

Abstract
In ovoid-shaped Gram-positive bacteria, MapZ guides FtsZ-ring positioning at cell equators. The cell wall of the ovococcus Streptococcus mutans contains peptidoglycan decorated with Serotype c Carbohydrates (SCCs). In this study, we identify the major cell separation autolysin AtlA as an SCC binding protein. AtlA binding to SCC is attenuated by the glycerol phosphate (GroP) modification. Using fluorescently-labeled AtlA constructs, we mapped SCC distribution on the streptococcal surface revealing enrichment of GroP-deficient immature SCCs at the cell poles and equators. The immature SCCs co-localize with MapZ at the equatorial rings throughout the cell cycle. In GroP-deficient mutants, AtlA is mislocalized resulting in dysregulated cellular autolysis. These mutants display morphological abnormalities associated with MapZ mislocalization leading to FtsZ-ring misplacement. Altogether, our data support a model in which maturation of a cell wall polysaccharide provides the molecular cues for the recruitment of cell division machinery, ensuring proper daughter cell separation and FtsZ-ring positioning.
Keywords: Streptococcus, autolysin, AtlA, glycerol phosphate, cell wall polysaccharide, FtsZ, MapZ
Graphical Abstract
Introduction
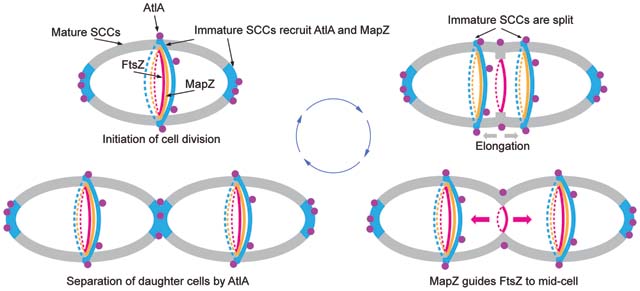
The cell wall of Gram-positive bacteria consists of peptidoglycan with covalently attached anionic glycopolymers known as wall teichoic acids (WTAs)1. This rigid structure confers the characteristic cell shape. The morphogenesis of ovoid-shaped Gram-positive bacteria, such as streptococci, arises from a combination of septal and lateral peptidoglycan synthesis, orchestrated by the divisome and elongasome multiprotein complexes, respectively2–4. Cell division is initiated by the recruitment and polymerization of FtsZ to form a ring (Z-ring) at mid-cell marked by a microscopically visible “equatorial ring”2,5,6. The Z-ring serves as a scaffold to assemble the cell division machinery4,6. In Streptococcus pneumoniae, synthesis of septal and lateral walls occurs almost simultaneously5. Early in division, the parental equatorial ring splits forming new equatorial rings in the daughter cells2. The newly formed rings migrate toward the equators of the daughter cells powered by splitting of the septal ingrowth and synthesis of the lateral wall2. When the equatorial ring approaches the mid-cell region of the daughter cell, elongation is halted. After completion, the septum is split by peptidoglycan hydrolases, so-called autolysins, to allow the proper separation of the daughter cells2,5,6.
Correct Z-ring placement depends on the chromosomal origin of replication7 and the FtsZ-binding protein MapZ8,9. In S. pneumoniae, MapZ forms a stable protein ring that co-migrates with the equatorial ring8,9, facilitating the alignment of the Z-ring perpendicular to the long axis of the cell7 and guiding the migration of FtsZ throughout the cell cycle10. MapZ localization and assembly into the ring depend on the association of the MapZ extracellular domain with an unknown cell wall component residing in the equatorial ring8,11.
WTA-deficient mutants exhibit morphological abnormalities, defects in daughter cell separation, and increased autolysis12–16, indicating a functional connection between WTA and the cell division machinery. In many streptococci, peptidoglycan carries rhamnose (Rha)-containing polysaccharides with repeating →3)α-Rha(1→2)α-Rha(1→ disaccharide backbone modified with species-specific and serotype-specific glycosyl side-chains17. Streptococcus mutans serotype c-specific carbohydrate termed SCC contains α-glucose (Glc) side-chains attached to polyrhamnose18. The homologous polysaccharide in Group A Streptococcus (GAS), the group A carbohydrate (GAC), carries β-N-acetylglucosamine (GlcNAc) side-chains19,20. SCC and GAC are anionic polysaccharides, through decoration with glycerol phosphate (GroP) moieties (Fig. 1a)21. GroP is attached to the GlcNAc side-chains of GAC at the C6 hydroxyl group21.
Fig. 1. Modification of SCC with Glc side-chains and GroP.
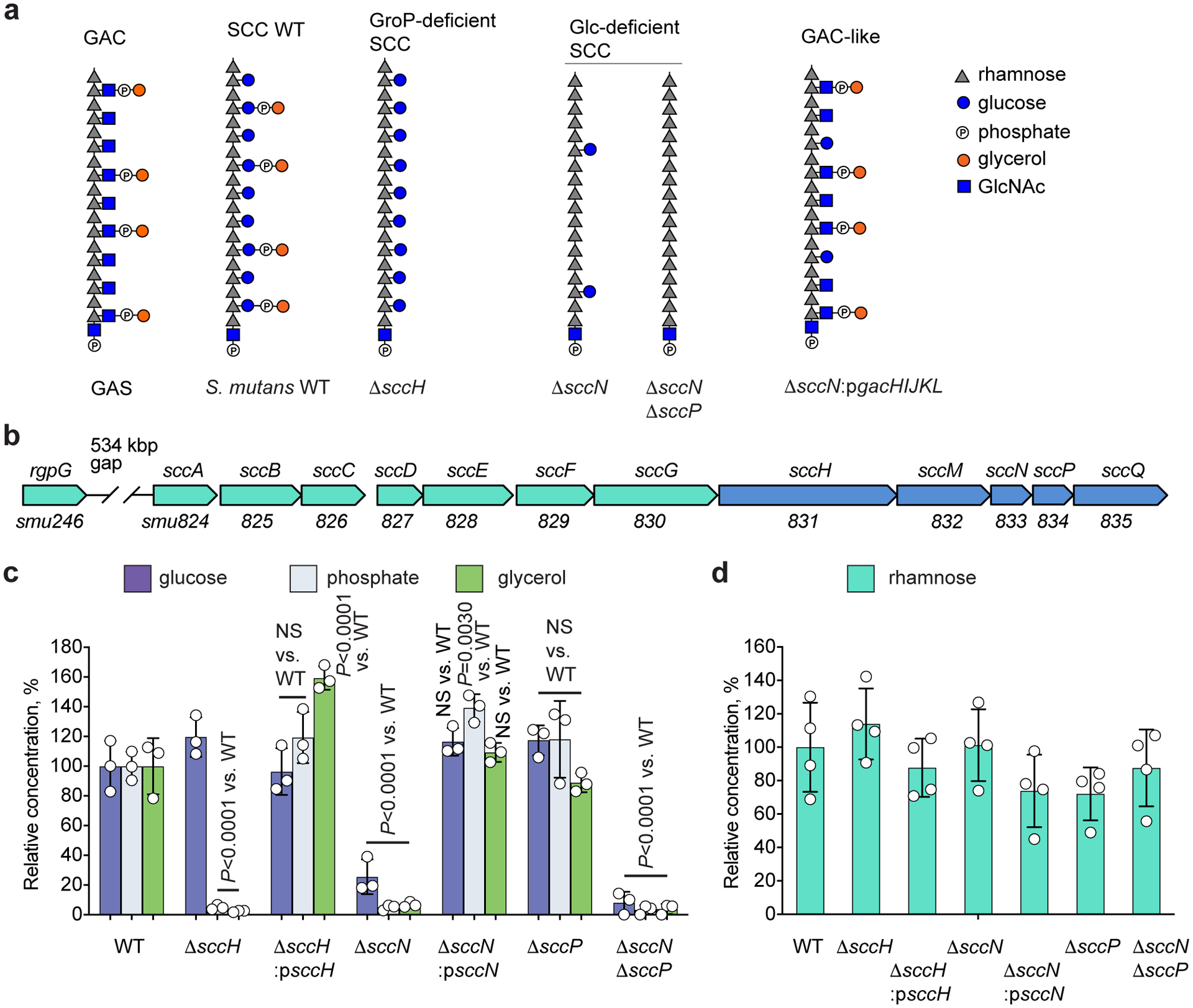
(a) Molecular model of cell wall polysaccharides isolated from GAS and S. mutans strains. Phosphate groups in the GAC and SCC structures are involved in the phosphodiester bond linking glycerol to the glycosyl side-chain. (b) Schematic representation of the SCC biosynthetic gene cluster. SCC gene cluster smu.824–835 was renamed sccABCDEFGHMNPQ21. (c) Composition analysis of SCCs isolated from S. mutans WT, ΔsccH, ΔsccH:psccH, ΔsccN, ΔsccN:psccN, ΔsccP, and ΔsccNΔsccP. The concentrations of Glc, phosphate, and glycerol are normalized to Rha content, and presented as a percentage of the ratios in the WT strain. Glc was analyzed by GC-MS method. Phosphate and glycerol were analyzed by colorimetric assays. Data are presented as mean values ± S.D., n = 3 biologically independent experiments. P values were calculated by 2-way ANOVA with Bonferroni’s multiple comparison test. (d) Rhamnose content of S. mutans strains. Values expressed as a percentage of the WT values. The concentration of Rha was analyzed by GC-MS method. Data are presented as mean values ± S.D., n = 4 biologically independent experiments.
Our study links GroP modification of SCC to spatial regulation of cell division in S. mutans. We show that ‘immature SCCs’, which lack the GroP modification, inform the proper positioning of MapZ and the major cell separation autolysin AtlA.
Results
GroP decorates the Glc side-chains of SCC.
In the SCC and GAC biosynthetic pathways encoded by 12-gene loci sccABCDEFGHMNPQ (Fig. 1b), and gacABCDEFGHIJKL, respectively, SccH/GacH catalyzes GroP transfer to respective polysaccharides21, and GacI is the GtrB-type glycosyltransferase involved in GAC GlcNAc side-chain modification22. The SCC gene cluster contains two putative GtrB-type glycosyltransferases encoded by sccN and sccP. To identify a sugar residue of SCC modified with GroP, we deleted sccN and sccP in S. mutans c serotype strain, creating ΔsccN, ΔsccP, and the double mutant ΔsccNΔsccP. SCCs from ΔsccN and ΔsccNΔsccP, but not ΔsccP, were highly deficient in the Glc content, and the Glc content in the ΔsccNΔsccP SCC was lower than in the ΔsccN SCC (Fig. 1c), supporting a major role for SccN and a minor role for SccP in Glc side-chain formation (Supplementary Fig. 1a). Furthermore, the glycerol and phosphate content in the ΔsccN and ΔsccNΔsccP SCCs was significantly reduced, similar to the ΔsccH SCC (Fig. 1c). The deficiency of Glc, glycerol and phosphate in ΔsccN was reversed in the complemented strain ΔsccN:psccN (Fig. 1c). The Rha content of the analyzed strains was similar (Fig. 1d). These data support the conclusion that the Glc-side chains of SCC are modified with GroP by SccH.
Modifications of SCC control S. mutans morphology.
We observed that planktonic ΔsccH and ΔsccN have a strong tendency to sediment after overnight growth, as compared to the wild type (WT) and complemented strains, ΔsccH:psccH and ΔsccN:psccN (Extended Data Fig. 1a). Microscopic analysis of bacteria revealed that ΔsccH and ΔsccN, but not WT, ΔsccH:psccH or ΔsccN:psccN, formed short chains that clump together, and are not dispersed by exogenously added DNAse (Fig. 2a and Supplementary Fig. 2a, b). Furthermore, the sedimentation rate of the WT sacculi was slower than the mutant sacculi, and ΔsccN sacculi sediment fastest (Extended Data Fig. 1b). Sedimentation was not affected by addition of salt, but was increased in the WT sacculi at lowered pH (Supplementary Fig. 2c). These data suggest that self-aggregation of ΔsccH and ΔsccN is due to increased intermolecular association of SCCs deficient in side-chain substituents.
Fig. 2. Modifications of SCC control S. mutans cell aggregation and morphology.
