

Abstract
Background
MEDI6012 is recombinant human lecithin cholesterol acyltransferase, the rate‐limiting enzyme in reverse cholesterol transport. Infusions of lecithin cholesterol acyltransferase have the potential to enhance reverse cholesterol transport and benefit patients with coronary heart disease. The purpose of this study was to test the safety, pharmacokinetic, and pharmacodynamic profile of MEDI6012.
Methods and Results
This phase 2a double‐blind study randomized 48 subjects with stable coronary heart disease on a statin to a single dose of MEDI6012 or placebo (6:2) (NCT02601560) with ascending doses administered intravenously (24, 80, 240, and 800 mg) and subcutaneously (80 and 600 mg). MEDI6012 demonstrated rates of treatment‐emergent adverse events that were similar to those of placebo. Dose‐dependent increases in high‐density lipoprotein cholesterol were observed with area under the concentration‐time curves from 0 to 96 hours of 728, 1640, 3035, and 5318 should be: mg·h/mL in the intravenous dose groups and 422 and 2845 mg·h/mL in the subcutaneous dose groups. Peak mean high‐density lipoprotein cholesterol percent change was 31.4%, 71.4%, 125%, and 177.8% in the intravenous dose groups and 18.3% and 111.2% in the subcutaneous dose groups, and was accompanied by increases in endogenous apoA1 (apolipoprotein A1) and non‐ATP‐binding cassette transporter A1 cholesterol efflux capacity. Decreases in apoB (apolipoprotein B) were observed across all dose levels and decreases in atherogenic small low‐density lipoprotein particles by 41%, 88%, and 79% at the 80‐, 240‐, and 800‐mg IV doses, respectively.
Conclusions
MEDI6012 demonstrated an acceptable safety profile and increased high‐density lipoprotein cholesterol, endogenous apoA1, and non‐ATP‐binding cassette transporter A1 cholesterol efflux capacity while reducing the number of atherogenic low‐density lipoprotein particles. These findings are supportive of enhanced reverse cholesterol transport and a functional high‐density lipoprotein phenotype.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: NCT02601560.
Keywords: atherosclerosis, cholesterol efflux capacity, cholesterol homeostasis, cholesterol reverse cholesterol transport, high‐density lipoprotein cholesterol
Subject Categories: Secondary Prevention, Clinical Studies, Lipids and Cholesterol, Atherosclerosis, Cardiovascular Disease
Nonstandard Abbreviations and Acronyms
- ABCA1
ATP‐binding cassette transporter A1
- ADA
anti‐drug antibodies
- AUC0‐96h
area under the curve from 0–96 hours
- CE
cholesterol ester
- CEC
cholesterol efflux capacity
- CETP
cholesteryl ester transfer protein
- HDL‐CE
high‐density lipoprotein cholesteryl ester
- LCAT
lecithin cholesterol acyltransferase
- RCT
reverse cholesterol transport
- REAL
Reduction of Infarct Size in Acute MI With LCAT
Clinical Perspective
What Is New?
Intravenous administration of MEDI6012, recombinant human lecithin cholesterol acyltransferase, to patients with coronary artery disease results in increases in high‐density lipoprotein cholesteryl ester and apoA1 (apolipoprotein A1), improvements in cholesterol efflux capacity, and concomitant decreases in apoB (apolipoprotein B) and total and small low‐density lipoprotein particles.
What Are the Clinical Implications?
The lipid, apolipoprotein, and cholesterol efflux changes induced by MEDI6012 support the hypothesis that infusions of MEDI6012 can enhance reverse cholesterol transport and possibly regress or reduce the progression of atherosclerotic plaque and lower major adverse cardiovascular events.
High‐density lipoprotein (HDL) has long been a therapeutic target for the prevention and treatment of atherosclerotic cardiovascular disease. Epidemiologic studies have demonstrated a consistent reduced risk of cardiovascular events with higher levels of HDL cholesterol (HDL‐C).1, 2, 3 However, pharmacologic therapies aiming to increase HDL‐C with niacin and cholesteryl ester (CE) transfer protein (CETP) inhibitors have, for the most part, had no4, 5, 6, 7 or just a modest8 effect in reducing major adverse cardiovascular events. Furthermore, trials using apoA1 (apolipoprotein A1) mimetics have not demonstrated reductions in atherosclerotic plaque progression.9, 10 Explanations include the fact that CETP inhibitors block low‐density lipoprotein (LDL) receptor–mediated reverse cholesterol transport (RCT), resulting in no significant change in fecal sterol excretion,11 and the finding that some apoA1 mimetics (apoA1‐Milano) inhibit cholesterol esterification.12 On the basis of these observations, some have suggested that increasing cholesterol esterification or activating CETP could be an effective way of enhancing LDL receptor–mediated RCT.13
MEDI6012 is recombinant human lecithin‐cholesterol acyltransferase (LCAT), a plasma enzyme secreted by the liver that is rate limiting in the maturation of HDL particles as it catalyzes the esterification of unesterified cholesterol to CE. LCAT has long been proposed to play a role in RCT,14 and hence may also be important in the development of atherosclerosis.15 Two prior open‐label studies, without placebo control, of recombinant human LCAT (ACP‐501) have been reported in coronary heart disease and familial LCAT deficiency.16, 17 The purpose of this study was to test the safety, pharmacokinetic, and pharmacodynamic profiles of MEDI6012 in subjects with stable coronary heart disease on statin therapy in a placebo‐controlled manner using MEDI6012, which has a higher specific activity compared with the former ACP‐501.
Methods
Study Design
This is a phase 2a, randomized, blinded (subject/investigator blinded, MedImmune/Astrazeneca unblinded), placebo‐controlled dose escalation trial (NCT02601560) to evaluate the safety, pharmacokinetics, and pharmacodynamics of single doses of MEDI6012. The trial was conducted in 8 sites in the United States and randomized a total of 48 subjects to MEDI6012 or placebo (6:2). MEDI6012 dose levels included 24, 80, 240, and 800 mg via intravenous infusion and 80 and 600 mg via subcutaneous injection (Figure 1). The study was conducted according to guidelines for Good Clinical Practice protocol, informed‐consent documents were approved by the appropriate institutional review boards, and study participants provided informed consent before any study‐related procedures were performed. Data underlying the findings described in this article may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. All relevant data are within the article and its supporting information files.
Figure 1. Study flow diagram and additional design details.
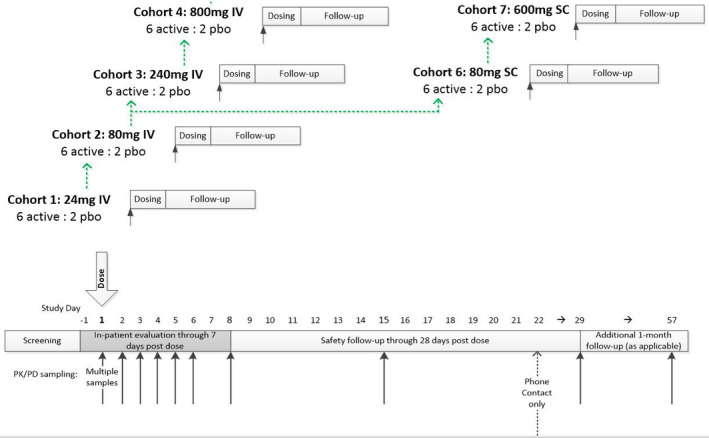
pbo indicates placebo; PD, pharmacodynamics; and PK, pharmacokinetics.
