

Summary
Glycosylation is one of the most common protein modifications in living organisms and has important regulatory roles in animal tissue development and homeostasis. Here, we present a protocol for generation of 3D organotypic skin models using CRISPR-Cas9 genetically engineered human keratinocytes (N/TERT-1) to study the role of glycans in epithelial tissue formation. This strategy is also applicable to other gene targets and organotypic tissue models. Careful handling of the cell cultures is critical for the successful formation of the organoids.
For complete details on the use and execution of this protocol, please refer to Dabelsteen et al. (2020).
Subject areas: Cell Biology, CRISPR, Organoids
Graphical abstract
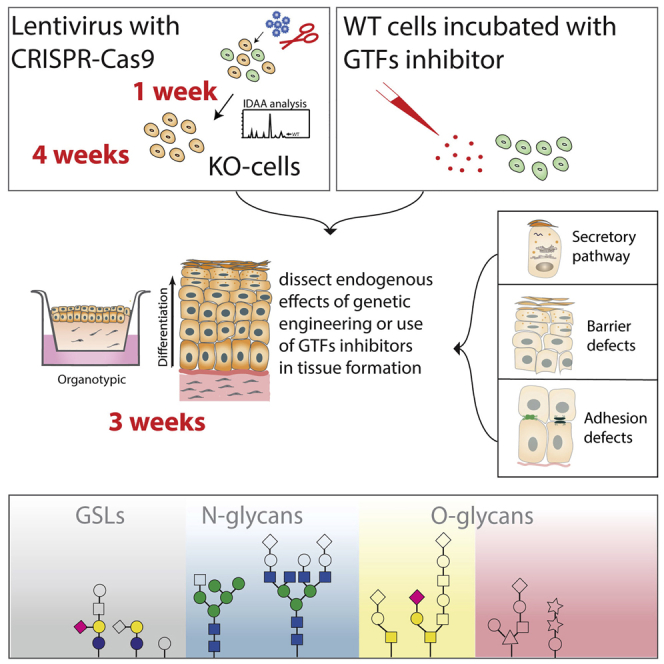
Highlights
-
•
CRISPR-Cas9 gene targeting to generate a library of 3D organotypic skin tissues
-
•
Approach can be used to study the role of glycans in epithelial tissue formation
-
•
Strategy applicable to other targets and organotypic tissue models
Glycosylation is one of the most common protein modifications in living organisms and has important regulatory roles in animal tissue development and homeostasis. Here, we present a protocol for generation of 3D organotypic skin models using CRISPR-Cas9 genetically engineered human keratinocytes (N/TERT-1) to study the role of glycans in epithelial tissue formation. This strategy is also applicable to other gene targets and organotypic tissue models. Careful handling of the cell cultures is critical for the successful formation of the organoids.
Before you begin
Human organotypic skin cultures build with genetically engineered cell lines are attractive models that offer a well-defined system to visualize the importance of select genes in cellular differentiation and histogenesis (Ridky et al., 2010). Keratinocytes have the ability to undergo complete histogenesis in three-dimensional (3D) organotypic cultures, faithfully reflecting the normal human epidermis (Parenteau et al., 1991, Gangatirkar et al., 2007). We recently combined human organotypic skin cultures with genetically engineered keratinocytes to create a library of 3D organotypic skin tissues that differ in their capacity to produce select glycan structures (Dabelsteen et al., 2020). We used this model system to address the importance of specific glycan structures for tissue formation and homeostasis (Dabelsteen et al., 2020). Here we describe a protocol that uses the N/TERT-1 keratinocyte cell line grown on a collagen gel with embedded fibroblasts (Dickson et al., 2000). Several other keratinocyte cell lines can be used (Ridky et al., 2010, Bagdonaite et al., 2020, Radhakrishnan et al., 2014), and similarly collagen can be exchanged with other scaffolds that support epidermal growth, including matrix based dermal compartments, de-epidermalised dermal tissue, or fibrinogen coated membranes (Stark et al., 2004, Prunieras et al., 1983, Garlick, 2007, El Ghalbzouri et al., 2009, Black et al., 2005, Berning et al., 2015, Auxenfans et al., 2009). The general genetic engineering strategy is applicable to other organotypic tissue models and can be combined with small compound inhibitors (Figure 1).
Figure 1.

