

Abstract
Cervical cancer (CC) is a type of pelvic malignant tumor that severely threatens women's health. Current evidence suggests that IER5, as a potential radiosensitizer, promotes irradiation-induced apoptosis in CC tissues in patients undergoing chemoradiotherapy. IER5 has been shown to be involved in the G2/M-phase transition. In the present study, we used Cdc25B as the breakthrough point to explore the underlying mechanism of IER5 in the cell cycle regulation of radiation-damaged HeLa cells. IER5 was evidently upregulated after irradiation, but Cdc25B was significantly downregulated. In monoclonal IER5-silenced HeLa cells, irradiation-induced downregulation of Cdc25B was attenuated. The effect of irradiation on Cdc25B promoter activity was determined by dual-luciferase reporter assays. The response elements on the Cdc25B promoter related to irradiation were predicted by JASPAR. These conserved sequences were mutated individually or in combination by splicing-by-overlap extension PCR, and their function was confirmed by dual-luciferase reporter assays. The enrichment efficiency of transcription factors after irradiation was determined by chromatin immunoprecipitation (ChIP) assay. Both Sp1/Sp3 and NF-YB binding sites were involved in irradiation-mediated regulation of Cdc25B. IER5 was involved in irradiation-mediated regulation of Cdc25B through the NF-YB binding site. Furthermore, ChIP assays showed that IER5 bound to the Cdc25B promoter, and the binding of IER5 to the Cdc25B promoter region in irradiation-induced HeLa cells induced the release of the coactivator p300 through interaction with NF-YB. Taken together, these findings indicate that IER5 is the transcriptional repressor that accelerates the downregulation of Cdc25B expression after irradiation.
Keywords: IER5, Cdc25B, transcription regulation, NF-YB, cervical cancer
Cervical cancer (CC) is the most common gynecological malignant tumor of the reproductive system worldwide. According to the latest global cancer incidence and mortality data, it is estimated that there were approximately 570 000 new cases and 311 000 deaths related to CC in 2018 worldwide. CC ranks as the fourth most commonly diagnosed cancer and the fourth leading cause of cancer-related death among women [1, 2]. Persistent infection with high-risk human papillomavirus (HR-HPV) plays a crucial etiological role in the occurrence and development of CC. However, HPV infection alone is not enough to cause malignant tumors. The occurrence of CC requires additional genetic and environmental cofactors [3]. With the effective application of cervical cytology screening in recent decades, CC and precancerous lesions can now be detected and treated early, and their morbidity and mortality rates have dropped significantly worldwide. However, in clinical practice, many women do not receive regular medical check-ups, which means that a large number of CC patients are already at an advanced stage when they are diagnosed. The treatment of CC is mainly pelvic external radiotherapy combined with afterloading intracavitary brachytherapy plus platinum-based concurrent chemotherapy [4, 5]. The 10-year survival rate of CC patients undergoing radiotherapy decreases with increasing FIGO stage. The poor treatment effect is mostly observed in stage II and III cases. Cervical cancer, especially squamous cell carcinoma, is a radiosensitive tumor, and radical radiotherapy is the main treatment for patients with stage IIa2 and earlier disease. However, as far as radiotherapy itself is concerned, there are large individual differences in the sensitivity of cervical cancer patients to radiotherapy. Therefore, how to improve the efficacy of radiotherapy for CC patients and further reduce the recurrence rate and mortality rate after CC surgery have become urgent problems for gynecological oncologists.
The biological effects of ionizing radiation are mainly caused by DNA damage, including DNA single-strand or double-strand breaks, which in turn cause changes in cell signaling pathways and lead to a series of radiation biological effects, such as apoptosis, cycle arrest, and mutation. Along with the continued development of modern molecular biology and gene therapy, a large number of gene expression screening and functional approaches have been used to explore the pathogenesis of CC, and these studies revealed that multiple genes may play an important role in the development and carcinogenesis of cervical cancer. It is also closely related to the radiotherapy response [6]. Thus, researchers are trying to seek optimal radiotherapy targets that are sensitive to ionizing radiation at the genetic level to destroy tumors while sparing normal tissues as much as possible, with the aim of reducing the side effects caused by radiotherapy and improving the efficacy of radiotherapy. Specific molecules related to radiosensitivity have been the focus of research in recent years. A study detected one of the molecules upregulated by ionizing radiation (IR) by cDNA microarray technology, immediate early response gene 5 (IER5) [7, 8].