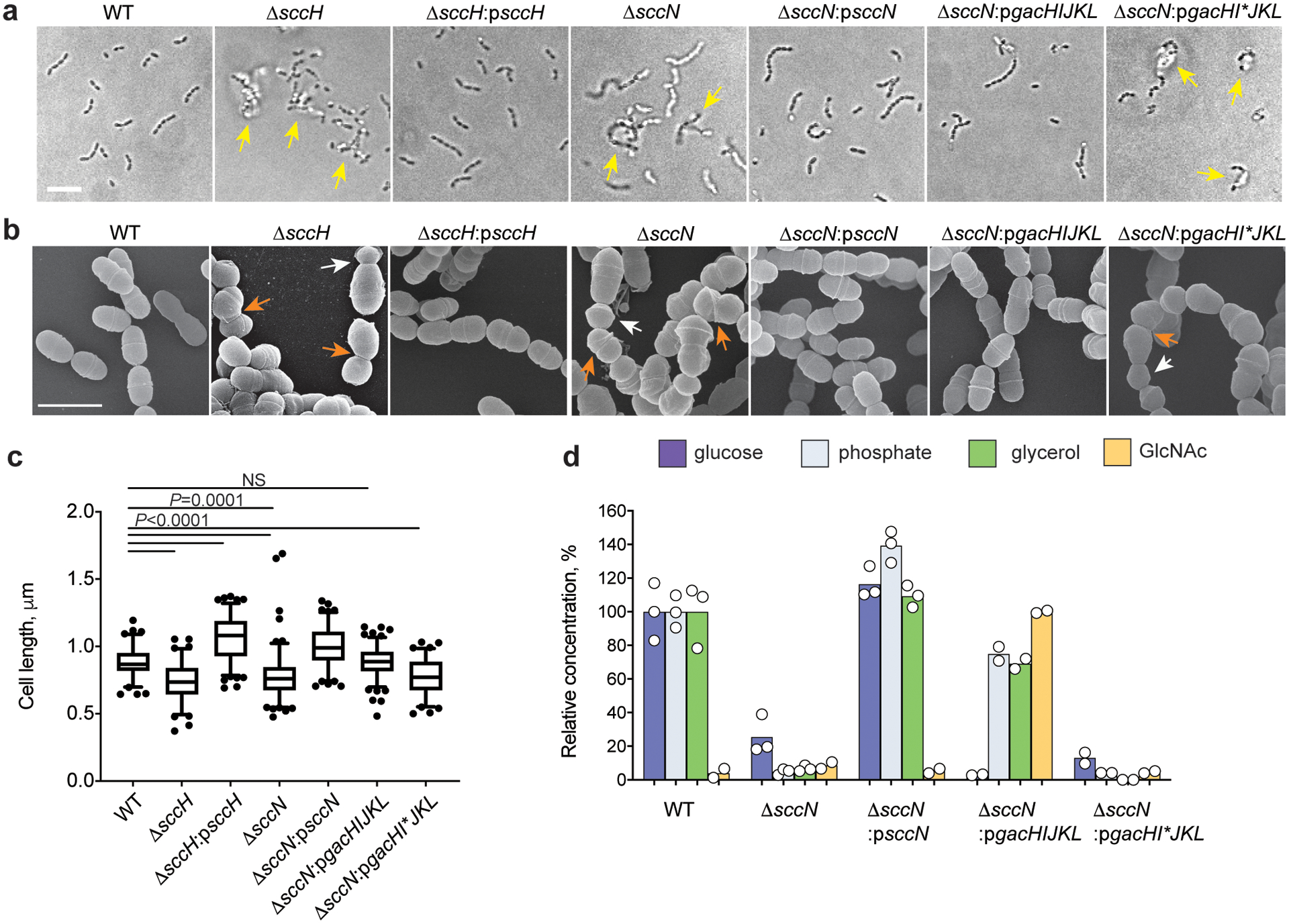
(a) Differential interference contrast (DIC) images of bacteria grown in THY broth overnight. Yellow arrows indicate bacterial aggregates. Scale bar is 5 μm. (b) Scanning electron micrographs of bacteria taken from mid-log growth phase. Bacteria were viewed by scanning electron microscopy (SEM). White and orange arrows denote small round cells and the cells with skewed division planes, respectively. Scale bar is 1 μm. Representative images from at least three independent experiments are shown in a and b. (c) Distribution of cell length of cell population at mid-log growth phase. DIC images of bacterial cells were analyzed using the software ImageJ to quantify cell length. Total number of cells was n = 85 (WT), n = 94 (ΔsccH), n = 136 (ΔsccH:psccH), n = 132 (ΔsccN), n = 113 (ΔsccN:psccN), n = 133 (ΔsccH:pgacHIJKL), and n = 82 (ΔsccH:pgacHI*JKL). Box plots show the median value (middle line), and 25%, 75% quartiles (boxes), and whiskers represent 5 – 95 percentile. P-values were determined by one-way Kruskal – Wallis non-parametric test. (d) Composition analysis of polysaccharides isolated from S. mutans WT, ΔsccN, ΔsccN:psccN, ΔsccN:pgacHIJKL and ΔsccN:pgacHI*JKL. The concentrations of Glc, phosphate, and glycerol are normalized to Rha content and presented relative to the WT strain. The concentration of GlcNAc is normalized to Rha content and presented relative to ΔsccN:pgacHIJKL. Glc, GlcNAc, phosphate and glycerol were analyzed as described in Methods. Data are presented as mean values; n = 2 biologically independent experiments (Glc, phosphate, and glycerol content) for ΔsccN:pgacHIJKL and ΔsccN:pgacHI*JKL, and GlcNAc content in all analyzed strains; n = 3 biologically independent experiments (Glc, phosphate, and glycerol content) for WT, ΔsccN and ΔsccN:psccN as in Fig 1 c.
Microscopic analysis of WT, ΔsccH, and ΔsccN revealed that the mutant cells have severe cell division defects (Fig. 2b and Extended Data Fig. 1c). We observed the ΔsccH and ΔsccN cells with asymmetrical and non-perpendicular planes of cell division (Fig. 2b). Furthermore, the mutant cells were significantly shorter than the WT cells, and the ΔsccN cells were significantly wider than the WT and ΔsccH cells (Fig. 2c, Supplementary Fig. 3a, c, d, 4, and Supplementary Tables 1 and 2). While only 2% of the WT cells displayed the minimal cell length, 21% of the ΔsccH cells and 14% of the ΔsccN cells were abnormally small (Supplementary Table 1). When WT, ΔsccH and ΔsccN were imaged using DAPI, few anucleated cells were detected in the mutants (Supplementary Fig. 3b and Supplementary Table 2), indicating that the morphological abnormalities rarely lead to a defect in chromosome replication or segregation into daughter cells. The morphological phenotypes correlated with a significant decrease in cell viability of ΔsccH in comparison to WT (Supplementary Fig. 5). The observed phenotypes were complemented in ΔsccH:psccH and ΔsccN:psccN except that the ΔsccH:psccH cells were longer than the WT cells (Fig. 2b, c and Supplementary Table 1). We concluded that the SCC side-chain substituents are required for proper S. mutans morphogenesis.
To dissect the structural elements controlling S. mutans morphology, we replaced the SCC Glc side-chains with GlcNAc in S. mutans by plasmid-based expression of gacHIJKL22 in ΔsccN creating ΔsccN:pgacHIJKL (Supplementary Fig. 1b, c). As a negative control, we generated ΔsccN:pgacHI*JKL which carries a stop codon in gacI. The GlcNAc and GroP content was increased in the ΔsccN:pgacHIJKL SCC, but not in the ΔsccN:pgacHI*JKL SCC (Fig. 2d). The morphological phenotypes of ΔsccN were reversed to WT in ΔsccN:pgacHIJKL, but not in ΔsccN:pgacHI*JKL (Fig. 2a, b, c, Extended Data Fig. 1a, Supplementary Fig. 4 and Supplementary Table 1), indicating that the underlying molecular mechanisms for self-aggregation and the morphological abnormalities are GroP-dependent and independent of the specific glycosyl side chain.
AtlA promotes autolysis of ΔsccH and ΔsccN.
We compared autolysis of WT with the ΔsccH and ΔsccN mutants and complemented strains by measuring the change in OD600 in liquid cultures after the addition of Triton-X100. In comparison to WT, ΔsccH and ΔsccN were sensitive to autolysis, with cellular lysis more pronounced in ΔsccN, than in ΔsccH (Fig. 3a, Supplementary Table 3). These phenotypes were reversed to WT in ΔsccH:psccH, ΔsccN:psccN and ΔsccN:pgacHIJKL, but not in ΔsccN:pgacHI*JKL (Fig. 3a).
Fig. 3. Mislocalization of AtlA promotes autolysis of ΔsccH and ΔsccN.
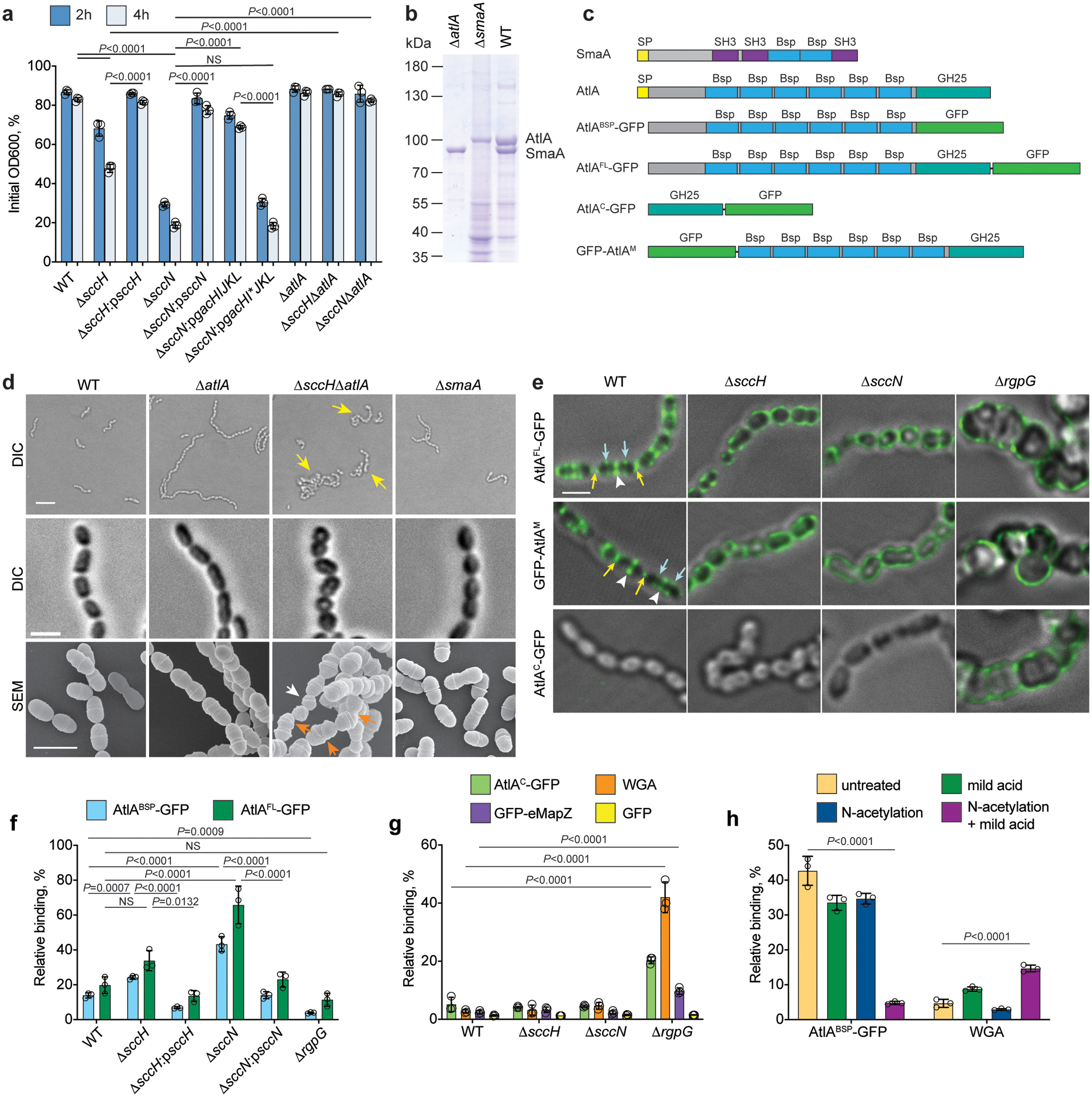
(a) Autolysis of S. mutans strains in 0.1% Triton X-100. Autolysis was monitored after 2 and 4 hours as the decrease in OD600 and normalized to the OD600 at time zero. Data are means ± S.D., n=3 biologically independent experiments. P-values were determined by two-way ANOVA, Tukey’s multiple comparisons. Supplementary Table 3 shows the raw data. (b) Pulldown of WT, ΔatlA, and ΔsmaA cell wall-bound proteins identified AtlA and SmaA. Proteins were separated on 10% SDS-PAGE. (c) The domain structure of SmaA, AtlA, AtlABSP-GFP, AtlAFL-GFP, AtlAC-GFP, and GFP-AtlAM. SP (yellow box) denotes signal peptide. (d) DIC images of bacteria in stationary (top panels, scale bar is 5 μm) and mid-log growth phases (middle panels, scale bar is 1 μm). Yellow arrows indicate bacterial aggregates. SEM images of WT, ΔatlA, ΔsccHΔatlA and ΔsmaA in mid-log growth phase (bottom panels, scale bar is 1 μm). White and orange arrows denote small cells and cells with skewed division plane, respectively. (e) The WT, ΔsccH, ΔsccN and ΔrgpG bacteria were incubated with AtlAFL-GFP, GFP-AtlAM, or AtlAC-GFP and examined by DIC and fluorescence microscopy. An overlay of fluorescence and DIC images is shown. Yellow and blue arrows, and white arrowheads show poles, equatorial rings and septa, respectively, labeled by protein fusions. Scale bar is 1 μm. Representative image from three independent experiments is shown in b, d and e. (f) Co-sedimentation of AtlAFL-GFP or AtlABSP-GFP with sacculi purified from S. mutans strains. (g) Co-sedimentation of AtlAC-GFP, eMapZ-GFP, WGA or GFP with sacculi purified from S. mutans strains. (h) Co-sedimentation of AtlABSP-GFP or WGA with the SCC-depleted ΔsccN sacculi. AtlABSP-GFP or WGA was incubated with intact sacculi (untreated), or subjected to mild acid hydrolysis (mild acid), or chemically N-acetylated (N-acetylation), or chemically N-acetylated followed by mild acid hydrolysis (N-acetylation + mild acid). In f, g, and h, data are means ± S.D., n=3 biologically independent experiments. Data are a percentage of fluorescence of the pellet normalized to the total fluorescence of the sample. P values were calculated by one-way ANOVA, Tukey’s multiple comparisons.