Glycosyltransferases and their respective biosynthetic pathways
Genes marked with bold are knocked out as described in the paper (Dabelsteen et al., 2020) and respective glycan moiety is shown in gray. Glycan symbols are drawn according to the SNFG format (Varki et al., 2015). Inhibition of sialylation by the Ac5SiaFEtoc inhibitor is designated by crosses in gray.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermo Fisher | Cat#C7373-03 |
| Chemicals, peptides, and recombinant proteins | ||
| Opti-MEM | Thermo Fisher (GIBCO) | Cat#31985062 |
| Polybrene | Sigma Aldrich | Cat#TR-1003-G |
| Puromycin | Thermo Fisher (GIBCO) | Cat#A1113803 |
| Blasticidin S HCl | Thermo Fisher (GIBCO) | Cat#A1113903 |
| Pen Strep | Thermo Fisher (GIBCO) | Cat#15140122 |
| DMEM | Thermo Fisher (GIBCO) | Cat#41966029 |
| Keratinocyte-SFM | Thermo Fisher (GIBCO) | Cat#17005034 |
| Dimethyl Sulfoxide | Sigma Aldrich | Cat#D2650 |
| TrypLE | Thermo Fisher (GIBCO) | Cat#12605028 |
| Bovine Pituitary Extract | Thermo Fisher (GIBCO) | Cat#13028014 |
| Fetal Bovine Serum | HyClone | Cat#SH30071.03HI |
| EGF | Thermo Fisher (GIBCO) | Cat#PHG0314 |
| Insulin | Sigma Aldrich | Cat#I6634 |
| Hydrocortisone | Sigma Aldrich | Cat# H0888 |
| Triidothryonine “T3” | Sigma Aldrich | Cat# T2752 |
| Rat tail collagen I | Made in lab | N/A |
| L-Glutamine | Thermo Fisher (GIBCO) | Cat#A2916801 |
| Hams F12 | Thermo Fisher (GIBCO) | Cat#11765-054 |
| Adenine | Sigma Aldrich | Cat#A8626 |
| Cholera Enterotoxin | Sigma Aldrich | Cat#C8180 |
| 10X MEM | Thermo Fisher (GIBCO) | Cat#11430-030 |
| Sodium Bicarbonate | Merck | Cat#6392 |
| Gentamycine (50mg/mL) | GIBCO | Cat#15750037 |
| Methyl (3-(R)-Fluoro-5-N-ethyloxycarbonyl- 2,4,7,8,9-penta-O-acetyl-3,5-dideoxy-D-a/ b-glycero-D-galacto-2-nonulopyranose) onate(Ac5SiaFEtoc) |
Thomas J. Boltje, Cluster for Molecular Chemistry, Institute for Molecules and Materials, Radboud University Nijmegen, the Netherlands |
N/A |
| BsmBI | NEB | Cat#R0580 |
| T4 DNA ligase Buffer (10x) | Thermo Fisher | Cat#B69 |
| Critical commercial assays | ||
| NucleoBond Xtra Midi EF | Macherey-Nagel | Cat#740420.50 |
| CoboXtract | Cobo Technologies | Cat#C20101 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| MRC-5 | ATCC | Cat# CCL-171, RRID:CVCL_0440 |
| N/TERT-1 | James G. Rheinwald, Harvard Institute of Medicine | N/A |
| Recombinant DNA | ||
| pCMV-VSV-G |
Addgene | RRID:Addgene_845 4 |
| lentiCRISPR v2 |
Addgene | RRID:Addgene_529 61 |
| psPAX2 | Addgene | RRID:Addgene_122 60 |
| Software and algorithms | ||
| Peak Scanner Software V1.0 | Thermo Fischer | Cat#: 4381867 |
| Other | ||
| Syringe filter 0.45-μm | Sartorius | Cat#17598-K |
| Deep well plates | Corning | Cat#355467 |
| Cell culture insert, PET membrane, 3.0 μm pore size | Corning | Cat#353092 |
| Airlift pads (cut in circles and autoclaved) | Whatman, GE Healthcare |
Cat# 10382461 |
Materials and equipment
Adenine (100× stock):
| 24 mg/mL adenine | Dissolve 243 mg of adenine in 100 mL of 0.05 M HCl |
-
•
Stir for about an hour at 20°C–22°C
-
•
Filter-sterilize with 0.2-μm filters
-
•
Divide into 5 mL aliquots
-
•
Store at −20°C for up to 2 years
Cholera enterotoxin (CT) – this is a 1000× stock solution for 100 pM
| 10 μM CT | 1 mg vial of cholera enterotoxin, add 1.18 mL of MiliQ water Dilute this in 110 mL PBS |
-
•
Filter-sterilize with 0.2-μm filters
-
•
Aliquot at 2 mL/cryovial
-
•
Store at −20°C for up to 2 years
Note: MWCT = 84.000 Da
Hydrocortisone (HC) - 500× stock for 0.4 μg/mL
-
•
First prepare 5 mg/mL HC, which is 12.500× for 0.4 μg/mL. This can be stored at –20°C indefinitely.
