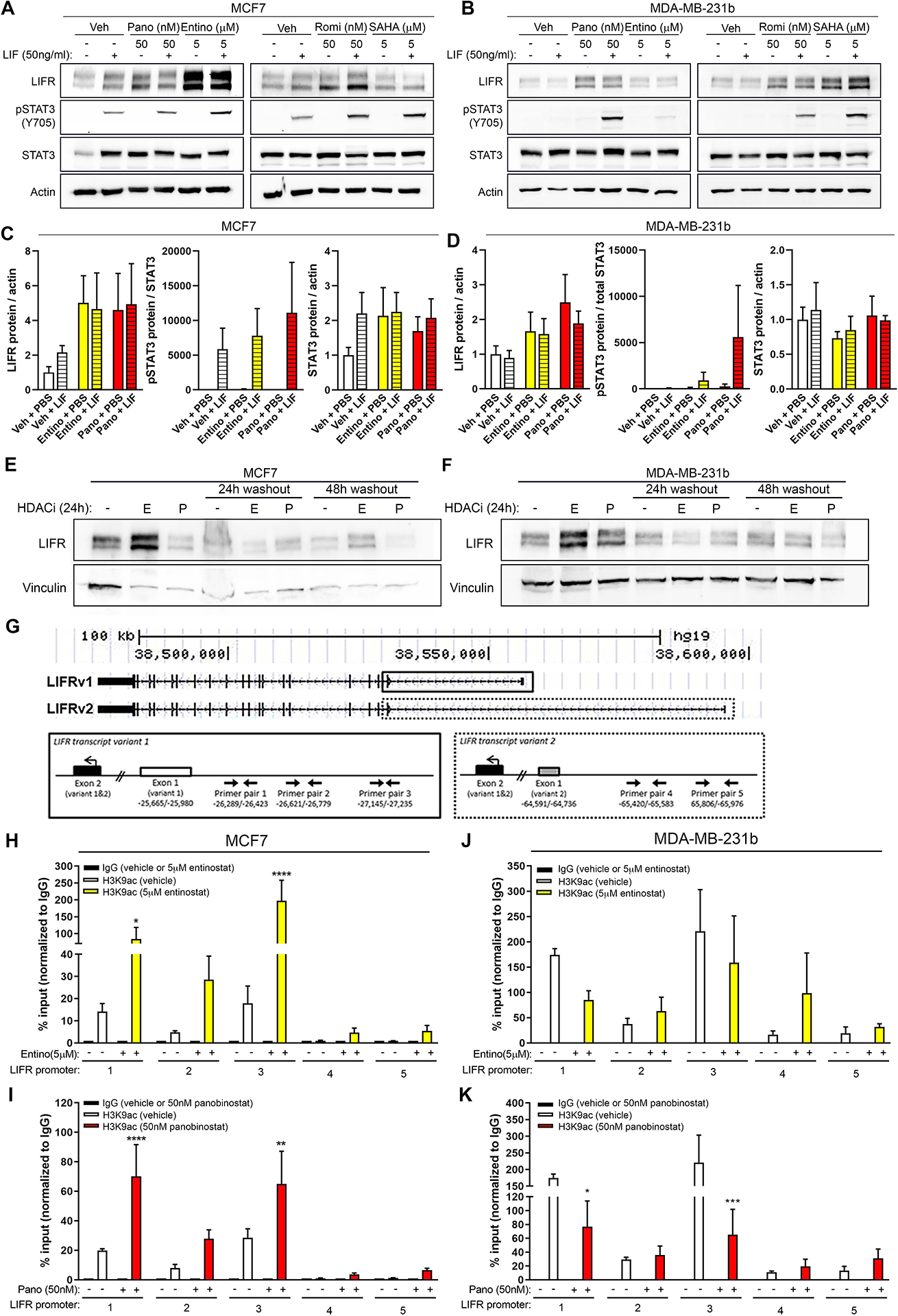
(A, B) Representative western blots for LIFR, pSTAT3 (Y705), total STAT3, and β-actin (loading control) in (A) MCF7 and (B) MDA-MB-231b cells after treatment with 5μM entinostat, 50nM panobinostat, 50nM romidepsin, 5μM vorinostat, or DMSO (vehicle control) for 24 hours followed by 15 minute treatment with PBS (vehicle control) or recombinant LIF (50ng/ml). (C, D) Quantitation of LIFR, pSTAT3/total STAT3, and STAT3 protein levels in (C) MCF7 and (D) MDA-MB-231b cells from western blots shown in (A, B). (E, F) Representative western blots for LIFR and vinculin (loading control) in (E) MCF7 and (F) MDA-MB-231b cells treated with 5μM entinostat (“E”), 50nM panobinostat (“P”), or DMSO (vehicle control) for 24 hours followed by drug washout and collection 24 or 48 hours later. (G) UCSC genome browser tracks for LIFR variant 1 and 2 and primer pairs used to evaluate promoter acetylation by ChIP-qPCR. Solid lined box indicates primer pairs designed to LIFRv1 and dashed lined box indicates primer pairs designed to LIFRv2. (H, I) ChIP-qPCR showing acetylated histone H3 lysine 9 (H3K9ac) enrichment (% ChIP/input) along the LIFR promoter region in MCF7 cells treated with (H) 5μM entinostat, (I) 50nM panobinostat, or DMSO (vehicle control). (J, K) ChIP-qPCR showing acetylated histone H3 lysine 9 (H3K9ac) enrichment (% ChIP/input) along the LIFR promoter region in MDA-MB-231b cells treated with (J) 5μM entinostat, (K) 50nM panobinostat, or DMSO (vehicle control). A-F: n=two independent biological replicates. G-K: n=three independent biological replicates. Bar graphs = mean +/− standard error of the mean. H-K: One-way ANOVA with Sidak’s multiple comparisons test, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Following HDACi withdrawal, LIFR protein rapidly returned to basal levels, suggesting dynamic and reversible regulation (Fig. 2E, F). We investigated whether HDACi directly promote LIFR transcriptional activation by altering LIFR promoter acetylation. MCF7 cells treated with HDACi showed significant enrichment of acetylated histone H3 lysine 9 (H3K9ac), a marker of active promoters, along the LIFRv1 promoter (Fig. 2H, I). High basal H3K9ac of the LIFRv1 promoter was observed in MDA-MB-231b cells, which was not enhanced with HDACi treatment (Fig. 2J, K). These findings are consistent with the high LIFR protein expression in these cells; however, it is unclear why H3K9ac is modestly reduced with HDACi. Peak promoter acetylation may not occur at the time points we tested or non-histone mechanisms such as LIFR protein acetylation/phosphorylation [18] may play an important role in stimulating LIFR expression in MDA-MB-231b cells. Notably, basal LIFRv2 mRNA expression was very low or undetectable in both cell lines and was not significantly induced with HDACi (Fig. S3C–E), explaining the lack of promoter acetylation observed for LIFRv2 (Fig. 2H–K, LIFR promoter regions 4 and 5).
