

Abstract
Paraquat (PQ) and diquat (DQ), two highly efficient herbicides sharing similar chemical backbone, both induce reactive oxygen species and are highly toxic to humans and livestock, however, PQ but not DQ poisoning result in pulmonary fibrosis, the leading cause of high mortality rate in patients suffering PQ toxicity. Understanding the unique mechanism of PQ different from DQ therefore would provide potential strategies to reduce PQ-induced pulmonary fibrosis. Here, we identified that PQ but not DQ continuously upregulates TGF-β expression in alveolar type II (AT II) cells. Importantly, such high expression of TGF-β increases cytosolic calcium levels and further promotes the activation of calcineurin-NFAT axis. TGF-β mainly activates NFATc1 and NFATc2, but not NFATc3 or NFATc4. Administration of the inhibitors targeting cytosolic calcium or calcineurin largely reverses PQ-induced epithelial–mesenchymal transition (EMT), whereas DQ has little effects on activation of NFAT and EMT. Ultimately, PQ poisoned patients exhibit significantly reduced blood calcium levels compared to DQ poisoning, possibly via the large usage of calcium by AT II cells. All in all, we found a vicious cycle that the upregulated TGF-β in PQ-induced EMT further aggravates EMT via promotion of the calcium–calcineurin axis, which could be potential drug targets for treating PQ-induced pulmonary fibrosis.
Keywords: Paraquat, diquat, TGF-β, calcium-NFAT axis, EMT
Introduction
Paraquat (PQ) is a highly efficient nonselective contact herbicide commonly used worldwide. So far, >140 countries and regions were reported to utilize PQ for agricultural production [1]. However, PQ is extremely toxic in human and livestock through self-administration or accidental ingestion. PQ can be absorbed by digestive tract, respiratory tract and skin, which ultimately results in severe damage to multiple organs, especially pulmonary fibrosis, one of the leading causes for PQ-induced mortality [2]. Due to its toxicity, PQ has been replaced by diquat (DQ), another dipyridylium herbicide with structural similarity of PQ, in some countries [3]. Though DQ is also toxic to humans, the toxicity of DQ has been reported to be less severe than that of PQ. DQ has been reported to mainly affect the liver or the kidney [4]. Unlike PQ, pulmonary injury is less prominent with DQ administration and no progressive pulmonary fibrosis has been observed in DQ poisoning [5]. It has been reported that DQ cannot be absorbed by alveolar type II (AT II) cells, leading to its low toxicity to the lung [6, 7]. However, both PQ and DQ treatment result in cell toxicity in A549 cells, a cell line of AT II cells, indicating that DQ toxicity can also affect AT II cells but not lead to pulmonary fibrosis, probably with a different mechanism compared to PQ poisoning. Therefore, understanding that how PQ and DQ behave differently in AT II cells might provide potential strategies to treat PQ poisoning and related pulmonary fibrosis.
Patients of pulmonary fibrosis suffer progressive deterioration. Some of them progress slowly whereas others decline quickly. TGF-β signaling has been proposed as an important initiator. Numerous studies have addressed the functions and mechanisms of TGF-β signaling in modulation of pulmonary fibrosis [8]. TGF-β induces multiple types of cell transition, including epithelial-to-mesenchymal transition (EMT), endothelial-to-mesenchymal transition, fibroblast-to-myofibroblast transition [9–11]. Importantly, EMT has been recognized as the pivotal event for the malignant progression of pulmonary fibrosis, from which TGF-β signaling plays a central role in EMT in AT II cells [12]. All of these observations raise the possibility that TGF-β is a potential important signal for PQ poisoning, which has been indeed clarified in several studies. However, little has been addressed whether DQ poisoning affects TGF-β signaling, which might be the key factor for the difference between PQ and DQ.
Activation of TGF-β downstream cascades are important process to promote EMT and pulmonary fibrosis, including the TGF-β/SMADs downstream axis as well as the crosstalk with the WNT signaling and the MAPK pathway [13]. Interestingly, DQ has also been recognized to activate WNT signaling or MAPK [14, 15], making us to address other potential cascades activated by TGF-β in modulation of EMT process. Calcium signaling has been indicated as another important modulator together with TGF-β signaling for the induction of EMT in tumor cells, such as breast cancer cells [16]. However, little has been addressed that whether PQ but not DQ modulates TGF-β raised calcium signaling and thus EMT in AT II cells that facilitates pulmonary fibrosis.
Calcium signaling is one of the major routes modulating multiple biological functions as calcium ions are the second messenger that exhibits almost omnipotent functions [17]. Unlike other second messengers, calcium ions could not be generated by cells but all from extracellular calcium entry [18]. The calcium influx could then increase cytosolic calcium levels that activates calcineurin and dephosphorylates NFAT family members, including NFATc1, NFATc2, NFATc3, and NFATc4 [19]. The dephosphorylated NFATs translocate into the nuclear and act as a transcriptional factor for multiple genes expression, including E-cadherin and Vimentin, the EMT markers [20]. Importantly, it is unclear that by which NFATs does TGF-β induce calcium signals and thus EMT in AT II cells.
