

The cardiometabolic landscape was transformed by the unexpected cardiovascular benefits seen in the landmark EMPA‐REG OUTCOME trial (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients), in which sodium glucose cotransporter 2 (SGLT2) inhibition with empagliflozin reduced major adverse cardiovascular events, cardiovascular death, all‐cause mortality, and heart failure hospitalizations.1 The benefit of SGLT2 inhibition was extended to heart failure outcomes in both people with and without diabetes mellitus in CANVAS (Canagliflozin in Diabetics with Cardiovascular Disease), DECLARE‐TIMI 58 (Dapagliflozin in Diabetics with Cardiovascular Disease or at High Cardiovascular Risk), DAPA‐HF (Dapagliflozin in Heart Failure with Reduced Ejection Fraction [HFrEF]), EMPEROR‐Reduced (Empagliflozin in HFrEF), and VERTIS‐CV (Ertugliflozin in Diabetics with Cardiovascular Disease).2, 3, 4, 5, 6 Ongoing trials are now evaluating SGLT2 inhibition on outcomes in heart failure with preserved ejection fraction: EMPEROR‐Preserved (NCT03057951; empagliflozin) and DELIVER (Dapagliflozin Evaluation to Improve the Lives of Patients with Preserved Ejection Fraction Heart Failure; NCT03619213; dapagliflozin).
Although the benefit of SGLT2 inhibition on cardiovascular outcomes including heart failure is clear, the mechanisms by which this benefit is realized are debated. SGLT2 inhibitors lower glucose by acting on the SLC5A2 gene encoding the SGLT2 to promote glucosuria and natriuresis. Although SLC5A2 gene expression is highly restricted to the kidney, the magnitude of cardiovascular benefit with SGLT2 inhibitors supports action beyond this kidney. Moreover historically, glucose‐lowering therapies on their own have not improved cardiovascular outcomes. Furthermore, specific agents such as thiazolidinediones have worsened outcomes.7, 8 Although the diuretic effect of SGLT2 inhibitors is also thought to contribute to its cardiovascular effectiveness, diuretics for volume unloading do not provide a cardiovascular mortality benefit.9 Several additional mechanisms of action for SGLT2 inhibitors are now being explored at the myocardial level (Figure), and one very intriguing area is the modulation of cardiac mitochondrial function and metabolism.
Figure 1. Effects of SGLT2 inhibition at the myocardial level.
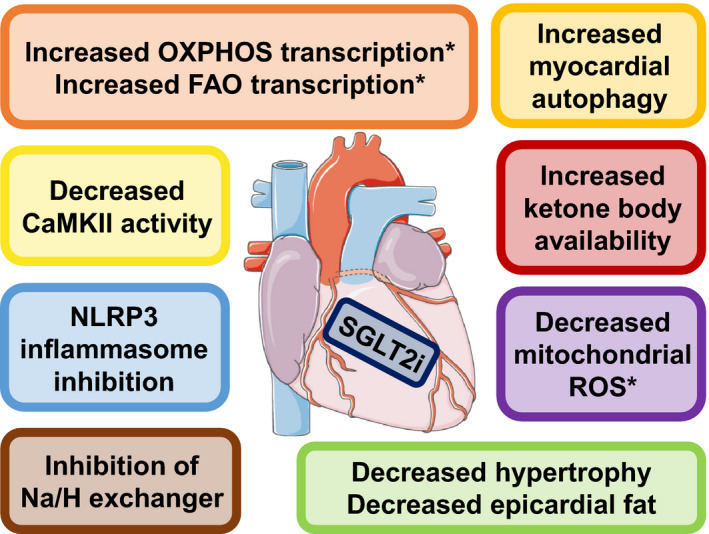
SGLT2 inhibitors act on the heart to exert potential cardioprotective effects. Asterisks (*) mark myocardial processes highlighted in the accompanying article. CaMKII indicates calcium/calmodulin‐dependent protein kinase II; FAO, fatty acid oxidation; Na/H, sodium/hydrogen; NLRP3, NLR family pyrin domain‐containing 3; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; and SGLT2i, sodium glucose cotransporter 2 inhibition. The art pieces used in this figure were provided by Servier Medical Art (http://servier.com) under a Creative Commons Attribution license.