HDAC inhibitors promote a pro-dormancy program
To determine whether HDACi promote a pro-dormancy program, we investigated expression of thirteen dormancy-associated genes following HDACi treatment (Fig. S4A) [19–25]. In contrast to consistent LIFR induction by all HDACi in all cell lines tested, we observed cell line-specific and drug-specific stimulation of the other dormancy-associated genes (Fig. 3A–D). For example, panobinostat stimulated AMOT and MSK1 expression in MCF7 cells, but THBS1 and P4HA1 in MDA-MB-231b cells (Fig. 3A–D). Notably, HDACi treatment stimulated these dormancy-associated genes in the absence of LIF stimulation and did not significantly reduce the expression of any genes (Fig. 3A, B, red indicates no change). Interestingly, AMOT expression was increased by multiple HDACi in both cell lines. Further, entinostat-stimulated TGFB2 expression was intriguing given its role in stem cell reprogramming [26] and promoting dormancy in the bone [20, 27]. Surprisingly, promoter H3K9ac was not greatly enriched following HDACi treatment (Fig. S4B–D), suggesting an indirect mechanism. These results led us to explore whether LIFR is required for induction of other dormancy-associated factors. LIFR knockdown cells (MCF7 shLIFR#6), which showed ~65% decrease in LIFR mRNA and protein expression, exhibited downregulation (>40% reduction) of 9 out of the 13 pro-dormancy genes (Fig. 3E, Fig. S4E, F). Similar results were observed using the shLIFR#3 line by RNA sequencing despite having only a ~40% decrease in LIFR expression (Fig. S4G). Further, LIFR knockdown partially blunted the induction of AMOT and MSK1 and completely blocked the induction of TGFB2 following HDACi treatment (Fig. 3F, G). Thus, we next sought to explore whether LIFR alone mediates induction of these dormancy-promoting genes and found that LIFR overexpression alone only modestly increased expression of two genes, CDKN1B (p27) and SELENBP1 (Fig. S4J) and did not significantly increase those genes induced by HDACi treatment (e.g., AMOT, TGFB2, MSK1). Notably, LIFR overexpression did not alter MCF7 cell proliferation (Fig. S4H, I). LIF stimulation alone did not significantly alter the expression of any dormancy-associated genes (Fig. S4K, L). Further, those dormancy genes induced by HDACi remained elevated regardless of LIF stimulation (Fig. S4K, L). Knockdown of STAT3 did not significantly alter any of the dormancy genes or blunt HDACi-mediated induction (Fig. S4M, N). Our results suggest that, in part, LIFR may mediate induction of some dormancy genes (AMOT and TGFB2) but that LIFR alone is not sufficient for induction. Importantly, given that HDACi induce LIFR in the shLIFR cell line, it is difficult to distinguish the contribution of LIFR versus HDACi to the stimulation of other pro-dormancy genes. Despite our efforts, the mechanism(s) by which LIFR may regulate expression of these genes remains unclear. A currently unknown factor(s) may work in conjunction with LIFR or HDACi potentially acetylate this factor(s) or LIFR at the protein level, resulting in enhanced dormancy gene expression that does not occur in the absence of HDACi treatment. Regardless of the specific mechanisms elicited by HDACi, which are known to be extensive, and whether these are LIFR-dependent, our findings indicate that HDACi effectively promote a pro-dormancy gene program.
Figure 3. HDACi stimulate a pro-dormancy gene program.
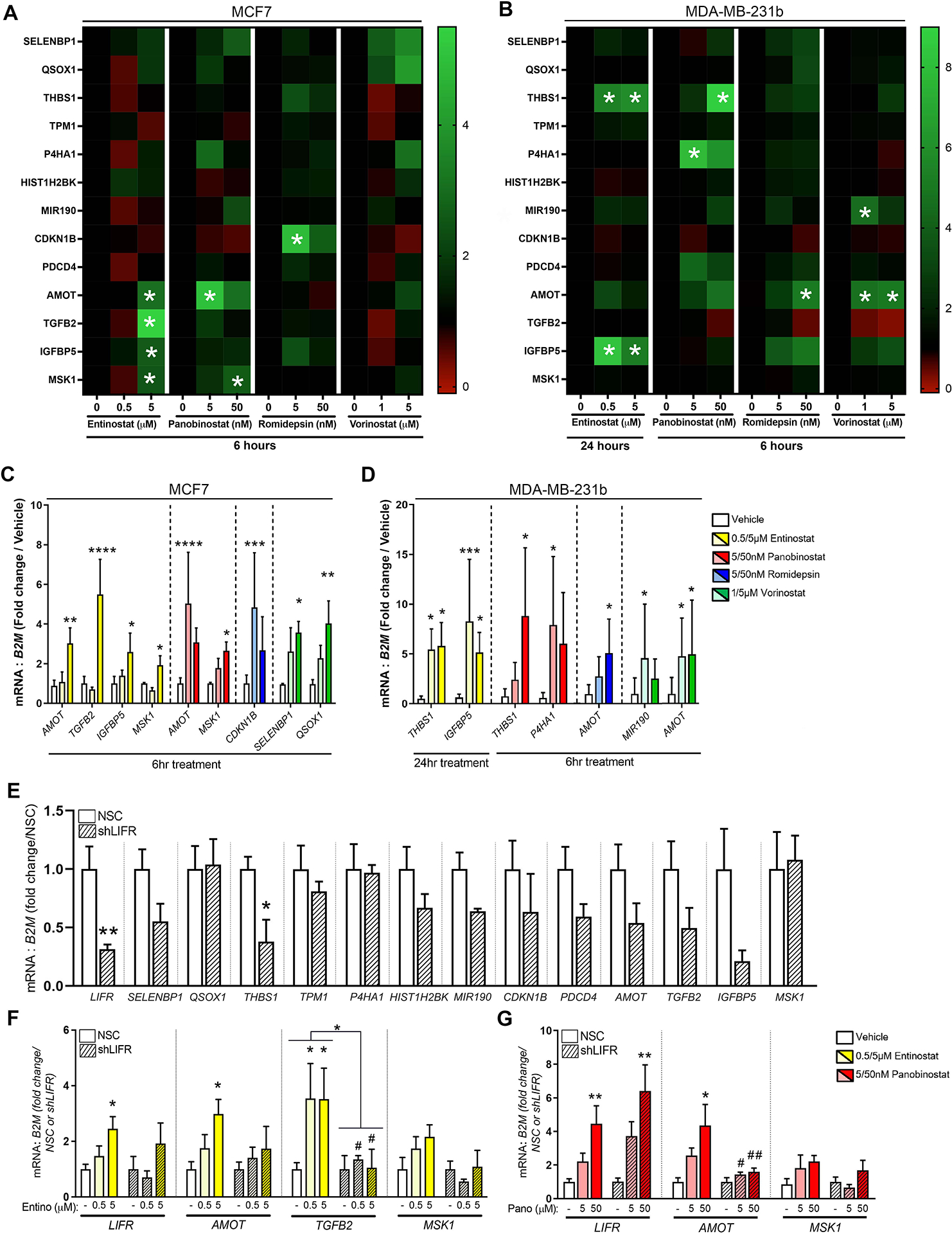
(A, B) mRNA levels of full dormancy-associated gene panel in (A) MCF7 and (B) MDA-MB-231b cells treated with 0.5μM or 5μM entinostat, 5nM or 50nM panobinostat, 5nM or 50nM romidepsin, 1μM or 5μM vorinostat, or DMSO (vehicle control) for 6 or 24 hours. (C, D) mRNA levels of significantly altered dormancy genes in (C) MCF7 and (D) MDA-MB-231b cells treated with 0.5μM or 5μM entinostat, 5nM or 50nM panobinostat, 5nM or 50nM romidepsin, 1μM or 5μM vorinostat, or DMSO (vehicle control) for 6 or 24 hours. (G) mRNA levels of dormancy associated genes in MCF7 NSC (control) and MCF7 LIFR knockdown (shLIFR) cells. (H, I) mRNA levels of dormancy associated genes in MCF7 NSC or MCF7 shLIFR cells treated with (H) 0.5μM or 5μM entinostat or (I) 5nM or 50nM panobinostat. B-I: n=three independent biological replicates. Bar graphs = mean +/− standard error of the mean. B-F, H, I: One-way ANOVA with Sidak’s multiple comparisons test. G: Mann-Whitney t-test. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Treatment with HDAC inhibitors slows tumor cell proliferation
We next sought to determine whether HDACi could promote functional outcomes of dormancy. We monitored tumor cell proliferation in the presence of low-dose HDACi over 48-hour increments for eight days. MCF7 cell proliferation was minimally affected during the first 48 hours but was substantially slowed by >3-fold with HDACi treatment between day 2 and day 8 (Fig. 4A). During the final 48 hours, the fold-change in proliferation of HDACi-treated cells fell below one suggesting a subset of cells underwent cell death (Fig. 4A). These results are further supported by a small but significant increase in the sub-G0/G1 population, characteristic of apoptotic cells, with HDACi on day 6 and day 8 (Fig. S5A). Entinostat significantly increased the G0/G1 population at day 2, but no dramatic cell cycle changes were observed with HDACi treatment (Fig. S5A, B). Long-term HDACi treatment significantly slowed MDA-MB-231b cell proliferation, albeit to a lesser extent than the MCF7 cell line but did not appear to induce cell death (Fig. 4B).