The current study was designed to answer whether and how PQ but not DQ modulates TGF-β induced calcium-NFAT axis and thus EMT in AT II cells. Understanding the precise mechanisms between PQ and DQ in modulation of TGF-β induced calcium signaling would provide specific efficient strategies for treating PQ-induced pulmonary fibrosis.
Material and Methods
Reagents
PQ (Cat. No: 36541) and DQ (Cat. No: 45422) were purchased from Sigma Chemical Co (St. Louis, MO, USA). The enhanced chemo-luminescence (ECL) reagent was acquired from Millipore (Billerica, MA, USA). The cell culture dishes or plates were purchased from CORNING (Tewksbury, MA, USA).
Cell culture
A549 cells were purchased from Stem Cell Bank, Chinese Academy of Sciences and cultured in Ham’s F-12K Medium (Genom, GNM21127) supplemented with 10% fetal bovine serum (GIBCO,42Q1095K), penicillin (Genom, GNM15140) and streptomycin (Genom, GNM15140). The cells were grown at 37°C in a 5% carbon dioxide incubator.
Quantitative RT-PCR analysis
A549 cells were treated with 10 ng/ml TGF-β, 50 μM BAPTA, 1 μM CsA, or 800 μM PQ as indicated. The treated cells were lysed in TRIZOL (Vazyme Biotech Co, R701-02), after which total RNA was extracted for reverse transcription by HiScript III RT SuperMix kit (Vazyme Biotech Co, R323). The mRNA expression of TGF-β, NFATc1, NFATc2, NFATc3, NFATc4, E-Cadherin, Vimentin was monitored by semiquantification analysis in QuantStudio 7 Flex (Thermo Fisher Scientific, CA, USA) with utilizing the ChamQ SYBR Color qPCR Master Mix (Vazyme Biotech Co, R323, Q411–02). Data are normalized to GAPDH and calculated by 2[−(Ct target gene−Ct GAPDH)]. The sequence of the PCR primers utilized in this study is shown below: GAPDH (F: 5′ ACCCAGAAGACTGTGGATGG 3′; R: 5′ TTCAGCTCAGGGATGACCTT 3′), E-cadherin (F: 5′ TTCCTCCCAATACATCTCCC 3′; R: 5′ TTGATTTTGTAGTCACCCACC 3′), Vimentin (F: 5′ CTCTTCCAAACTTTTCCTCCC 3′; R: 5′ AGTTTCGTTGATAACCTGTCC 3′), NFATc1 (F: 5′ TGTGCCGGAATCCTGAAACTC 3′; R: 5′ GAGCATTCGATGGGGTTGGAG 3′), NFATc2 (F: 5′ GAGCCGAATGCACATAAGGTC 3′; R: 5′ CCAGAGAGACTAGCAAGGGG 3′), NFATc3 (F: 5′ GCTCGACTTCAAACTCGTCTT 3′; R: 5′ GATGCACAATCATCTGGCTCA 3′), NFATC4 (F: 5′ ACCCTACAGATGTTCATCGGC 3′; R: 5′ ATTCCCGCGCAGTCAATGT 3′).
Western blot
A549 cells were treated with or without 800 μM PQ or DQ for 24 hours, after which the cells were lysed in 1× SDS loading buffer. The lysates were separated by 10% SDS-PAGE gels and transferred onto a nitrocellulose filter membrane (NC) membrane (Pall, 66 485). The membrane was blocked in 5% nonfat milk for 1 hour and primary antibodies targeting TGF-β (Affinity, AF1027, 1:1000), E-Cadherin (Cell Signaling Technology, 3195S, 1:1000), Vimentin (Cell Signaling Technology, 5741S, 1:1000), or GAPDH (Proteintech, 60 004-1-Ig, 1:1000) were utilized. The blots were imaging in Amersham Imager 600 (Cytiva, MA, USA).
Immunofluorescence
A549 cells were plated on glass coverslips in a 24-well plate. After attachment, the cells were stimulated with 800 μM PQ for 24 hours, fixed in 4% paraformaldehyde, permeabilized with 0.1% TritonX-100 (Biosharp, BS100) and blocked with 0.2% BSA. The cells were then incubated with primary antibodies for TGF-β (Affinity, AF1027, 1:100) at RT for 1 hour, followed by incubation with 594-conjugated goat antirabbit secondary antibody (Thermo, A32723, 1:2000) for 1 hour. The stained cells were imaged and recorded by laser scanning confocal microscopy (Leica).
Flow cytometric analysis
A549 cells were plated in a six-well plate at a concentration of 1 × 106 cells/dish in the complete growth medium. The attached cells were stimulated with 10 ng/ml TGF-β for 24 hours. The stimulated cells were then washed twice with Hanks’ Balanced Salt Solution (Beyotime, C0218) and incubated with 2 μM Fluo-3/AM (Beyotime, S1056) diluted in Hanks’ buffer for 30 minutes at 37°C in the dark. The stained cells were then washed and collected to detect the presence of [Ca2+]i using a FACScan flow cytometer (BD FACS Fortessa, USA).