| 5 mg/mL HC | 25 g vial (Calbiochem), dissolve in 5 mL of 100% ethanol |
-
•
Prepare 0.2 mg/mL HC from the 5 mg/mL stock
| 0.2 mg/mL HC | 2 mL 5 mg/mL HC, fill up with 48 mL sterile (PBS + 0.1% BSA) |
-
•
Filter-sterilize with 0.2-μm filters
-
•
Aliquot 2 mL into cryovials
-
•
Store at −20°C for up to 2 years
Epidermal growth factor (EGF) – this is a 50000× stock for 0.2 ng/mL
| 10 μg/mL EGF | Dissolve 100 μg EGF in 10 mL of (PBS + 0.1% BSA) |
-
•
Filter-sterilize with 0.2-μm filters
-
•
Aliquot at 1 mL into cryovials
-
•
Store at −20°C for up to 2 years
Triidothryonine (T3) - this is a 1000× stock for 20 pM (2 × 10−11 M)
-
•
First prepare 2 μM T3, which is a 100,000× for 20 pM T3
| 2 μM T3 | 6.8 mg of T3, dissolve in 7.5 mL of 0.02 N NaOH Add 42.5 mL sterile PBS |
-
•
Store at −20°C in 10 mL aliquots
-
•
Prepare 20 nM T3 to make the 1000× stock for 20 pM (2 × 10−11 M)
| 20 nM T3 | 0.4 mL 2 μM T3, dilute in 39.6 mL sterile PBS |
-
•
Filter-sterilize with 0.2-μm filters
-
•
Divide into 2 mL aliquots
-
•
Store at −20°C for up to 2 years
Calcium chloride (CaCl2) - this is a 1000× stock for 0.3 mM
| 300 mM CaCl2 | 4.4 g CaCl2.2H2O in 100 mL of MiliQ water |
-
•
Stir with magnetic bar to dissolve completely at 20°C–22°C
-
•
Filter-sterilize with 0.2-μm filters
-
•
Aliquot at 2 mL per cryovial
-
•
Store at −20°C indefinitely
Insulin – this is a 1000× for 5 μg/mL
Note: Use Insulin from Bovine Pancreas
| 5 mg/mL Insulin | 100 mg insulin in 20 mL of 0.005 M HCl |
-
•
Filter-sterilize through a cellulose-acetate, low-protein-binding 0.2 μm filter
-
•
Aliquot at 500 μL
-
•
Store at −20°C for up to 2 years
Keratinocyte Serum Free Medium (K-SFM) complete
| Reagent | Final concentration | Amount |
|---|---|---|
| Keratinocyte-SFM | n/a | 500 mL |
| CaCl2 (1000×) | 0.3 mM | 500 μL |
| BPE (16.8 mg/mL) | 25 μg/mL | 750 μL |
| EGF (10 μg/mL) | 0.2 ng/mL | 10 μL |
| Total | n/a | 500.91 mL |
Store at 4°C for up to 3 months
Organoid culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM | n/a | 375 mL |
| F12 | n/a | 125 mL |
| Pen/Strep | 100 U/mL | 5.14 mL |
| Adenine (2.4 mg/mL stock) | 24 μg/mL | 5.14 mL |
| HC (0.2 mg/mL stock) | 0.4 μg/mL | 1 mL |
| CT (10 μM stock) | 100 pM | 0.514 mL |
| T3 (1000×) | 20 pM | 0.514 mL |
| Insulin (1000×) | 5 μg/mL | 500 μL |
| FCS | 0.3% | 1.54 mL |
Store at 4°C for up to 3 months
DMEM 10% FBS
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM | n/a | 545mL |
| FBS | 10% | 50 mL |
| Pen/Strep | 100 U/mL | 5 mL |
| Total | 500 mL |
Store at 4°C for up to 3 months
Optional: Penicillin/Streptomycin can be added to all media
Step-by-step method details
Culturing of human N/TERT-1 keratinocytes
Timing: ∼1 week
This step describes a detailed protocol for culturing human N/TERT-1 keratinocytes.
CRITICAL: All procedures described are performed in a Class II biological hood following standard aseptic techniques. Work is performed within laboratories rated at biosafety level 1 or 2 according to the local regulations. All cell cultures are maintained in a humidified incubator at 37°C and 5% CO2. All media are pre-warmed in a 37°C water bath before use.
-
1.Reviving human N/TERT-1 keratinocytes from frozen stock
-
a.Remove a vial of cryopreserved N/TERT-1 cells containing ∼1 × 106 cells from liquid nitrogen and thaw for 1–2 min in a 37°C water bath.
-
b.Disinfect the cryovial with 70% EtOH.
-
c.Transfer the content of the vial to a sterile canonical 50 mL tube, and slowly add 12 mL DMEM 10% FCS to the cells.CRITICAL: Cells are very sensitive at this stage and should be handled with care.
-
d.Centrifuge at 300 × g for 5 min at 20°C–22°C.
-
e.Aspirate supernatant, and carefully resuspend cell pellet in 10 mL complete K-SFM
-
f.Transfer cell suspension to a 100 mm culture dish and place at 37°C with 5% CO2 in a humidified incubator.
-
g.The next day, aspirate medium and replace with 10 mL of fresh pre-warmed complete K-SFM. Examine cell survival and morphology under microscope.
-
h.Change growth medium every other day. Cells are ready for passaging when they reach ∼30% confluence (to keep confluency low which is preferred with N/TERT-1. Other keratinocyte cell lines may tolerate to be grown to a higher confluency before differentiating). Generally, this takes ∼3–4 days after thawing.
-
a.
-
2.Passaging human N/TERT-1 keratinocytes
-
a.When cells reach ∼30% confluence, remove the culture medium and add 0.5 mL warm TrypLE per 100 mm dish. Incubate the dish for approximately 5 min at 37°C until the cells have detached.
-
b.Add 1.5 mL DMEM 10% FBS, resuspend the cells and take 20 μL suspension for counting cell number. Transfer cell suspension to a 15 mL conical tube and centrifuge for 5 min, 300 × g, at 20°C–22°C.
-
c.Aspirate supernatant and resuspend in warm complete K-SFM. Seed 2–3 × 105 cells into a new 100 mm dish. Rock the plate gently to distribute cells evenly. Place plates at 37°C in a humidified incubator and monitor growth every day. Change growth medium every second day by removing medium from the dish and adding 10 mL warm complete K-SF medium.
-
a.
Figure 2.

DIC pictures of keratinocyte colonies
(A) represents a good colony.
(B) represents differentiating colonies not suited for organotypic cultures. Scale bar equals 20 μm.
Culturing human fibroblasts and HEK293T cells
This step provides a general protocol for culturing human fibroblasts and HEK293T cells.
-
3.Culturing human fibroblasts and HEK293T cells
-
a.Human fibroblasts (we use MRC-5, but other dermal fibroblasts can be used) and HEK293T cells are grown in DMEM supplemented with 10% fetal bovine serum.