Figure 4. HDACi-stimulated pro-dormancy phenotype results in slowed tumor cell proliferation and reduced primary tumor growth.
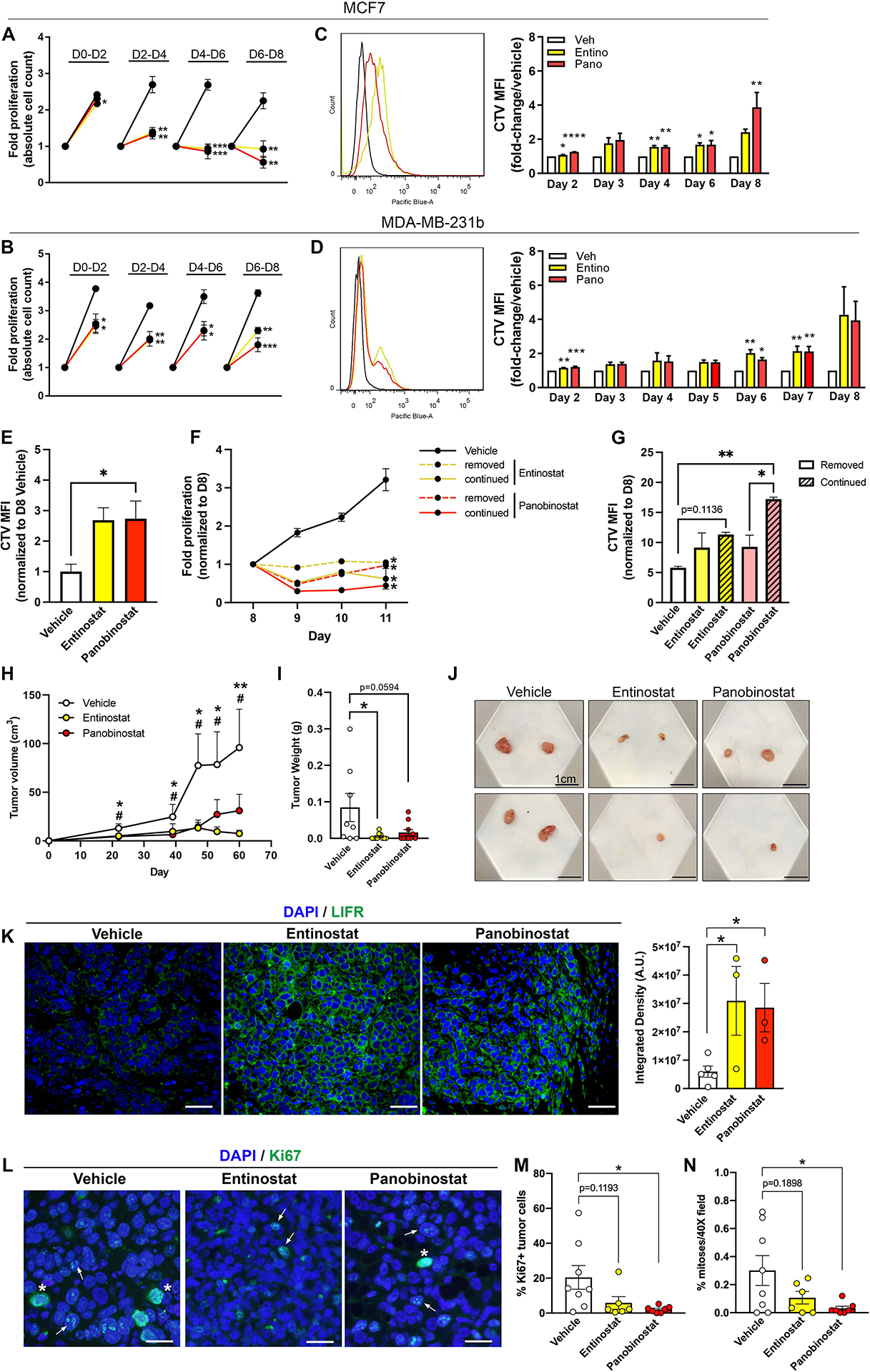
(A, B) Trypan blue exclusion assay to assess fold proliferation in (A) MCF7 and (B) MDA-MB-231b cells treated with 0.5μM entinostat, 5nM panobinostat, or vehicle for a total of eight days. On day 2, 4, 6, and 8, cells were trypsinized and counted followed by reseeding of an equal number of cells per treatment group. Data are presented as fold-proliferation during each 48-hour increment. (C, D) CellTrace Violet proliferation dye was loaded into (C) MCF7 and (D) MDA-MB-231b cells followed by treatment with 0.5μM entinostat, 5nM panobinostat, or vehicle for a total of eight days. Mean fluorescence intensity (MFI) was tracked over eight days by flow cytometry to assess proliferation. (E) CellTrace Violet proliferation dye was loaded into MCF7 cells followed by treatment with 0.5μM entinostat, 5nM panobinostat, or vehicle for a total of eight days. Mean fluorescence intensity (MFI) was determined on day eight by flow cytometry to assess proliferation. (F) Trypan blue exclusion assay to assess fold proliferation in MCF7 cells treated with .5μM entinostat, 5nM panobinostat, or vehicle for a total of eight days followed by drug removal (dashed lines) or continuation (solid lines) for an addition 3 days. (G) CellTrace Violet proliferation dye was loaded into MCF7 cells following treatment with 0.5μM entinostat, 5nM panobinostat, or vehicle for a total of eight days. On day 8, cells were separated into groups whereby HDACi treatment was removed or continued for an additional 3 days. Mean fluorescence intensity (MFI) was tracked over these 3 days by flow cytometry to assess proliferation. (H) Tumor volume by caliper measurements over 60 days following orthotopic injection of MCF7 cells into mice and treatment with entinostat (10mg/kg; n=8 mice), panobinostat (5mg/kg; n=10 mice) or vehicle (n=10 mice). * = significance between vehicle and entinostat. # = significance between vehicle and panobinostat. (I) Final tumor weight at sacrifice of mice described in (H). (J) Representative images of primary tumors collected from mice described in (H). (K) Representative LIFR (green) and DAPI (blue) staining and quantitation of primary tumors from mice described in (H). All panels = 40X and scale bars = 100μm. (L) Representative Ki67 (green) and DAPI (blue) staining of primary tumors from mice described in (H). All panels = 40X and scale bars = 20μm. Arrows indicate Ki67+ tumor cells and asterisks indicate mitotic figures. (M) Quantitation of % Ki67+ tumor cells/total tumor cells from images described in (L). (N) Quantitation of mitoses (# mitotic figures/total cells in 40X field) by DAPI staining from images described in (L). Bar graphs = mean +/− standard error of the mean. A-D: n=three independent biological replicates. Graphs represent mean +/− standard error of the mean. A-D, K: Unpaired t-test. E, G, H, I, M, N: One-way ANOVA with Sidak’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
We sought to determine whether this slowed proliferation was a result of the entire population entering a dormant-like state or equal rates of proliferation and cell death. We used the proliferation dye, CellTrace Violet (CTV), which becomes diluted with subsequent generations and thus would be retained longer in dormant cells compared to proliferating cells. CTV retention was increased in both cell lines treated with HDACi (Fig. 4C, D). MCF7 cells displayed consistent CTV retention indicating that HDACi slowed proliferation of the entire cell population. Interestingly, MDA-MB-231b cells showed two retention peaks on day 8, suggesting that HDACi may differentially affect the proliferation of two subpopulations (Fig. 4D).