Statistical analysis
Data are represented as mean ± SD. Experiments with multiple comparisons were performed using one-way or two-way ANOVA followed by Bonferroni post-tests, as indicated. N.S., not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
Results
PQ but not DQ raises TGF-β expression in AT II cells
TGF-β is well recognized as a secreted protein that acts in an autocrine or a paracrine pattern [21]. We therefore firstly observed whether PQ treatment could induce TGF-β mRNA expression in A549 cells, a cell line of AT II cells. Consistent with other reports, PQ treatment significantly induces TGF-β expression at 24 hours (Fig. 1B). TGF-β expression shows a trend of increase after PQ treatment at 6 hours, yet no statistical significance has been observed, indicating that the induced expression of TGF-β by PQ might continuously aggravate pulmonary fibrosis, probably via an autocrine pattern. We then further observed the distribution of TGF-β via immunofluorescence after 24 hours treatment of PQ. Consistent with the mRNA expression of TGF-β, the overall TGF-β signal is largely enhanced in PQ treated cells compared to the vehicle control (Fig. 1C). Importantly, multiple signals of TGF-β are evenly distributed in whole cells, indicating that TGF-β is under post-modification and ready to secret out of the cells. In addition, some of the TGF-β signals seem to exhibit membrane-localized pattern, raising the possibility that TGF-β secreted by A549 cells could act in an autocrine pattern to modulate AT II cells functions.
Figure 1.
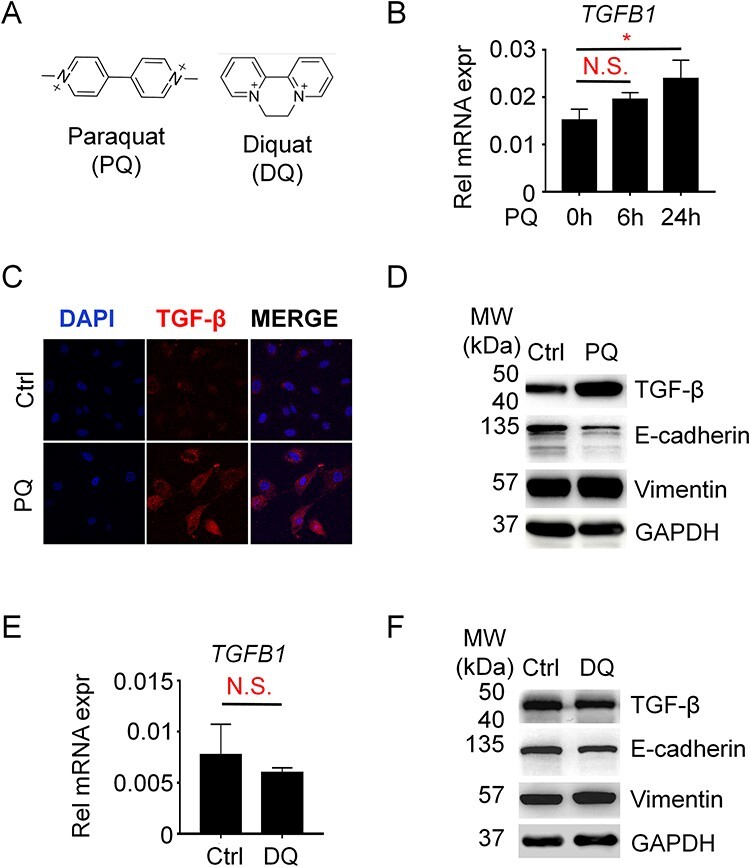
TGF-β is upregulated in PQ but not DQ-treated A549 cells. (A) Chemical structure of paraquat and diquat. (B) QPCR analysis of tgf-β expression. A549 cells were treated with 800 μM PQ for 0, 6, or 24 hours as indicated. One-way ANOVA was performed for analyzing statistical significance. N.S., not significant; *P < 0.05. (C) Immunofluorescence assay for observation of TGF-β abundance and distribution in A549 cells treated with or without 800 μM PQ. (D) Western blotting for the expression level of TGF-β in A549 cells treated with or without 800 μM PQ. E-Cadherin and Vimentin are makers for EMT. (E) A549 cells were treated with or without 800 μM DQ for 24 hours as indicated. Student’s t-test was performed for analyzing statistical significance. N.S., not significant. (F) Western blotting for the expression level of TGF-β in A549 cells treated with or without 800 μM DQ for 24 hours.
To further confirm the abundance of TGF-β proteins raised by PQ, we performed WB analysis and again found that PQ strikingly increases the expression of TGF-β accompanied with a robust decline of E-Cadherin expression and increase of Vimentin expression (Fig. 1D), biomarkers of epithelial cells or mesenchymal cells, respectively. Next, we wonder whether DQ would also affect TGF-β expression. Interestingly, unlike PQ, DQ exhibits little effects on the expression of TGF-β both in mRNA level (Fig. 1E) or protein level (Fig. 1F), indicating that the increased expression of TGF-β and followed activation of TGF-β signaling would be the potential reason for the difference between PQ and DQ poisoning.