-
b.Passaging - both cell lines are trypsinized (TrypLE) at ∼80–90% confluence. Trypsin is neutralized with DMEM/FBS and cells are seeded at ∼25%–30% confluency in a new T75 or 100 mm petri dish.
-
a.
Generation of lentiCRISPR-v2 puro/blast vector with inserted gRNA sequence
We use validated (Narimatsu et al., 2018) or GPP predicted (Doench et al., 2016) sgRNA targeting the exons of particular glycogenes. This step describes how to insert sgRNA coding sequence into lentiCRIPSR-v2 vector containing a puromycin or blasticidin resistance cassette.
-
4.Anneal the forward and reverse oligos:
-
a.Prepare the following solution in a clean PCR tube:
Reagent Final concentration Amount T4 Ligase buffer (10×) n/a 1 μL Oligo F (100 mM) 10 mM 1 μL Oligo R (100 mM) 10 mM 1 μL MiliQ water n/a 7 μL -
b.Place the tube in a PCR machine and use the following program to anneal the two oligos: 95°C for 5 min; ramp down to 25°C at 5°C per min
-
c.Dilute the annealed oligos 1:200 with MiliQ water and use in the ligation reaction (see step 6a)
-
a.
-
5.Digestion of lentiCRISPR-v2 puro/blast vector with Bsmb1 restriction enzyme
-
a.Set up the following digestion reaction:
Reagent Final concentration Amount lentiCRISPR-v2 vector 50 ng/μL 1 μg Buffer3 NEB (10×) n/a 1 μL DTT (10 mM) 1 mM 2 μL Bsmb1 NEB (10000 U/mL) 10 units 1 μL MiliQ water n/a up to 20 μL -
b.Incubate at 55°C for 30 minAlternatives: Longer incubation time at lower temperature is also possible, such as 2 h at 37°C
-
c.Check digestion by running 2–3 μL of the digested product on 0.8% agarose gelNote: Digested lentiCRISPR-v2 vector results in two bands with the size of 1885 bp and 12990 bp
-
d.Measure concentration of the digested product on nanodrop
-
a.
-
6.Ligate the annealed and phosphorylated oligo duplexes (from step 4) into the digested lentiCRISPR-v2 vector (from step 5)
-
a.Set up the following ligation reaction
Reagent Final concentration Amount Digested lentiCRISPR-v2 vector 5 ng/μL 100 ng Diluted oligo duplexes (1:200) 2.5 μM 1 μL T4 ligase buffer (10×) n/a 2 μL T4 ligase 1 Weiss unit 1 μL MiliQ water n/a up to 20 μL -
b.Incubate at 20°C–22°C for 2 h 30 min. Ligation mixture can now be used for bacterial transformation
-
a.
-
7.Transformation of Stbl3 E. coli cellsNote: (1) Set a water bath to heat to 42°C. (2) Pre-warm SOC medium to 20°C–22°C.
-
a.Thaw chemical competent Stbl3 E. coli cells on ice for 10 min
-
b.Add 2 μL ligation mixture from step 6b
-
c.Keep on ice for 15–20 min
-
d.Heat-shock in a water bath at 42°C for 30 s
-
e.Place on ice for 2 min
-
f.Add 200 μL room-temperature SOC medium and shake (300 rpm) at 37°C for 1 h
-
g.Plate cells on agar plates containing 100 μg/mL carbenicillin and incubate at 37°C for 16–18 h
-
a.
-
8.Perform colony PCR to test for positive transformants
-
a.Prepare the following mastermix for colony PCR
Reagent Final concentration Amount 2× MM buffer n/a 6.25 μL QCG F primer (25 mM) 250 μM 0.125 μL Reverse primer (25 mM) 250 μM 0.125 μL Colony lysate n/a 1 μL MiliQ water n/a 5 μL -
b.Pick up and lyse single bacterial colonies:
-
i.Transfer 10 μL 0.5 M NaOH to clean eppendorf tubes
-
ii.Scrape a colony with a clean stick and rinse the stick in the tube containing 0.5 M NaOH
-
iii.Incubate for 5 min at 20°C–22°C
-
iv.Add 5 μL 1 M Tris
-
v.Incubate for 5 min at 20°C–22°C
-
vi.Add 85 μL MiliQ water
-
vii.Add 1 μL of this lysate to the mastermix (step 8a) to perform colony PCR
-
i.
-
c.Run the following PCR program
PCR cycling conditions Steps Temperature Time Cycles Initial denaturation 95°C 15 min 1 Denaturation 95°C 30 s 15 Annealing 72°C 30 s Extension 72°C 30 s Denaturation 95°C 30 s 25 Annealing 58°C 30 s Extension 72°C 30 s Final extension 72°C 30 s 1 Hold 4°C Forever Note: Touch down 72°C-1°/cycle from cycle one -
d.Run the PCR product on a 2.5% agarose gel. Positive colonies should give a product of 267 bp.
-
a.
-
9.Purify plasmid DNA by Midiprep
-
a.Choose a positive colony and inoculate in 200 mL LB medium containing 100 μg/mL carbenicillin
-
b.Incubate at 37°C in a shaking incubator for 16–18 h
-
c.We use NucleoBond Xtra Midi EF (Macherey-Nagel) kit to isolate plasmid DNA.
-
d.Test the plasmid DNA with suitable restriction enzymes
-
a.