Mitochondrial dysfunction occurs in both the diabetic and failing heart, but no current therapeutics directly target cardiac mitochondria.10 There is evidence that SGLT2 inhibition improves cardiac mitochondrial function in animal models unrelated to diabetes mellitus status. In nondiabetic pigs with ischemic cardiomyopathy, empagliflozin reduced pathological cardiac remodeling and associated with increased myocardial oxidation of fatty acids, ketone bodies, and branched‐chain amino acids.11 In a genetic mouse model of diabetes mellitus, empagliflozin increased overall cardiac ATP production by 30% and associated with an increase in the rates of cardiac glucose and fatty acid oxidation, but with no change in the rate of ketone oxidation. Interestingly, empagliflozin‐treated diabetic mice demonstrated elevated circulating ketone levels, suggesting that the increase in ketone body availability with empagliflozin may serve as an additional source of cardiac ATP production without affecting myocardial ketone oxidation rates.12
In this issue of the Journal of the American Heart Association (JAHA), Croteau and colleagues add to this growing evidence base by demonstrating that SGLT2 inhibition with ertugliflozin improves maximal systolic function and reprograms cardiac mitochondrial metabolism regardless of diabetes mellitus status.13 The authors used a well‐characterized high‐fat high‐sucrose (HFHS) diet in adult mice to induce a diabetic cardiomyopathy phenotype of left ventricular hypertrophy, myocyte hypertrophy and fibrosis, and diastolic dysfunction. At the time of HFHS or control diet initiation, adult mice were randomized to ertugliflozin or control treatment groups. After 4 months, cardiac function was assessed by echocardiography and in working hearts. Ventricular tissue was also harvested to study mitochondrial function and gene expression.
As expected, ertugliflozin prevented the development of insulin resistance in HFHS‐fed mice with lower fasting glucose and insulin levels. Ertugliflozin also prevented left ventricular hypertrophy, myocyte hypertrophy and fibrosis, and diastolic dysfunction in HFHS‐fed mice. Systolic contractile reserve was severely impaired in beating hearts isolated from HFHS‐fed mice, but this improved to supra‐normal levels in ertugliflozin‐treated HFHS mice. Mitochondria from HFHS‐fed hearts showed reduced overall energy production and increased oxidative stress compared with control mitochondria, and these markers of mitochondrial function were normalized in ertugliflozin‐treated HFHS‐fed mitochondria. Gene set enrichment analysis of RNA sequence analyses of left ventricular tissue showed that the top 20 gene ontology sets enriched by ertugliflozin were all related to mitochondrial structure or function. Oxidative phosphorylation was the highest‐ranking hallmark gene set enriched when analyzed by diet or ertugliflozin treatment—whereas oxidative phosphorylation was significantly downregulated by HFHS diet, it was upregulated by a similar magnitude in ertugliflozin‐treated mice on the HFHS diet.
These results are particularly interesting in the context of primary prevention of heart failure. SGLT2 inhibitors reduce the risk of heart failure among patients with diabetes mellitus, and the 2020 American College of Cardiology Expert Consensus Decision Pathway recommends a patient–clinician discussion about initiating SGLT2 inhibitors for patients with diabetes mellitus who are at very high risk of developing heart failure or atherosclerotic cardiovascular disease.14 In this study in mice, ertugliflozin and HFHS diet were initiated concurrently starting at 8 weeks of age, consistent with initiating SGLT2 inhibition before the onset of the diabetes mellitus‐inducing diet. Ertugliflozin not only prevented diastolic dysfunction in HFHS‐fed mice but also augmented maximal systolic function as measured by contractile reserve. These results suggest that SGLT2 inhibitors may also play a role in the primary prevention of heart failure and mitochondrial dysfunction in normoglycemic individuals who are at very high risk of developing diabetes mellitus and diabetic cardiomyopathy.
Because ertugliflozin was administered in mice before the onset of the diabetic cardiomyopathy phenotype, it remains to be seen if SGLT2 inhibition can rewire mitochondrial metabolism in mice with established diabetic cardiomyopathy. Diabetic cardiomyopathy is associated with extensive mitochondrial metabolic reprogramming, characterized by an overreliance on fatty acids as a fuel source coupled with inefficient fatty acid oxidation, leading to the accumulation of toxic lipid intermediates.15 Interestingly, the second‐most upregulated hallmark gene set in the ertugliflozin‐treated mice regardless of diet status was fatty acid metabolism. Future research is needed to assess if SGLT2 inhibition can transcriptionally upregulate cardiac fatty acid oxidation in established diabetic cardiomyopathy to better match the increase in cellular fatty acid uptake, prevent the accumulation of lipotoxic molecules, and potentially attenuate cardiac dysfunction.