IER5 is a member of the slow-kinetics immediate-early gene family and encodes a highly proline-rich protein with homology to the amino terminus of the immediate-early gene PIP92/IER2/ETR10 [9, 10]. IER5 is an intranuclear protein that is highly conserved during evolution [10]. IER5 is a growth factor-inducible gene that can be induced by gamma-ray radiation, heat shock, and various growth-promoting stimuli, such as PMA, serum and ionomycin [11–13]. Heat treatment induces significant expression of IER5 in an HSF1-dependent manner, leading to upregulation of chaperone gene expression and to an increase in refolding of heat-denatured proteins [13]. IER5 forms a ternary complex with HSF1 and PP2A and promotes the dephosphorylation of HSF1 to generate a novel hypophosphorylated active form of HSF1, which contributes to tumorigenesis. IER5 is highly expressed in various solid tumors, such as cervical, liver, bladder, and ovarian cancer [11]. Pan et al. presented that miR-UL148D, an miRNA that regulates viral latency, directly targets IER5 and inhibits its expression, which rescues the expression level of Cdc25B during the establishment of viral latency [14]. Cdc25B is a key regulator of entry into mitosis and stringently controls Cdc25B expression. Activation is essential for normal G2/M progression and exit from G2 phase checkpoint arrest [15]. Our previous studies identified that IER5 enhances radiation-induced G2 stage arrest and increases radioresistance by disturbing cell cycle checkpoints [12]. Importantly, recent studies have demonstrated that IER5 binds to Cdc25B, and stress-induced IER5 expression strengthens G2/M arrest [16]. However, the potential mechanism by which IER5 modulates the radiosensitivity of CC has never been investigated.
In the present study, we used Cdc25B as the breakthrough point to explore the underlying mechanism of IER5 in the cell cycle regulation of irradiation-damaged HeLa cells. We found that the expression of IER5 was upregulated after irradiation, which further caused the release of the coactivator p300 through interaction with NF-YB, leading to the downregulation of Cdc25B expression.
Materials and Methods
Cell culture
The normal human cervical cancer cell line HeLa, IER5-silenced HeLa cells and human renal epithelial cells (293 T cells) were frozen in our laboratory. All cell lines used for this study were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, USA) containing 10% fetal bovine serum (FBS, Sigma, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 humidified incubator at 37 °C. The culture medium was renewed every 2–3 days. When the cells grew to a confluence of approximately 80%–90%, they were passaged. The cells used in one experiment were the same batch of resuscitated cells, and the cells used in repeated experiments were from different batches of resuscitated cells.
Ionizing radiation treatment
Cells were irradiated by using γ-rays generated by 60Co. The radiation source was a 60Co source device of the Beijing Institute of Radiation Medicine. HeLa cells were replaced with fresh medium 30 min before irradiation and were exposed to 4-Gy radiation at a dose rate of 68.26 cGy/min. After irradiation, the cells were returned to the incubator for further culture. The cells were collected at different time points according to the experimental protocol.
RNA interference and cell transfection
siRNA targeting IER5, NF-YB, and negative control siRNA were chemically synthesized by GenePharma Co., Ltd (Shanghai, China). Plasmid DNA and siRNA transfections were carried out using Lipofectamine 2000 Reagent (Invitrogen, USA) according to the manufacturer’s instructions. Briefly, siRNA was diluted in DMEM without FBS, and then the transfection reagent was diluted, mixed gently, and incubated for 5 min at room temperature. Subsequently, the diluted plasmid DNA was combined with the diluted transfection reagent, mixed gently, and incubated for 20 min at room temperature. The transfection complex was added to the well directly. The medium was replaced with fresh medium after 6 h of transfection. For silencing IER5 or NF-YB, we used the following sequences: IER5 siRNA, forward 5′- CCU CAU CAG CAU CUU CGG UUU -3′, reverse 5′- ACC GAA GAU GCU GAU GAG GUU -3′; NF-YB siRNA, forward 5′- GGC AUU UAC UAA CCA GUU ATT −3′, reverse 5′- UAA CUG GUU AGU AAA UGC CTT −3′; and NC siRNA, forward 5′- UUC UCC GAA CGU GUC ACG UTT −3′, reverse 5′- ACG UGA CAC GUU CGG AGA ATT −3′.
In addition, IER5-specific shRNA and NC shRNA were synthesized and cloned into the BamH I/Hind III sites of the pSilencer 3.1-H1 hygro vector (Ambion, USA) to form IER5-silencing and negative control plasmids, respectively, and were verified by sequencing alignment. HeLa cells were transfected with a successfully constructed plasmid. After 48 h, the HeLa cells were treated with medium containing G418 at a concentration of 1000 μg/mL for 2 weeks. Positive clones were picked out using a sterile pipet tip, and then the cells were maintained in medium containing G418 at a concentration of 500 μg/mL to generate stable IER5-silenced HeLa and control HeLa cells.
Quantitative real-time PCR
Total RNA was extracted from the collected cells using TRIzol reagent (Invitrogen, USA). Total RNA (1 μg) was reverse transcribed to cDNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Japan). Then, 1 μL of cDNA reaction solution was used for qPCR analysis with THUNDERBIRD SYBR qPCR Mix (TOYOBO, Japan) on a CFX96 real-time PCR detection system (BIO-RAD, USA). The expression of the target gene was normalized to that of GAPDH, and the fold change was calculated using the 2-ΔΔCt method. All primers in this study were synthesized by Taihe Biotechnology Co., Ltd (Beijing, China). Quantitative real-time PCR (RT-qPCR) was performed using the following primers: IER5, forward 5’-CCG GGA ACG TGG CTA ACC-3′, reverse 5’-CCG GGA ACG TGG CTA ACC-3′; Cdc25B, forward 5’-ACG CAC CTA TCC CTG TCT C-3′, reverse 5’-CTG GAA GCG TCT GAT GGC AA-3′; and GAPDH, forward 5’-AAC GTG TCA GTG GTG GAC CT-3′, reverse 5’-TGC TGT AGC CAA ATT CGT TG-3′. For the PCR cycle reaction program, we chose the two-step method as follows: initial denaturation at 95 °C for 60 s, followed by 40 cycles of denaturation at 95 °C for 15 s and extension at 60 °C for 60 s.