Patient Population
The study included both men and postmenopausal or surgically sterile women aged 40 through 75 years who had stable coronary heart disease, were diagnosed ≥3 months before screening, and were receiving statin therapy for ≥6 weeks. Other requirements included body mass index of 18 to 45 kg/m2, systolic blood pressure of ≥100 and <160 mm Hg and diastolic blood pressure of <90 mm Hg, and heart rate of ≥45 and <85 beats per minute. Exclusion criteria were severe angina (Canadian Cardiovascular Society Class III or higher) or heart failure (New York Association Class III or higher); severe peripheral artery disease; left ventricular ejection fraction of <35% or left ventricular aneurysm; high‐risk coronary or carotid disease; recent (<3 months) stroke, transient ischemic attack, or major surgery; recent (<6 months) acute coronary syndrome or hospitalization for heart failure; clinically significant ECG abnormalities that may interfere with the interpretation of serial ECG and Q‐T interval changes; presence of an intracardiac device; recent (<12 months) myocarditis or restrictive pericarditis; severe valvular disease; history of acute aortic syndrome or aneurysm; disturbances in cholesterol metabolism; uncontrolled endocrine disorder or use of systemic corticosteroids; ongoing renal or liver disease; fasting triglycerides of >500 mg/dL, LDL cholesterol (LDL‐C) of >150 mg/dL and HDL‐C of >60 mg/dL for men and >65 mg/dL for women; use of nonstatin lipid‐lowering medications or supplements; chronic infection or recent febrile illness; and history of severe allergy or hypersensitivity, including any component of the investigational product formulation or other biological agent.
Study Procedures
Following a 28‐day screening period (or up to 42 days for subjects who required a washout of a concomitant medication), subjects were admitted before randomization (day −1) and dose administration (day 1) and remained at the study center for 8 days. Subjects were followed through 28 days after the dose of investigational product (day 29 visit). Timing of pharmacokinetic and pharmacodynamic sampling are noted in Figure 1. Additional follow‐up was done in subjects with positive anti‐drug antibody (ADA) immunogenicity results on day 29.
For intravenous cohorts, MEDI6012 or placebo was administered over 60 minutes. For subcutaneous cohorts, MEDI6012 or placebo was administered as subcutaneous injections in the lower abdomen. Safety monitoring included vital signs, ECG assessments, telemetry (for intravenous administration), and laboratory safety tests.
The primary pharmacodynamic end point was serum concentration of HDL‐C; secondary pharmacodynamic end points were serum concentrations of other key lipids, lipoproteins, and apolipoproteins (Medpace Research Laboratories, Cincinnati, OH): total cholesterol, unesterified cholesterol, CE, HDL‐CE, HDL unesterified cholesterol, non–HDL‐C, non–HDL‐CE, non–HDL unesterified cholesterol, LDL‐C (direct measure), apoB, apoA1, and pre‐beta HDL. For samples requiring fasting, a minimum of 6 hours was required. Subjects were also asked to refrain from strenuous exercise and alcohol consumption for ≈48 hours before any study visit. The serum concentration of MEDI6012 was measured by enzymatic method (MPI Research, Mattawan, MI).
Exploratory pharmacodynamic end points investigated in this study included lipoprotein size and composition assessed by nuclear magnetic resonance spectroscopy (LipoScience, Morrisville, NC) and ex vivo non–ATP binding cassette 1 (non‐ABCA1) and ABCA1 cholesterol efflux capacity (CEC) (Vascular Strategies, Plymouth Meeting, PA). In addition, an inflammation suppression assay was used to test the anti‐inflammatory properties of HDL from patients before and after treatment (Data S1).
Adverse events and serious adverse events were collected from the time of informed consent until the end of the follow‐up period (day 29 or longer, if applicable) and were graded by severity and relationship to investigational product.
The immunogenicity potential of MEDI6012 was assessed with an ADA screening assay, followed by a confirmatory assay and titer evaluations when applicable. Samples positive for ADA were further characterized as either neutralizing or nonneutralizing with an enzymatic assay (MPI Research).
Statistical Analysis
A total of 48 subjects were randomized in a 6:2 ratio to receive MEDI6012 or placebo. The sample size was empirically determined to provide adequate safety, tolerability, and pharmacokinetic/pharmacodynamic data to achieve study objectives while exposing as few subjects as possible to the investigational product and study procedures.
Categorical data were summarized by the number and percentage of subjects in each category. Continuous variables were summarized by descriptive statistics, including mean, standard deviation, median, and minimum and maximum, as appropriate. Data analyses were conducted with SAS software version 9.3 or higher (SAS Institute, Cary, NC).
The primary analysis of the safety and pharmacodynamic end point was performed on the basis of the as‐treated population. The pharmacokinetic and immunogenicity data were analyzed in the pharmacokinetic population and the immunogenicity population, respectively (Table S1).
The primary pharmacodynamic variable of interest was the baseline‐adjusted area under the curve from 0 to 96 hours (AUC0–96h) for HDL‐C, calculated using the trapezoidal rule. Area under the curve between‐treatment group comparisons were determined by using ANCOVA, with adjustment of the baseline values and the treatment group.
For pharmacokinetic parameters, the actual time of pharmacokinetic sampling, rather than the nominal (planned) sampling time, was used to derive pharmacokinetic parameters with noncompartmental model 200 to 202 for extravascular administration, using Phoenix WinNonlin 6.3 (Pharsight, Mountain View, CA) according to the standard operating procedures described in Standard Methods for the Non‐Compartmental Analysis of Pharmacokinetic Data.18 The pharmacokinetic parameters were determined from the concentration‐time data for MEDI6012 if data allowed.
Nominal P values are presented in this article, and no multiplicity adjustment was performed.
Results
Baseline Characteristics and Treatment Allocation
The study randomized 48 individuals to MEDI6012 (n=36) or placebo (n=12) at MEDI6012 doses of 24, 80, 240, or 800 mg IV (n=24) or doses of 80 or 600 mg SC (n=12).
Baseline characteristics were generally balanced and reflected a population with stable coronary heart disease on statin therapy (Table 1). The mean age of trial participants was 64 years, and 81.3% were men. Baseline mean levels of HDL‐C were 34.7±7.4 mg/dL and 39.1±9.2 mg/dL in the MEDI6012 intravenous and subcutaneous groups, and 35.0±12.8 mg/dL and 33.8±7.1 mg/dL in the placebo intravenous and subcutaneous groups. Baseline mean levels of LDL‐C (direct assay) were 77.9±24.3 mg/dL and 79.2±16.6 mg/dL in the MEDI6012 intravenous and subcutaneous groups, and 79.4±23.2 mg/dL and 84.3±21.5 mg/dL in the placebo intravenous and subcutaneous groups.
Table 1.