To identify autolysins associated with SCCs, we stripped cell surface-bound proteins from WT cells, and used the re-folded proteins for pulldown experiments with S. mutans cell wall material. Two major proteins were identified as AtlA and SmaA by LC-MS/MS analysis (Fig. 3b). AtlA is the major autolysin involved in the separation of daughter cells after division23–26, consisting of an N-terminal domain with six Bsp repeats and a C-terminal catalytic domain. SmaA has two Bsp repeats and three SH3 domains (Fig. 3c).
To confirm the identities of the proteins and investigate their roles in the observed phenotypes of ΔsccH and ΔsccN, we generated ΔatlA and ΔsmaA mutants, and the double mutants, ΔsmaAΔatlA, ΔsccHΔatlA, ΔsccNΔatlA, and ΔsccNΔsmaA. AtlA and SmaA were not recovered from pulldown experiments using surface proteins stripped from ΔatlA and ΔsmaA, respectively (Fig. 3b). Deletion of SmaA had no effect on detergent-induced autolysis of the mutants (Supplementary Fig. 6). However, the increased sensitivity of ΔsccH and ΔsccN to autolysis was lost in the AtlA-deleted double mutants (Fig. 3a), indicating a major role for AtlA in autolysis.
The ΔsmaA cells remained in suspension after overnight growth, and showed WT cell morphology (Fig. 3d, Supplementary Fig. 4, 7, and Supplementary Table 1). The ΔatlA mutant displayed a chaining phenotype, but the cells retained the normal morphology and did not self-aggregate. The phenotype of ΔsccHΔatlA combined the phenotypes of the single mutants, as ΔsccHΔatlA cells formed long chains that self-aggregate and demonstrated aberrant morphology (Fig. 3d, Supplementary Fig. 4, 7, and Supplementary Table 1). These observations reveal that the morphological phenotypes of ΔsccH and ΔsccN are not linked to either SmaA or AtlA.
Modifications of SCC coordinate AtlA localization.
Enhanced autolysis of ΔsccH and ΔsccN suggests either mislocalization or increased expression of AtlA in the mutants. Using western blot analysis we examined the amount of AtlA secreted into the growth medium and attached to WT and the mutants (Supplementary Fig. 8). We observed no differences in the amount of the 90-kDa mature form of AtlA recovered from the analyzed bacteria. A significantly reduced amount of the 70-kDa proteolytic fragment of AtlA was determined in the ΔsccN growth medium.
To provide a detailed picture of the bacterial regions targeted by AtlA, we generated protein fusions, AtlAFL-GFP and GFP-AtlAM, in which green fluorescent protein (GFP) is fused with the full-length form of AtlA at the C-terminus, and the 90-kDa mature form of AtlA at the N-terminus, respectively (Fig. 3c and Supplementary Fig. 9). The fusions were added exogenously to the WT, ΔsccH, and ΔsccN cells. Fluorescence microscopy imaging revealed that the binding pattern of the fusions is similar, demonstrating that GFP-tagging does not compromise the binding of AtlA to its surface receptor (Fig. 3e and Supplementary Fig. 10a). GFP alone failed to bind to the bacteria (Supplementary Fig. 10b). In the WT newborn cells, the fusions predominantly targeted the poles and the mid-cell zones. As the WT cells elongate, the GFP-labeled mid-cell zones split into three parallel rings corresponding to the septa and equatorial rings (Fig. 3e and Supplementary Fig. 10a). When AtlAFL-GFP was titrated downward (at 50 and 100-fold dilutions), the fusion labeled predominantly the septa and poles, suggesting tighter binding of AtlA to these regions (Supplementary Fig. 11). In contrast to WT, AtlAFL-GFP was uniformly distributed over the surface of the ΔsccH (56 %) and ΔsccN (82 %) cells (Fig. 3e and Supplementary Table 2). To investigate the participation of SCCs in the localization of AtlA, we constructed a SCC-deficient mutant by deletion of rgpG whose product is essential for SCC biosynthesis27. The mutant was devoid of SCC (<0.5% WT level), and demonstrated aberrant morphology (Supplementary Fig. 12a, b). AtlAFL-GFP and GFP-AtlAM were evenly distributed over the ΔrgpG cell surface (Fig. 3e).
To identify the AtlA domain mediating the binding to bacteria, we generated two protein fusions, AtlABSP-GFP and AtlAC-GFP, in which the Bsp repeat domain (hereafter Bsp domain) and the catalytic domain, respectively, are fused with GFP (Fig. 3c and Supplementary Fig. 9). AtlAC-GFP did not associate with WT, ΔsccH and ΔsccN, but labeled ΔrgpG in a pattern similar to full-length AtlA (Fig. 3e). In contrast, AtlABSP-GFP did not bind ΔrgpG cells (Supplementary Fig. 12c). The binding pattern of AtlABSP-GFP to WT, ΔsccH and ΔsccN cells was identical to AtlAFL-GFP with one exception — during early elongation stage, AtlABSP-GFP did not associate with the WT septa (Extended Data Fig. 2a and Supplementary Table 2). The complemented strains ΔsccH:psccH and ΔsccN:psccN bound AtlABSP-GFP similar to the WT strain (Extended Data Fig. 2a). Furthermore, AtlABSP-GFP, but not GFP alone, associated with the sacculi purified from WT, ΔsccH and ΔsccH:psccH indistinguishably from the intact cells (Extended Data Fig. 2b and Supplementary Fig. 10c), suggesting that the Bsp domain recognizes a cell wall component, either SCC or peptidoglycan.
To study the binding of AtlA to sacculi purified from S. mutans strains in more detail, we employed a co-sedimentation assay. Consistent with the microscopy, the binding of AtlAFL-GFP and AtlABSP-GFP to ΔsccN was tightest, ΔsccH was second tightest, and WT and complemented strains were the weakest (Fig. 3f). AtlABSP-GFP did not associate with ΔrgpG, but AtlAFL-GFP and AtlAC-GFP bound weakly to these sacculi (Fig. 3f, g). Lastly, neither AtlAC-GFP nor GFP alone bound to WT, ΔsccH and ΔsccN sacculi (Fig. 3g). Collectively, these results indicate that (1) SCCs are required for the Bsp domain binding to bacteria; (2) the AtlA catalytic domain likely interacts with peptidoglycan; (3) the Bsp domain recognizes a cell wall component present at the poles and equatorial rings, and mediates the catalytic domain binding to peptidoglycan; (4) the absence of GroP and side-chain decorations in SCC promotes mislocalized binding of AtlA to the entire surface of S. mutans.
AtlA binds to SCC polyrhamnose.
To examine directly the role of SCCs in the recruitment of AtlA, we chemically removed the polysaccharides from peptidoglycan of ΔsccN sacculi using either HF-treatment or mild acid hydrolysis (Supplementary Fig. 13a). The efficient release of SCCs from peptidoglycan by mild acid requires prior chemical N-acetylation. The GlcNAc and MurNAc content were not depleted by mild acid hydrolysis, demonstrating that peptidoglycan remains largely intact (Supplementary Fig. 13b). The SCC-depleted sacculi displayed a significant reduction in AtlABSP-GFP binding (Fig. 3h), indicating that the Bsp domain interacts directly with SCCs. Importantly, neither N-acetylation nor mild acid treatment alone affected AtlABSP-GFP binding to the sacculi, supporting the notion that these conditions do not induce breakdown of sacculi.
Our observation that AtlAC-GFP binds to ΔrgpG sacculi, but fails to associate with WT, ΔsccH and ΔsccN sacculi, suggests that SCCs conceal peptidoglycan from molecular interactions with proteins. To test this, we employed wheat germ agglutinin (WGA), a GlcNAc-specific lectin, in a co-sedimentation assay with sacculi. WGA did not associate strongly with the WT, ΔsccH and ΔsccN sacculi, but efficiently labeled the ΔrgpG and SCC-depleted ΔsccN sacculi, indicating that SCCs obscure peptidoglycan from molecular interactions with WGA (Fig. 3g, h).
The strongest binding of AtlABSP-GFP to ΔsccN suggests interaction of AtlA with SCC polyrhamnose. Hence, we compared the binding of AtlABSP-GFP to bacterial cell walls carrying the polysaccharides containing →3)α-Rha(1→2)α-Rha(1→ disaccharide repeats (S. mutans serotypes c, f, e and k, GAS and Streptococcus equi)17 with bacterial cell walls lacking these repeats (GBS, Enterococcus faecalis) (Supplementary Fig. 14). As compared to GFP control, AtlABSP-GFP strongly binds to cell walls from the first group of strains. No binding was detected to ΔrgpG, GBS or E. faecalis cell walls (Fig. 4a), suggesting that AtlA recognizes →3)α-Rha(1→2)α-Rha(1→ disaccharide repeats.
Fig. 4. AtlA binds to the polyrhamnose backbone of SCC.
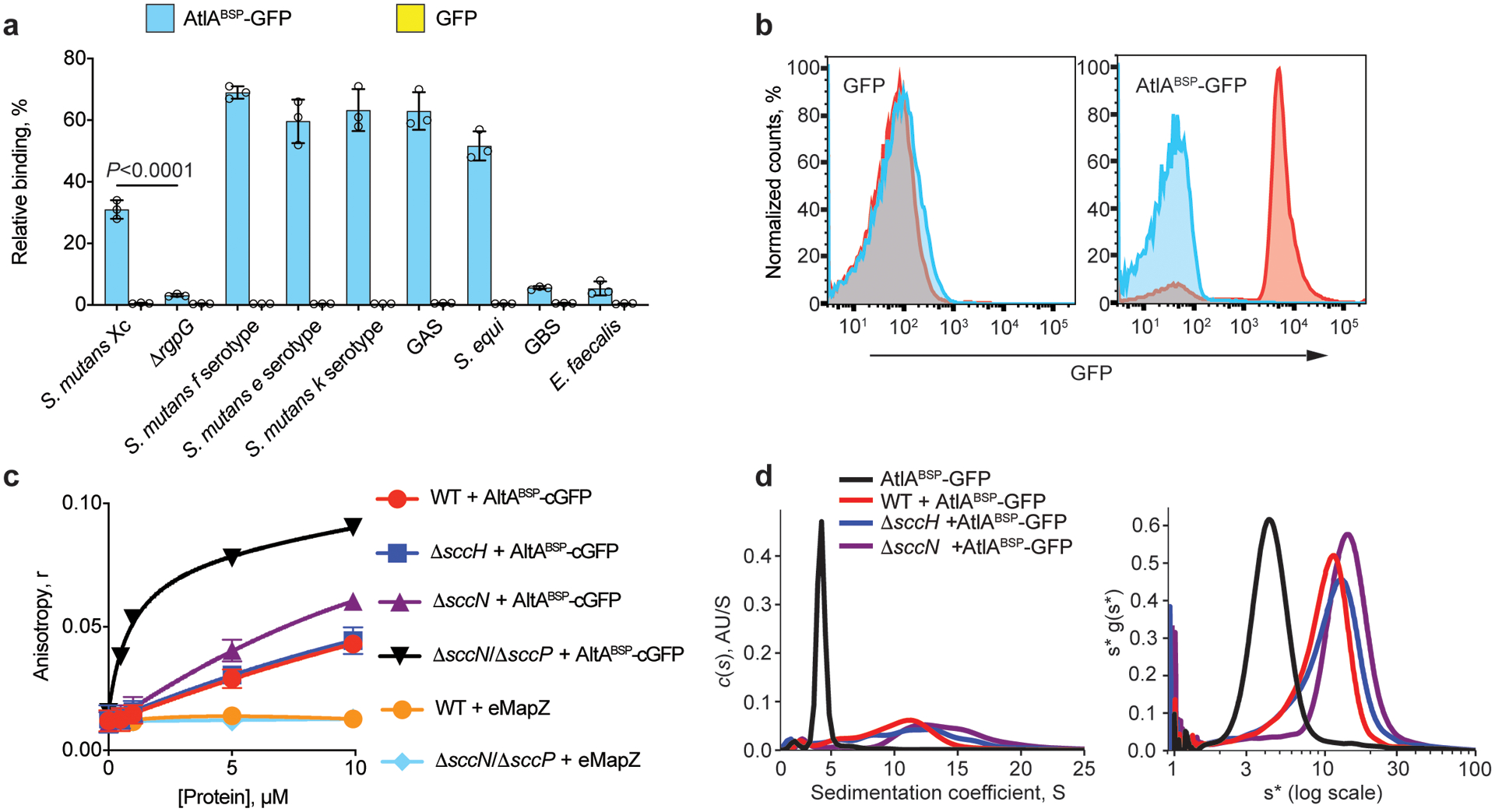
(a) Co-sedimentation assay of AtlABSP-GFP and GFP with cell walls purified from S. mutans serotypes c, f, e and k, ΔrgpG, GAS, S. equi, GBS and E. faecalis. Data are presented as a percentage of fluorescence of the pellet normalized to the total fluorescence of the sample. Data are means ± S.D., n = 3 biologically independent experiments. P values were calculated by a one-way ANOVA, Tukey’s multiple comparisons. (b) AtlABSP-GFP binds to E. coli expressing polyrhamnose on the cell surface. Flow cytometry analysis of AtlABSP-GFP (right panel) and GFP (left panel) binding to the polyrhamnose-expressing E. coli (red) and its parental strain (blue). For flow cytometric analysis, at least 10,000 events were collected. Experiments depicted in b were performed independently three times and yielded the same results. Histograms of representative results are shown. (c) Fluorescence anisotropy of 0.5 μM ANDS-labeled SCC variants incubated with titrations of AtlABSP-cGFP or GFP-eMapZ. Binding of the ΔsccNΔsccP SCC to AtlABSP-GFP was fitted to a one-site total-binding model. Symbols and error bars represent the means ± S.D., n = 3 biologically independent experiments. (d) Analytical ultracentrifugation analysis of AtlABSP-GFP binding to SCCs purified from WT, ΔsccH and ΔsccN. The continuous c(s) distributions for 2.5 μM AtlABSP-GFP combined with 25 μM SCC variants (left panel). Plot of s*g(s*) vs. log(s*) using wide distribution analysis in SedAnal (right panel). Experiments depicted in d was performed independently twice and yielded the same results.