Western blot analysis
The cells were washed twice with ice-cold PBS and lyzed on ice by adding a protein extraction reagent (Thermo, USA) to prepare whole-cell lysates. The protein supernatant was collected by centrifugation and quantified by a BCA protein assay kit (TIANGEN, China). Then, 5× loading buffer was added, and the sample was boiled in a water bath for 5 min. Equal amounts of total protein (40 μg/lane) were separated by 10% SDS-PAGE and then transferred onto nitrocellulose filter membranes (PALL, USA). The membranes were blocked with 5% skim milk powder dissolved in 1 × TBST and then incubated with primary antibodies against IER5 (1:1000, HPA029894, Sigma, USA), Cdc25B (1:1000, ab124819, Abcam, UK), NF-YB (1:1000, sc-376 546, Santa Cruz, USA), and β-actin (1:1000, TA-09, ZSGB-BIO, China) at 4 °C overnight. The membranes were then washed three times with 1 × TBST for 10 min each time and then incubated with the following secondary antibodies: horseradish peroxidase-conjugated goat anti-mouse antibody (1:4000, ZB-2305, ZSGB-BIO, China) and horseradish peroxidase-conjugated goat anti-rabbit antibody (1:4000, ZB-2301, ZSGB-BIO, China) for 1 h at RT. The membranes were washed three times once more with 1 × TBST for 10 min each time. The desired protein bands were detected using a chemiluminescence imager (LAS500, GE, USA) in the presence of ECL Western blotting Substrate (Thermo, USA). The autoradiographic intensity of each band was quantified using ImageJ software.
Construction of the Cdc25B promoter-luciferase reporter gene vector
The Eukaryotic Promoter Database (EPD) online site (http://epd.vital-it.ch/) was applied to search for the human Cdc25B gene promoter sequence. The region 2000 bp to 100 bp upstream of the transcription start site (TSS) of the Cdc25B gene was selected as the basic promoter region. The transcription factor binding site of the Cdc25B promoter region was analyzed using the JASPAR online database (http://jaspar.genereg.net/). Primers used in PCR amplification of Cdc25B promoter sequences were designed using Primer Premier 5.0 software. The 5′ end of the primer includes a protected base and a restriction site (Kpn I/Hind III). The target fragment was amplified by PCR using Q5 High-Fidelity DNA Polymerases (NEB, USA) with primers. Subsequently, the Cdc25B promoter-luciferase reporter construct was generated by inserting the sequence of interest into the Kpn I/Hind III sites of the pGL3-Basic vector (Promega, USA) upstream of the luciferase gene. Plasmid manipulations in E. coli were performed according to standard procedures. The amplified plasmids were isolated from overnight cultured E. coli. The inserted sequence of interest was verified by sequencing alignment. By designing different primers, a series of Cdc25B promoter deletion constructs were further constructed. A total of seven different lengths of luciferase reporter gene vectors were obtained. Mutation of the site of interest in the Cdc25B promoter region was completed by Taihe Biotechnology (Beijing) Co., Ltd using splicing-by-overlap extension PCR.
Luciferase reporter assay
One day before transfection, the cells were seeded at a density of 2 × 105 cells/well in a 24-well plate and cultured until the cell confluence reached approximately 50%. Cell transfection was performed using Lipofectamine 2000 (Invitrogen, USA) at a ratio (μL reagent:μL DNA) of 2.5:1, DMEM and 180 ng DNA/well, including 9 ng/well of pRP-TK internal control vector. The cells were lyzed 48-h post-transfection using passive lysis buffer, and the luciferase activity was detected using a dual-luciferase reporter assay system (Promega, USA). Luminescence was measured using a Modulus Single Tube Fluorometer (Promega, USA).