Baseline Characteristics
| Characteristics | Placebo Intravenous, n=8 | MEDI6012, 24 mg IV, n=6 | MEDI6012, 80 mg IV, n=6 | MEDI6012, 240 mg IV, n=6 | MEDI6012, 800 mg IV, n=6 | Placebo SC, n=4 | MEDI6012, 80 mg SC, n=6 | MEDI6012, 600 mg SC, n=6 |
|---|---|---|---|---|---|---|---|---|
| Age, y, median (range) | 63.5 (52–73) | 62.5 (48–66) | 58.0 (54–71) | 63.5 (54–70) | 69.5 (55–73) | 64.0 (49–71) | 72.0 (65–74) | 67.0 (59–73) |
| Men | 7 (87.5) | 6 (100) | 3 (50.0) | 4 (66.7) | 5 (83.3) | 3 (75.0) | 6 (100) | 5 (83.3) |
| BMI, median (range) | 28.59 (21.3–39.9) | 35.83 (34.5–39.1) | 32.40 (27.1–36.6) | 35.04 (21.8–40.2) | 32.50 (27.3–39.3) | 33.48 (29.0–36.0) | 29.35 (25.7–35.0) | 31.79 (28.1–36.5) |
| Race | ||||||||
| Asian | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 |
| Black | 2 (25.0) | 1 (16.7) | 2 (33.3) | 0 | 2 (33.3) | 1 (25.0) | 0 | 0 |
| White | 6 (75.0) | 5 (83.3) | 3 (50.0) | 6 (100) | 4 (66.7) | 3 (75.0) | 6 (100) | 6 (100) |
| Prior MI | 4 (50.0) | 5 (83.3) | 2 (33.3) | 5 (83.3) | 2 (33.3) | 1 (25.0) | 3 (50.0) | 4 (66.7) |
| Prior PCI | 6 (75.0) | 5 (83.3) | 4 (66.7) | 6 (100) | 5 (83.3) | 2 (50.0) | 1 (16.7) | 3 (50.0) |
| Prior CABG | 4 (50.0) | 0 | 3 (50.0) | 1 (16.7) | 2 (33.3) | 2 (50.0) | 3 (50.0) | 3 (50.0) |
| Current/prior smoker | 6 (75.0) | 5 (83.3) | 5 (83.3) | 4 (66.7) | 4 (66.7) | 4 (100) | 5 (83.3) | 4 (66.7) |
| Diabetes mellitus | 5 (62.5) | 2 (33.3) | 3 (50.0) | 3 (50.0) | 3 (50.0) | 1 (25.0) | 3 (50.0) | 3 (50.0) |
| Hypertension | 8 (100) | 6 (100) | 6 (100) | 4 (66.7) | 6 (100) | 4 (100) | 6 (100) | 5 (83.3) |
| Dyslipidemia | 8 (100) | 6 (100) | 6 (100) | 5 (83.3) | 6 (100) | 4 (100) | 6 (100) | 6 (100) |
| Baseline HDL‐C, median (range) | 31.0 (20–52) | 35.0 (29–49) | 37.0* (28–40) | 29.0 (25–43) | 35.0 (22–49) | 35.0 (24–41) | 37.0* (32–52) | 37.0* (28–53) |
| Baseline LDL‐C, median (range) | 84.5 (45–106) | 104.5 (66–120) | 80.0* (72–116) | 62.0 (44–118) | 65.5 (37–70) | 81.5 (62–112) | 75.0* (60–105) | 72.0* (64–110) |
Values are number (%) unless otherwise indicated. Ranges are shown as minimum–maximum. BMI indicates body mass index (kg/m2); CABG, coronary artery bypass graft; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; MI, myocardial infarction; and PCI, percutaneous coronary intervention.
N=5.
HDL‐C and Other Key Pharmacodynamic End Points
Single doses of MEDI6012 resulted in dose‐dependent increases in the primary pharmacodynamic end point: the baseline‐adjusted AUC0–96h for HDL‐C across all dose levels, with peak concentrations occurring between days 2 and 4 (Figures 2A and 3A). Consistent with the mechanism of action of MEDI6012, the baseline‐adjusted AUC0–96h for HDL‐CE and CE increased in a dose‐dependent manner across all intravenous and subcutaneous treatment groups (Figures 2B, 2C, 3B, and 3C), reaching statistical significance at the 80‐, 240‐, and 800‐mg IV doses and 600‐mg SC doses. Baseline‐adjusted area under the curve from time 0 to 168 hours demonstrated similar increases (data not shown). Percent change from baseline in peak mean HDL‐C was 31.4%, 71.4%, 125%, and 178% in the 24‐, 80‐, 240‐, and 800‐mg IV dose groups, respectively, and 18.3% and 111% in the 80‐ and 600‐mg SC dose groups, respectively. Percent CE increased in a dose‐dependent manner when referenced to HDL‐C, total cholesterol, and non–HDL‐C (Figure S1). In addition, the baseline‐adjusted AUC0–96h of endogenous apoA1 increased significantly across all intravenous doses and with the 600‐mg SC dose (Figures 2D and 3D). AUC0–96h for select lipid parameters can be found in Table S2.
Figure 2. Change from baseline (milligrams per deciliter) in serum concentration over time for HDL‐C (A), HDL‐CE (B), CE (C), ApoA1 (D), LDL‐C (E), and ApoB (F).

Mean baseline levels are listed in the following order: placebo intravenous, MEDI6012 intravenous group, placebo subcutaneous, MEDI6012 SC group, respectively. HDL‐C: 35, 34.7, 33.8, and 39.1 mg/dL; HDL‐CE: 28.9, 29, 28.8, and 33 mg/dL; CE: 108.3, 103.3, 114, and 110.4 mg/dL; apoA1: 127.0, 121.3, 116.3, and 129.5 mg/dL. LDL‐C: 82.7, 78, 80.5, and 78.2 mg/dL; apoB: 79.1, 74.1, 80.8, and 74.4 mg/dL. Error bars show standard error of the mean. ApoA1 indicates apolipoprotein A1; ApoB, apolipoprotein B; CE, cholesteryl ester; HDL‐C, high‐density lipoprotein cholesterol; HDL‐CE, high‐density lipoprotein cholesteryl ester; and LDL‐C, low‐density lipoprotein cholesterol.
Figure 3. Box and whisker plots for area under the curve (AUC) from 0 to 96 hours (milligrams per hour per deciliter) for high‐density lipoprotein cholesterol (HDL‐C) (A), high‐density lipoprotein cholesteryl ester (HDL‐CE) (B), cholesteryl ester (C), apolipoprotein A1 (D), low‐density lipoprotein cholesterol (LDL‐C) (E), and apolipoprotein B (F).

Results for other lipid measurements can be found in Table S2. Error bars show standard error of the mean. P‐IV indicates placebo (intravenous); and P‐SC, placebo (subcutaneous).
LDL‐C, ApoB, and LDL Particle Number
We hypothesized that enhancement of LDL receptor–mediated RCT would result in the transfer of cholesterol and CE from HDL particles to LDL particles, resulting in an increase in LDL‐C content but no increase in LDL particle number. There was no change in baseline‐adjusted AUC0–96h for LDL‐C in the 24‐ and 80‐mg IV or the 80‐mg SC dose groups (Figures 2E and 3E). However, at higher doses, LDL‐C increased in the 240‐mg IV group (P=0.096) and the 800‐mg IV group (P=0.002). The peak in LDL‐C occurred on day 4, following the peak in HDL‐CE on days 2 to 3 (Figure 2B). Baseline‐adjusted AUC0–96h for apoB significantly decreased across all intravenous dose cohorts of MEDI6012 (Figures 2F and 3F) and was not dose dependent.