Next, we treated MCF7 cells with HDACi for eight days followed by HDACi removal or continuation. As previously observed, CTV retention was enhanced in MCF7 cells treated with HDACi for eight days (Fig. 4E). MCF7 cell proliferation was significantly reduced in both HDACi removed and continued treatment groups and cells remained dormant since the cell number on days 9–11 did not exceed the initial seeding density. (Fig. 4F). HDACi continuation and removal resulted in a ~2.5-fold and ~1.6-fold increase in CTV retention, respectively (Fig. 4G). CTV retention was significantly increased with continued panobinostat treatment and modestly increased (p=0.1136) with continued entinostat treatment (Fig. 4G). These results suggest that HDACi treatment induces a semi-permanent dormancy phenotype.
Given the ability of HDACi to reprogram cells and the unfavorable association of cancer stem cells (CSC) with poor prognosis and therapy resistance, we also investigated whether HDACi alter the CSC phenotype, here characterized as CD44High/CD24Low [28]. While there was a significant increase in the CD44Low/CD24High population with HDACi treatment, there was no change in the percentage of CSC-like cells (Fig. S5C–F).
We next sought to determine whether HDACi treatment reduces primary tumor growth in vivo. MCF7 cells were orthotopically inoculated and HDACi treatment was initiated 24 hours later and given 5 days/week until sacrifice. Strikingly, entinostat and panobinostat significantly reduced MCF7 primary tumor growth as indicated by decreased tumor volume and final tumor weight (Fig. 4H–J). Primary tumors from HDACi treated mice showed significantly increased LIFR expression and fewer Ki67+ tumor cells and mitoses (Fig. 4K–N). Together, these findings further support the ability of HDACi to induce LIFR and a pro-dormancy phenotype and slow tumor cell proliferation.
HDACi-induced dormancy phenotype is inversely associated with proliferation and recurrence in patients with breast cancer
To determine the clinical relevance of these findings, we examined the association of LIFR, AMOT, and TGFB2 expression, which were dramatically stimulated with HDACi, with clinical prognosis and recurrence in two independent patient cohorts of breast cancer. In the first cohort (METABRIC, Nature 2012 & Nat Commun 2016), tumor proliferation was assessed in ER+ tumors as part of the molecular subtyping classifications. Using this data subset, mRNA expression of these pro-dormancy genes was significantly reduced in highly proliferative tumors compared to those with low proliferation (Fig. 5A). Analysis of all primary tumors, regardless of subtype, revealed a significant increase in disease-free survival in patients with high LIFR expression (Fig. 5B, C). Further, patients with tumor recurrence had significantly lower LIFR expression in their primary tumor suggesting an association with metastatic progression (Fig. 5D). These data are consistent with previously reported reductions in overall survival in patients with down-regulated LIFR signaling [10, 12]. There was no significant association of AMOT or TGFB2 expression with these clinical parameters (data not shown). Additionally, decreased LIFR expression correlated with increased Nottingham prognostic index (NPI) scores, which incorporates tumor size, lymph node involvement, and tumor grade to predict patient prognosis following surgery (Fig. 5E). The second patient cohort (Bos et al. [29]) also revealed a significant reduction in LIFR mRNA expression and modest decreases in AMOT and TGFB2 in highly Ki67+ proliferative tumors (Fig. 5F–H). Indeed, low expression of LIFR, AMOT, and TGFB2 was associated with decreased recurrence-free survival (Fig. 5I–K). Analysis of the TCGA patient cohort revealed a similar correlation of low LIFR with poor overall survival and reduced LIFR and TGFB2 expression in primary tumors with high proliferation rates (Fig. S6A–F).
Figure 5. HDACi-induced dormancy phenotype is inversely associated with proliferation and recurrence in breast cancer patients.
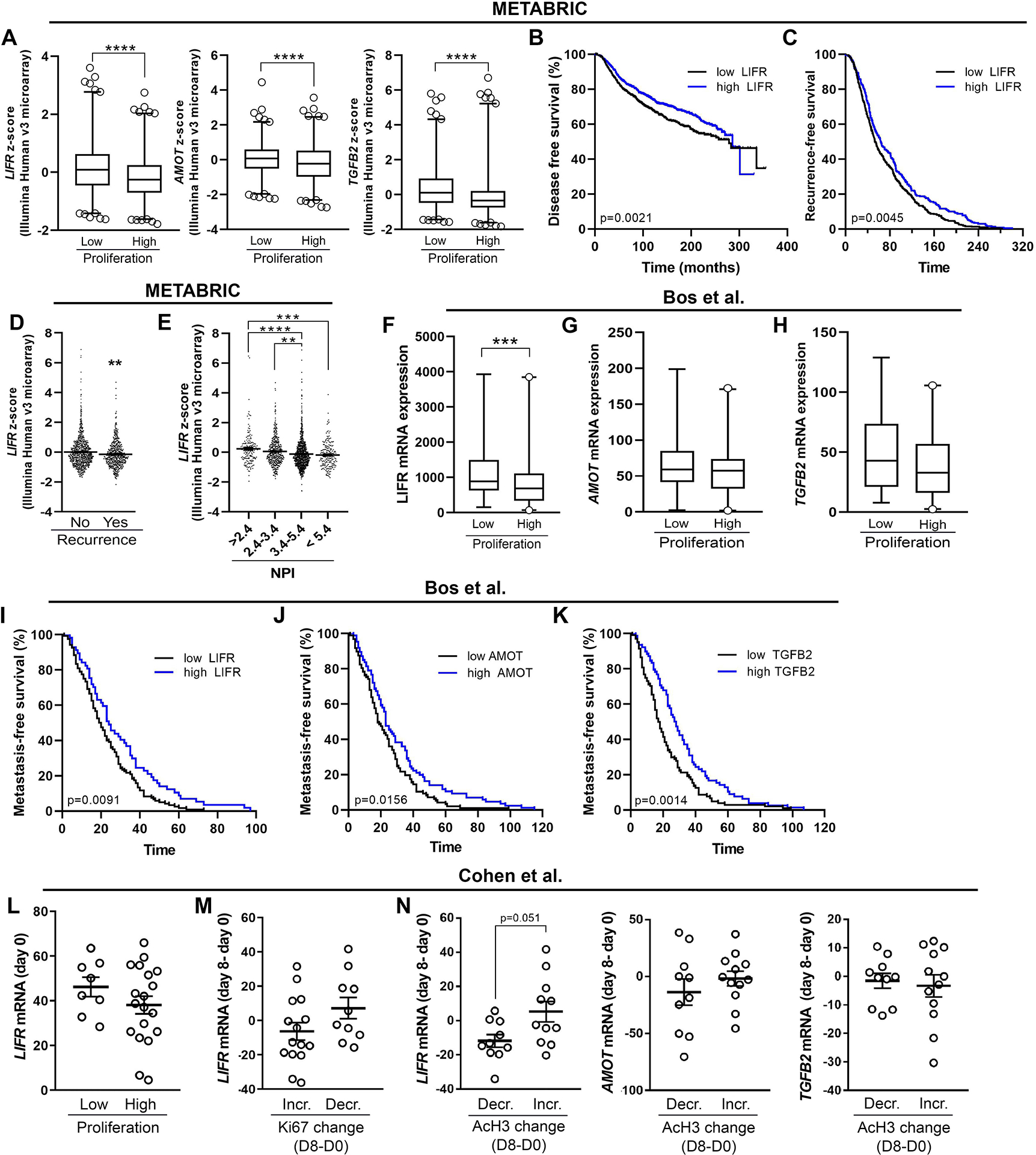
(A) mRNA levels of LIFR, AMOT, and TGFB2 in ER+ breast tumors displaying low or high proliferation (low n=623, high n=603). The data are displayed as z-score values from Illumina Human v3 microarray data (METABRIC, Nature 2012 & Nat Commun 2016). (B) Survival analysis representing the proportion of disease-free patients stratified according to LIFR mRNA levels in samples from breast cancer patients (low n=979, high = 999; HR = 1.273; 95% CI: 1.091–1.486; METABRIC, Nature 2012 & Nat Commun 2016 dataset). (C) Analysis of time to recurrence in patients (HR = 1.262, 95% CI: 1.075–1.483) described in (B). (D) LIFR mRNA levels in breast cancer patients stratified by recurrence (no n=1278, yes n=622). The data are displayed as z-score values from Illumina Human v3 microarray data (METABRIC, Nature 2012 & Nat Commun 2016). (E) LIFR mRNA levels in breast tumors displaying low or high proliferation (low n= 86, high = 106; all breast cancer subtypes; Bos et al. dataset (GSE12276). (F, G) mRNA levels of (B) AMOT and (C) TGFB2 tumors displaying low or high proliferation. (H) LIFR mRNA levels in breast cancer patients stratified by Nottingham prognostic index (NPI) scores. The data are displayed as z-score values from Illumina Human v3 microarray data (METABRIC, Nature 2012 & Nat Commun 2016). (I-K) Survival analysis representing the proportion of relapse-free patients stratified according to (I) LIFR, (J) AMOT, or (K) TGFB2 mRNA levels in samples from breast cancer patients (LIFR (low n= 120, high = 57), (HR=1.486, 95% CI: 1.102–2.004)); AMOT (low = 97, high = 86), (HR=1.409, 95% CI: 1.052–1.885)); TGFB2 (low = 104, high = 78), (HR=1.583, 95% CI: 1.182–2.119); all breast cancer subtypes; Bos et al. dataset (GSE12276). (L) LIFR mRNA levels pre-treatment (day 0) in tumors displaying low and high proliferation (low n=8, high n=19). (M) LIFR mRNA expression change (day 8 – day 0) in tumors that displayed increased or decreased Ki67 levels post-treatment (day 8 – day 0) (low n=15, high = 10). (N) LIFR, AMOT, or TGFB2 mRNA expression change (day 8 – day 0) in patients that displayed increased or decreased acetylated histone H3 (AcH3) post-treatment (day 8 – day 0) (low n= 10, high n=12). A, D, E, N: Mann-Whitney t-test, H: One-way ANOVA with Sidak’s multiple comparisons test. *p<0.01, **p<0.01, ***p<0.001 and ****p<0.0001.