Although several studies have examined the cardiac effects of SGLT2 inhibition in animal models of heart failure, this paper explores the effects of SGLT2 inhibition on cardiac function in working hearts using metabolic transcriptomics in nondiabetic control animals without heart failure. Surprisingly, Croteau and colleagues showed that the effects of ertugliflozin on contractile reserve and metabolic gene expression were not limited to mice with diabetes mellitus. Ertugliflozin treatment impressively increased contractile reserve in beating hearts from mice fed a normal diet by nearly 25% compared with untreated control mice. No significant change was seen in phosphocreatine or overall myocardial ATP concentrations, suggesting that ertugliflozin improves overall myocardial energy balance or contractile efficiency. Myocardial transcriptomics also showed that ertugliflozin upregulated the oxidative phosphorylation gene set to a similar extent in normal chow‐fed hearts compared with HFHS‐fed hearts, demonstrating that the effect of ertugliflozin on oxidative phosphorylation gene expression is independent of diabetes mellitus. Interestingly, ertugliflozin increased left ventricular systolic pressure in control mice. This is in contrast to human studies that have shown that SGLT2 inhibitors have a mild blood pressure lowering effect, suggesting that the myocardial metabolic and hemodynamic effects of SGLT2 inhibitors in mice may not be directly applicable to humans.
Although the authors do not identify a unifying mechanism for the beneficial effects of ertugliflozin on systolic contractile function in nondiabetic and diabetic mice, the results are hypothesis generating. Genes encoding the oxidoreductase NADH (nicotinamide adenine dinucleotide + hydrogen) dehydrogenase are among the top gene ontology terms enriched by ertugliflozin after controlling for diet. This enzyme is part of complex I of the mitochondrial electron transport chain and converts nicotinamide adenine dinucleotide (NAD) from its reduced form (NADH) to its oxidized form (NAD+). NADH dehydrogenase controls the balance of NAD+/NADH redox, which in turn regulates substrate oxidation, NAD+ dependent protein deacetylation, and the mitochondrial inflammatory response. Low NAD+ levels and reduced NAD+/NADH ratio are seen in the failing heart, which impair overall metabolic flux and mitochondrial function. Additionally, prior studies have shown that NAD+ repletion improves mitochondrial and cardiac function in mouse models of HFrEF and heart failure with preserved ejection fraction.16, 17 Given the lack of SGLT2 expression in the heart, ertugliflozin may indirectly reprogram cardiac mitochondrial NAD+/NADH balance through extracardiac plasma metabolites or intermediates. Alternatively, SGLT2 inhibitors may act directly on cardiac mitochondria through off‐target effects, such as inhibition of NHE1 (cardiac sodium/hydrogen exchanger).18 Further mechanistic studies are needed to assess the potential role of SGLT2 inhibition on cardiac NAD+ homeostasis. Additional studies are needed to more comprehensively assess the possible effects of SGLT2 inhibition on cardiac mitochondrial function, such as mitochondrial protein levels, post‐translational modifications, oxidative capacity, metabolic flux, and dynamics. Metabolic studies in both animals and humans are also needed to better understand the cardioprotective effects of SGLT2 inhibition and who will benefit the most from this therapy.
Broad questions regarding the cardioprotective effects of SGLT2 inhibition remain unanswered in the field. Can we identify a priori those most likely to have cardiovascular benefits from SGLT2 inhibition? Is there a role for SGLT2 inhibitors in preventing cardiovascular events in prediabetes mellitus? Who is at the highest risk for side effects from SGLT2 inhibition, such as genital infections and euglycemic ketoacidosis? Do SGLT2 inhibitors confer cardioprotection through similar mechanisms in heart failure, diabetes mellitus, and chronic kidney disease? At the population‐health level, should SGLT2 inhibitors supplant metformin as first‐line therapy for diabetes mellitus? Basic and clinical investigation will need to address these and many other questions over the next several years.