Nuclear magnetic resonance spectroscopy was used to measure the effect of MEDI6012 on lipoprotein particle size and number. An increase in LDL particle size was observed at all dose levels except the 80‐mg SC group. A decrease in total LDL particle number was observed at the 80‐ and 240‐mg IV doses. Marked maximum decreases in small LDL particles of 41±26%, 88±7%, and 79±21% were seen at the 80‐, 240‐, and 800‐mg IV dose levels, respectively (Figure 4A), and an increase in large LDL particles was seen in the 240‐ and 800‐mg IV dose groups (data not shown).
Figure 4. Change from baseline in small low‐density lipoprotein (LDL) particle concentration overall (A) and apolipoprotein B (ApoB) area under the curve from 0 to 96 hours (AUC0–96h) (B) according to statin intensity.

Statin intensity was classified according to the American College of Cardiology/American Heart Association guidelines on treatment of blood cholesterol.19 P values were calculated using the Mann‐Whitney nonparametric 2‐tailed t test.
Post hoc, we hypothesized that the reduction in apoB may be related to the intensity of statin therapy,19 because statins would increase the number of LDL receptors expressed on hepatocytes. If MEDI6012 is enhancing LDL receptor–mediated RCT, this would be more evident in subjects taking high‐intensity statins. Subjects treated with MEDI6012 who were on high‐intensity statins had significantly lower apoB levels than those taking placebo (mean AUC0–96h, −740.3 versus 104.7; P=0.005) and MEDI6012 patients receiving low‐intensity statins (mean AUC0–96h, −740.3 versus 378.2; P=0.01) (Figure 4B).
Pre‐Beta HDL and Ex Vivo CEC
Administration of LCAT resulted in dose‐dependent decreases in pre‐beta HDL (Figure 5A). No significant change was seen in ABCA1 efflux across all doses except the 240‐mg dose (Figure 5B). However, dose‐dependent increases in global and non‐ABCA1 CEC were observed (Figure 5C and 5D). Ex vivo inflammation suppression assays performed in the 600‐mg SC cohort demonstrated that MEDI6012 resulted in HDL that was capable of suppressing the secretion of proinflammatory cytokines interferon‐γ, interleukin‐1β, interleukin‐8, and tumor necrosis factor‐α within 7 days (see Data S1 and Figure S2).
Figure 5. Change from baseline in pre–beta‐1 high‐density lipoprotein (preB1‐HDL) (A), ATP‐binding cassette A1 (ABCA1) (B), Global (C), and non‐ABCA1 (D) cholesterol efflux capacity (CEC).

Pharmacokinetics of MEDI6012
The MEDI6012 concentration‐time profile and pharmacokinetic parameters are shown in Table 2 and Figure S3. In summary, MEDI6012 concentrations increased rapidly in a dose‐proportional manner (Figure S3) and declined, with a mean half‐life of 17.6 hours for the 24‐mg IV dose and mean half‐life of 45.7 to 55.4 hours for the 80‐ to 800‐mg IV doses, and exhibited linear pharmacokinetics. Peak exposure and systemic plasma exposure (area under the curve to infinite time) increased proportionally between the 80‐ and 800‐mg IV doses (Table 2). Mean clearance appeared to be dose independent and was 199 mL/h for the 24‐mg dose and 79.9–103 mL/h at the 80‐ to 800‐mg IV doses. At 24 mg, the high clearance rate (199 mL/h) and short half‐life (17.6 hours) were associated with the low concentration of MEDI6012 at this dose and the assay sensitivity (below the limit of measurement, <2.5 μg/mL).
Table 2.
Summary of Noncompartmental Analysis Pharmacokinetic Parameter Estimates
| MEDI6012, 24 mg IV, n=6 | MEDI6012, 80 mg IV, n=6 | MEDI6012, 240 mg IV, n=6 | MEDI6012, 800 mg IV, n=6 | MEDI6012, 600 mg SC, n=6 | |
|---|---|---|---|---|---|
| Cmax, µg/mL, mean (SD) | 5.02 (2.25) | 20.2 (4.15) | 73.5 (9.40) | 229 (55.4) | 22.8 (9.75) |
| Tmax, h, mean (SD)* | 1.00–1.00 | 1.00–1.50 | 1.00–2.00 | 1.00–1.50 | 48.0–72.0 |
| t1/2, h, mean (SD)† | 17.6 (5.47) | 46.9 (17.6) | 45.7 (3.67) | 55.4 (14.2) | 89.5 (46.7) |
| AUC0–168, µg·h/mL, mean (SD)† | 137 (55.0) | 805 (265) | 2760 (249) | 8470 (2000) | 2460 (824) |
| AUC0‐last, µg·h/mL, mean (SD) | 46.8 (42.7) | 683 (267) | 2760 (249) | 9020 (2760) | 2460 (824) |
| AUC0‐inf, µg·h/mL, mean (SD)† | 137 (56.1) | 887 (308) | 3030 (298) | 9510 (2720) | 3560 (732) |
| CL or CL/F, mL/h, mean (SD)† | 199 (80.7) | 103 (47.1) | 79.9 (7.47) | 89.9 (24.6) | 176 (46.8) |
| Vss, L, mean (SD)† | 4.73 (1.14) | 5.85 (1.53) | 4.97 (0.442) | 5.41 (0.603) | NR |
AUC0–168 indicates area under the concentration‑time curve to from 0 to 168 hours; AUC0‐inf, area under the concentration‑time curve to infinite time; AUC0‐last, area under the concentration‑time curve to the last measurable timepoint; CL, clearance; Cmax, first occurrence of the maximum observed plasma concentration determined directly from the raw concentration‑time data; F, fraction of the subcutaneous dose absorbed; NR, not reported; t1/2, elimination half‑life; Tmax, time to which Cmax was determined directly from raw concentration‑time data; and Vss, volume of distribution at steady state.
Range, as minimum–maximum, is provided instead of mean.
n=4.
For subscuaneous administration, MEDI6012 levels from the 80‐mg SC dose group were predominantly below the limit of quantification (<2.5 µg/mL). After the 600‐mg SC administration, MEDI6012 was absorbed with a mean time to maximum concentration of 56.0 hours (Table 2). MEDI6012 concentrations declined after reaching peak exposure, with a mean half‐life of 89.5 hours. Mean peak exposure, area under the curve to infinite time, and clearance of the fraction of subcutaneous dose absorbed are shown in Table 2. The relative bioavailability derived based on mean dose‐normalized area under the curve to infinite time between the 600‐mg SC to the 80‐mg IV, 240‐mg IV, and 800‐mg IV doses was 53.5%, 47.0%, and 49.8%, respectively.
LCAT mass and activity showed a strong linear correlation (R=0.98, P<0.001), and there was no plateau at higher doses (Figure S4).
Safety and Adverse Events
A single dose of MEDI6012 was generally safe and well tolerated in this trial. There were no treatment‐emergent serious adverse events or deaths. No significant changes in safety laboratory parameters, vital signs, or ECG parameters were noted after either intravenous or subcutaneous dosing.