TGF-β stimulation enhances cytosolic calcium burden in AT II cells
The increased expression of TGF-β and its partial membrane-localized pattern after PQ treatment raises us to further clarify the followed activated signaling. As mentioned in the Introduction, we are specifically interested in understanding whether TGF-β could enhance calcium signals in AT II cells. Therefore, we measured the cytosolic calcium levels via Fluo-3 AM staining in A549 cells treated with TGF-β for 24 hours. Compared to the blank group, the Control group shows a strongly enhanced signal, suggesting that the calcium measurement system we established in this study works well. Compared to the control cells, the TGF-β treated cells exhibit further enhanced signals (increased 40.6% compared to the control cells) (Fig. 2A–C). Consistently, the analysis of the Geometric mean further confirms the observations. We repeatedly observed the increase of cytosolic calcium levels in TGF-β treated cells with a consistent increase trend (Fig. 2D), suggesting that the TGF-β signal is critical for raising cytosolic calcium levels in AT II cells.
Figure 2.
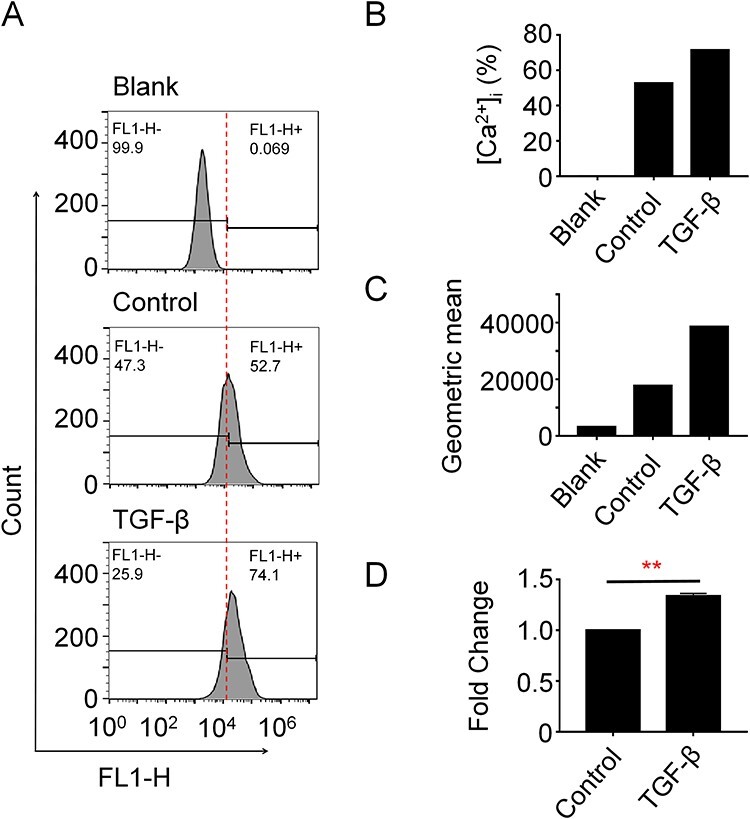
TGF-β increases cytosolic calcium concentrations in A549 cells. (A–D) A549 cells were treated with 10 ng/ml human recombinant TGF-β for 24 hours. Cells were subjected to Fluo-3 AM staining and flow cytometry measurement. Representative data (A) and quantification analysis of the positive percentage of fluorescence signals (B) or geometric mean (C) were shown. (D) The experiment was biologically replicated and fold change of the replicate experiment was calculated. Student’s t-test was performed for analyzing statistical significance. **P < 0.01.
TGF-β promotes the expression of NFATc1, NFATc2 and NFATc3, but not NFATc4
NFAT activation is well characterized as the major downstream event raised by increased cytosolic calcium concentrations [22]. Different cell types exhibit their unique calcium-NFAT axis by activating specific NFAT family members. To clarify the major NFAT activation in response to TGF-β stimulation in A549 cells, we analyzed the NFAT expression profile w/wo TGF-β stimulation in A549 cells. As shown in Fig. 3A, we have been able to detect the expression of NFATc1, NFATc2, and NFATc3 in A549 cells while little NFATc4 could be observed (Fig. 3A), suggesting that NFATc4 plays little roles in A549 cells. Moreover, all of the three NFATs are highly upregulated with TGF-β stimulation, indicating TGF-β is a nonselective inducer for NFAT activation.
Figure 3.