Production of lentiviral particles in HEK293T cells
This section describes the production of lentiviral particles using pCMV-VSV-G envelope and psPAX2 packaging plasmid, which can be used with 2nd and 3rd generation lentiviral vectors. Transfected HEK293T cells will produce lentiviral particles which accumulate in the culture medium supernatant (Ferreira et al., 2020) and can be used directly for transduction of N/TERT-1 keratinocytes
-
10.
Seed 5 × 105 cells per well in a 6-well plate the day before transfection.
Alternatives: Seed 1 × 105 HEK293T cells per well in a 6-well plate and incubate for 2–3 days. Grow cells in 2 mL DMEM 10% FBS.
-
11.
On the day of transfection, aspirate off culture medium and carefully add 1.5 mL fresh growth medium containing serum.
-
12.For each transfection sample prepare DNA-PEI complexes as follows:
-
a.Mix the following components in a sterile 1.5 mL Eppendorf tube:
Reagent Amount Opti-MEM (100 μM) 200 μL PEI (1 mg/mL) 8 μL pCMV-VSV-G plasmid 0.6 μg psPAX2 plasmid 0.6 μg gRNA in lentiCRISPR-v2 plasmid 0.8 μg -
b.Vortex the tube vigorously for 15–20 s and leave for 20 min at 20°C–22°C.
-
a.
-
13.
Add the Opti-MEM mixture dropwise to each well. Mix gently and incubate cells for 24 h at 37°C with 5% CO2 in a humidified incubator.
-
14.
On the following day, carefully remove the culture medium and add warm complete K-SFM. Incubate cells for 24 h at 37°C with 5% CO2 in a humidified incubator.
-
15.
Harvest lentivirus-containing medium and filter through a 0.45-μm microfilter. The filtrate is used directly for transduction of N/TERT-1 keratinocytes (see steps 16 and 17).
Alternatives: The filtered lentivirus-containing medium from step 15 can be placed immediately at -80°C for later use.
Lentiviral transduction of N/TERT-1 keratinocytes
This step provides a detailed protocol for the lentiviral transduction of N/TERT-1 keratinocytes necessary for the generation of knockout clones.
-
16.
Two days before transduction, seed N/TERT-1 keratinocytes in 6 well plates. Seed 3 × 104 cells per well and include an extra well with cells to serve as a negative control.
Note: It usually takes 2 days for the cells to reach 30% confluence and be ready for transduction.
-
17.
On the day of transduction, mix 1 mL of complete K-SFM and 1 mL of the filtered lentivirus-containing medium produced by HEK293T cells (see previous section). Add 4 μg/mL polybrene and mix carefully by inverting the tube. Do not vortex.
-
18.
Remove the culture medium of the N/TERT-1 cells. Add the lentivirus-containing medium from step 15 to the cells. Incubate cells overnight at 37°C with 5% CO2 in a humidified incubator.
-
19.
24 h post-transduction, remove medium containing virus and replace with complete K-SFM.
-
20.Start selection of knockout clones 48 h post-transduction: remove grow medium and add complete KSFM containing the suitable antibiotic for selection.
-
a.Puromycin selection - use 1 μg/mL puromycin. Non-transduced WT N/TERT-1 cells should die within 3 days of selection.
-
b.Blasticidin selection - use 5 μg/mL blasticidin. Non-transduced WT N/TERT-1 cells should die within 4–5 days of selection.
-
a.
Note: Change media containing antibiotic every second day
-
21.
When cells reach ∼70% confluence, trypsinize the cells and take an aliquot for IDAA (Yang et al., 2015) (see step 22). Seed 1/5 of the cells into a new well of a 6-well plate and freeze the rest of the bulk. After 3 days, passage cells again and expand to a 100 dish if necessary.
Isolation of knockout clones
This step describes how we perform isolation and validation of isogenic knockout clones.
-
22.Quick DNA extract for INDELs detection by IDAA
-
a.Take an aliquot of bulk cells and transfer to a well in a 96 well plate. Fill up with 50 μL grow medium
-
b.On the next day, remove medium and add 30 μL CoboXtract buffer
-
c.Incubate at 70°C for 20 min
-
d.Incubate at 98°C for 10 min
-
a.
-
23.
Use this crude DNA extract for setting up a PCR reaction with primers flanking the DNA cut size yielding a product of >200 bp. Analyze the presence of out of frame INDELs by performing IDAA (Yang et al., 2015, Bennett et al., 2020). We use ABI PRISMTM 3010 Genetic Analyzer (Thermo Fisher) and Peak Scanner Software V1.0 (Thermo Fisher).
-
24.Once you have validated the presence of knockout clones in the bulk, you can perform serial dilution of the bulk cells to isolate knockout clones.
-
a.Harvest the bulk and take an aliquot to count cell number
-
b.Dilute cells in order to seed 0.4 cells per well in a 96-well plateNote: For each KO, we recommend you seed 4–5 96-well plates
-
c.Incubate plates at 37°C in a humidified incubator
-
d.After 4–5 days single colonies should become visible under the microscope. Mark the wells with single colonies and change grow medium with complete K-SFM
-
e.When cells become large enough (1/3 of the well covered), cells are trypsinized and expanded to a 24-well plate. Take out and aliquote for IDAA and DNA sequencing to select for positive KO colonies
-
f.When cells reach 30% confluence, expand them to a 12-well plate
-
g.Positive candidates are subjected to another round of single cell cloning
-
a.
Casting collagen/fibroblast gels for organotypic cultures
This step provides a recipe for casting collagen/fibroblast gels for organotypic cultures (Methods video S1).
-
25.