Treatment‐emergent adverse events were reported by 55.6% (20/36 subjects) of all MEDI6012‐treated subjects and 41.7% (5/12 subjects) of placebo‐treated subjects (Table S3). For the MEDI6012‐treated subjects, the most frequent treatment‐emergent adverse events in the intravenous cohorts were headache (2/24 subjects, 8.3%) and irritation at the site of the medical device (2/24 subjects, 8.3%). In the subcutaneous cohorts, the most frequent treatment‐emergent adverse events for MEDI6012‐treated subjects were injection site reaction (3/12 subjects, 25.0%), injection site erythema, headache, and constipation (each 2/12 subjects, 16.7%). Most treatment‐emergent adverse events were mild or moderate in severity. One grade 3 treatment‐emergent adverse event of presyncope was considered unrelated to MEDI6012 by the investigator. The incidence of treatment‐related, treatment‐emergent adverse events was not greater with higher doses of MEDI6012.
Immunogenicity
ADAs were detected in 5 of 36 subjects treated with MEDI6012, 3 from the 800‐mg IV group and 1 each from the 80‐ and 600‐mg SC groups. In 4 of the 5 subjects, the ADA titer was <1, with no notable changes in HDL‐C and no neutralizing antibody. In the remaining subject (80‐mg SC group), HDL‐C declined from 35 mg/dL at baseline to 17 mg/dL on day 57, increased to 28 mg/dL on day 128, and returned to an above‐baseline level of 38 mg/dL on day 247 despite continued elevated ADA titers of 64:1 to 256:1. There was no change in percent CE, and an enzymatic assay that determined the ability of LCAT to esterify a fluorescent analog of cholesterol was negative for neutralizing antibody.
DISCUSSION
This phase 2a, single‐ascending‐dose study testing MEDI6012 in patients with stable coronary heart disease on statin therapy demonstrated that (1) a single dose of MEDI6012 is safe and well tolerated and that MEDI6012 results in (2) dose‐dependent increases in HDL‐C driven by increases in CE in the HDL particle, consistent with the mechanism of action of LCAT; (3) dose‐dependent increases in endogenous apoA1; (4) improvements in HDL function via improvements in non‐ABCA1 CEC; (5) a favorable profile of LDL particles with an increase in LDL‐C and concomitant decreases in apoB and total and small LDL particle numbers; and that (6) MEDI6012 may work synergistically with statins with greater decreases in apoB with high‐intensity statin therapy. Together, these findings support the concept that treatment with MEDI6012 may enhance LDL receptor–mediated RCT.
MEDI6012 Mechanism of Action
MEDI6012 is human recombinant LCAT, the rate‐limiting enzyme in RCT that esterifies unesterified cholesterol in HDL particles (Figure 6). LCAT works by binding to apoA1 on pre‐beta HDL and larger HDL particles. Unesterified cholesterol in HDL is esterified to become CE, which is pulled into the HDL core and creates a unidirectional gradient of cholesterol movement from cholesterol‐loaded cells to HDL. The majority of CE in HDL will be transferred to apoB‐containing particles via CETP. The vast majority of CE transferred from HDL to apoB‐containing particles is destined for uptake by the LDL receptor on hepatocytes (major route), completing the cycle of LDL receptor–mediated RCT.20 A minority of these HDL particles will be taken up by the SRB1 (scavenger receptor class B type 1) receptor on the liver (minor route).
Figure 6. Effect of MEDI6012 on biomarkers of reverse cholesterol transport.

αHDL indicates alpha high‐density lipoprotein; ABCA1, ATP‐binding cassette A1; ABCG1, ATP‐binding cassette transporter G1; ApoA1, indicates apolipoprotein A1; ApoB, apolipoprotein B; CE, cholesteryl ester; CETP, cholesteryl ester transfer protein; FC, free cholesterol; HDL, high‐density lipoprotein; HDL‐C, high‐density lipoprotein cholesterol; HDL‐CE, high‐density lipoprotein cholesteryl ester; LCAT, lecithin cholesterol acyltransferase; LDL, low‐density lipoprotein; LDL‐C, low‐density lipoprotein cholesterol; LDLR, low‐density lipoprotein receptor; pre‐β‐HDL, pre‐beta high‐density lipoprotein; SRB1, Scavenger Receptor Class B type 1; TG, triglyceride; and VLDL, very low‐density lipoprotein.
The results from this phase 2a study support the mechanism of action of LCAT and show that infusions of MEDI6012 result in rapid increases in HDL‐C, HDL‐CE, and CE. The transfer of CE to LDL via CETP is evident, with peaks in LDL‐C that follow the peak of HDL‐C and HDL‐CE. The apoB levels decreased with infusions of MEDI6012, indicating that larger CE‐rich LDL particles may be more readily taken up by the LDL receptor. Indirectly, the greater decrease in apoB‐containing particles with higher‐intensity statin therapy suggests that apoB‐containing particle uptake is enhanced in the presence of MEDI6012 and with the higher LDL receptor expression that occurs with more intense statin therapies. These findings together support the concept that MEDI6012 may enhance LDL receptor–mediated RCT.
This phase 2a study follows a phase 1b, open‐label, single‐dose, dose‐escalation study with recombinant human LCAT, formerly known as ACP‐501.17 Although both studies infused LCAT in patients with coronary heart disease, the specific activity of the MEDI6012 used in this study is higher, as demonstrated by the much lower doses used and the much higher HDL‐C levels achieved in the current study. For example, the 3.0‐mg/kg dose of ACP‐501 equated to a mean dose of 279 mg and achieved a 19% maximum increase in HDL‐C. In comparison, a 240‐mg dose of MEDI6012 achieved a 115% maximum increase in HDL‐C, a 6‐fold difference. Similarly, responses were less for ACP‐501, compared with MEDI6012, for CE, apoA1, and total cholesterol. The time course of changes in lipids was similar between the two LCAT preparations in regard to the onset of changes in total cholesterol, HDL‐C, and LDL‐C.
LCAT, CETP, and LDL Receptor–Mediated RCT
The failures of the CETP inhibitors torcetrapib, dalcetrapib, and evacetrapib, as well as modest reductions in major cardiovascular events with anacetrapib, have demonstrated that increasing HDL‐C by blocking the transfer of CE to the LDL particle is not an effective method for reducing major adverse cardiovascular events.4, 6, 7, 8 CETP inhibitors and MEDI6012 share some similarities in the first step of reverse cholesterol transport, CEC, which is inversely associated with cardiovascular events.21 MEDI6012 increased global CEC mainly driven by an increase in ATP‐binding cassette transporter G1 CEC. The lack of an increase in ABCA1 CEC is likely secondary to the decrease seen in pre‐beta1. This is indirect evidence that efflux may have occurred in vivo with the consumption of pre‐beta1 particles that are no longer available to show their effects in an ex vivo assay. In contrast, evacetrapib increases both ABCA1 and ATP‐binding cassette transporter G1 efflux capacity and increases pre‐beta1 particles.22 Similarly, dalcetrapib also increases CEC, but this depends on genotype.23, 24 However, CETP inhibitors raise HDL levels by placing a roadblock in LDL receptor–mediated RCT, blocking the transfer of CE to apoB‐containing particles. This has led to some calling for CETP activators, not inhibitors.13 Although MEDI6012 is not a direct CETP activator, it does provide additional substrate for CETP to transfer to the LDL particle, a completely different method of increasing HDL‐C than CETP inhibitors.