To gather additional evidence that AtlA interacts with polyrhamnose, we expressed sccABCDEFG in E. coli resulting in decoration of lipopolysaccharide with polyrhamnose28,29 as confirmed by anti-GAC antibodies recognizing the GAC polyrhamnose backbone (Supplementary Fig. 15). Flow cytometric analysis revealed that AtlABSP-GFP, but not GFP alone, binds to polyrhamnose-producing E. coli and not to the parental strain (Fig. 4b and Extended Data Fig. 3), confirming that polyrhamnose is sufficient to confer AtlABSP-GFP binding.
Modifications of SCC hinder AtlA binding to polyrhamnose.
To investigate how SCC side-chain substituents affect AtlA recognition of polyrhamnose, we employed fluorescence polarization anisotropy to compare the binding affinity of the colorless AtlABSP-GFP variant protein, AtlABSP-cGFP, to the fluorescent ANDS-conjugated SCC variants prepared from WT, ΔsccH, ΔsccN, and ΔsccNΔsccP (Fig. 1a). The binding of AtlABSP-cGFP to ΔsccNΔsccP SCC was the strongest, with an apparent Kd = 0.9 μM. WT, ΔsccH, and ΔsccN SCCs demonstrated clear association with AtlABSP-cGFP, but the binding was unable to reach saturation due to protein solubility constraints (Fig. 4c). The affinity of ΔsccN SCC was at least 12-fold weaker than ΔsccNΔsccP, with WT and ΔsccH SCCs being indistinguishable and at least 25-fold lower affinity than ΔsccNΔsccP SCC. These data suggest that the primary recognition site of AtlA is unmodified polyrhamnose, and the addition of branching structures decreases binding affinity, presumably due to steric hindrance. The differences in binding between the ΔsccNΔsccP and ΔsccN SCCs further reinforces our hypothesis that both SccN and SccP participate in modification of SCC with the side-chains.
To explore further the Bsp domain binding to SCCs, we employed analytical ultracentrifugation. Continuous c(s) distribution analysis confirmed that AtlABSP-GFP remains mostly monomeric at the concentration range being studied. Upon addition of WT, ΔsccH, or ΔsccN SCCs, higher-order complexation was observed between 10–20 S (Fig. 4d, left panel). WT SCCs produced lower-sized complexes than ΔsccH and ΔsccN SCCs. To understand overall complexation better, the same data were analyzed using wide distribution analysis (Fig. 4d, right panel). Each sample resolved into a more Gaussian distribution, with a weight-averaged sedimentation coefficient of 5.5 S for AtlABSP-GFP alone, and 10.6 S, 12.3 S and 14.5 S for AtlABSP-GFP complexes with WT, ΔsccH and ΔsccN SCCs, respectively. Hence, wide distribution analysis confirms the formation of smaller complexes in the presence of WT SCCs compared to ΔsccH and ΔsccN SCCs, and further demonstrates that SCC side-chain substituents obstruct the binding of AtlA to polyrhamnose.
SCC is highly heterogeneous with regard to GroP.
AtlA localization studies together with the binding affinity analysis imply that S. mutans equatorial sites and poles contain SCCs deficient in either GroP or Glc-GroP, whereas the lateral walls carry the mature SCCs decorated with Glc-GroP. Importantly, we established that deletion of either sccH or sccN has no effect on total cellular Rha content (Fig. 1d). This fact excludes the possibility that the mislocalized binding of AtlABSP-GFP along the entire surface of the mutant cells is due to altered expression of SCC within the cell wall. To determine whether S. mutans expresses SCCs deficient in side-chain substituents, we separated the WT and ΔsccH SCCs using anion-exchange chromatography. As compared to the ΔsccH SCC which elutes as a single peak of neutral polysaccharide (Extended Data Fig. 4a), the WT SCC is composed largely of negatively charged material binding tightly to the anion-exchange column, and a substantial portion of neutral polysaccharide with decreased Glc and phosphate content (Extended Data Fig. 4b, c). As we previously reported, the phosphate content in GAC and SCC is an indication of the presence of the GroP modification in the polysaccharide21.
We next examined the electrophoretic mobility of ANDS-conjugated WT, ΔsccH, ΔsccN, ΔsccP and ΔsccNΔsccP SCCs. ANDS introduces a negative charge to SCCs conferring electrophoretic mobility to neutral polysaccharides extracted from the GroP-deficient mutants, ΔsccH, ΔsccN and ΔsccNΔsccP. As expected, these SCCs migrated as a single band on SDS-PAGE (Extended Data Fig. 4d). However, consistent with the anion-exchange chromatography analysis, we observed a distinctive “laddering” of bands in the WT and ΔsccP SCCs, demonstrating that S. mutans produces SCCs with different degrees of GroP modification.
Modifications of SCC control MapZ-ring positioning.
The morphological abnormalities of ΔsccH and ΔsccN suggest that GroP modification affects either the assembly or correct positioning of the Z-ring. To test this, we expressed FtsZ fused with tagRFP from its chromosomal locus in the WT and mutant cells. AtlABSP-GFP was added exogenously to locate the GroP-deficient SCCs on the surface of bacteria. We observed both FtsZ-tagRFP and AtlABSP-GFP at the mid-cell in the newborn WT cells (Fig. 5a, stage 1). As the cells elongate, the AtlABSP-GFP signal splits into two parallel bands that move away from the division site where a single FtsZ-ring is present (Fig. 5a, stages 2 and 3). During middle-to-late cell division stage, a part of FtsZ splits into two rings and migrates to the mid-cell regions of the newly formed daughter cells, resulting in a distinctive three-band pattern (Fig. 5a, stage 4). AtlABSP-GFP was detected at the constricting septum at a later stage (Fig. 5a, stage 5 and Supplementary Fig. 16), indicating that immature SCCs lacking the GroP modification are inserted into septal walls during the pole maturation. In 46% of the ΔsccH and ΔsccN cells, Z-rings were displaced from the cell center or not perpendicular to the cell’s long axis (Fig. 5b, Supplementary Table 4), suggesting that asymmetrical cell division observed in the mutants arises from this defect. As expected, in the majority of the mutant cells expressing FtsZ-tagRFP, the AtlABSP-GFP signal was evenly distributed over the cell surface.
Fig. 5. Modifications of SCC guide the positioning of FtsZ- and MapZ-rings.

(a) Localization of FtsZ (middle panel, red) in WT cells expressing FtsZ-tagRFP labeled with AtlABSP-GFP (left panel, green) at different stages of the cell cycle (designated 1 to 5). The percentages of cells in each division stage (total 498 cells counted) are indicated in the far-left panel. (b) Localization of FtsZ (middle panel, red) in the ΔsccH and ΔsccN cells expressing FtsZ-tagRFP labeled with AtlABSP-GFP (left panel, green). Blue arrows indicate mislocalized Z-rings. (c) Localization of MapZ (left panels, green) and FtsZ (middle panels, red) in the WT cells expressing iGFP-mapZ/FtsZ-tagRFP at different stages of cell cycle designated 1 to 4. Schematic pictures (far-left panel) illustrate MapZ- and FtsZ-ring positions during different stages of the cell cycle. The percentages of cells in each division stage (total 468 cells counted) are indicated in the far-left panel. (d) Localization of MapZ (left panels, green) and SCCs labeled with AtlABSP-tagRFP (middle panels, red) in the WT, ΔsccH, and ΔsccN cells expressing iGFP-mapZ. White arrows show mislocalized MapZ. (e) Localization of MapZ (left panels, green) and FtsZ (middle panels, red) in the ΔsccH and ΔsccN cells expressing iGFP-mapZ/FtsZ-tagRFP. White arrows indicate mislocalized MapZ. Representative images from at least three independent experiments are shown in a, b, c, d and e. Images were deconvolved using Huygens Professional software, and represent DIC and fluorescence overlays except right panels in d (merge) that show an overlay of GFP and tagRFP signals. Right panels in a, b, c, and e (merge) show an overlay of DIC, GFP, and tagRFP signals. Supplementary Fig. 18 demonstrates representative raw and data-processed images of the WT, ΔsccH and ΔsccN cells expressing iGFP-mapZ/FtsZ-tagRFP. Scale bar is 1 μm in a, c, d and e, and 2 μm in b.
Since MapZ guides Z-ring positioning, we next examined the localization of MapZ in WT, ΔsccH and ΔsccN expressing FtsZ-tagRFP and a fusion of MapZ with GFP (iGFP-MapZ). In agreement with previous studies7–10,30, MapZ co-localizes with the FtsZ-ring at mid-cell in the WT newborn cells (Fig. 5c, stage 1). As cell division progresses, MapZ migrates as a pair of rings, while a single FtsZ-ring remains at the mid-cell of the parental cell (Fig. 5c, stages 2 and 3). At a later cell division stage, a portion of FtsZ co-localizes with MapZ at the future division sites in the daughter cells (Fig. 5c, stage 4).
MapZ did not form the characteristic two ring-like structures in the dividing ΔsccH and ΔsccN cells. Instead, MapZ was either randomly distributed on the cell periphery or assembled into a ring along with the dispersed MapZ signal present on the cell periphery (Fig. 5e, Supplementary Table 4). MapZ was mislocalized in >80% of the ΔsccH and ΔsccN cells, whereas only 22% of the WT cells displayed this defect. AtlABSP-tagRFP (the tagRFP fusion with the Bsp domain) labeled the WT, ΔsccH and ΔsccN cells expressing iGFP-MapZ in a pattern similar to AtlABSP-GFP (Fig. 5d). Importantly, MapZ co-localizes with AtlABSP-tagRFP at mid-cell regions in the WT cells, supporting the idea that immature SCCs are present in the equatorial rings.
To identify a cell wall structure targeted by MapZ, we produced the extracellular domain of MapZ, eMapZ, and an N-terminal fusion of eMapZ with GFP, GFP-eMapZ. Fluorescence polarization anisotropy revealed that eMapZ does not bind to ANDS-conjugated WT and ΔsccNΔsccP SCCs (Fig. 4c). Co-sedimentation of GFP-eMapZ with WT, ΔsccH, ΔsccN and ΔrgpG sacculi showed that GFP-eMapZ binds to ΔrgpG sacculi only (Fig. 3g), suggesting the association of MapZ with peptidoglycan. Altogether, these results support a mechanism in which MapZ localization relies on an exclusion strategy whereby decoration of SCC with GroP provides the molecular signal to exclude recruitment of MapZ to the peptidoglycan of lateral walls in favor of the peptidoglycan of equatorial rings.
Discussion
Despite years of intensive research, it remains unclear how oval-shaped Gram-positive bacteria generate equally sized daughter cells after division. This work proposes a mechanism of molecular exclusion to position the cell division machinery in S. mutans. We observed that the Glc side-chains of S. mutans SCC are decorated by GroP, and the GroP- and side-chain-deficient mutants display severely impaired cell division. The morphological changes are accompanied by reduced viability and enhanced susceptibility to autolysis attributable to mislocalization of autolysin AtlA. Fluorescence anisotropy and analytical ultracentrifugation experiments conclusively demonstrated that the AtlA Bsp domain binds to all SCCs, but the binding affinity for polysaccharides, containing GroP-Glc side-chains, is significantly lower than for SCCs lacking the side-chain substituents. We further showed that the Bsp domain targets specifically the SCC polyrhamnose backbone and the catalytic domain likely binds to peptidoglycan.
Using fluorescence microscopy, we demonstrated that GFP fusions with either the AtlA Bsp domain, or full-length AtlA, associate with the cell poles and equators in the WT cells. These fusions bind evenly along the cell surface of the majority of mutant cells lacking either GroP or Glc-GroP modifications, indicating that peptidoglycan is decorated with SCCs throughout the cell surface. Importantly, the absence of binding of AtlABSP-GFP to the SCC-deficient cells eliminates the possibility that the Bsp domain recognizes additional structures on the bacterial surface. Finally, surface expression of SCC polyrhamnose in E. coli is sufficient to confer binding of AtlABSP-GFP. These observations lead to the conclusion that the mislocalization of AtlA causes the enhanced cell lysis of the mutants.
In ovococci, peptidoglycan architecture at the equatorial rings has been proposed to direct the positioning of the cell division machinery through MapZ8,9. Our findings that in S. mutans, a significant portion of SCC contains low or no GroP, and MapZ co-localizes with the GroP-deficient SCCs labeled by AtlABSP-GFP support the idea that peptidoglycan in the equatorial rings and cell poles is decorated with newly synthesized SCCs lacking the modifications. The analysis of MapZ localization in SCC side-chain deficient mutants revealed major defects in MapZ recruitment to the equatorial rings. The mislocalization of MapZ causes Z-rings to assemble apart from the mid‐cell, strongly suggesting that the cell shape deformations observed in the mutants arise from septum misplacement. These findings highlight the pivotal role of SCC modifications in cell division, leading to a model wherein immature SCCs serve as landmarks for the recruitment of MapZ to equatorial rings, and AtlA to equatorial rings and cell poles (Fig. 6). Thus, this simple mechanism unifies septum positioning with its subsequent splitting.
Fig. 6. A schematic model of cell division in S. mutans.