Make acellular gel: prepare a deepwell plate (6 well size), by adding one cell culture insert into each well. Use sterile forceps.
-
26.
Mix in a 50 mL tube on ice the following components, without making air bubbles:
| Reagent | Amount |
|---|---|
| 10× MEM with Earle’s salts | 650 μL |
| L-glutamine (100× stock of 200 mM) | 70 μL |
| Gentamicin Sulfate (40 mg/mL) | 10 μL |
| Fetal calf serum | 725 μL |
| Sodium Bicarbonate (71.2 mg/mL) | 725 μL |
| rat tail collagen (4 mg/mL in 0.05% acetic acid) (homemade, see (Rajan et al., 2006) | 5.5 mL |
| 10% calf serum DMEM | 850 μL |
Mix gently and add a few drops of 1 M NaOH to the solution (the color should change to orange/light pink.
-
27.
Carefully pipet 1 mL of the acellular gel mixture into the culture insert and make sure it covers the whole bottom of the insert. Leave to polymerize in the incubator while preparing the fibroblasts and the cellular gel.
-
28.
Trypsinize fibroblasts with TrypLE. Neutralize and spin down. Count the cells and resuspend the fibroblasts in 10% calf serum DMEM at a concentration of 1 X 106 cells/mL. You will need 3 X 105 fibroblasts for each 6 well tray.
-
29.Making of cellular gel
-
a.Mix in a 50 mL tube on ice the following components without making air bubbles:
Reagent Amount 10× MEM with Earle’s salts (without L-glutamine or bicarbonate (NaHCO3)) 1.7 mL L-glutamine (100× stock of 200 mM) 170 μL Gentamicin Sulfate (40 mg/mL) 25 μL Fetal calf serum 1.9 mL Sodium Bicarbonate (71.2 mg/mL) 525 μL Rat tail collagen (4 mg/mL in 0.05% acetic acid) (homemade, see (Rajan et al., 2006) 14.4 10% calf serum DMEM 850 μL Human fibroblasts suspended in 10% calf serum DMEM 2.1 mL
-
a.
-
30.
Mix gently and add a few drops of 1 M NaOH to neutralize the solution (the color should change to orange/light pink
-
31.
Quickly add 3 mL to the culture inserts on top of the acellular gel. Avoid air bubbles.
-
32.
Place the tray in the incubator for approximately 30 min and the gels will be fully polymerized.
-
33.
Fill DMEM 10% FCS in the well (18 mL) and 2 mL on top of the gel. Do not spray.
-
34.
After 4–6 days the gels are contracted and appear macroscopically as a slightly shrunken, white disk. They are now ready for keratinocytes. In our hands and for our specific modeling format, this action results in successful and reproducible organotypic cultures, possibly due to active deposition and re-organization of the ECM by fibroblasts.
Seeding keratinocytes on the casted gels
This step describes how to seed keratinocytes on the contracted collagen/fibroblast gels.
-
35.
Trypsinize N/TERT-1 cells to be seeded (WT and validated knockouts) and adjust concentration to 1 × 107 cells/mL.
Note: 3 × 105 cells need to be plated for each gel
-
36.
Aspirate medium from the bottom of each well in the deepwell plate.
-
37.
Remove medium from the culture insert with a p1000 pipette.
-
38.
Add 30 μL cell suspension (from step 35) in the center of the gel, in the concave area of the shrunken gel.
-
39.
Add organoid culture medium at the bottom of the well in the deepwell plate until it reaches the bottom of the culture insert (∼9 mL).
-
40.
Incubate at 37°C for 20 min to let the cells adhere.
-
41.
Add 2 mL organoid culture medium on top of the adhered cells carefully and place in the incubator. Do not spray as the keratinocytes will then detach.
-
42.
Incubate for 4 days before lifting the gels.
Air-liquid interface lifting of the keratinocytes
This step describes how to perform air-liquid interface lifting of the 3D organoid culture.
-
43.
On day 4 after seeding the keratinocytes, aspirate medium from the culture insert and from the plate wells carefully.
-
44.
Place the insert into a sterile 100 mm dish using sterile forceps.
-
45.
Add two sterile airlift pads to the base of each well in the deepwell plate and place back the insert into the well over the pads.
-
46.
Add approximately 9 mL organoid culture medium in the deep wells until the pads are completely wet.
Note: Level of the medium should cover the pads and not be higher.
Alternatives: For experiments using the sialyltransferase inhibitor Ac5SiaFEtoc, culture medium is supplemented with 1 mM Ac5SiaFEtoc when the inserts were raised to the air-liquid interface. Concentration of the inhibitor is replenished with subsequent medium changes.
-
47.
Incubate the plate at 37°C in humidified incubator and change media in the deep wells every other day. Gels are ready to harvest in 10 days. The models are stable for 3 weeks.
Processing of skin organoids for histology
This step describes how we harvest and process the organotypic skin cultures for histology.
For PFA fixation:
-
48.
Collect the insert with forceps and cut the bottom of the insert with a scalpel.
-
49.
Place the skin organoid in an embedding cassette
-
50.
Close the cassette tightly and dip it in 4% PFA. Incubate for 18 h.
-
51.
Embed the organotypic cultures in paraffin like normal tissue for histology
-
52.
Sections are cut in 3–5 μm thickness and put onto slides ready for staining
-
53.
Sections can be stained with hematoxyline-eosine for phenotypic visualization or used for immunohistochemistry or immunofluorescence
For frozen sections:
-
54.
Cut the insert like above
-
55.