It has been known for some time that LCAT has the potential to enhance LDL receptor–mediated RCT,25 possibly enabling regression of atherosclerotic plaque. Hoeg and colleagues demonstrated that overexpression of LCAT in transgenic rabbits prevents diet‐induced atherosclerosis.26 In nonhuman primates, overexpression of human LCAT reduced apoB levels because of an increase in LDL catabolism.27 Furthermore, it was demonstrated that the mechanism of action of LCAT is via the LDL receptor when it was shown that human LCAT increased the catabolism of LDL particles only when the LDL receptor is present, again in transgenic rabbits.11 Similar results were shown in nonhuman primates that express human LCAT.27 Together, these studies demonstrate that overexpression of LCAT enhances LDL receptor–mediated RCT, resulting in atheroma regression. In the current study, total number of LDL particles, especially small LDL particles, and levels of apoB decreased. The decrease in apoB appeared to be enhanced by high‐intensity statins, the human equivalent of increasing LDL receptors, as has been done in prior preclinical studies. These findings support the hypothesis that LCAT may enhance RCT via the LDL receptor.
One question that does arise from this study is whether LDL receptor–mediated RCT could be maxed out at higher doses. This is unclear from the current data. However, LDL‐C rises significantly with the highest dose of MEDI6012, and at this dose, total LDL particle numbers are not significantly decreased. However, apoB does decrease significantly across all intravenous doses in a non–dose‐dependent manner.
LCAT, ApoA1, and ApoB
Reductions in cardiovascular risk seen with increased levels of HDL‐C track with apoA1 levels,1 making apoA1 a target for the prevention of cardiovascular diseases. Furthermore, the ratio of apoB to apoA1 is an independent predictor of cardiovascular events, because higher ratios are associated with higher event rates. In the current study, infusions of MEDI6012 resulted in decreases in apoB and increases in endogenous apoA1, making this ratio more favorable for potential cardiovascular risk reduction.28 We did observe that although higher doses of MEDI6012 continued to increase HDL cholesterol content, going from the 240‐mg to the 800‐mg dose did not further increase the level of apoA1. The reason for this cannot be answered by this study but may suggest there may be less return in regard to apoA1 with higher doses. Several attempts have been made to increase apoA1 via infusions of apoA1 particles. Synthetic apoA1 particles packaged with phospholipids have failed to demonstrate atheroma regression on invasive imaging studies.9, 10, 29 The lack of efficacy of these therapies may be related to evidence that these particles result in the inhibition of LCAT, as seen with recombinant apoA1‐Milano complexed with palmitoyl‐oleoyl‐phosphatidylcholine30 or sphingomyelin complexed with recombinant HDL.31 The inhibition of LCAT probably accounted for the decreases in HDL‐C and LDL‐C seen in the MDCO‐216 Infusions Leading to Changes in Atherosclerosis: a Novel Therapy in Development to Improve Cardiovascular Outcomes—Proof of Concept IVUS, Lipids, and Other Surrogate Biomarkers (MILANO‐PILOT) trial,9 as opposed to the findings from infusions of LCAT in this study.
Limitations and Future Clinical Development of MEDI6012
The results from this phase 2a, single‐ascending‐dose study support further development of MEDI6012 as a therapy in patients at risk for cardiovascular events. However, there are several limitations that need to be taken into consideration. First, this is a small study with a single dose of MEDI6012, and further studies with multiple doses are warranted. Second, although the changes in lipid levels demonstrated in this study support the concept of enhanced LDL receptor–mediated RCT, these measurements are once‐in‐time evaluations and do not adequately describe the kinetics of lipid metabolism changes with MEDI6012. A tracer kinetics study is being finalized that will further evaluate these findings. Finally, MEDI6012 is a recombinant enzyme with a half‐life of ≈2 days, and the bioavailability was limited to ≈50% with subcutaneous dosing. Furthermore, injection site reactions and injection site erythema occurred in a third of subjects receiving a single dose of MEDI6012. Taken together, these attributes favor MEDI6012 to be a once‐weekly intravenous therapy that will be best suited for a short‐term and perhaps acute therapy.
One potential option is acute myocardial infarction, for which there is emerging evidence that apoA1 and HDL may have a role in cardioprotection. Epidemiologic and preclinical studies have established that higher levels of HDL‐C are cardioprotective in patients after myocardial infarction, and infusions of HDL or apoA1 mimetics reduce myocardial infarct size and improve left ventricular systolic function in animal models of acute myocardial infarction.32, 33, 34, 35, 36 This cardioprotective mechanism is supported by the presence of HDL and its constituent sphingosine 1 phosphate and occurs via the RISK/SAFE (reperfusion injury salvage kinase/survivor activating factor enhancement) pro‐survival kinase pathways.37 The increases in apoA1 seen in these studies are similar to the increases seen with MEDI6012.
In addition, although HDL has anti‐inflammatory properties in healthy individuals, the systemic oxidative stress and inflammation that are present in chronic disease states like heart disease and diabetes mellitus leads to alterations in HDL that render it dysfunctional and proinflammatory.38, 39, 40 This dysfunctional HDL displays impaired CEC,41 impaired ability to prevent oxidation of LDL,42 and impaired ability to protect against endothelial dysfunction.43
In these disease states, abnormal HDL can become proinflammatory and contribute to oxidative damage. The present study demonstrated that the anti‐inflammatory properties of HDL from a single dose of LCAT treatment in study participants was improved over baseline in terms of the ability to suppress cytokine production in response to a proinflammatory stimulus. Interestingly, the extent of anti‐inflammation was seen at 7 days after treatment, just past the peak in HDL‐C levels in patients receiving LCAT (see Data S1). This suggests that HDL generated by this treatment mimics the physiological properties of healthy HDL.
On the basis of the potential of MEDI6012 for atheroma regression and post–myocardial infarction cardioprotection and its pharmacokinetic/pharmacodynamic profile, MEDI6012 is being tested in the REAL‐TIMI (Reduction of Infarct Size in Acute MI With LCAT‐Thrombolysis in Myocardial Infarction) 63B phase 2b trial (NCT03578809).44 This trial has completed enrolling patients with acute ST‐segment–elevation myocardial infarction, where we will test the potential for MEDI6012 to be a first‐in‐class therapy for myocardial and atheroprotection. This proof‐of‐concept study will determine whether MEDI6012 can reduce infarct size, improve ejection fraction, and regress coronary atheroma, using state‐of‐the‐art, noninvasive imaging.
In summary, this phase 2a, single‐ascending‐dose study demonstrates that infusions of MEDI6012 result in dose‐dependent increases in HDL‐C, HDL‐CE, and CE, followed by increases in LDL‐C content and decreases in apoB levels and LDL particle numbers. Together, these findings indirectly support the concept of enhanced LDL receptor–mediated RCT by MEDI6012. Future clinical development of MEDI6012 will leverage its unique mechanism of action and pharmacokinetic/pharmacodynamic profile to rapidly increase functional HDL‐C in the setting of acute myocardial infarction in the REAL‐TIMI 63B phase 2b trial.