We sought to investigate whether HDACi could be used to stimulate pro-dormancy genes in patients with breast cancer. While few published studies exist, one publicly available dataset investigated gene expression changes in primary breast tumors before and after 8 days of HDACi treatment (valproic acid; Cohen et al. [30]). We previously demonstrated that the HDACi valproic acid stimulates LIFR expression [12], however it is a relatively weak HDACi [31, 32]. In this cohort, LIFR mRNA expression was modestly lower in highly proliferative primary tumors before HDACi treatment (Fig. 5L). Tumors with reduced proliferation rates had higher LIFR expression changes after treatment (Fig. 5M). Further, patients with increased peripheral blood acetylation levels, an indicator of effective treatment, had higher LIFR expression and a modest increase in AMOT, but not TGFB2, levels post-treatment (Fig. 5N).
We next interrogated pro-dormancy gene expression in a microarray dataset (Table S1) from a Phase II clinical trial of entinostat and azacitidine, a DNA methyltransferase inhibitor and hypomethylating agent, in patients with metastatic breast cancer [33]. Paired metastatic tumor biopsies were available pre- and 8 weeks post- epigenetic therapy for ER+ hormone-resistant (n=14) and TNBC (n= 5) patients. Notably, most post-treatment samples were collected 13–21 days following the last therapy dose. Despite the small cohort and relatively few significantly altered genes [33], we observed modest induction of several pro-dormancy genes in a patient-specific manner that was consistent with our in vitro data (Fig. 6A). We observed a significant change in CDKN1B and LIFR expression and a modest increase in AMOT following treatment (Fig. 6B–D). The three most induced dormancy genes post-treatment (>1.3 fold-change) were CDKN1B (58% patients), AMOT (42% patients), and TPM1 (47% patients), which were induced in both ER+ and TNBC patients (Fig. 6E). Additionally, IGFBP5, LIFR, P4HA1, and SELENBP1 were induced in a smaller number of patients, but in a subtype-specific manner (Fig. 6E). Interestingly, ER+ patients showed a slightly higher number of pro-dormancy genes stimulated (average=4.3 genes) compared to TNBC patients (average=3 genes) following treatment (Fig. 6F). Pro-dormancy gene induction showed no significant association with progressive or stable disease (Fig. 6G, H). Those patients who had more prolonged survival (>10.5 months) showed a modest increase in the number of pro-dormancy genes induced compared to patients with <10.5 months survival (Fig. 6I). This trend was predominately observed in ER+ patients, however clear trends for TNBC are difficult to define given the small patient cohort (Fig. 6J). Notably, patients with 5+ dormancy genes induced showed significantly prolonged survival (15.7 vs 9.8 months) (Fig. 6K). Interestingly, CDKN1B induction alone was significantly associated with improved patient survival (Fig. 6L). Excluding CDKN1B induction from our original analysis revealed that having 4+ induced dormancy genes was still associated with improved survival (Fig. 6M). Combined, these patient data from multiple studies suggest that high expression of these pro-dormancy genes, which can be therapeutically increased with HDACi, correlates with reduced tumor proliferation, relapse, and improved patient survival. Thus, HDACi therapy may represent an effective strategy to induce a pro-dormancy gene program in DTCs to delay breast cancer recurrence and improve patient survival.
Figure 6. Induction of pro-dormancy genes following entinostat and azacitidine treatment is associated with longer breast cancer patient survival.