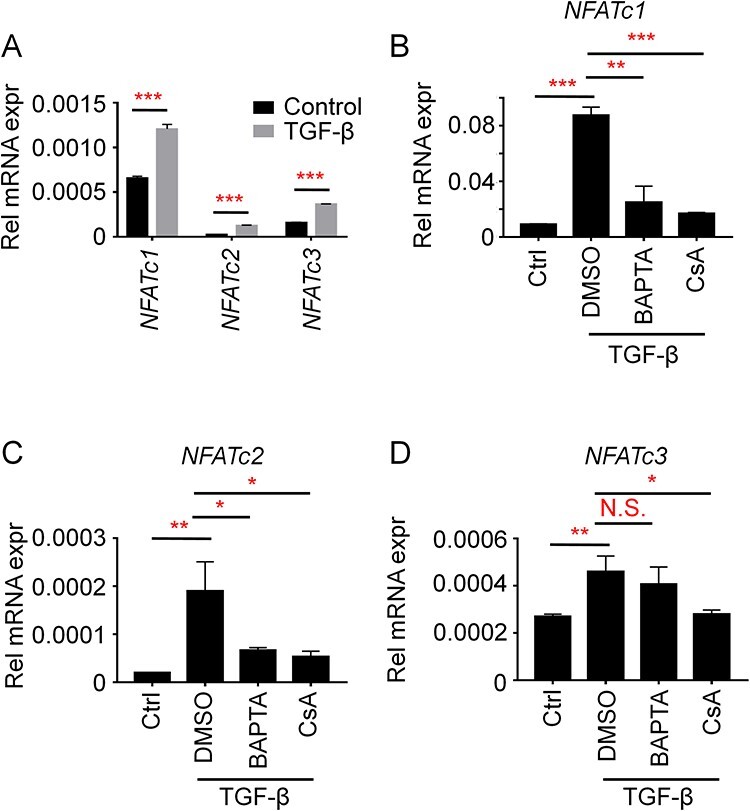
TGF-β induces NFATc1 and NFATc2 activation via promotion of the calcium–calcineurin axis. (A) A549 cells were treated with 10 ng/ml TGF-β for 24 hours. The cells were then subjected to QPCR analysis for NFATc1, NFATc2, or NFATc3. GAPDH was utilized as the housekeeping gene. (B–D) A549 cells were treated with 10 ng/ml TGF-β, together with 50 μM BAPTA or 1 μM CsA as indicated. The cells were then subjected to QPCR analysis for NFATc1 (B), NFATc2 (C), or NFATc3 (D). GAPDH was utilized as housekeeping gene. The statistical significance in all of the panels was analyzed by Two-way ANOVA. N.S., not significant; *P < 0.05, **P < 0.01, ***P < 0.001.
TGF-β activates NFATc1 and NFATc2 via the calcium–calcineurin axis
To further clarify whether the expression of NFATs is raised by TGF-β induced calcium cascades, we then introduced cyclosporine A (CsA) and BAPTA, two inhibitors targeting calcineurin or cytosolic calcium ions, respectively [23, 24]. As shown in Fig. 3B–D, treatment of CsA and BAPTA largely reduces TGF-β induced NFATc1 and NFATc2 expression (Fig. 3B and C). Interestingly, CsA but not BAPTA exhibits effective inhibition on TGF-β induced NFATc3 expression (Fig. 3D), indicating alternative pathways independent of cytosolic calcium signals exist for activation of calcineurin and thus NFATc3 raised by TGF-β. Nevertheless, these results clearly show that TGF-β mainly activates NFATc1 and NFATc2 through calcium–calcineurin axis in A549 cells. Moreover, as NFATc1 is much strongly expressed compare to NFATc2 or NFATc3, it would be highly possible that NFATc1 plays a major role in A549 cells in response to extracellular stimulator induced calcium signals, such as TGF-β.
PQ but not DQ promotes EMT via the calcium–calcineurin axis in AT II cells
The activation of calcium-NFAT axis by TGF-β in A549 cells arouse us to further understand the relevant biological functions. Many studies have already shown TGF-β induces EMT via the calcium signaling in tumor cells [25]. We have clarified that PQ induces TGF-β expression in A549 cells in Fig. 1. Therefore, we speculated that PQ induced EMT could be a result of increased TGF-β expression induced calcium-NFAT activation. To address this possibility, we examined the expression of E-Cadherin and Vimentin. Consistent with others and our previous reports, PQ treatment significantly reduces the expression of E-Cadherin while increases the expression of Vimentin in A549 cells, suggesting that PQ induces EMT in AT II cells. Administration of CsA or BAPTA significantly reduces PQ-induced EMT. Specifically, treatment of BAPTA almost normalizes PQ-reduced expression of E-Cadherin (Fig. 4A) while inhibits PQ-induced expression of Vimentin (Fig. 4B). Similar trends were observed in CsA treated cells. These results suggest that PQ-induced EMT is largely dependent on calcium-NFAT signaling.
Figure 4.