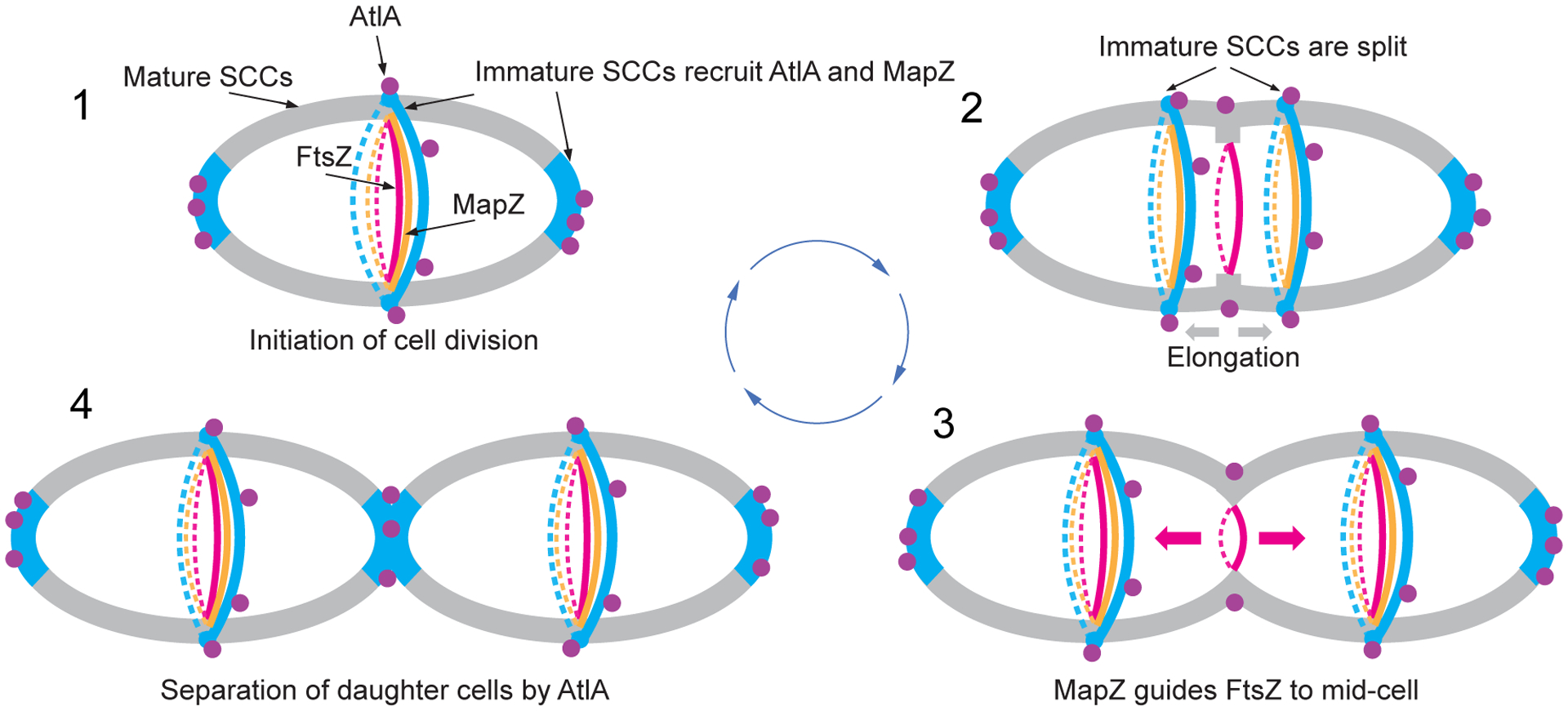
1. Cell division is initiated at mid-cell by recruitment and alignment of the FtsZ-ring with the equatorial ring. Alignment of the FtsZ-ring is guided by MapZ which binds to peptidoglycan carrying immature SCCs. The AtlA Bsp domain targets the autolysin to the cell poles and equatorial ring populated by immature SCCs. 2. Cell division machinery assembled at mid-cell initiates synthesis of septal and lateral walls. Concomitantly with this process, the equatorial ring is split in the middle presumably under turgor pressure and due to AtlA action. Splitting of the septal wall and synthesis of the lateral wall leads to migration of new equatorial rings apart. The MapZ-rings follow the equatorial ring. The Bsp and catalytic domains of AtlA target the autolysin to parental septum to cleave the septal wall. 3. The equatorial rings approach the equator of new daughter cells. MapZ guides alignment of FtsZ with new equatorial rings. 4. Cell division machinery synthesizes the polar septal wall decorated with immature SCCs. The Bsp domain of AtlA mediates recruitment of the autolysin to newly forming poles to separate the daughter cells. Cell walls decorated with mature and immature SCCs are shown in gray and blue, respectively. Purple circles indicate AtlA. FtsZ- and MapZ-rings are shown in red and orange colors, respectively.
The mechanism of recognition of immature SCCs by MapZ is unknown but under investigation. The MapZ extracellular domain does not interact directly with SCCs, but binds to SCC-deficient sacculi, implicating MapZ in peptidoglycan recognition. Phosphate groups in WTA have been proposed to affect the packing density and rigidity of the cell wall due to electrostatic repulsion between the polysaccharides31. Furthermore, addition of side-chain modifications to linear polysaccharides gives rise to less densely packed polysaccharide by affecting the overall structural architecture of the polysaccharide and decreasing intermolecular association between the polysaccharide chains32,33. This is consistent with our observation that the intact bacteria and sacculi deficient in in SCC side-chains are prone to aggregation. Thus, it is possible that a cue for recognition of equatorial rings by MapZ is an alteration in peptidoglycan density due to absence of the SCC side-chain substituents.
The Rha-containing cell wall polysaccharides are the functional homologs of canonical WTAs in streptococci. Much like GroP modification, WTAs restrict the binding of autolysins from the entire bacterial surface, directing them to the specific regions where they are enzymatically active12,14,16. Considering the similar function of WTAs and GroP modification of SCC, it is plausible that the here-described mechanism of the positioning of cell division proteins is widespread in Gram-positive bacteria. However, cell separation autolysins and the proteins bridging the Z-ring to the cell wall likely differ among species.
Methods
Bacterial strains, growth conditions and media
All strains and plasmids used in this study are listed in Supplementary Table 5. Streptococci and E. faecalis were grown in BD Bacto Todd-Hewitt broth supplemented with 1% yeast extract (THY) without aeration at 37°C. S. mutans strains were grown either on THY or brain heart infusion (BHI) agar at 37°C with 5% CO2. E. coli strains were grown in Lysogeny Broth (LB) medium or on LB agar plates at 37°C. Antibiotics were included at the following concentrations: ampicillin at 100 μg mL−1 for E. coli; erythromycin at 5 μg mL−1 for S. mutans; chloramphenicol at 10 μg mL−1 for E. coli and 2 μg mL−1 for S. mutans; spectinomycin at 500 μg mL−1 for S. mutans; kanamycin at 300 μg mL−1 for S. mutans.
Construction of mutant strains
All primers used in this study are listed in Supplementary Table 6. To delete sccN, S. mutans chromosomal DNA was used as a template for the amplification of two DNA fragments using two primers pairs: sccNup-BglII-f/sccNup-SalI-r and sccNdown-BamHI-f/sccNdown-XhoI-r. The first PCR product was digested with BglII/SalI and ligated into BglII/SalI-digested pUC19BXspec22. The resultant plasmid was digested with BamHI/XhoI and used for ligation with the second PCR product that was digested with BamHI/XhoI. The resultant plasmid, pUC19BXspec-sccN, was digested with BglII and XhoI to obtain a DNA fragment containing the nonpolar spectinomycin resistance cassette flanked with the sccN upstream and downstream regions. The DNA fragment was transformed into S. mutans by electroporation. The mutants were isolated as described below. The plasmids for the deletion of sccP and atlA were constructed using the same strategy described for sccN deletion with primer pairs listed in Supplementary Table 6.
To delete rgpG and smaA in the WT strain, smaA in the ΔatlA and ΔsccN backgrounds, sccP in the ΔsccN background, and atlA in the ΔsccN and ΔsccH backgrounds, we used a PCR overlapping mutagenesis approach21. Briefly, 600–700 bp fragments both upstream and downstream of the gene of interest were amplified with designed primers that contained 16–20 bp extensions complementary to the nonpolar antibiotic resistance cassette. Spectinomycin, kanamycin, and erythromycin resistance cassettes were PCR-amplified from pLR16T, pOSKAR, and pHY304, respectively. The two fragments of the gene of interest and the fragment with the antibiotic resistance cassette were purified using the QIAquick PCR purification kit (Qiagen) and fused by Gibson Assembly (SGA-DNA). A PCR was then performed on the Gibson Assembly sample amplify the fused fragments. The assembled PCR fragments were transformed into corresponding S. mutans strains by electroporation. The transformants were selected either on THY or BHI agar containing the corresponding antibiotic. Double-crossover recombination was confirmed by PCR and Sanger sequencing using the specific primers.
To construct S. mutans strains expressing FtsZ fused at its C-terminus with a monomeric red fluorescent protein (tagRFP)34 using GSGS linker, we replaced ftsZ with ftsZ-tagRFP at its native chromosome locus. A nonpolar kanamycin resistance cassette was inserted downstream of ftsZ-tagRFP to allow the selection of recombinant bacteria. The fragment encoding tagRFP was PCR-amplified from pTagRFP-N (Evrogen). The fragments of ftsZ, tagRFP, kanamycin resistance cassette, and the ftsZ downstream region were PCR-amplified, purified and assembled as described above. S. mutans strains expressing iGFP-MapZ in which MapZ is fused at its N-terminus with a superfolder green fluorescent protein (GFP)35 via a LEGSGQGPGSGQGSG linker, were constructed similarly, except that a nonpolar spectinomycin resistance cassette followed by the GAS mapZ promoter was inserted upstream of iGFP-MapZ to confer mapZ expression and facilitate selection of recombinant bacteria. GFP was amplified from pHR-scFv-GCN4-sfGFP-GB1-NLS-dWPRE plasmid. To enhance expression of this protein fusion, an i-tag sequence10,36 plus an additional serine were added to the N-terminus of the GFP sequence (iGFP). A fragment of the mapZ upstream region was amplified using primers mapZ-F/spec-mapZ-R1. Spectinomycin resistance cassette was sequentially amplified using primer pairs mapZ-spec-F1/iGFP-Spec-R2a and mapZ-spec-F1/iGFP-Spec-R2b. iGFP-encoding fragment was sequentially amplified using primer pairs Spec-iGFP-F2a/mapZ-lnGFP-R3 and Spec-iGFP-F2b/mapZ-lnGFP-R3. 3’ fragment encoding MapZ was amplified using primers GFPln-mapZ-F3/mapZ-r. The fragments were assembled and transformed into S. mutans strains as outlined above. The WT cells expressing iGFP-MapZ show no apparent morphological defects (Supplementary Fig. 17), and have the characteristic oval shape with the average cell length and width of 0.92±0.07 μm (80 cells) and 0.58±0.04 μm (80 cells), respectively, according to fluorescent microscopy of the Nile Red-stained cells.
Complementation of ΔsccN
To complement ΔsccN, primers sccN-HindIII-f/sccN-BglII-r were used to amplify sccN from S. mutans DNA. The HindIII/BglII-digested PCR product was ligated into pDC123, yielding psccN. To replace the SCC Glc side-chains with the GAC GlcNAc side-chains, a part of the GAC operon, the gacHIJKL region (Supplementary Fig. 1c), required for the addition of the GlcNAc side-chains and GroP22 was expressed on pDC123 in ΔsccN. GAS 5448 DNA was used to amplify the gacHIJKL region using primers A109-f/A101-r. The PCR fragment was digested using XhoI/BamHI, and ligated into pDC123, yielding pgacHIJKL. All plasmids were confirmed by sequencing analysis (Eurofins MWG Operon) before electroporation into S. mutans. We found that one E. coli clone carrying pgacHIJKL acquired a frameshift resulting in a stop codon at leucine 49 in GacI. The plasmid was designated pgacHI*JKL and used as a negative control in our experiments. Plasmids were transformed into ΔsccN by electroporation. Chloramphenicol resistant single colonies were picked and checked for the presence of psccN or pgacHIJKL by PCR, yielding strains ΔsccN:psccN, ΔsccN:pgacHIJKL and ΔsccN:pgacHI*JKL.