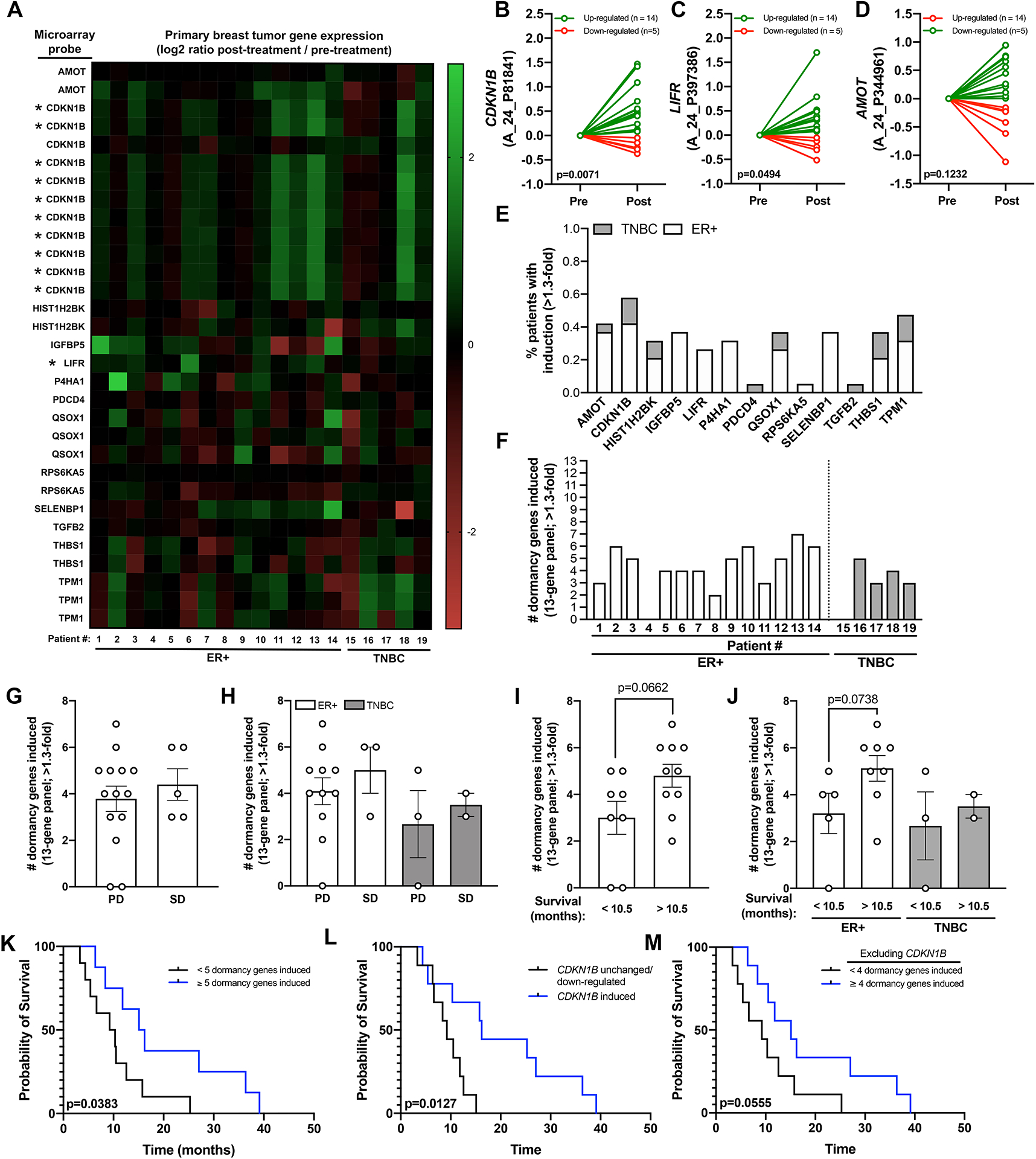
(A) Heatmap showing mRNA levels of pro-dormancy genes in metastatic tumor biopsies (n=15 ER+ and n=4 TNBC) following 8 weeks of treatment with entinostat and azacitidine. Several genes had multiple microarray probes all of which are displayed individually. Asterisk indicates those probes that were significantly altered post-treatment. (B-D) mRNA levels of (B) CDKN1B, (C) LIFR, and (D) AMOT pre- and post-treatment separated by whether expression was down-regulated (red) or up-regulated (green) post-treatment. (E) Percent patients with induction (>1.3-fold change) of each pro-dormancy gene. (F) Number of dormancy genes induced (>1.3-fold change) per patient. (G, H) Association of the number of dormancy genes induced with progressive disease (PD) and stable disease (SD) in (G) all patients and (H) patients separated by ER+ and TNBC subtype. (I, J) Association of the number of dormancy genes upregulated with patient survival in (I) all patients and (J) patients separated by ER+ and TNBC subtype. Patients were separated into 2 cohorts based on the median survival of 10.5 months. (K) Survival curve showing overall survival of patients with <5 (black line) or ≥5 (blue line) dormancy genes induced. (L) Survival curve showing overall survival of patients with CDKN1B unchanged or down-regulated (black line) or CDKN1B induced (blue line). (M) Survival curve showing overall survival of patients with <4 (black line) or ≥4 (blue line) dormancy genes induced excluding CDKN1B. A-D: Wilcoxon matched-pairs signed rank test. I, J: Mann-Whitney U-test. K-M: Log-rank Mantel-Cox test.
Discussion
Significant progress has been made in the mechanistic understanding of tumor dormancy in various tissues, but clinical options remain limited. It remains controversial whether therapies should aim to mobilize cells out of their niche, thereby forcing them into a proliferative state and sensitizing them to chemotherapy, or to maintain tumor cells in a chronic dormant state. An ideal anti-tumor therapy would eliminate all dormant, residual tumor cells from a patient, but the clinical likelihood of this is low given early dissemination of tumor cells [1–4] and persistence due to therapeutic resistance [34]. Signaling molecules including MERTK and high ERK/p38 activity ratio have been suggested to promote dormancy escape [20, 35, 36] and thus could be exploited to promote chemosensitization. However, recent work showed that while targeting integrin signaling in the bone metastatic niche re-sensitizes disseminated tumor cells (DTCs) to chemotherapy and reduces DTC burden, some DTCs persist and ~20–30% of mice relapsed [37]. Conversely, studies have implicated inhibition of Src [38, 39] or ERK [20, 36, 39, 40] signaling and DNA demethylating agents [41] as a means to maintain tumor dormancy and reprogram breast cancer into a chronic treatable disease. HDACi offer another potential therapeutic approach to maintain persistent tumor cells in a chronic state of dormancy. This approach is not without hazards, and our data indicate that withdrawal of HDACi rapidly reverses the effects on LIFR; however, HDACi treatment reprograms tumor cells into a semi-permanent dormant state as proliferation does not dramatically increase following short-term HDACi removal. HDACi effects on residual tumor cells and the molecular mechanisms involved warrant investigation given that these therapies are currently being tested in clinical trials.
High LIFR expression and signaling are associated with low metastatic potential in breast cancer cells and are negatively correlated with tumor cell proliferation and patient survival [12]. Our results indicate that HDACi stimulate LIFR expression and restore LIF signaling in aggressive cancer cells (MDA-MB-231b). Previous findings identified STAT3 in a dormancy gene signature [19] and demonstrated that STAT3 loss increased tumor-induced osteolytic bone destruction [12]. While these studies suggest that STAT3 acts as a mediator of dormancy, recent work demonstrated that LIFR-induced STAT3 activation results in anti-apoptotic signaling including enhanced BCL-2 and MCL-1 expression, leading to therapeutic resistance [13]. Combined, these data suggest that the ability to re-sensitize tumor cells to LIF, which inhibits tumor growth [42, 43], likely represents another pro-dormancy response initiated by HDACi treatment but requires further investigation.
Additionally, our data show direct epigenetic induction of LIFR by HDACi and stimulation of TGFB2 and MSK1, which may be partially LIFR-mediated, in breast cancer. The mechanism by which LIFR may contribute to this enhanced expression remains unclear as STAT3 does not appear to be involved. Expression of pro-dormancy genes was not altered with ERK or PI3K inhibitors [12], other LIFR-activated signaling pathways, or LIFR overexpression alone. Despite our efforts, the exact mechanism by which LIFR may mediate induction of TGFB2 and, to a lesser extent, AMOT remains unclear. Other factors that work in conjunction with LIFR or non-histone-mediated mechanisms may be involved in the induction of these genes. HDACs also regulate the acetylation status and often subsequent protein activity of many non-histone proteins involved in transcription, replication, and DNA repair [44]. LIFR acetylation and phosphorylation enhances or suppresses downstream STAT3 activation, respectively [18]. We sought to investigate the role of LIFR protein acetylation/phosphorylation following HDACi treatment; however, we were unable to find antibodies suitable for detecting these changes. Thus, currently we are unable to determine if these post-translational modifications are important for induction of AMOT and TGFB2 or the restored LIF response of MDA-MB-231b cells with HDACi treatment. Previous studies have also shown enhanced STAT3 protein acetylation and consequent transcriptional activity following treatment with vorinostat [13]. However, as previously mentioned, our data indicate that AMOT and TGFB2 induction is not dependent on STAT3 since knockdown did not alter basal or HDACi-mediated expression. Given our results and the known abundant transcriptional and post-translational alterations induced by HDACi, it is likely that numerous mechanisms, both LIFR-dependent and independent, are involved in promoting this pro-dormancy phenotype. While the precise mechanisms for HDACi-induced dormancy remains unclear, our findings demonstrate that HDACi effectively induce a dormancy phenotype in vitro and in vivo. High expression of pro-dormancy genes is associated with lower tumor proliferation and prolonged relapse-free survival in patients with breast cancer further demonstrating the potential impact of our findings that HDACi induce a pro-dormancy gene program.
In summary, these data provide mechanistic insight into the epigenetic regulation of LIFR by HDACi and the induction of a dormancy program in breast cancer cells. In patients with breast cancer, dormancy-associated factors are stimulated following HDACi treatment and inversely correlated with tumor proliferation and relapse-free survival. Combined, these findings offer HDACi as a potential therapeutic avenue to promote dormancy in breast cancer, prevent recurrence, and potentially improve patient survival.