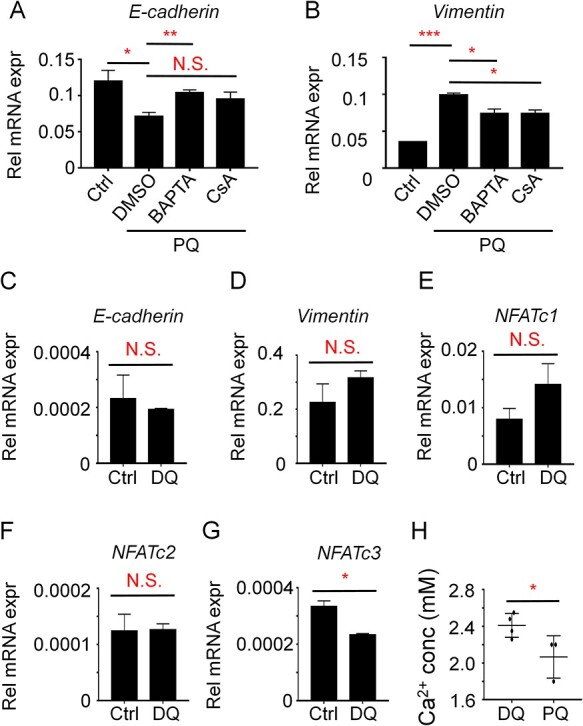
PQ but not DQ drives EMT and is largely reversed by blockage of the calcium–calcineurin axis in A549 cells. (A–B) A549 cells were treated with 800 μM PQ, together with 50 μM BAPTA or 1 μM CsA as indicated, for 24 hours. The cells were then subjected to QPCR analysis for E-Cadherin (A) and Vimentin (B). (C–G) A549 cells were treated with 800 μM DQ for 24 hours. The cells were then subjected to QPCR analysis for E-Cadherin (C), Vimentin (D), NFATc1 (E), NFATc2 (F), and NFATc3 (G). GAPDH was utilized as the housekeeping gene. The statistical significance was analyzed by Two-way ANOVA. N.S., not significant; *P < 0.05, **P < 0.01, ***P < 0.001. (H) Retrospectively analysis of the blood calcium levels in patients suffering PQ or DQ poisoning. Student’s t-test was performed for analyzing statistical significance. *P < 0.05.
On the contrary, DQ plays little effects on EMT process as DQ treatment in A549 cells does not affect the expression of E-Cadherin or Vimentin (Fig. 4C and D). Also, DQ does not induce NFAT family members expression (Fig. 4E–G), even reduces the expression of NFATc3. These results together suggest DQ plays little effects on calcium-NFAT activation as well as EMT.
Patients suffering PQ poisoning exhibits relatively low blood calcium levels compared to DQ poisoning
The different response of calcium-NFAT activation between PQ and DQ treatment in A549 cells raises us to further explore the clinical relevance. Since calcium ions in cells all come from extracellular calcium entry, the continuous increased cytosolic calcium levels by the PQ-TGF-β axis might slightly reduce extracellular calcium pools. We therefore speculate that PQ poisoning might exhibit reduced blood calcium levels in patients. By retrospectively analyzing the results of blood tests in patients suffering PQ or DQ poisoning in our hospital in recent years, we found that patients suffering PQ poisoning exhibit significantly reduced blood calcium levels compared to DQ poisoning in the initial term (Fig. 4H). More importantly, blood calcium levels in DQ poisoning patients are kept in normal range. These results indicate that reduced blood calcium levels and intracellular calcium overburden are important features of PQ poisoning that differs from DQ poisoning, which might be the potential drug targets for treating PQ poisoning and related pulmonary fibrosis.
Discussion
Both PQ and DQ have been recognized to promote reactive oxygen species (ROS) and therefore lead to cell toxicity and organs failure [26, 27]. However, little has been addressed that how PQ and DQ show largely different toxicity in AT II cells thus pulmonary fibrosis. Here, by using the in vitro analysis of A549 cells exposure to PQ or DQ, we revealed that PQ but not DQ raises the expression of TGF-β in A549 cells and the expressed TGF-β further activates calcium–calcineurin-NFATc1 axis, leading to EMT process in AT II cells, which is responsible for pulmonary fibrosis in PQ poisoning. Our study provides a potential reason why PQ and DQ exhibit totally different functions in pulmonary fibrosis, yet sharing a similar chemical backbone.
ROS has been recognized as one of the major signals that aggravates pulmonary fibrosis [28, 29]. In the scenario of PQ treatment, multiple studies have revealed that ROS is the major signal for pulmonary fibrosis and numerous strategies developed to eradicate ROS have been reported to treat PQ induced pulmonary fibrosis [30], however, the output is limited as expected, leading to the possibility that signals upstream of ROS exist in response to PQ poisoning. It has been reported that abnormal calcium signals would promote ROS accumulation and such accumulation could act as a vicious cycle that further aggravates intracellular calcium burdens [31]. Here we found that PQ and TGF-β induced intracellular calcium signals are essential for EMT, raising the possibility that TGF-β could be an important mediator linking PQ and ROS accumulation via TGF-β induced intracellular calcium burdens in AT II cells. In addition, considering DQ is also well characterized to induce ROS, one would expect to observe similar functions of DQ as PQ in TGF-β expression and EMT. However, our study clearly showed that DQ cannot induce TGF-β expression as well as EMT, raising the possibility that other critical mechanisms but not ROS exist for PQ poisoning and result in major toxicity in lungs. Further studies are required to deeply elucidate that how PQ modulates intracellular calcium signaling and therefore pulmonary fibrosis.