Embed OCT in tissue tech and freeze in hexapentane cooled on dry ice.
-
56.
Keep in −80°C freezer until cutting sections on cryomicrotome.
-
57.
Sections can be stained with hematoxyline-eosine for phenotypic visualization or used for immunohistochemistry or immunofluorescence
Expected outcomes
The human epidermal keratinocyte cell line N/TERT-1 has the ability to undergo complete differentiation and stratification in 3D organotypic cultures reflecting differentiation, morphogenesis, cell differentiation, cell-cell interactions, and cell-matrix interactions (Figure 3) (Dickson et al., 2000, Dabelsteen et al., 2020). The creation of tissue models with genetically engineered cells allows the generation of libraries of tissues that selectively differ in for example, their capacity to produce glycan structures on the main types of N- and O-linked glycoproteins and glycolipids. This tissue library revealed distinct changes in skin formation and provided phenotypic cues that serve as a resource for further genetic dissection and identification of the specific structural features involved.
Figure 3.

Comparison of N/TERT-1 organotypic skin models and normal human skin
HE stain of (A) Organotypic culture with N/TERT-1 cells, and (B) of normal human skin. Immunohistochemical staining for the proliferation marker Ki67 of (C) N/TERT-1 organotypic culture, and (D) normal human skin. Immunofluorescence staining of (E) N/TERT-1 WT organotypic tissue and (F) normal human skin for differentiation marker keratin 10 in green and nuclei stained with DAPI in blue. (G) Diagram illustrating equal number of basal Ki67 positive cells in human skin sections and sections of N/TERT-1 organotypic cultures. Data are presented as mean ± SEM. No significant difference was observed between human skin and N/TERT-1 tissues using unpaired T-test. Technical replicates = 3-4, biological replicates = 3. Scale bar represents 50 μm.
Limitations
The protocol is time and cost consuming, and the effect of potential technical problems is diagnosed at the end of the procedure. The model is not suitable for long term cultures (more than 3 months) and primary keratinocytes will have to be immortalized to make CRISPR-Cas9 KO cell lines for longer studies.
Troubleshooting
Problem 1
Failure to generate KO clones (steps 12–15).
Potential solution
Make new virus. You might have too low titer.
Re-design your guide.
Problem 2
Problem to obtain pure KO clones (step 24a–d).
Potential solution
Make sure your cell suspension is single cells before seeding in 96 wells.
Seed only 0.4 cells per well.
Feed the single cell clones with 1:1 KSFM and conditioned KSFM from WT keratinocytes.
Problem 3
Collagen gels do not contract (a pre-requisite for our modeling technique) (steps 26–34).
Potential solution
Good quality collagen and fresh growing fibroblasts are pivotal for establishing good quality organoids.
Collagen gels may be left for up to 7 days in the incubator before seeding keratinocytes, giving the fibroblasts more time to reorganize the ECM.
Problem 4
Keratinocytes do not form stratified layer (steps 43–47).
Potential solution
Keratinocytes should be fresh and actively dividing. Do not shake the culture dishes during incubations especially within the first four hours after seeding the cells.
Do not dry out the gel while lifting to the air-liquid interphase. Do not add too much media at this step as well (9 mL is enough).
The organotypic cultures are stable for up to 3 weeks after lifting.
Too much proteolysis in the collagen gel will inhibit epidermal stratification. Protease inhibitors can in some cases be added to the dermal compartment.
Problem 5
Organotypic culture sectioning fails (step 48–52).
Potential solution
Take care not to harm the epithelia or wrinkle the gel during fixation.
Leave sections in water bath after cutting until they completely unfold.
Make sure to cut perpendicular. This is best done if gels are cut in half when embedded in paraffin and mounted on the edge.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hans H. Wandall (hhw@sund.ku.dk).
Materials availability
All KO cell lines are available on request under a standard MTA with the University of Copenhagen for academic research purposes.
Data and code availability
This study did not generate any datasets or code.
Acknowledgments
This work was supported by the European Commission (GlycoSkin H2020-ERC), European Commission (Imgene H2O20), European Commission (Remodel), Lundbeck Foundation, the Danish Research Councils (Sapere Aude Research Leader grant to H.H.W.), Danish National Research Foundation (DNRF107), the Friis Foundation, the Michelsen Foundation, A.P. Møller og Hustru Chastine Mc-Kinney Møllers Fond til Almene Formaal, Danish Strategic Research Council, Lundbeck Foundation, and the program of excellence from the University of Copenhagen (CDO2016). We thank Karin Uch Hansen, Birgit Poulsen, Karen Biré, and Louise Rosgaard Duus for the excellent technical assistance and Johan H. Wandall for the music for the video. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen. We would like to thank James G. Rheinwald, Harvard Institute of Medicine, for providing the N/TERT-1 cells.
Author contributions
I.N.M. and S.D. wrote the manuscript, and all authors edited and approved the final version.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100668.
Contributor Information
Hans H. Wandall, Email: hhw@sund.ku.dk.
Sally Dabelsteen, Email: sdab@sund.ku.dk.