ChIP assay
HeLa cells were seeded at a density of 2 × 106 cells/dish in 10-cm diameter petri dishes in DMEM containing 10% FBS for 24 h and then treated with 4-Gy radiation. After irradiation, the cells were cultured for 12 h in an incubator. A total of 3 × 106 or more cells were collected for ChIP assays using a ChIP kit (Abcam, USA) according to the manufacturer’s protocols. Briefly, the cells were resuspended and incubated in Buffer A/Formaldehyde/PBS mix for 10 min at RT, and then glycine was added to quench the formaldehyde and end cross-linking. The cells were washed with ice cold PBS. The cells were resuspended and incubated in Buffer B for 10 min at RT and then centrifuged, and deposits were collected. The deposits were resuspended and incubated in ice-cold Buffer C for 10 min on ice and then centrifuged to obtain nuclei. The nuclei were suspended in 100 μL Buffer D/PI mix and then subjected to sonication for 8 cycles of 30″/30″ (ON/OFF cycles) in high power during an ambient ice-cold water bathing using Bioruptor Plus (Diagenode, Belgium) to shear DNA into an optimal DNA fragment size of 200–1000 bp. The cell debris was centrifuged to collect the supernatant containing sheared chromatin. Then, 1× ChIP buffer/PI mix was added to the sheared chromatin and vortexed for 5 s. Then, 280 μL of diluted chromatin was aliquoted, and 5 μg of antibodies against IER5 (HPA029894, Sigma, USA), SP1 (sc-59, Santa Cruz), SP3 (sc-644, Santa Cruz), NF-YB (sc-376 546, Santa Cruz), and p300 (sc-585, Santa Cruz) were added to the appropriate sample and incubated overnight with rotation at 4 °C. The remaining chromatin from each sample was frozen to be used for later real-time PCR analysis as input extracts for the ChIP assay. The samples were centrifuged to remove insoluble material. Then, 250 μL of the supernatant was removed and transferred to the bead/ChIP buffer mix and rotated for 60 min at 4 °C. Thereafter, the beads were washed three times more with 1× ChIP buffer. The washed bead samples and input extracts were incubated for 10 min at 98 °C with 100 μL of DNA purifying slurry. The samples were treated with proteinase K, incubated for 30 min at 55 °C, and incubated for 10 min at 98 °C again. The supernatant was collected by centrifugation to obtain DNA solutions. One microlitre of the obtained DNA solution was analyzed by real-time PCR to quantitate immunoprecipitated promoter fragments with primer set 2 designed to amplify the −174 to −8 bp region of the Cdc25B promoter. Similarly, we used primer set 2 designed to amplify −369 to −210 bp as a control. For ChIP assays, the primers were as follows: set 1, forward 5′-GGG AGC CAG TTG GAG CCT-3′, reverse 5′-GAT CCC GGC CTC CCA AAA −3′; and set 2, forward 5’-CTT AAT TCC TCC GGC CCA CC-3′, reverse 5’-GCT GCC ACT TCC ACC TCC TT-3′.
Statistical analysis
All experimental data were analyzed using SPSS 17.0 statistical software (SPSS Inc., USA), and graphs were generated using GraphPad Prism 6.0 software (GraphPad Software Inc., USA). Differences between two groups were analyzed using Student’s t-test. Differences among multiple groups were assessed using one-way ANOVA. Data are expressed as the mean ± SD. P values <.05 were considered to indicate statistical significance.
Results
The expression levels of IER5 and Cdc25B changed inversely after the irradiation of HeLa cells
To characterize the relationship between IER5 and Cdc25B under irradiation, we investigated the expression levels of IER5 and Cdc25B in HeLa cells irradiated with 4 Gy γ-ray radiation. The cells were harvested at .5, 4, 8, 12 and 24 h after irradiation. Western blot analyses indicated that IER5 was evidently upregulated in HeLa cells after 4 hours. Instead, Cdc25B was significantly downregulated beginning at the 4th hour (Fig. 1A). The results of RT-qPCR analyses were consistent with the Western blot analysis results (Fig. 1B). Meanwhile, we detected the expression of Cdc25B when IER5 was silenced and found that IER5 silencing significantly increased the expression of Cdc25B compared with that in NC cells (Fig. 1C and D). The results suggest that there are inverse changes in the expression levels of IER5 and Cdc25B in HeLa cells after irradiation.
Figure 1.

IER5 was involved in the inhibition of Cdc25B expression after irradiation. (A) Western blot analysis of the protein expression levels of IER5 and Cdc25B in samples harvested at various time points (0, .5, 4, 8, 12 and 24 h) after 4-Gy radiation of normal HeLa cells. (B) RT-qPCR analysis of mRNA expression levels of IER5 and Cdc25B in samples harvested at various time points (0, .5, 4, 8, 12 and 24 h) after 4-Gy radiation of normal HeLa cells. (C) Western blot analysis of the protein expression levels of IER5 and Cdc25B in si-NC HeLa cells and si-IER5 HeLa cells. (D) RT-qPCR analysis of the mRNA expression levels of Cdc25B in si-IER5-silenced HeLa cells compared with si-NC HeLa cells. (E) Western blot analysis of the protein expression levels of IER5 in NC HeLa cells and IER5-silenced HeLa cells. (F) RT-qPCR analysis of the mRNA expression levels of IER5 in IER5-silenced HeLa cells compared with NC HeLa cells. (G) Western blot analysis of the protein expression levels of IER5 and Cdc25B in samples harvested at various time points (0, .5, 4, 8, 12 and 24 h) after 4-Gy radiation of IER5-silenced HeLa cells. (H) RT-qPCR analysis of mRNA expression levels of IER5 and Cdc25B in samples harvested at various time points (0, .5, 4, 8, 12 and 24 h) after 4-Gy radiation of IER5-silenced HeLa cells. **P < .01.