Materials and Methods
Cells.
Human MCF7 cells were bought from ATCC. Murine D2.0R [45], D2.A1 [45], and bone-metastatic 4T1BM2 [46] cells were gifted as previously described [12]. A bone-metastatic clone of human MDA-MB-231 cells (MDA-MD-231b) [14, 15] was generated by the Mundy laboratory and were recently re-selected and shared by Dr. Julie (Sterling) Rhoades. All cell lines were cultured as previously described [12], regularly tested for mycoplasma contamination, and recently re-authenticated by ATCC.
shRNA and siRNA.
MCF7 NSC, LIFR shRNA#3 and STAT3 shRNA were previously generated [12]. Additional LIFR knockdown lines (Dharmacon, shLIFR#6: V3LHS_347496 and shLIFR#8: V3LHS_347498) were generated as previously described [12]. For LIFR overexpression studies, MCF7 cells were stably transfected with a LIFR- or GFP-NeoR pGL3-Basic plasmid. The LIFR plasmid was designed by our lab using full LIFR (1339bp) sequence with EGFP fused to the C-terminal end, synthesized by Genscript, and validated by sequencing.
HDAC inhibitor treatment.
Cells were seeded in a 6-well plate (2×105 cells/well) or 10cm plate (1.5×106 cells/plate) for RNA and protein analysis, respectively. The following day, cells were treated with vehicle (DMSO), entinostat (0.5μM, 5μM; SelleckChem, S1053), panobinostat (5nM, 50nM; SelleckChem, S1030), romidepsin (5nM, 50nM; SelleckChem, S3020), or vorinostat (1μM, 5μM; SelleckChem, S1047) for 1–24 hours in full serum medium. Cells were harvested for RNA in TRIzol (Thermo Fisher) or protein in RIPA buffer (Sigma) as discussed below. For short-term HDACi washout experiments, cells were treated with HDACi for 24 hours, washed, and incubated for another 24–48 hours in medium without drug.
RNA extraction and real-time qPCR.
Cells were harvested for real-time qPCR analysis as previously described [12]. See the Supplementary Materials for detailed experimental procedures.
Western blotting.
Protein expression was assessed as previously described [12]. See the Supplementary Materials for detailed experimental procedures.
Flow cytometry.
Cells were treated with vehicle (DMSO), entinostat (0.5μM) or panobinostat (5nM) for 8 days. Cells were fixed in 10% formalin for 20 minutes at room temperature, washed with PBS, and stored in PBS at 4°C before staining. Cells were stained in 1% BSA+PBS with CD24 (BD Biosciences, 563371, 1:300) and CD44 (BD Biosciences, 550989, 1:150) for 30 minutes on ice, washed, and resuspended in 1% BSA+PBS for analysis. Cells were analyzed in the VMC Flow Cytometry Shared Resource using the BD Fortessa cytometer and analyzed using FlowJo software.
Proliferation assays.
Trypan blue exclusion and CellTrace Violet proliferation assays were performed as previously described [47]. See the Supplementary Materials for detailed experimental procedures.
Cytokine treatment.
Cells were treated with vehicle (DMSO), entinostat (5μM) or panobinostat (50nM) for a total of 6 hours (MCF7) or 24 hours (MDA-MB-231b). Recombinant LIF (R&D Systems, 50 ng/ml−1) or vehicle (0.1% BSA+PBS) was added to the medium for the final 15 minutes of HDACi treatment and protein harvested as discussed above. All treatments were performed in full serum medium.
RNA-sequencing, microarray, and bioinformatics.
The NCI-60 Human Tumor Cell Lines Screen dataset was analyzed using the NCI Transcriptional Pharmacodynamics Workbench (https://tpwb.nci.nih.gov/GeneExpressionNCI60/index.html). MCF7 NSC and shLIFR RNA (n=3 independent replicates/group) was sequenced by the Stanford Functional Genomics Facility and analyzed by the VANGARD core at Vanderbilt University Medical Center as previously described [48]. The EdgeR package was used to compute log2 fold change values. These data have been deposited in the GEO database under accession number GSE121677. The patient datasets presented in Figure 5 were chosen given their relatively large patient numbers and availability of mRNA expression, tumor proliferation status, and survival outcome data. The Phase II clinical trial microarray data was partially published previously [33]. The full microarray dataset can be found in Table S1. For survival analysis, the median expression of LIFR, AMOT, and TGFB2 were calculated, and patients classified based on expression being below (“low”) or above (“high”) the median.
Chromatin Immunoprecipitation and qPCR.
Cells were plated onto 500cm2 plates (~20–25 million cells/plate) and cultured overnight before treatment with vehicle (DMSO), entinostat (5μM) or panobinostat (50nM) for 6 hours (MCF7) or 24 hours (MDA-MB-231b). Chromatin was prepared as previously described (144). See the Supplementary Materials for detailed experimental procedures. The primers used for LIFR, AMOT, TGFB2, and STAT3 promoters are listed in Table S2. Primers for LIFRprom4 and LIFRprom5 were previously published [13].
Animals.
All experiments were performed in accordance with the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals and were approved by the Vanderbilt University Institutional Animal Care and Use Committee. Orthotopic tumor experiments were performed as previously described [47]. See the Supplementary Materials for detailed experimental procedures. Treatment with vehicle (7.5% DMSO+10% HPBCD+water), entinostat (10mg/kg), or panobinostat (5mg/kg) was initiated 24-hours post-tumor cell inoculation and given 5 days/week until sacrifice. Mice were randomized for treatment and investigator blinded for histological analysis.
Histology and Immunostaining.
Primary mammary fat pad tumors were dissected and fixed in 10% formalin for 48hr. Tumors were embedded in paraffin, 5-μM thick sections were cut, and immunostaining performed as previously described [47]. See the Supplementary Materials for detailed experimental procedures.
Statistical methods.
For all studies, n per group is as indicated in the figure legend and the scatter dot plots indicate the mean of each group and error bars indicate the standard error of the mean. All graphs and statistical analyses were generated using Prism software (Graphpad). All in vitro and in vivo assays were analyzed for statistical significance using an unpaired t-test, Mann-Whitney U-test or ANOVA with Sidak’s multiple comparisons test. For all analyses P<0.05 was statistically significant, and *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplementary Material
Significance.
This work demonstrates that HDAC inhibitors stimulate a tumor dormancy gene program in breast cancer and provides new insights into the use of HDAC inhibitors to promote dormancy and improve patient outcomes.
Acknowledgements
The authors wish to acknowledge Mr. Joshua Johnson for histological processing and sectioning, Dr. Vered Stearns and her team at Johns Hopkins for their assistance in accessing the clinical patient data. Samples from Phase II clinical trial of entinostat and azacitidine [33] were obtained thanks to the Cancer Therapy Evaluation Program, National Cancer Institute (MCR-0019-P2C, U01CA070095 and UM1CA186691). The authors wish to thank the NCI Cancer Therapy Evaluation Program (CTEP), Stand Up to Cancer and the American Association for Cancer Research, Celgene Corporation and Syndax Pharmaceuticals, Lee Jeans and the Entertainment Industry Foundation (EIF), and the research teams and physicians at participating sites. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt-Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Competing Interests
The authors declare no competing financial interests. M.E.C., L.H., C.E., and R.W.J. are supported by DoD Breakthrough Award W81XWH-18-1-0029 (R.W.J.) and V.T. and R.W.J. are supported by NIH award R00CA194198 (R.W.J.). This project was also supported by scholarship funds from NIH award P30CA068485 Vanderbilt-Ingram Cancer Center Support Grant. R.M.C has received research grants to institution from Novartis, Puma Biotechnology, Merck, Genentech, and Macrogenics.
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest.