In summary, this study demonstrates that SGLT2 inhibition with ertugliflozin improves not only gross cardiac function but also cardiomyocyte function at the level of the mitochondria in mice independent of diabetes mellitus. It also adds to the compelling evidence base that modulation of mitochondrial metabolism may serve as a promising cardiovascular therapeutic target, both in people with and without diabetes mellitus. There is still much to be learned about the cardiometabolic mechanisms of SGLT2 inhibitors, and this study takes an important step in demonstrating their effects on the heart.
Disclosures
Dr McNally serves as a consultant to Amgen, AstraZeneca, Avidity, 4D Molecular Therapeutics, Cytokinetics, Janssen, Pfizer, PepGen, Invitae, and Tenaya Therapeutics. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2021;10:e021949. DOI: 10.1161/JAHA.121.021949.)
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
For Disclosures, see page 4.
See Article by Croteau et al.
References
- 1.Zinman B, Lachin JM, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2016;374:1094. DOI: 10.1056/NEJMc1600827. [DOI] [PubMed] [Google Scholar]
- 2.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–657. DOI: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 3.Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–357. DOI: 10.1056/NEJMoa1812389. [DOI] [PubMed] [Google Scholar]
- 4.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. DOI: 10.1056/NEJMoa1911303. [DOI] [PubMed] [Google Scholar]
- 5.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383:1413–1424. DOI: 10.1056/NEJMoa2022190. [DOI] [PubMed] [Google Scholar]
- 6.Cannon CP, Pratley R, Dagogo‐Jack S, Mancuso J, Huyck S, Masiukiewicz U, Charbonnel B, Frederich R, Gallo S, Cosentino F, et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N Engl J Med. 2020;383:1425–1435. DOI: 10.1056/NEJMoa2004967. [DOI] [PubMed] [Google Scholar]
- 7.Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail‐Beigi F, Grimm RH Jr, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. DOI: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dormandy JA, Charbonnel B, Eckland DJA, Erdmann E, Massi‐Benedetti M, Moules IK, Skene AM, Tan MH, Lefèbvre PJ, Murray GD, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. DOI: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed A, Husain A, Love TE, Gambassi G, Dell'Italia LJ, Francis GS, Gheorghiade M, Allman RM, Meleth S, Bourge RC. Heart failure, chronic diuretic use, and increase in mortality and hospitalization: an observational study using propensity score methods. Eur Heart J. 2006;27:1431–1439. DOI: 10.1093/eurheartj/ehi890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–3726. DOI: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos‐Gallego CG, Requena‐Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E, Garcia‐Ropero A, Sanz J, Hajjar RJ, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019;73:1931–1944. DOI: 10.1016/j.jacc.2019.01.056. [DOI] [PubMed] [Google Scholar]
- 12.Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci. 2018;3:575–587. DOI: 10.1016/j.jacbts.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croteau D, Luptak I, Chambers JM, Hobai I, Panagia M, Pimentel DR, Siwik DA, Qin F, Colucci WS. Effects of sodium‐glucose linked transporter 2 inhibition with ertugliflozin on mitochondrial function, energetics and metabolic gene expression in the presence and absence of diabetes in mice. J Am Heart Assoc. 2021;10:e019995. DOI: 10.1161/JAHA.120.019995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das SR, Everett BM, Birtcher KK, Brown JM, Januzzi JL Jr, Kalyani RR, Kosiborod M, Magwire M, Morris PB, Neumiller JJ, et al. 2020 expert consensus decision pathway on novel therapies for cardiovascular risk reduction in patients with type 2 diabetes: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2020;76:1117–1145. DOI: 10.1016/j.jacc.2020.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart–deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. 2013;2:e000433. DOI: 10.1161/JAHA.113.000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. 2018;137:2256–2273. DOI: 10.1161/CIRCULATIONAHA.116.026099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdellatif M, Trummer‐Herbst V, Koser F, Durand S, Adão R, Vasques‐Nóvoa F, Freundt JK, Voglhuber J, Pricolo M‐R, Kasa M, et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med. 2021;13:eabd7064. DOI: 10.1126/scitranslmed.abd7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. 2018;61:722–726. DOI: 10.1007/s00125-017-4509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]