Construction of the plasmids for expression of AtlAFL-GFP, GFP-AtlAM, AtlAC-GFP, AtlABSP-GFP, AtlABSP-cGFP, AtlABSP-tagRFP, GFP, eMapZ and GFP-eMapZ
GFP and tagRFP fusions were constructed using a PCR overlapping mutagenesis approach. The fragments were purified and assembled as described above. pRSF-NT plasmid was used for cloning and expression of all recombinant proteins. To create pKV1527 plasmid for AtlABSP-GFP expression, 5’ fragment encoding the Bsp domain was amplified from S. mutans DNA using the primers atlA_fus-F/atlA_fus-R, and 3’ fragment encoding GFP was amplified from pHR-scFv-GCN4-sfGFP-GB1-NLS-dWPRE using the primers gfp_fus-F/gfp_fus-R. To create pKV1532 plasmid for expression of GFP, a PCR product was amplified from pKV1527 using primers sfGFP_BspH/gfp_fus-R. To create pKV1556 plasmid for expression of the Bsp domain fused with colorless GFP (AtlABSP-cGFP), two PCR products were PCR-amplified from pKV1527 using primer pairs atlA_fus-F/Y66L_R and Y66L_F/gfp_fus-R to introduce Y66L mutation into GFP37. To create pKV1572 plasmid for expression of AtlABSP-tagRFP, 5’ fragment encoding the Bsp domain was amplified from S. mutans DNA using primers atlA_fus-F/atl_tRFP_R, and 3’ fragment encoding tagRFP was amplified from pTagRFP-N using primers tRFP_fusF/tRFP_fusR. To create pKV1641 plasmid for expression of AtlAFL-GFP, two 5’ fragments encoding the 104-kDa full-length form of AtlA (residues 24–979) carrying a catalytic site mutation E871A were PCR-amplified from S. mutans DNA using primer pairs atlA_fusF/E871A-R and E871A-F/GFP-atlA-R. GFP (3’ fragment) was PCR-amplified as described above using primers atlA-GFP-F/GFP-Not-R. To create pKV1642 plasmid for expression of AtlAC-GFP fusion, a DNA fragment encoding the catalytic domain of AtlA fused at the C-terminus with GFP was PCR-amplified from pKV1641 using primers atlA_770Nco/GFP-Not-Rm. To create pKV1643 plasmid for expression of GFP-AtlAM, 5’ fragment encoding GFP was PCR-amplified as described above using primerssfGFP_Nco/atlA-gfp-R, and 3’ fragment encoding the 90-kDa mature form of AtlA (residue 167–979) carrying a catalytic site mutation E871A was PCR-amplified from pKV1641 using primers gfp-atlA-F/atlA-Not. To create pKV1604 plasmid for expression of GFP-eMapZ, 5’ fragment encoding GFP was PCR-amplified as described above using primerssfGFP_Nco/gfp_map1_R, and two 3’ fragments encoding the extracellular domain of MapZ were PCR-amplified from S. mutans DNA using primer pairs gfp_map1_F/map1_dHind_R and map1_dHind_F/map_Hind_R. To create pKV1638 plasmid for expression of eMapZ, a DNA fragment encoding the extracellular domain of MapZ was PCR-amplified from pKV1604 using primers mapZ1-Nco/map_Hind_R.
Protein expression and purification
To purify the recombinant proteins, E. coli Rosetta (DE3) cells carrying the respective plasmids were grown in LB at 37°C to OD600 = 0.4–0.6 and induced with 0.25 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 27°C for approximately 4 hours. Bacteria were lysed in 20 mM Tris-HCl pH 7.5, 300 mM NaCl by a microfluidizer cell disrupter. AtlABSP-GFP, AtlABSP-cGFP, AtlABSP-tagRFP, GFP, eMapZ and GFP-eMapZ were purified by Ni-NTA chromatography followed by size exclusion chromatography (SEC) on a Superdex 200 column in 20 mM HEPES pH 7.5, 100 mM NaCl. AtlAFL-GFP, GFP-AtlAM and AtlAC-GFP were purified using Ni-NTA chromatography followed by rapid desalting to remove imidazole using PD-10 column.
Viability assay
S. mutans were grown to an OD600 of 0.5 and pushed ten times through a 26G 3/8 syringe to break bacterial clumps. Bacteria were serially diluted in phosphate-buffered saline (PBS) and plated on THY agar for enumeration.
Triton X-100-induced autolysis assay
Overnight cultures of S. mutans were diluted (1:100) into fresh THY broth and grown to an OD600 of 0.5. The autolysis assay was performed as outlined38 with one exception — cells were allowed to autolyze in PBS containing 0.2% Triton X-100. The autolysis was monitored after 2, 4, and 21 hours as a decrease in OD600. Results were normalized to the OD600 at time zero (OD600 of 0.5).
Isolation of cell walls and sacculi
The cell walls were isolated from mid-log phase cultures (OD600 of 0.5) by the SDS-boiling procedure and lyophilized, as previously described22. The sacculi were obtained by the SDS-boiling procedure followed by 4 washes each with 1 M NaCl and distilled water. The sacculi for fluorescence microscopy imaging were purified using the protocol for cell wall isolation with one exception — the bead beating step was omitted.
Co-sedimentation assay of fluorescent proteins with sacculi and cell wall material
To examine the binding of fluorescent proteins to sacculi, 0.5 mL of sacculi resuspended in PBS (OD600 of 3.4) were incubated with 48 μg mL−1 AtlABSP-GFP, or 67 μg mL−1 AtlAFL-GFP, or 45 μg mL−1 AtlAC-GFP, or 46 μg mL−1 GFP-eMapZ, or 28 μg mL−1 GFP, or 10 μg mL−1 succinylated wheat germ agglutinin (Vectorlabs) for 1 hour with agitation. Sample aliquots were assayed to determine the total fluorescence. Then samples were centrifuged (16,000 g, 3 min), followed by two washes with PBS. The pellet was resuspended in 0.5 mL of PBS, and sample aliquots were assayed to determine the pellet fluorescence. Data are presented as a percentage of fluorescence of the pellet normalized to the total fluorescence of the sample.
To examine the binding of AtlABSP-GFP to cell wall material, 0.5 mg of lyophilized cell wall was incubated with 48 μg mL−1 AtlABSP-GFP in 0.5 mL of PBS. The experiment was conducted as described above.
Pulldown of cell wall-associated proteins
S. mutans (1 L) were grown to an OD600 of 1.0, collected by centrifugation (5,000 g, 10 min), washed three times with PBS, and resuspended in 25 mL of urea solution (8 M urea, 20 mM Tris-HCl pH 7.5, 150 mM NaCl). The sample was rotated at room temperature for 1 hour, and then centrifuged (3,200 g, 10 min). The supernatant was dialyzed overnight at 4°C to remove urea and centrifuged again (3,200 g, 10 min). The supernatant was transferred to a fresh centrifuge tube and incubated with 10 mg of lyophilized cell wall with rotation for two hours. The cell wall was collected by centrifugation (3,200 g, 10 min) and washed three times with PBS. The proteins, retained in the cell wall, were dissolved in 0.5 mL of SDS sample buffer, and separated on 10% SDS-PAGE. Protein identification was performed at the Proteomics Core Facility (University of Kentucky) by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled with an Eksigent Nanoflex cHiPLC system (Eksigent) through a nanoelectrospray ionization source. The LC-MS/MS data were subjected to database searches for protein identification using Proteome Discoverer software V. 1.3 (Thermo Fisher Scientific) with a local MASCOT search engine.
Western blot analysis of AtlA expression in S. mutans strains
All strains were grown to an OD600 of 0.5. Bacteria (10 ml) were collected by centrifugation (5,000 g, 10 min). 1 ml of the supernatant was reserved for chloroform/methanol precipitation of proteins secreted in the growth medium. The pellet was washed two times with PBS and resuspended in 1 ml of 4% SDS. The sample was rotated at room temperature for 1 hour, and then centrifuged (3,200 g, 10 min). The supernatant (SDS-wash) and the chloroform/methanol precipitated proteins were separated on 10% SDS-PAGE. AtlA was detected by Western immunoblotting using rabbit anti-AtlA antibodies (a gift from Dr. Sang-Joon Ahn, University of Florida). Goat anti-Rabbit IgG (H+L) conjugated with HRP (ThermoFisher scientific) secondary antibody was used in Western blotting.
Glycerol and phosphate assays
The total phosphate content of SCCs was determined by the malachite green method following digestion with perchloric acid, as previously described21. To determine glycerol, SCCs were incubated with 2 N HCl at 100°C for 2 hours. The samples were neutralized with NaOH in the presence of 62.5 mM HEPES pH 7.5. Glycerol concentration was measured using the Glycerol Colorimetric assay kit (Cayman Chemical) according to the manufacturer’s protocol.
Modified anthrone assay
The Rha and Glc content was estimated by the anthrone procedure as described21 with a minor modification — the absorbance of the samples was measured at 580 nm and 700 nm. The chromophore produced from Rha has a relatively discreet absorbance maximum and is essentially zero at 700 nm. The absorbance of the Glc chromophore at 700 nm is approximately equal to its absorbance at 580 nm. Therefore, the contribution of Glc to the absorbance at 580 nm can be estimated from its absorbance at 700 nm and subtracted from the absorbance at 580 nm to obtain the Rha-specific signal. Rha and Glc concentrations were estimated using l-Rha and D-Glc standard curves, respectively.
Glycosyl composition analysis
Glycosyl composition analysis of SCC samples was performed at the Complex Carbohydrate Research Center (University of Georgia, Athens, GA) by combined gas chromatography/mass spectrometry (GC-MS) as alditol acetates as described previously39, following strong acid hydrolysis (4 N HCl, 100°C) to insure efficient hydrolysis of peptidoglycan22. Alternatively, glycosyl analysis as per-O-trimethylsilyl derivatives of O-methyl glycosides was performed following in-house pretreatment with HF, as described below. Appropriate aliquots were supplemented with 2 nmol inositol (internal standard), taken to dryness, and incubated with 25 μL 51% HF, 4°C, 16 hours. Following HF treatment, the samples were evaporated under a stream of air (the evaporating HF was captured by bubbling through a trap containing 1 M KOH), dissolved in water, transferred to screw-cap tubes equipped with Teflon lined caps and dried again. 0.2 mL 1 N HCl in methanol (formed by the drop-wise addition of acetyl chloride into rapidly-stirring anhydrous methanol) was added, the tubes were tightly sealed and incubated at 80°C, 16 hours. Following methanolysis, the reactions were neutralized with ~3–5 mg AgCO3, centrifuged, and the supernatant transferred to a fresh tube. To re-N-acetylate amino sugars, the samples were taken to dryness under a stream of air, evaporated out of 0.5 mL of methanol, and re-dissolved in 0.1 mL of methanol containing 10 % pyridine and 10 % acetic anhydride. The reactions were dried, trimethylsilylated with 25 μL Tri-Sil TH (Sigma Aldrich), and analyzed by methane chemical ionization GC-MS using a Thermo ISQ mass spectrometer interfaced with a gas chromatograph, equipped with a 15 m Equity 1701 glass capillary column and helium carrier gas.
GlcNAc analysis
GlcNAc content was assayed using the Megazyme Glucosamine Assay Kit according to the manufacturer’s instructions with some minor modifications. Partially purified polysaccharide (~40–60 nmol Rha) was hydrolyzed in 40 μL 2 N HCl, 100°C, 2 hours, neutralized with 10 N NaOH (to ~pH 7.0 by pH paper) and adjusted to a final volume of 50 μL with water. The acid hydrolysis de-acetylates GlcNAc to generate glucosamine. An aliquot (5 μL) of the neutralized hydrolysate was diluted with 171 μL water and mixed with a total of 24 μL of Megazyme Glucosamine Assay Kit Enzyme Mix. Absorbance at 340 nm was recorded after incubation at room temperature for 40 min. Glucosamine content was determined by comparison with a glucosamine standard curve and verified by comparison with GlcNAc standards treated similarly as the polysaccharide.
Analysis of Rha content in the intact S. mutans
S. mutans strains were grown to an OD600 of 0.5, collected by centrifugation (5,000 g, 10 min), washed one time with PBS, and resuspended in PBS. The Rha content was estimated by GC-MS method following methanolysis as described above. The concentration was expressed as a percentage of the WT values.
Release of SCCs from cell wall and sacculi by mild acid hydrolysis
SCCs were released from purified cell walls and sacculi by mild acid hydrolysis following N-acetylation, as previously described for WTA of Lactobacillus plantarum40 with some modifications. Lyophilized cell wall material (4 mg) or sacculi (0.5 mL, OD600 of 3.4) were N-acetylated using 2 % acetic anhydride in 1 mL of saturated NaHCO3. After incubation at room temperature overnight, the reactions were diluted with 2 vol water, and sedimented at 50,000 g, 10 min. The cell walls/sacculi were washed three times by water followed by sedimentation at 50,000 g, 10 min. N-acetylated cell walls/sacculi were resuspended with 0.02 N HCl (0.2 mL), and heated to 100°C for 20 min. The reactions were cooled on ice, neutralized by the addition of 4 μL 1 N NaOH, and sedimented at 50,000 g, 10 min. The supernatant fraction was removed and reserved. The pellet was resuspended in 0.2 mL water and re-sedimented. The supernatant fractions were combined and either analyzed or purified further by a combination of SEC and ion-exchange chromatography. The pellet fractions were assayed for Rha content and either discarded or assayed for binding of AtlABSP-GFP and WGA.
Release of SCCs from sacculi by hydrofluoric acid hydrolysis
Sacculi (0.5 mL, OD600 of 3.4) prepared from ΔsccN were sedimented (50,000 × g, 10 min), resuspended in ice-cold 0.1 mL HF (48–51%, Axios Chemicals), and incubated at 4°C for 144 h. The reactions were neutralized on ice by the addition of 10 N KOH followed by centrifugation (50,000 × g, 10 min). The pellet was washed extensively with water and used for analysis of AtlABSP-GFP binding. The supernatant and washes were combined, desalted by centrifugation through Amicon Ultra 0.5 mL Centrifugal Filter (3,000 NMWL), and analyzed for Rha using anthrone assay. The efficiency of removal of Rha polysaccharide was determined from identical parallel incubations.
Fractionation of SCCs on DEAE-Toyopearl
SCCs were released from purified cells walls by mild acid hydrolysis and fractionated on BioGel P150 equilibrated in 0.2 N sodium acetate, pH 3.7, 0.15 M NaCl. The fractions containing SCCs were combined and concentrated using an Amicon Ultra - 15 Centrifugal Filter (KUltracel -3K). The retentate was desalted by repeated cycles of dilution with water and centrifugation. The concentrated SCCs (~0.5 mL) were applied to a 1×18 cm column of TOYOPEARL DEAE-650M (TOSOH Bioscience), equilibrated in 10 mM Tris-Cl, pH 7.4, and fractions of 2 mL were collected. After 20 fractions, the column was eluted with an 80 mL gradient of NaCl (0–0.5 M). Appropriate aliquots were analyzed for Rha and Glc by anthrone assay.