Suppression of IER5 prevented irradiation-induced downregulation of Cdc25B
Our previous studies identified that IER5 enhanced irradiation-induced G2 stage arrest and increased radioresistance by disturbing cell cycle checkpoints [13]. Based on the idea that Cdc25B acts as a “trigger” in the G2/M-phase transition, we further investigated whether IER5 was involved in the inhibition of Cdc25B expression after irradiation. Here, we used successfully screened monoclonal IER5-silenced HeLa cells. The silencing efficiency was detected (Fig. 1E and F). Next, we explored whether silencing IER5 affected the change in Cdc25B expression caused by radiation. Similarly, IER5-silenced HeLa cells were exposed to 4-Gy radiation. The cells were harvested at .5, 4, 8, 12 and 24 h after irradiation. Western blot analyses showed that IER5 had increased expression after 8 h. However, Cdc25B was distinctly decreased after 8 h (Fig. 1G). The RT-qPCR results indicated that the downregulation of Cdc25B caused by radiation in IER5-silenced HeLa cells was attenuated compared to that in normal HeLa cells (Fig. 1B and H). Therefore, the above results indicate that inhibition of IER5 blocks irradiation-induced downregulation of Cdc25B.
Radiation decreased Cdc25B promoter activity
It has previously been revealed that Cdc25B is a predictor of tumor radiosensitivity [17]. We speculated that the Cdc25B promoter region contains many elements related to radiation that regulate its expression at the transcription level. To identify promoter elements involved in irradiation-mediated regulation of Cdc25B, we linked stepwise-deleted fragments of the Cdc25B promoter to the luciferase reporter gene (Supplementary Fig. S1). The recombinant constructs were transfected into HeLa cells. Twenty-four hours post-transfection, the cells were treated with 4-Gy radiation. The cells were collected 12 h after irradiation treatment, and transcriptional activity was evaluated by a dual-luciferase reporter assay system. The luciferase reporter assay results showed that radiation decreased the transcriptional activity of the −1930, −1546, −1044, −655, −516, and − 160 luciferase constructs but not that of the −53 luciferase construct, suggesting that there are response elements regulated by radiation in the −160 to −53 region of the Cdc25B promoter (Fig. 2A). The results indicated that radiation can reduce the transcriptional activity of the Cdc25B promoter.
Figure 2.
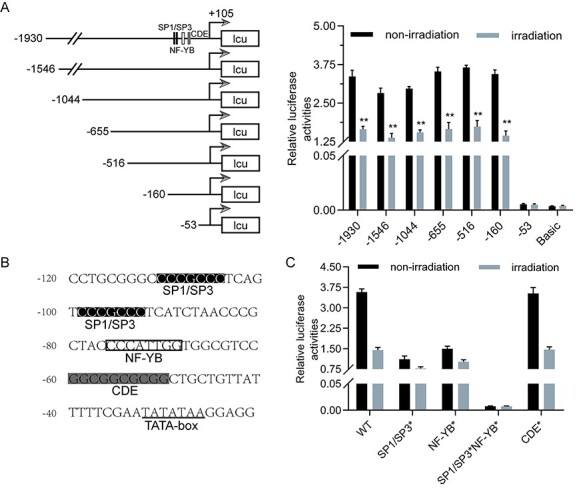
Radiation decreased Cdc25B promoter activity through GC-rich and CCAAT box motifs. (A) The promoter sequence of Cdc25B gene was cloned into pGL3-basic vector. Luciferase reporter assays were performed to determine the effect of radiation on Cdc25B promoter activity. HeLa cells were co-transfected with stepwise Cdc25B promoter-luciferase constructs and pRL-TK (an internal control plasmid), and 24 h later, the cells were treated with 4-Gy radiation. The cells were harvested 12 h post-irradiation for dual-luciferase assays. **P < .01, non-irradiation vs. irradiation. (B) Cdc25B promoter sequence including the two GC-rich motifs for binding Sp1/Sp3 and one CCAAT box motif for binding NF-YB, along with two paratactic CDE motifs. (C) Both Sp1/Sp3 and NF-YB binding sites are involved in irradiation-mediated regulation of Cdc25B. The −160 luciferase constructs with WT or mutated (*)binding sites for Sp1/Sp3 (replace CGCC with ACAT), NF-YB (replace CAAT with TGAA), or the CDE motifs (replace GGCG with CTAT) were analyzed for response to as in (A).
Both Sp1/Sp3 and NF-YB binding sites are involved in irradiation-mediated regulation of Cdc25B
The transcription factor binding spectrum database JASPAR was used to analyze this region. The results revealed that there were two GC-rich motifs for binding Sp1/Sp3 and one CCAAT box motif for binding NF-YB, both of which were important for the transcriptional activity of the Cdc25B promoter [18]; there were also two paratactic cell cycle-dependent element (CDE) motifs (Fig. 2B). To elucidate the function of these motifs in the irradiation-mediated control of Cdc25B, we mutated these conserved sequences individually or in combination in the −160 luciferase construct. The recombinant mutant constructs were transfected into HeLa cells that were also treated with radiation. The luciferase reporter assay results showed that the cell cycle-dependent element mutant (CDE*) and the wild type (WT) promoters performed at comparable levels, but the Sp1/Sp3 mutant (Sp1/Sp3*) and NF-YB mutant (NF-YB*) promoters were affected to a lesser extent (Fig. 2C). However, when the Sp1/Sp3 binding site was mutated together with the NF-YB site, radiation had little effect on the Cdc25B mutant promoter (Fig. 2C). The results indicated that both Sp1/Sp3 and NF-YB binding sites are involved in the irradiation-mediated regulation of Cdc25B. Combined with the above data, these results suggest that radiation decreases Cdc25B promoter activity through GC-rich and CCAAT box motifs.