Sources of Funding
This study was sponsored by MedImmune (One MedImmune Way, Gaithersburg, MD), a wholly owned subsidiary of AstraZeneca. The sponsor was responsible for the design and conduct of the study; the collection, analysis, and interpretation of the data; and the preparation, review, and approval of the article.
Disclosures
Dr George is an employee of AstraZeneca. Dr George, Dr Karathanasis, Dr She, Dr Buss, C. Jin, and Dr Hirshberg have stock ownership and/or stock options in AstraZeneca. Dr Abuhatzira and Dr Stoughton had stock ownership/options in AstraZeneca at the time the research was performed. Drs She and Abuhatzira are current employees of Viela Bio. Dr Karathanasis is an employee of NeoProgen and is a volunteer for the National Heart, Lung, and Blood Institute, National Institutes of Health. Dr Buss is an employee of REGENXBIO Inc. Dr Stoughton is an employee of C4 Medical Writing, LLC. C. Jin is an employee of Gilead Sciences. R. Bakker‐Arkema was a paid consultant for MedImmune (AstraZeneca) at the time the research was performed and is currently at BIA Clinical Group. Dr Koren received research funding from AstraZeneca to enroll subjects whose data are reported in this article.
Supporting information
Data S1
Tables S1–S3
Figures S1–S4
Acknowledgments
Editorial support was provided by D. Shuman of AstraZeneca.
(J Am Heart Assoc. 2021;10:e014572. DOI: 10.1161/JAHA.119.014572.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.119.014572
For Sources of Funding and Disclosures, see page 13.
REFERENCES
- 1.Collaboration ERF , Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. DOI: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pekkanen J, Linn S, Heiss G, Suchindran CM, Leon A, Rifkind BM, Tyroler HA. Ten‐year mortality from cardiovascular disease in relation to cholesterol level among men with and without preexisting cardiovascular disease. N Engl J Med. 1990;322:1700–1707. DOI: 10.1056/NEJM199006143222403. [DOI] [PubMed] [Google Scholar]
- 3.Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988;8:737–741. DOI: 10.1161/01.atv.8.6.737. [DOI] [PubMed] [Google Scholar]
- 4.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, Lopez‐Sendon J, Mosca L, Tardif J‐C, Waters DD, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. DOI: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 5.Investigators A‐H , Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes‐Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. DOI: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 6.Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, Gibson CM, Granger C, Menon V, Montalescot G, et al. Evacetrapib and cardiovascular outcomes in high‐risk vascular disease. N Engl J Med. 2017;376:1933–1942. DOI: 10.1056/NEJMoa1609581. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. DOI: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 8.Group HT‐RC , Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, Sammons E, et al. Effects of anacetrapib in ptients with atherosclerotic vascular disease. N Engl J Med. 2017;377:1217–1227. DOI: 10.1056/NEJMoa1706444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls SJ, Puri R, Ballantyne CM, Jukema JW, Kastelein JJP, Koenig W, Wright RS, Kallend D, Wijngaard P, Borgman M, et al. Effect of infusion of high‐density lipoprotein mimetic containing recombinant apolipoprotein A‐I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO‐PILOT trial: a randomized clinical trial. JAMA Cardiol. 2018;3:806–814. DOI: 10.1001/jamacardio.2018.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tardif J‐C, Ballantyne CM, Barter P, Dasseux J‐L, Fayad ZA, Guertin M‐C, Kastelein JJP, Keyserling C, Klepp H, Koenig W, et al. Effects of the high‐density lipoprotein mimetic agent CER‐001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. 2014;35:3277–3286. DOI: 10.1093/eurheartj/ehu171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brousseau ME, Diffenderfer MR, Millar JS, Nartsupha C, Asztalos BF, Welty FK, Wolfe ML, Rudling M, Björkhem I, Angelin BO, et al. Effects of cholesteryl ester transfer protein inhibition on high‐density lipoprotein subspecies, apolipoprotein A‐I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25:1057–1064. DOI: 10.1161/01.ATV.0000161928.16334.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kempen HJ, Gomaraschi M, Simonelli S, Calabresi L, Moerland M, Otvos J, Jeyarajah E, Kallend D, Wijngaard PLJ. Persistent changes in lipoprotein lipids after a single infusion of ascending doses of MDCO‐216 (apoA‐IMilano/POPC) in healthy volunteers and stable coronary artery disease patients. Atherosclerosis. 2016;255:17–24. DOI: 10.1016/j.atherosclerosis.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita S, Matsuzawa Y. Re‐evaluation of cholesteryl ester transfer protein function in atherosclerosis based upon genetics and pharmacological manipulation. Curr Opin Lipidol. 2016;27:459–472. DOI: 10.1097/MOL.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 14.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 15.Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang X‐C, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. DOI: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shamburek RD, Bakker‐Arkema R, Auerbach BJ, Krause BR, Homan R, Amar MJ, Freeman LA, Remaley AT. Familial lecithin:cholesterol acyltransferase deficiency: first‐in‐human treatment with enzyme replacement. J Clin Lipidol. 2016;10:356–367. DOI: 10.1016/j.jacl.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shamburek RD, Bakker‐Arkema R, Shamburek AM, Freeman LA, Amar MJ, Auerbach B, Krause BR, Homan R, Adelman SJ, Collins HL, et al. Safety and tolerability of ACP‐501, a recombinant human lecithin: cholesterol acyltransferase, in a phase 1 single‐dose escalation study. Circ Res. 2016;118:73–82. DOI: 10.1161/CIRCRESAHA.115.306223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veng‐Pedersen P. Theorems and implications of a model independent elimination/distribution function decomposition of linear and some nonlinear drug dispositions. I. Derivations and theoretical analysis. J Pharmacokinet Biopharm. 1984;12:627–648. DOI: 10.1007/BF01059557. [DOI] [PubMed] [Google Scholar]