Derivatization of SCCs with 7-amino-1,3-naphthalenedisulfonic acid (ANDS)
SCCs purified by a combination of SEC and ion-exchange chromatography were fluorescently tagged at the reducing end by reductive amination with ANDS as previously described41. Reaction mixtures contained 30–100 nmol of SCCs (lyophilized), 0.75 mM ANDS and 0.5 M NaCNBH4 in 0.05 mL acetic acid/DMSO at 7.5/50 (%). Following overnight incubation at 37°C, the reactions were desalted by centrifugation on an Amicon Ultra Centrifugal Filter (3,000 NMWL). Derivatized SCCs were further purified by SEC over a Superdex 75 10/300 GL column (GE Healthcare Bio-Sciences AB).
FACS analysis
AtlABSP-GFP (5 μL, 3.8 mg mL−1) or GFP (5 μL, 3.5 mg mL−1) were added to 100 μL of E. coli CS277528 or PHD136 [E. coli CS2775 harboring pRGP1 plasmid28] resuspended to an OD600 of 0.4 in PBS. After 20 minutes of incubation on ice, the cells were centrifuged (20,800 g, 5 min). The pellet was washed twice with PBS, resuspended in PBS, incubated with paraformaldehyde (4% final concentration) at 4°C, 20 min, and then washed once with PBS. The cells were resuspended in PBS with 0.3% BSA, and immediately analyzed by flow cytometry (BD LSRFortessa). Anti-GAC antibodies conjugated with FITC (ABIN238144, antibodies-online, titer 1:50) were used as a positive control to confirm polyrhamnose expression in E. coli strain PHD136. The FACS data were analyzed using flowJo version 10.
Scanning electron microscopy (SEM)
S. mutans were grown to an OD600 of 0.7, harvested by centrifugation (3,200 g, 10 min), washed once with 20 mM HEPES, 0.5% BSA buffer (pH 8.0), resuspended in PBS, incubated with paraformaldehyde (4% final concentration) at 4°C, 15 min, and then pipetted onto microscope slide cover glasses coated with poly-l-lysine. Following one hour incubation, the cover glasses were washed 3 times with PBS. Bacteria were dehydrated stepwise in a gradient series of ethanol (35%, 50%, 70%, and 96% for 20 min each and then 100% overnight), followed by critical point drying with liquid CO2 in a Leica EM CPD300. Samples were coated with about 5 nm of platinum controlled by a film-thickness monitor. SEM images were performed in the immersion mode of an FEI Helios Nanolab 660 dual beam system.
Fluorescent and differential interference contrast (DIC) microscopy
S. mutans were grown to an OD600 of 0.3, incubated with paraformaldehyde (4% final concentration) at 4°C, 15 min, pipetted onto microscope slide cover glasses (high performance, D=0.17 mm, Zeiss) coated with poly-l-lysine, and allowed to settle for one hour at room temperature. Bacteria were incubated with 15.6 μg mL−1 AtlAFL-GFP, or 15.5 μg mL−1 GFP-AtlAM, or 15.7 μg mL−1 AtlABSP-GFP, or AtlAC-GFP, or AtlABSP-tagRFP, or GFP for 15 min at room temperature. Additionally, 10, 50 and 100x dilutions of AtlAFL-GFP were used for some experiments. The samples were washed four times with PBS, dried at room temperature, and mounted on a microscope slide with ProLong Diamond Antifade Kit with DAPI (Invitrogen). Samples were imaged on a Zeiss LSM 880 using Airyscan mode and a Leica SP8 equipped with 100X, 1.44 N.A. objective, DIC optics, and “lightning” post-processing. Images were processed with Airyscan processing and the “lightning” processing tool, respectively. Samples with S. mutans cells expressing iGFP-MapZ or iGFP-MapZ/FtsZ-tagRFP were prepared similarly except that the high refractive index coverslips (HIGHINDEX-CG, N4247800, Olympus) were used for imaging on a Leica SP8 equipped with APON100XHOTIRF. Images were deconvolved using Huygens Professional software. Raw and data-processed images of S. mutans are shown in Supplementary Fig. 18.
Two approaches were used to determine the sizes of cells. The images of S. mutans (grown to an OD600 of 0.3) in DIC were obtained on a Leica SP8 confocal microscope and analyzed using ImageJ software (ObjectJ plug-in). Alternatively, the bacteria (OD600 of 0.3) were incubated with 1 μg mL−1 Nile red prior to fixation. Cells were imaged and analyzed as described above. To visualize cells with DNA, the bacteria were incubated with 0.1 μg mL−1 4’,6-Diamidino-2-Phenylindole (DAPI) prior to fixation.
Fluorescence anisotropy
Reactions (150 μL) containing 0.5 μM ANDS-labeled polysaccharide and 0–10 μM AtlABSP-cGFP or GFP-eMapZ were incubated at room temperature for 30 minutes in 20 mM Tris 7.5, 300 mM NaCl. The anisotropy was then measured on a Fluoromax-4 (Horiba) photon-counting steady-state fluorometer at 25°C using an excitation wavelength of 310 nm and the emission at 450 nm with slit widths of 5 nm and an integration time of 1 second. Curves terminate at 10 μM of AtlABSP-cGFP due to aggregation issues at higher protein concentration. The single-site total binding equation in Graphpad Prism 8.4 was used to fit the binding of the ΔsccNΔsccP SCC. Lower-bound estimates of the Kd for the other three SCC species were determined by fitting to the binding equation after fixing the maximal binding limit, Bmax, to that of ΔsccNΔsccP.
Analytical ultracentrifugation
Sedimentation velocity (SV) experiments were performed in a Beckman ProteomeLab XL-I at 20°C using absorbance optics at 278 nm in an An-60Ti rotor at 32,000 rpm until complete sedimentation of sample occurred. The analysis was conducted using Sedfit 16.1c42,43 using the c(s) data model and expressed using sedimentation coefficient distributions. Each fit rmsd was 0.005 or lower. GUSSI 1.4.144 was used for data visualization. SV data were also fitted using Wide Distribution Analysis (WDA) in SedAnal v7.1145. WDA distributions were computed from 6.10 cm to 7.00 cm with an increment of 0.01 cm. The radial range plotted was 6.40 – 6.60 cm in 0.01 cm increments, with a 2% smoothing algorithm applied (equivalent to 4 scans). The weight-average sedimentation coefficient (sw) was computed using a range of 2–100 S. Partial specific volume (v-bar) was 0.725 ml g−1, and the solution density (ρ) was 0.998 g mL−1.
Statistical analysis
Unless otherwise indicated, statistical analysis was carried out on pooled data from at least three independent biological repeats. Statistical analysis of data was performed using one-way ANOVA, 2-way ANOVA, two-tailed Student’s t-test, Kruskal – Wallis non-parametric test and Friedman’s test as described for individual experiments. A P-value equal to or less than 0.05 was considered statistically significant.
Extended Data
Extended Data Fig. 1 |. Modifications of SCC control S. mutans cell aggregation and morphology.
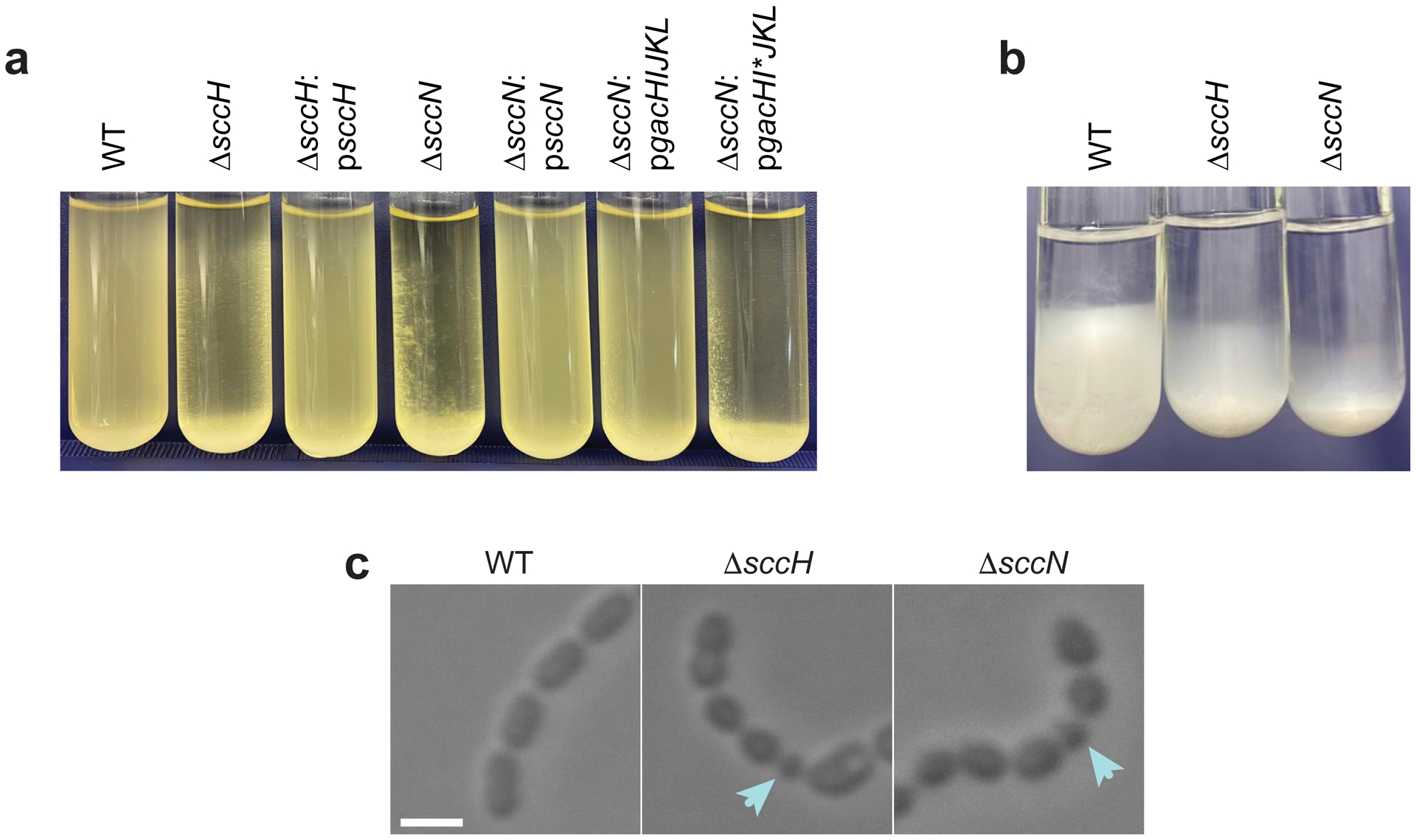
a, Sedimentation phenotype of S. mutans strains after overnight growth in THY broth. b, Sedimentation phenotype of sacculi purified from S. mutans strains. Sacculi were resuspended in PBS (OD600 of 8), and allowed to sediment for 72 hours at 4 °C. c, DIC images of S. mutans strains taken from mid-log growth phase. Blue arrows denote small round cells. Scale bar is 1 μm. Representative images from at least three independent experiments are shown in a, b and c.
Extended Data Fig. 2 |. Fluorescent microscopy of S. mutans intact cells and sacculi labeled by AtlABSP-GFP.
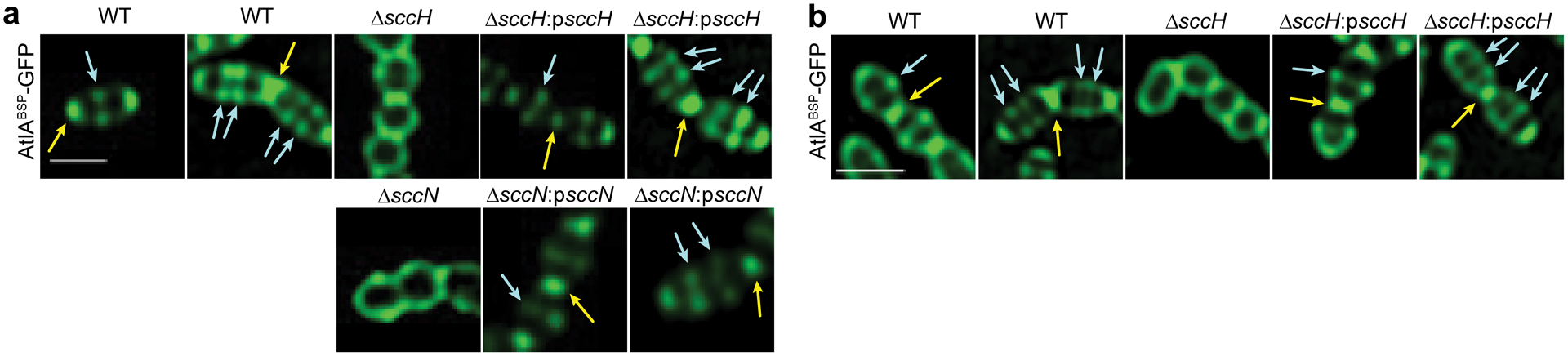
a, Binding of AtlABSP-GFP to the intact WT, ΔsccH, ΔsccH:psccH, ΔsccN and ΔsccN:psccN bacterial cells. b, Binding of AtlABSP-GFP to the sacculi purified from WT, ΔsccH and ΔsccH:psccH. In a and b, yellow and blue arrows show polar and equatorial sites labeled by AtlABSP-GFP, respectively. Scale bar is 1 μm in a and b. The experiments depicted in a and b were performed independently three times and yielded the same results.
Extended Data Fig. 3 |. Gating Strategy for Flow Cytometry (Fig. 4b).