References
- 1.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. , Systemic spread is an early step in breast cancer. Cancer Cell, 2008. 13(1): p. 58–68. [DOI] [PubMed] [Google Scholar]
- 2.Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. , Early dissemination seeds metastasis in breast cancer. Nature, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguirre-Ghiso JA, Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer, 2007. 7: p. 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomis RR and Gawrzak S, Tumor cell dormancy. Molecular oncology, 2017. 11(1): p. 62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, et al. , Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer, 2011. 129(10): p. 2522–6. [DOI] [PubMed] [Google Scholar]
- 6.Falkenberg KJ and Johnstone RW, Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov, 2014. 13(9): p. 673–91. [DOI] [PubMed] [Google Scholar]
- 7.West AC and Johnstone RW, New and emerging HDAC inhibitors for cancer treatment. J Clin Invest, 2014. 124(1): p. 30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suraweera A, O’Byrne KJ, and Richard DJ, Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Frontiers in oncology, 2018. 8: p. 92–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckschlager T, Plch J, Stiborova M, and Hrabeta J, Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci, 2017. 18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, et al. , LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med, 2012. 18(10): p. 1511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iorns E, Ward TM, Dean S, Jegg A, Thomas D, Murugaesu N, et al. , Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Research and Treatment, 2012. 135(1): p. 79–91. [DOI] [PubMed] [Google Scholar]
- 12.Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, et al. , Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol, 2016. 18(10): p. 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng H, Qu J, Jin N, Xu J, Lin C, Chen Y, et al. , Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell, 2016. 30(3): p. 459–473. [DOI] [PubMed] [Google Scholar]
- 14.Yoneda T, Sasaki A, and Mundy GR, Osteolytic bone metastasis in breast cancer. Breast Cancer Research and Treatment, 1994. 32(1): p. 73–84. [DOI] [PubMed] [Google Scholar]
- 15.Campbell JP, Merkel AR, Masood-Campbell SK, Elefteriou F, and Sterling JA, Models of bone metastasis. J Vis Exp, 2012(67): p. e4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang G, Yan H, Ye S, Tong C, and Ying Q-L, STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem cells (Dayton, Ohio), 2014. 32(5): p. 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harhous Z, Booz GW, Ovize M, Bidaux G, and Kurdi M, An Update on the Multifaceted Roles of STAT3 in the Heart. Frontiers in Cardiovascular Medicine, 2019. 6(150). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X. j., Qiao Y, Xiao MM, Wang L, Chen J, Lv W, et al. , Opposing Roles of Acetylation and Phosphorylation in LIFR-Dependent Self-Renewal Growth Signaling in Mouse Embryonic Stem Cells. Cell Reports, 2017. 18(4): p. 933–946. [DOI] [PubMed] [Google Scholar]
- 19.Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, et al. , Dormancy Signatures and Metastasis in Estrogen Receptor Positive and Negative Breast Cancer. PLOS ONE, 2012. 7(4): p. e35569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. , TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol, 2013. 15(11): p. 1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, et al. , Transcriptional Switch of Dormant Tumors to Fast-Growing Angiogenic Phenotype. Cancer Research, 2009. 69(3): p. 836–844. [DOI] [PubMed] [Google Scholar]
- 22.Almog N, Briggs C, Beheshti A, Ma L, Wilkie KP, Rietman E, et al. , Transcriptional changes induced by the tumor dormancy-associated microRNA-190. Transcription, 2013. 4(4): p. 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, et al. , A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0–G1 transition. Scientific reports, 2014. 4: p. 4012–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pernodet N, Hermetet F, Adami P, Vejux A, Descotes F, Borg C, et al. , High expression of QSOX1 reduces tumorogenesis, and is associated with a better outcome for breast cancer patients. Breast Cancer Research, 2012. 14(5): p. R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, et al. , MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nature Cell Biology, 2018. 20(2): p. 211–221. [DOI] [PubMed] [Google Scholar]
- 26.Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, et al. , Chromatin-modifying enzymes as modulators of reprogramming. Nature, 2012. 483(7391): p. 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, et al. , Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep, 2016. 6: p. 36520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, and Clarke MF, Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences, 2003. 100(7): p. 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. , Genes that mediate breast cancer metastasis to the brain. Nature, 2009. 459(7249): p. 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen AL, Neumayer L, Boucher K, Factor RE, Shrestha G, Wade M, et al. , Window-of-Opportunity Study of Valproic Acid in Breast Cancer Testing a Gene Expression Biomarker. JCO Precision Oncology, 2017(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckschlager T, Plch J, Stiborova M, and Hrabeta J, Histone Deacetylase Inhibitors as Anticancer Drugs. International journal of molecular sciences, 2017. 18(7): p. 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abaza M-SI, Bahman A-M, and Al-Attiyah R.a.J., Valproic acid, an anti-epileptic drug and a histone deacetylase inhibitor, in combination with proteasome inhibitors exerts antiproliferative, pro-apoptotic and chemosensitizing effects in human colorectal cancer cells: Underlying molecular mechanisms. Int J Mol Med, 2014. 34(2): p. 513–532. [DOI] [PubMed] [Google Scholar]
- 33.Connolly RM, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, et al. , Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clinical Cancer Research, 2017. 23(11): p. 2691–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clements ME and Johnson RW, Breast Cancer Dormancy in Bone. Current Osteoporosis Reports, 2019. 17(5): p. 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cackowski FC, Eber MR, Rhee J, Decker AM, Yumoto K, Berry JE, et al. , Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J Cell Biochem, 2017. 118(4): p. 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, et al. , Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res, 2018. 78(11): p. 2911–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. , Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nature Cell Biology, 2019. 21(2): p. 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. , Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell, 2009. 16(1): p. 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, and Green JE, Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. The Journal of Clinical Investigation, 2014. 124(1): p. 156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L, and Hunter T, Urokinase Receptor and Fibronectin Regulate the ERKMAPK to p38MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Molecular Biology of the Cell, 2001. 12(4): p. 863–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. , NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun, 2015. 6: p. 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douglas AM, Goss GA, Sutherland RL, Hilton DJ, Berndt MC, Nicola NA, et al. , Expression and function of members of the cytokine receptor superfamily on breast cancer cells. Oncogene, 1997. 14: p. 661. [DOI] [PubMed] [Google Scholar]
- 43.Douglas AM, Grant SL, Gross GA, Clouston DR, Sutherland RL, and Begley CG, Oncostatin M induces the differentiation of breast cancer cells. International Journal of Cancer, 1998. 75(1): p. 64–73. [DOI] [PubMed] [Google Scholar]
- 44.Glozak MA, Sengupta N, Zhang X, and Seto E, Acetylation and deacetylation of non-histone proteins. Gene, 2005. 363: p. 15–23. [DOI] [PubMed] [Google Scholar]
- 45.Morris VL, Tuck AB, Wilson SM, Percy D, and Chambers AF, Tumor progression and metastasis in murine D2 hyperplastic alveolar nodule mammary tumor cell lines. Clinical & Experimental Metastasis, 1993. 11(1): p. 103–112. [DOI] [PubMed] [Google Scholar]
- 46.Kusuma N, Burrows A, Ling X, Jupp L, Anderson RL, and Pouliot N, Bone-derived soluble factors and laminin-511 cooperate to promote migration, invasion and survival of bone-metastatic breast tumor cells AU - Denoyer, Delphine. Growth Factors, 2014. 32(2): p. 63–73. [DOI] [PubMed] [Google Scholar]
- 47.Clements ME and Johnson RW, PREX1 drives spontaneous bone dissemination of ER+ breast cancer cells. Oncogene, 2020. 39(6): p. 1318–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson RW, Sun Y, Ho PWM, Chan ASM, Johnson JA, Pavlos NJ, et al. , Parathyroid Hormone-Related Protein Negatively Regulates Tumor Cell Dormancy Genes in a PTHR1/Cyclic AMP-Independent Manner. Frontiers in Endocrinology, 2018. 9: p. 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.