Overexpression of the IER5 gene inhibited the promoter activity of the Cdc25B gene
Gene expression is regulated by transcription factors binding selectively to specific parts of the genome [19]. As described above, knockdown of the IER5 gene was able to upregulate Cdc25B expression. Next, we investigated whether Cdc25B is regulated by IER5 at the transcriptional level. The IER5 expression plasmid was donated by Dr Yumei Wu [20]. Here, we used 293 T cells, which have high transfection efficiency. IER5 expression was conspicuously improved, whereas the expression of Cdc25B was inhibited in IER5-transfected 293 T cells (Fig. 3A and B). The IER5 expression plasmid or control vector was transfected into 293 T cells. Twenty-four hours later, the stepwise-deleted Cdc25B promoter-luciferase constructs and internal control plasmid were co-transfected into the above 293 T cells. Cells were collected 48 h post-transfection for dual-luciferase assays. The results of the luciferase reporter assays revealed that overexpression of the IER5 protein reduced the transcriptional activities of the −1930, −1546, −1044, −655, −516, and − 160 luciferase constructs but not that of the −53 luciferase construct, which indicated that IER5 response elements are present in the −160 to −53 region of the Cdc25B promoter (Fig. 3C). Notably, both the IER5 response element and the radiation response element are located in the −160 to −53 region of the Cdc25B promoter. To further understand the role of this region in the regulation of Cdc25B mediated by IER5, we used the mutant promoter constructed above to explore the effect of IER5 on its transcriptional activity. Luciferase reporter assay results showed that the CDE* and WT promoters performed at comparable levels, whereas the Sp1/Sp3* promoter was affected to a lesser extent, and the NF-YB* promoter was hardly affected (Fig. 3D). The results indicated that exogenous expression of the IER5 gene could inhibit the promoter activity of the Cdc25B gene. We found that the NF-YB binding site might play an important role in this process.
Figure 3.

Exogenous expression of the IER5 gene inhibited the promoter activity of the Cdc25B gene. (A) Western blot analysis of the protein expression levels of IER5 and Cdc25B in IER5-transfected 293 T cells compared with NC 293 T cells. (B) RT-qPCR analysis of the mRNA expression levels of Cdc25B in IER5-transfected 293 T cells compared with NC 293 T cells. (C) Luciferase reporter assays were performed to determine the effect of exogenous IER5 gene expression on Cdc25B promoter activity. **P < .01, vector vs. IER5. (D) Luciferase reporter assay authenticated the role of these conserved motifs in the regulation of Cdc25B transcription activity by IER5.
IER5 is involved in irradiation-induced downregulation of Cdc25B promoter activity
Based on the above results, we hypothesized that irradiation-induced upregulation of IER5 could also participate in the regulation of Cdc25B expression through the NF-YB binding site. We used the mutant promoter constructed above to clarify this conjecture. HeLa cells were transfected with IER5 or NC siRNA and, 24 h later, they were co-transfected with mutant promoter plasmid and internal control plasmid. The cells were cultured for another 24 hours and treated with 4-Gy radiation. The cells were lyzed 12 h post-irradiation for dual-luciferase assays. The results showed that the CDE* and WT promoters presented comparable activity levels, whereas the Sp1/Sp3* promoter activity was affected to a lesser extent, and the NF-YB* promoter activity was hardly affected (Fig. 4). The results indicated that IER5 is involved in irradiation-mediated Cdc25B expression regulation through the NF-YB binding site. We hold the opinion that IER5 may be a transcriptional repressor to play a critical role in the negative regulation of Cdc25B gene transcription.
Figure 4.

IER5 is involved in Cdc25B expression regulation through the NF-YB binding site in irradiation-induced HeLa cells.