References
- Auxenfans C., Fradette J., Lequeux C., Germain L., Kinikoglu B., Bechetoille N., Braye F., Auger F.A., Damour O. Evolution of three dimensional skin equivalent models reconstructed in vitro by tissue engineering. Eur. J. Dermatol. 2009;19:107–113. doi: 10.1684/ejd.2008.0573. [DOI] [PubMed] [Google Scholar]
- Bagdonaite I., Pallesen E.M., Ye Z., Vakhrushev S.Y., Marinova I.N., Nielsen M.I., Kramer S.H., Pedersen S.F., Joshi H.J., Bennett E.P. O-glycan initiation directs distinct biological pathways and controls epithelial differentiation. EMBO Rep. 2020;21:e48885. doi: 10.15252/embr.201948885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E.P., Petersen B.L., Johansen I.E., Niu Y., Yang Z., Chamberlain C.A., Met Ö., Wandall H.H., Frödin M. INDEL detection, the 'Achilles heel' of precise genome editing: a survey of methods for accurate profiling of gene editing induced indels. Nucleic Acids Res. 2020;48:11958–11981. doi: 10.1093/nar/gkaa975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berning M., Prätzel-Wunder S., Bickenbach J.R., Boukamp P. Three-dimensional in vitro skin and skin cancer models based on human fibroblast-derived matrix. Tissue Eng. Part C Methods. 2015;21:958–970. doi: 10.1089/ten.TEC.2014.0698. [DOI] [PubMed] [Google Scholar]
- Black A.F., Bouez C., Perrier E., Schlotmann K., Chapuis F., Damour O. Optimization and characterization of an engineered human skin equivalent. Tissue Eng. 2005;11:723–733. doi: 10.1089/ten.2005.11.723. [DOI] [PubMed] [Google Scholar]
- Dabelsteen S., Pallesen E.M.H., Marinova I.N., Nielsen M.I., Adamopoulou M., Romer T.B., Levann A., Andersen M.M., Ye Z., Thein D. Essential functions of glycans in human epithelia dissected by a CRISPR-Cas9-engineered human organotypic skin model. Dev. Cell. 2020;54:669–684 e7. doi: 10.1016/j.devcel.2020.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson M.A., Hahn W.C., Ino Y., Ronfard V., Wu J.Y., Weinberg R.A., Louis D.N., Li F.P., Rheinwald J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghalbzouri A., Commandeur S., Rietveld M.H., Mulder A.A., Willemze R. Replacement of animal-derived collagen matrix by human fibroblast-derived dermal matrix for human skin equivalent products. Biomaterials. 2009;30:71–78. doi: 10.1016/j.biomaterials.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Ferreira M.V., Cabral E.T., Coroadinha A.S. progress and perspectives in the development of lentiviral vector producer cells. Biotechnol. J. 2020:e2000017. doi: 10.1002/biot.202000017. [DOI] [PubMed] [Google Scholar]
- Gangatirkar P., Paquet-Fifield S., Li A., Rossi R., Kaur P. Establishment of 3D organotypic cultures using human neonatal epidermal cells. Nat. Protoc. 2007;2:178–186. doi: 10.1038/nprot.2006.448. [DOI] [PubMed] [Google Scholar]
- Garlick J.A. Engineering skin to study human disease--tissue models for cancer biology and wound repair. Adv. Biochem. Eng. Biotechnol. 2007;103:207–239. doi: 10.1007/b137206. [DOI] [PubMed] [Google Scholar]
- Narimatsu Y., Joshi H.J., Yang Z., Gomes C., Chen Y.H., Lorenzetti F.C., Furukawa S., Schjoldager K.T., Hansen L., Clausen H. A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology. 2018;28:295–305. doi: 10.1093/glycob/cwx101. [DOI] [PubMed] [Google Scholar]
- Parenteau N.L., Nolte C.M., Bilbo P., Rosenberg M., Wilkins L.M., Johnson E.W., Watson S., Mason V.S., Bell E. Epidermis generated in vitro: practical considerations and applications. J. Cell. Biochem. 1991;45:245–251. doi: 10.1002/jcb.240450304. [DOI] [PubMed] [Google Scholar]
- Prunieras M., Regnier M., Woodley D. Methods for cultivation of keratinocytes with an air-liquid interface. J. Invest. Dermatol. 1983;81:28s–33s. doi: 10.1111/1523-1747.ep12540324. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan P., Dabelsteen S., Madsen F.B., Francavilla C., Kopp K.L., Steentoft C., Vakhrushev S.Y., Olsen J.V., Hansen L., Bennett E.P. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl. Acad. Sci. U S A. 2014;111:E4066–E4075. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan N., Habermehl J., Coté M.F., Doillon C.J., Mantovani D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006;1:2753–2758. doi: 10.1038/nprot.2006.430. [DOI] [PubMed] [Google Scholar]
- Ridky T.W., Chow J.M., Wong D.J., Khavari P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010;16:1450–1455. doi: 10.1038/nm.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark H.J., Willhauck M.J., Mirancea N., Boehnke K., Nord I., Breitkreutz D., Pavesio A., Boukamp P., Fusenig N.E. Authentic fibroblast matrix in dermal equivalents normalises epidermal histogenesis and dermoepidermal junction in organotypic co-culture. Eur. J. Cell Biol. 2004;83:631–645. doi: 10.1078/0171-9335-00435. [DOI] [PubMed] [Google Scholar]
- Varki A., Cummings R.D., Aebi M., Packer N.H., Seeberger P.H., Esko J.D., Stanley P., Hart G., Darvill A., Kinoshita T. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology. 2015;25:1323–1324. doi: 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Steentoft C., Hauge C., Hansen L., Thomsen A.L., Niola F., Vester-Christensen M.B., Frodin M., Clausen H., Wandall H.H. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015;43:e59. doi: 10.1093/nar/gkv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets or code.