- 19.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd‐Jones DM, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–S45. DOI: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–1607. DOI: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. DOI: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholls SJ, Ruotolo G, Brewer HB, Kane JP, Wang MD, Krueger KA, Adelman SJ, Nissen SE, Rader DJ. Cholesterol efflux capacity and pre‐beta‐1 HDL concentrations are increased in dyslipidemic patients treated with evacetrapib. J Am Coll Cardiol. 2015;66:2201–2210. DOI: 10.1016/j.jacc.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Ballantyne CM, Miller M, Niesor EJ, Burgess T, Kallend D, Stein EA. Effect of dalcetrapib plus pravastatin on lipoprotein metabolism and high‐density lipoprotein composition and function in dyslipidemic patients: results of a phase IIb dose‐ranging study. Am Heart J. 2012;163:515–521, 521.e1–3. DOI: 10.1016/j.ahj.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Tardif J‐C, Rhainds D, Brodeur M, Feroz Zada Y, Fouodjio R, Provost S, Boulé M, Alem S, Grégoire JC, L’Allier PL, et al. Genotype‐dependent effects of dalcetrapib on cholesterol efflux and inflammation: concordance with clinical outcomes. Circ Cardiovasc Genet. 2016;9:340–348. DOI: 10.1161/CIRCGENETICS.116.001405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norum KR, Remaley AT, Miettinen HE, Strøm EH, Balbo BEP, Sampaio CATL, Wiig I, Kuivenhoven JA, Calabresi L, Tesmer JJ, et al. Lecithin:cholesterol acyltransferase: symposium on 50 years of biomedical research from its discovery to latest findings. J Lipid Res. 2020;61:1142–1149. DOI: 10.1194/jlr.S120000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoeg JM, Santamarina‐Fojo S, Berard AM, Cornhill JF, Herderick EE, Feldman SH, Haudenschild CC, Vaisman BL, Hoyt RF Jr, Demosky SJ Jr, et al. Overexpression of lecithin:cholesterol acyltransferase in transgenic rabbits prevents diet‐induced atherosclerosis. Proc Natl Acad Sci USA. 1996;93:11448–11453. DOI: 10.1073/pnas.93.21.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amar MJ, Shamburek RD, Vaisman B, Knapper CL, Foger B, Hoyt RF Jr, Santamarina‐Fojo S, Brewer HB Jr, Remaley AT. Adenoviral expression of human lecithin‐cholesterol acyltransferase in nonhuman primates leads to an antiatherogenic lipoprotein phenotype by increasing high‐density lipoprotein and lowering low‐density lipoprotein. Metabolism. 2009;58:568–575. DOI: 10.1016/j.metabol.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walldius G, Jungner I. The apoB/apoA‐I ratio: a strong, new risk factor for cardiovascular disease and a target for lipid‐lowering therapy—a review of the evidence. J Intern Med. 2006;259:493–519. DOI: 10.1111/j.1365-2796.2006.01643.x. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls SJ, Andrews J, Kastelein JJP, Merkely B, Nissen SE, Ray KK, Schwartz GG, Worthley SG, Keyserling C, Dasseux JL, et al. Effect of serial infusions of CER‐001, a pre‐beta high‐density lipoprotein mimetic, on coronary atherosclerosis in patients following acute coronary syndromes in the CER‐001 atherosclerosis regression acute coronary syndrome trial: a randomized clinical trial. JAMA Cardiol. 2018;3:815–822. DOI: 10.1001/jamacardio.2018.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kempen HJ, Gomaraschi M, Bellibas SE, Plassmann S, Zerler B, Collins HL, Adelman SJ, Calabresi L, Wijngaard PL. Effect of repeated apoA‐IMilano/POPC infusion on lipids, (apo)lipoproteins, and serum cholesterol efflux capacity in cynomolgus monkeys. J Lipid Res. 2013;54:2341–2353. DOI: 10.1194/jlr.M033779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rye KA, Hime NJ, Barter PJ. The influence of sphingomyelin on the structure and function of reconstituted high density lipoproteins. J Biol Chem. 1996;271:4243–4250. DOI: 10.1074/jbc.271.8.4243. [DOI] [PubMed] [Google Scholar]
- 32.Gordts SC, Muthuramu I, Nefyodova E, Jacobs F, Van Craeyveld E, De Geest B. Beneficial effects of selective HDL‐raising gene transfer on survival, cardiac remodelling and cardiac function after myocardial infarction in mice. Gene Ther. 2013;20:1053–1061. DOI: 10.1038/gt.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heywood SE, Richart AL, Henstridge DC, Alt K, Kiriazis H, Zammit C, Carey AL, Kammoun HL, Delbridge LM, Reddy M, et al. High‐density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci Transl Med. 2017;9:eaam6084. DOI: 10.1126/scitranslmed.aam6084. [DOI] [PubMed] [Google Scholar]
- 34.Marchesi M, Booth EA, Davis T, Bisgaier CL, Lucchesi BR. Apolipoprotein A‐IMilano and 1‐palmitoyl‐2‐oleoyl phosphatidylcholine complex (ETC‐216) protects the in vivo rabbit heart from regional ischemia‐reperfusion injury. J Pharmacol Exp Ther. 2004;311:1023–1031. DOI: 10.1124/jpet.104.070789. [DOI] [PubMed] [Google Scholar]
- 35.Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, et al. High‐density lipoproteins and their constituent, sphingosine‐1‐phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. DOI: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- 36.Wang TD, Wu CC, Chen WJ, Lee CM, Chen MF, Liau CS, Sung FC, Lee YT. Dyslipidemias have a detrimental effect on left ventricular systolic function in patients with a first acute myocardial infarction. Am J Cardiol. 1998;81:531–537. DOI: 10.1016/S0002-9149(97)00974-0. [DOI] [PubMed] [Google Scholar]
- 37.Kalakech H, Hibert P, Prunier‐Mirebeau D, Tamareille S, Letournel F, Macchi L, Pinet F, Furber A, Prunier F. RISK and SAFE signaling pathway involvement in apolipoprotein A‐I‐induced cardioprotection. PLoS One. 2014;9:e107950. DOI: 10.1371/journal.pone.0107950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high‐density lipoprotein. Curr Opin Lipidol. 2007;18:427–434. DOI: 10.1097/MOL.0b013e3282364a17. [DOI] [PubMed] [Google Scholar]
- 39.Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST, et al. Inflammatory/antiinflammatory properties of high‐density lipoprotein distinguish patients from control subjects better than high‐density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. DOI: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 40.Ossoli A, Simonelli S, Varrenti M, Morici N, Oliva F, Stucchi M, Gomaraschi M, Strazzella A, Arnaboldi L, Thomas MJ, et al. Recombinant LCAT (lecithin:cholesterol acyltransferase) rescues defective HDL (high‐density lipoprotein)‐mediated endothelial protection in acute coronary syndrome. Arterioscler Thromb Vasc Biol. 2019;39:915–924. DOI: 10.1161/ATVBAHA.118.311987. [DOI] [PubMed] [Google Scholar]
- 41.Shao B, Tang C, Sinha A, Mayer PS, Davenport GD, Brot N, Oda MN, Zhao XQ, Heinecke JW. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high‐density lipoprotein oxidation by myeloperoxidase. Circ Res. 2014;114:1733–1742. DOI: 10.1161/CIRCRESAHA.114.303454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navab M, Reddy ST, Van Lenten BJ, Anantharamaiah GM, Fogelman AM. The role of dysfunctional HDL in atherosclerosis. J Lipid Res. 2009;50(suppl):S145–S149. DOI: 10.1194/jlr.R800036-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monette JS, Hutchins PM, Ronsein GE, Wimberger J, Irwin AD, Tang C, Sara JD, Shao B, Vaisar T, Lerman A, et al. Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced HDL particle concentration. Circ Res. 2016;119:83–90. DOI: 10.1161/CIRCRESAHA.116.308357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonaca MP, George RT, Morrow DA, Bergmark BA, Park J‐G, Abuhatzira L, Vavere AL, Karathanasis SK, Jin C, She D, et al. Recombinant human Lecithin‐Cholesterol acyltransferase in patients with atherosclerosis: phase 2a primary results and phase 2b design. Eur Heart J Cardiovasc Pharmacother. 2021:pvab001. Jan 25 [epub ahead of print]. DOI: 10.1093/ehjcvp/pvab001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S3
Figures S1–S4