Several studies have reported that TGF-β increases cytosolic calcium signaling and related biological functions, including the rapid increase of collagen A1 and fibronectin in pulmonary fibroblasts [32]. However, little has been addressed that how TGF-β modulates calcium signaling cascades. Here, we have identified that TGF-β activates NFATc1 and NFATc2 but not NFATc3 or NFATc4 via the calcium–calcineurin axis in AT II cells, which clarifies the major downstream targets of TGF-β induced calcium signaling. For the upstream cascades, cytosolic calcium signals could be modulated by two major routes. The intracellular calcium stores, especially the endoplasmic reticulum (ER), can increase cytosolic calcium levels via ER localized IP3Rs, RyRs, or SERCA pumps [33]. The extracellular calcium pools can affect intracellular calcium signals via extracellular calcium entry mediated by calcium release activated calcium channels (CRAC), voltage-gated calcium channels, P2Xs, etc [18]. Mukherjee S et al. reported that blockage of calcium release from ER by ryanodine totally depletes TGF-β induced calcium signaling in fibroblasts, suggesting that the ER calcium store is essential for TGF-β increased cytosolic calcium levels [34]. However, blockage of the ER calcium store might also reduce CRAC-mediated extracellular calcium entry. Therefore, it would be reasonable to speculate that the upstream of TGF-β induced calcium signaling could be ascribed to both the ER calcium store and CRAC activation. In the scenario of PQ-TGF-β axis, by which route for intracellular calcium overburden does PQ poisoning utilize to induce EMT in AT II cells requires further efforts to elucidate. Nevertheless, combinational strategies targeting TGF-β, calcium-NFATc1 axis and ROS together would be expected to interfere the vicious cycle of ROS-calcium signaling raised by PQ and thus ameliorate PQ-induced pulmonary fibrosis.
Overall, in this study we identified TGF-β driven calcium signaling as the unique downstream of PQ poisoning different from DQ poisoning. Considering the low toxicity of DQ in lung injury, strategies targeting TGF-β driven calcium signaling might be beneficial for treating PQ induced pulmonary fibrosis.
Conclusions
In conclusion, in this study we demonstrated that TGF-β is induced in PQ but not DQ-stressed AT II cells, which promotes intracellular calcium burdens and mainly activates NFATc1 for the promotion of EMT via enhancing the expression of mesenchymal markers while restricting the expression of epithelial makers. Our study provides a novel insight that how PQ activates a unique downstream pathway differently from DQ that drives EMT process in AT II cells, which would be beneficial for developing strategies to treat PQ poisoning and pulmonary fibrosis.
Contributor Information
Wenyu Yang, Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Xinrun Ma, Institute of clinical Immunology, Center for Translational Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Yong Zhu, Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Xiaoxiao Meng, Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Rui Tian, Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Zhengfeng Yang, Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China; Institute of clinical Immunology, Center for Translational Medicine, Shanghai General Hospital, Shanghai Jiaotong University, School of Medicine, 650 Xingsongjiang Road, Shanghai 201620, China.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81 971 555 to Z.Y.), Shanghai Pujiang Program (19PJ1408700 to Z.Y.), and Shanghai Sailing Program (19YF1440100 to Y.Z.).
Conflict of Interest
The authors declared that they have no conflict of interest to this study.
Reference
- 1.Dinis-Oliveira RJ, Duarte JA, Sánchez-Navarro A et al. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol 2008;38:13–71. [DOI] [PubMed] [Google Scholar]
- 2.Sun Z, Yang Z, Wang M et al. Paraquat induces pulmonary fibrosis through Wnt/beta-catenin signaling pathway and myofibroblast differentiation. Toxicol Lett 2020;333:170–83. [DOI] [PubMed] [Google Scholar]
- 3.Adler-Flindt S, Martin S. Comparative cytotoxicity of plant protection products and their active ingredients. Toxicol In Vitro 2019;54:354–66. [DOI] [PubMed] [Google Scholar]
- 4.Xing J, Chu Z, Han D et al. Lethal diquat poisoning manifesting as central pontine myelinolysis and acute kidney injury: A case report and literature review. J Int Med Res 2020;48:300060520943824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fussell KC, Udasin RG, Gray JP et al. Redox cycling and increased oxygen utilization contribute to diquat-induced oxidative stress and cytotoxicity in Chinese hamster ovary cells overexpressing NADPH-cytochrome P450 reductase. Free Radic Biol Med 2011;50:874–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witschi H, Kacew S, Hirai KI et al. In vivo oxidation of reduced nicotinamide-adenine dinucleotide phosphate by paraquat and diquat in rat lung. Chem Biol Interact 1977;19:143–60. [DOI] [PubMed] [Google Scholar]
- 7.Sun DZ, Song CQ, Xu YM et al. Involvement of PINK1/Parkin-mediated mitophagy in paraquat- induced apoptosis in human lung epithelial-like A549 cells. Toxicol In Vitro 2018;53:148–59. [DOI] [PubMed] [Google Scholar]