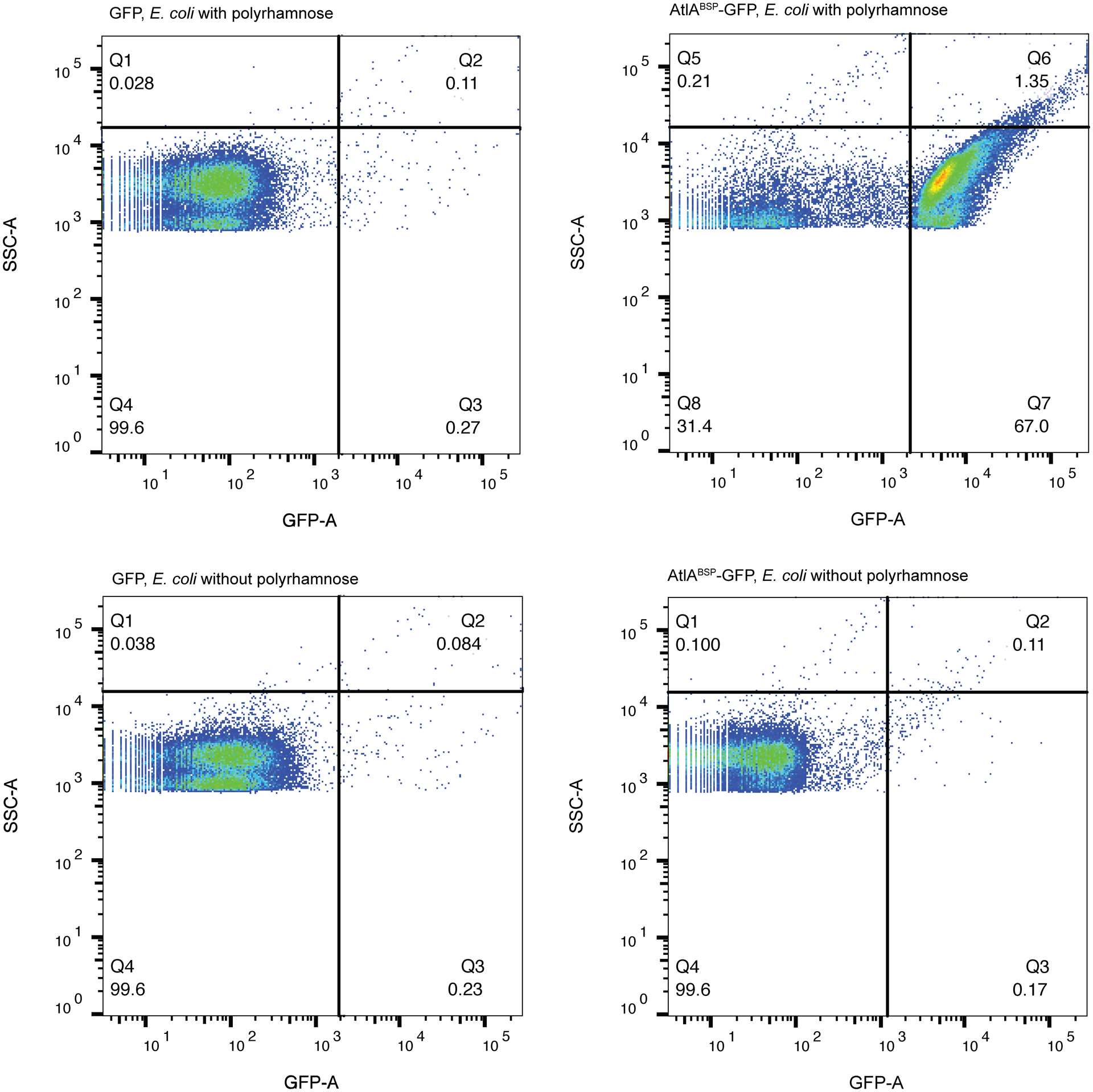
In this sample gating, E. coli expressing polyrhamnose on the cell surface and its parental strain (E. coli without polyrhamnose) were gated based on the presence of fluorescent signal (AtlABSP-GFP). Bacterial gating occurred at the GFP/SCC density plot omitting signals derived from bacteria stained with GFP alone (GFP). For flow cytometric analysis, at least 10,000 events were collected. Experiments were performed independently three times and yielded the same results. Dot plots of representative results are shown.
Extended Data Fig. 4 |. S. mutans WT produces SCCs with different degrees of GroP modification.

a, Ion exchange chromatography of SCCs purified from ΔsccH. b, Ion exchange chromatography of SCCs purified from WT S. mutans. SCC material was loaded onto Toyopearl DEAE-650M and eluted with a NaCl gradient (0–0.5 M). Fractions were analyzed for Rha and Glc contents by anthrone assay and total phosphate (Pi) content by malachite green assay following digestion with perchloric acid. c, Composition analysis of minor and major fractions from b. Fractions were pooled, concentrated, and desalted by spin column and analyzed by GC-MS to determine the Rha and Glc concentrations. The concentration of Glc is presented relative to the Rha concentration. Data are means ± S.D., n = 3 biologically independent experiments. P values were calculated by a two-tailed t-test. d, SDS-PAGE analysis of ANDS-labeled SCCs extracted from S. mutans mutants. In a, b and d, the experiments were performed at least three times and yielded the same results. Data from one representative experiment are shown.
Supplementary Material
Acknowledgments
The authors thank Dr. Sang-Joon Ahn (University of Florida) for the kind gift of anti-AtlA antibodies, Dr. Jacqueline Abranches (University of Florida) for providing S. mutans serotype e, f and k strains. Dr. John F. Timoney (University of Kentucky) and Dr. Johannes Huebner (von Hauner Children’s Hospital, LMU) for providing S. equi and E. faecalis, respectively, Dr. Jeffrey M. Bosken and Dr. Edward D. Hall (University of Kentucky) for the use of the Thermo Scientific GC-MS instrument and Dr. Catalina Velez-Ortega (University of Kentucky) for the access to Leica SP8 confocal microscope.
This work was supported by NIH grants R01 DE028916 from the NIDCR and R01 AI143690 from the NIAID (to NK), R01 GM094363 from the NIGMS (to ABH) and R01 DC014658 from the NIDCD (to GIF), Tenovus Scotland Large Research Grant T17/17 and University of Dundee Wellcome Fund 105606/Z/14/Z (to SAC and HCD), Wellcome and Royal Society Grant 109357/Z/15/Z (to HCD). Scanning electron microscopy was performed at the Electron Microscopy Center, which belongs to the National Science Foundation NNCI Kentucky Multiscale Manufacturing and Nano Integration Node, supported by ECCS-1542174. Carbohydrate composition analysis at the Complex Carbohydrate Research Center was supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, U.S. Department of Energy grant (DE-FG02-93ER20097). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
All data generated during this study are included in the article and supplementary information files.
References
- 1.Brown S, Santa Maria JP Jr. & Walker S Wall teichoic acids of Gram-positive bacteria. Annu Rev Microbiol 67, 313–336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higgins ML & Shockman GD Model for cell wall growth of Streptococcus faecalis. J Bacteriol 101, 643–648 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Land AD & Winkler ME The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. Journal of bacteriology 193, 4166–4179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sham LT, Tsui HC, Land AD, Barendt SM & Winkler ME Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opin Microbiol 15, 194–203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheeler R, Mesnage S, Boneca IG, Hobbs JK & Foster SJ Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Mol Microbiol 82, 1096–1109 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Massidda O, Novakova L & Vollmer W From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol 15, 3133–3157 (2013). [DOI] [PubMed] [Google Scholar]
- 7.van Raaphorst R, Kjos M & Veening JW Chromosome segregation drives division site selection in Streptococcus pneumoniae. Proc Natl Acad Sci U S A 114, E5959–E5968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleurie A et al. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature 516, 259–262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holeckova N et al. LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio 6, e01700–01714 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez AJ et al. Movement dynamics of divisome proteins and PBP2x:FtsW in cells of Streptococcus pneumoniae. Proc Natl Acad Sci U S A 116, 3211–3220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manuse S et al. Structure-function analysis of the extracellular domain of the pneumococcal cell division site positioning protein MapZ. Nat Commun 7, 12071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto H, Miyake Y, Hisaoka M, Kurosawa S & Sekiguchi J The major and minor wall teichoic acids prevent the sidewall localization of vegetative DL-endopeptidase LytF in Bacillus subtilis. Mol Microbiol 70, 297–310 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Schirner K, Marles-Wright J, Lewis RJ & Errington J Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J 28, 830–842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlag M et al. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol Microbiol 75, 864–873 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Lunderberg JM, Liszewski Zilla M, Missiakas D & Schneewind O Bacillus anthracis tagO Is Required for Vegetative Growth and Secondary Cell Wall Polysaccharide Synthesis. J Bacteriol 197, 3511–3520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnet J et al. Nascent teichoic acids insertion into the cell wall directs the localization and activity of the major pneumococcal autolysin LytA. The Cell Surface 2, 24–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mistou MY, Sutcliffe IC & van Sorge NM Bacterial glycobiology: rhamnose-containing cell wall polysaccharides in Gram-positive bacteria. FEMS Microbiol Rev 40, 464–479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St Michael F et al. Investigating the candidacy of the serotype specific rhamnan polysaccharide based glycoconjugates to prevent disease caused by the dental pathogen Streptococcus mutans. Glycoconj J 35, 53–64 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Coligan JE, Kindt TJ & Krause RM Structure of the streptococcal groups A, A-variant and C carbohydrates. Immunochemistry 15, 755–760 (1978). [DOI] [PubMed] [Google Scholar]
- 20.McCarty M Variation in the group-specific carbohydrate of group A streptococci. II. Studies on the chemical basis for serological specificity of the carbohydrates. J Exp Med 104, 629–643 (1956). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RJ et al. Discovery of glycerol phosphate modification on streptococcal rhamnose polysaccharides. Nat Chem Biol 15, 463–471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rush JS et al. The molecular mechanism of N-acetylglucosamine side-chain attachment to the Lancefield group A carbohydrate in Streptococcus pyogenes. J Biol Chem 292, 19441–19457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown TA Jr. et al. A hypothetical protein of Streptococcus mutans is critical for biofilm formation. Infect Immun 73, 3147–3151 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata Y, Kawada M, Nakano Y, Toyoshima K & Yamashita Y Identification and characterization of an autolysin-encoding gene of Streptococcus mutans. Infect Immun 73, 3512–3520 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn SJ & Burne RA The atlA operon of Streptococcus mutans: role in autolysin maturation and cell surface biogenesis. J Bacteriol 188, 6877–6888 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshimura G et al. Identification and molecular characterization of an N-Acetylmuraminidase, Aml, involved in Streptococcus mutans cell separation. Microbiol Immunol 50, 729–742 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Yamashita Y et al. A novel gene required for rhamnose-glucose polysaccharide synthesis in Streptococcus mutans. J Bacteriol 181, 6556–6559 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata Y, Yamashita Y, Ozaki K, Nakano Y & Koga T Expression and characterization of streptococcal rgp genes required for rhamnan synthesis in Escherichia coli. Infect Immun 70, 2891–2898 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zorzoli A et al. Group A, B, C, and G Streptococcus Lancefield antigen biosynthesis is initiated by a conserved alpha-d-GlcNAc-beta-1,4-l-rhamnosyltransferase. J Biol Chem 294, 15237–15256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y et al. MapZ Forms a Stable Ring Structure That Acts As a Nanotrack for FtsZ Treadmilling in Streptococcus mutans. ACS Nano 12, 6137–6146 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Neuhaus FC & Baddiley J A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in Gram-positive bacteria. Microbiol Mol Biol Rev 67, 686–723 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Y et al. Systematic Hydrogen-Bond Manipulations To Establish Polysaccharide Structure-Property Correlations. Angew Chem Int Ed Engl 58, 13127–13132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y & Delbianco M Conformational Studies of Oligosaccharides. Chemistry 26, 9814–9825 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merzlyak EM et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods 4, 555–557 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC & Waldo GS Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24, 79–88 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Catalao MJ, Figueiredo J, Henriques MX, Gomes JP & Filipe SR Optimization of fluorescent tools for cell biology studies in Gram-positive bacteria. PLoS One 9, e113796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenow MA, Huffman HA, Phail ME & Wachter RM The crystal structure of the Y66L variant of green fluorescent protein supports a cyclization-oxidation-dehydration mechanism for chromophore maturation. Biochemistry 43, 4464–4472 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Dufour D & Levesque CM Cell death of Streptococcus mutans induced by a quorum-sensing peptide occurs via a conserved streptococcal autolysin. J Bacteriol 195, 105–114 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Templeton DW, Quinn M, Van Wychen S, Hyman D & Laurens LM Separation and quantification of microalgal carbohydrates. J Chromatogr A 1270, 225–234 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Kojima N, Araki Y & Ito E Structural studies on the linkage unit of ribitol teichoic acid of Lactobacillus plantarum. Eur J Biochem 148, 29–34 (1985). [DOI] [PubMed] [Google Scholar]
- 41.Lehrman MA & Gao N Alternative and sources of reagents and supplies of fluorophore-assisted carbohydrate electrophoresis(FACE). Glycobiology 13, 1G–3G (2003). [DOI] [PubMed] [Google Scholar]
- 42.Chaturvedi SK, Ma J, Brown PH, Zhao H & Schuck P Measuring macromolecular size distributions and interactions at high concentrations by sedimentation velocity. Nat Commun 9, 4415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brautigam CA Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol 562, 109–133 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Stafford WF & Braswell EH Sedimentation velocity, multi-speed method for analyzing polydisperse solutions. Biophys Chem 108, 273–279 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during this study are included in the article and supplementary information files.