NF-YB is vital for irradiation-mediated Cdc25B downregulation by IER5
The literature reports that specific proteins interact with Sp1 and NF-YB transcription factors, pointing out that this interaction reduces the ability of these factors to bind to DNA, resulting in a lack of general transcription factors [21, 22]. To illustrate the underlying mechanisms of irradiation-mediated downregulation of Cdc25B by IER5, we conducted ChIP assays. To this end, we first designed two pairs of primers to perform ChIP analysis in HeLa cells (Fig. 5A). Chromatin fragments were immunoprecipitated using anti-IER5 antibody, anti-H3 antibody as positive control, and anti-IgG antibody as negative control. The precipitated DNA fragments were amplified by RT-qPCR. Agarose gel electrophoresis was used to detect DNA bands. The results showed that protein IER5 was recruited to the Cdc25B promoter region, demonstrating that Cdc25B was a direct target of IER5 (Fig. 5B). The well-growing HeLa cells were transfected with IER5 or NC siRNA, and 24 h later, they were treated with 4-Gy radiation. The cells were harvested 12 h post-irradiation for ChIP assays using anti-IER5, anti-Sp1, anti-Sp3, and anti-NF-YB antibodies. The ChIP results showed that the IER5 protein was replenished in irradiation-treated cells, and a marked change in the recruitment of this factor was detected following IER5 silencing. The NF-YB protein was absent in irradiation-treated cells, but a great increase in the recruitment of this factor was detected following IER5 silencing. Both Sp1 and Sp3 proteins were absent in irradiation-treated cells, and no change in the recruitment of either factor was detected following IER5 silencing (Fig. 5C-D). p300, a known co-activator that interacts with NF-YB, is a histone acetyltransferase [23]. To explore whether the reduced binding of NF-YB to the Cdc25B promoter was detected following IER5 silencing, we performed ChIP assays as described above using anti-p300. The results showed a great increase in the recruitment of this factor following IER5 silencing, while the p300 protein was also absent in irradiation-treated cells (Fig. 5E). To confirm that NF-YB was involved in irradiation-mediated Cdc25B regulation by IER5, we inhibited NF-YB expression by transfecting NF-YB siRNA into HeLa cells. We found that inhibiting the expression of NF-YB decreased Cdc25B mRNA and protein expression levels, but silencing IER5 in NF-YB siRNA-transfected HeLa cells increased Cdc25B expression compared with that in siNC-transfected HeLa cells (Fig. 5F-G). The ChIP assay results showed that the binding of IER5 to the Cdc25B promoter region in irradiation-induced HeLa cells induced the release of the coactivator p300 through interaction with NF-YB, leading to the downregulation of Cdc25B expression.
Figure 5.
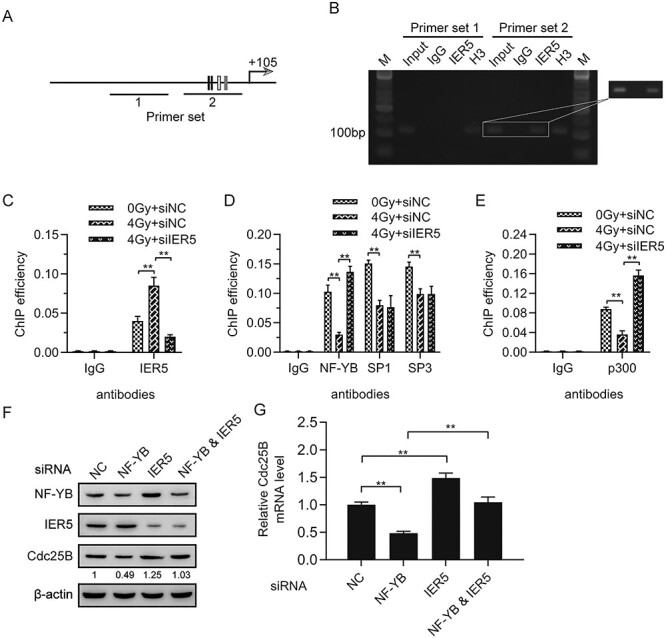
Importance of general transcription factors in irradiation-mediated Cdc25B regulation by IER5. (A) Schematic representation of the location of the primer we designed, pointing sites for Sp1, Sp3, and NF-YB motifs. (B) ChIP assays for IER5 in the Cdc25B promoter region amplified from the precipitated DNA by designed primer sets 1 and 2. Agarose gel electrophoresis was used to detect DNA bands, which were amplified from precipitated DNA fragments. (C) Radiation caused recruitment of protein IER5 to the Cdc25B promoter and a decrease in this factor following IER5 silencing. (D) Radiation led to the absence of general transcription factors on the Cdc25B promoter and no change in the recruitment of Sp1 and Sp3 but caused a great increase in the recruitment of NF-YB following IER5 silencing. (E) Radiation led to the absence of p300 and a great increase in this factor following IER5 silencing. (F and G) HeLa cells were transfected with NF-YB siRNA, IER5 siRNA, both, or NC siRNA, and 48 h later, the cells were collected for western blot and RT-qPCR analyses.
Discussion
Among females, CC is the fourth most common malignant tumor and the leading cause of cancer-related death in developing countries [2]. Radiotherapy is the main treatment for patients with CC, especially patients with advanced CC [24]. However, insensitivity to radiotherapy is still the biggest obstacle for its application. Yang et al. uncovered that the expression of DLL4 is significantly higher in radiotherapy-resistant CC cells, than in radiotherapy-sensitive cells, indicating that it is notably related to the radiosensitivity of CC [25]. Ni et al. showed that the expression of BRD4 is remarkably upregulated in radioresistant cervical cancer samples and that inhibition of BRD4 can sensitize CC cells to radiotherapy by weakening DNA repair mechanisms [26]. IER5 is one of the radiotherapy sensitivity related genes in CC [12, 20]; therefore, we aimed to understanding the mechanism and finding radiotherapy sensitization targets to improve the radiosensitivity of CC and achieve the best therapeutic effect with the lowest dose of radiotherapy. In this study, we explored the relationship between IER5 and Cdc25B under the influence of irradiation. We found that inhibition of IER5 expression blocked irradiation-induced downregulation of Cdc25B. These results combined with those of our previous studies indicate that inhibition of IER5 expression can enhance irradiation-induced G2 stage arrest [12]. This study provides powerful proof that IER5 participates in the cell cycle regulation of irradiation-treated cells.