- 8.Veith C, Hristova M, Danyal K et al. Profibrotic epithelial TGF-beta1 signaling involves NOX4-mitochondria cross-talk and redox-mediated activation of the tyrosine kinase FYN. Am J Physiol Lung Cell Mol Physiol 2020; 320:L356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.QIN W, Pan Y, Zheng X et al. MicroRNA-124 regulates TGF-alpha-induced epithelial-mesenchymal transition in human prostate cancer cells. Int J Oncol 2014;45:1225–31. [DOI] [PubMed] [Google Scholar]
- 10.Wu DM, Deng SH, Zhou J et al. PLEK2 mediates metastasis and vascular invasion via the ubiquitin-dependent degradation of SHIP2 in non-small cell lung cancer. Int J Cancer 2020;146:2563–75. [DOI] [PubMed] [Google Scholar]
- 11.Wnuk D, Paw M, Ryczek K et al. Enhanced asthma-related fibroblast to myofibroblast transition is the result of profibrotic TGF-beta/Smad2/3 pathway intensification and antifibrotic TGF-beta/Smad1/5/(8)9 pathway impairment. Sci Rep 2020;10:16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dopeso H, Jiao HK, Cuesta AM et al. PHD3 controls lung cancer metastasis and resistance to EGFR inhibitors through TGFalpha. Cancer Res 2018;78:1805–19. [DOI] [PubMed] [Google Scholar]
- 13.Borahay MA, al-Hendy A, Kilic GS et al. Signaling pathways in leiomyoma: Understanding pathobiology and implications for therapy. Mol Med 2015;21:242–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun LY, Bokov AF, Richardson A et al. Hepatic response to oxidative injury in long-lived Ames dwarf mice. FASEB J 2011;25:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jalil AS, Reddy SB, Plautz CZ. Cellular effects of diquat dibromide exposure: Interference with Wnt signaling and cytoskeletal development. Toxicol Res Appl 2019;3:1–13. [Google Scholar]
- 16.Davis FM, Azimi I, Faville RA et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014;33:2307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busti S, Mapelli V, Tripodi F et al. Respiratory metabolism and calorie restriction relieve persistent endoplasmic reticulum stress induced by calcium shortage in yeast. Sci Rep 2016;6:27942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Yue Z, Ma X et al. Calcium homeostasis: A potential vicious cycle of bone metastasis in breast cancers. Front Oncol 2020;10:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Liu RB, Cao Q et al. USP16-mediated deubiquitination of calcineurin A controls peripheral T cell maintenance. J Clin Invest 2019;129:2856–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sengupta S, Jana S, Bhattacharyya A. TGF-beta-Smad2 dependent activation of CDC 25A plays an important role in cell proliferation through NFAT activation in metastatic breast cancer cells. Cell Signal 2014;26:240–52. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H, Wei J, Sun J. Roles of TGF-beta signaling pathway in tumor microenvirionment and cancer therapy. Int Immunopharmacol 2020;89:107101. [DOI] [PubMed] [Google Scholar]
- 22.Kutschat AP, Hamdan FH, Wang X et al. STIM1 mediates calcium-dependent epigenetic reprogramming in pancreatic cancer. Cancer Res 2021;81(11):2943–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou ZH, Song JW, Li W et al. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J Exp Clin Cancer Res 2017;36:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aflaki M, Qi XY, Xiao L et al. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained beta-adrenergic activation in guinea pig hearts. Circ Res 2014;114:993–1003. [DOI] [PubMed] [Google Scholar]
- 25.Miao Y, Shen Q, Zhang S et al. Calcium-sensing stromal interaction molecule 2 upregulates nuclear factor of activated T cells 1 and transforming growth factor-beta signaling to promote breast cancer metastasis. Breast Cancer Res 2019;21:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng F, Liu T, Zhu J et al. FoxF1 protects rats from paraquat-evoked lung injury following HDAC2 inhibition via the microRNA-342/KLF5/IkappaB/NF-kappaB p65 axis. Exp Cell Res 2020;395:112208. [DOI] [PubMed] [Google Scholar]
- 27.Park A, Koh HC. NF-kappaB/mTOR-mediated autophagy can regulate diquat-induced apoptosis. Arch Toxicol 2019;93:1239–53. [DOI] [PubMed] [Google Scholar]
- 28.Ma J, Cai Q, Yang D et al. A Positive feed forward loop between Wnt/beta-Catenin and NOX4 promotes silicon dioxide-induced epithelial-mesenchymal transition of lung epithelial cells. Oxid Med Cell Longev 2020;2020:3404168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, Tian T, Wang Y et al. Paraquat-activated BV-2 microglia induces neuroinflammatory responses in the neuron model through NF-kappaB signaling pathway. Toxicol In Vitro 2021;72:105076. [DOI] [PubMed] [Google Scholar]
- 30.Liu MW, Su MX, Tang DY et al. Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression. BMC Pulm Med 2019;19:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gualdani R, de Clippele M, Ratbi I et al. Store-operated calcium entry contributes to cisplatin-induced cell death in non-small cell lung carcinoma. Cancers (Basel) 2019;11:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee S, Sheng W, Michkov A et al. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca(2+) signaling. Am J Physiol Lung Cell Mol Physiol 2019;316:L810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chami M, Checler F. Alterations of the endoplasmic reticulum (ER) calcium signaling molecular components in Alzheimer's disease. Cell 2020;9:2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee S, Sheng W, Sun R et al. Ca(2+)/calmodulin-dependent protein kinase IIbeta and IIdelta mediate TGFbeta-induced transduction of fibronectin and collagen in human pulmonary fibroblasts. Am J Physiol Lung Cell Mol Physiol 2017;312:L510–9. [DOI] [PubMed] [Google Scholar]