Immediate early response gene 5 (IER5) was first described as a radiation response gene in 2006 and decreases cell survival by promoting apoptosis [7, 8, 12]. External stimulation (such as insomnia or ovariectomy) or the application of chemicals (such as estrogen) can cause changes in the expression of the IER5 gene, and its response speed is still slower than that of the c-fos gene [8, 27]. IER5 can mediate the response of cells to mitotic signals through phosphorylation and/or dephosphorylation and can also play an important role in regulating transcription by binding to DNA or mediating protein–protein interactions [10]. Several studies have regarded IER5 as a potential radiation biomarker that responds to gamma rays, and the response is unrelated radiation dose [28]. DNA damage repair plays an important role in maintaining the stability of the genome and the survival of organisms. Double-strand breaks (DSBs) are the most serious form of DNA damage. Yu et al. reported that IER5 participates in NHEJ-mediated DSB repair [20]. IER5 is required for Notch-mediated induction of squamous cell differentiation [29]. IER5 was previously confirmed to be significantly expressed and served as a tumor promoter in HeLa cells [30]. In the present study, the data revealed inverse changes in the expression levels of IER5 and Cdc25B in HeLa cells after exposure to irradiation. Additionally, IER5 gene silencing significantly improved the expression of Cdc25B. Furthermore, we found that inhibition of IER5 blocked irradiation-induced downregulation of Cdc25B, suggesting that IER5 might be involved in the downregulation of Cdc25B expression after irradiation.
Cdc25B is a member of the Cdc25 family of phosphatases, and it plays an important role in cancer cell growth. Previous studies have found that Cdc25B is responsible for the initial activation of CDK1-cyclin B in the centrosome during the G2/M-phase transition [31], which is then followed by complete activation of CDK1-cyclin B by Cdc25C in the nucleus at the beginning of mitosis [32]. Cdc25 phosphatase activities are strictly regulated by multiple mechanisms, including changes in cell content, subcellular localization and phosphorylation status. The abnormal expression of Cdc25B has been described in various solid tumors, such as breast, gastric, prostate, and colon cancer [33]. The dysregulation of Cdc25B expression is thought to be linked to disease progression. However, the mechanism of Cdc25B in the response to cervical cancer cell radiotherapy remains elusive. Dalvai et al. reported that doxorubicin promotes the transcriptional upregulation of Cdc25B in cancer cells by releasing Sp1 from the promoter [34]. The results in our study are consistent with previous researches. In the current study, the results of our study are consistent with those of previous studies. In the current study, we constructed a Cdc25B promoter-luciferase reporter gene vector and detected promoter activity. We found that Cdc25B promoter activity was significantly reduced after irradiation, and response elements affected by radiation existed in the −160 ~ −53 region of the Cdc25B promoter. There are two GC-rich motifs, one CCAAT box motif, and two paratactic CDE motifs in this region. Sp1 and NF-YB, as sequence-specific DNA-binding proteins, can identify the GC-rich motif and the CCAAT box motif, respectively, and participate in the transcriptional regulation of multiple genes [35, 36]. This reminded us to investigate the effect of these motifs. We mutated these conserved sequences individually or in combination and found that both Sp1 and NF-YB binding sites were involved in irradiation-mediated regulation of Cdc25B.
The transcriptional activity of many genes involved in irradiation-induced damage is regulated by the c-fos, c-jun, egr-1, p53, and NF- B transcription factors [37–39]. NF-YB, as a ubiquitous heterotrimeric transcription factor, has binding affinity to the CCAAT consensus motif [40]. Mounting evidence suggests that NF-YB, which drives the transcription of a mass of cell cycle regulatory genes, is a key factor in regulating cell proliferation [40]. It has also been shown that NF-YB mediates transcriptional inhibition of mitotic cyclins and Cdc25C genes during p53-dependent G2 arrest caused by DNA damage [41]. In this study, we reported that NF-YB was absent in HeLa cells after exposure to irradiation, but a great increase in the recruitment of this factor was detected following IER5 silencing. p300, a known co-activator that interacts with NF-YB, is a histone acetyltransferase [23]. We found that p300 was also absent after exposure to irradiation. Taken together, these data suggest that both NF-YB and p300 are vital for irradiation-mediated Cdc25B downregulation by IER5.
B transcription factors [37–39]. NF-YB, as a ubiquitous heterotrimeric transcription factor, has binding affinity to the CCAAT consensus motif [40]. Mounting evidence suggests that NF-YB, which drives the transcription of a mass of cell cycle regulatory genes, is a key factor in regulating cell proliferation [40]. It has also been shown that NF-YB mediates transcriptional inhibition of mitotic cyclins and Cdc25C genes during p53-dependent G2 arrest caused by DNA damage [41]. In this study, we reported that NF-YB was absent in HeLa cells after exposure to irradiation, but a great increase in the recruitment of this factor was detected following IER5 silencing. p300, a known co-activator that interacts with NF-YB, is a histone acetyltransferase [23]. We found that p300 was also absent after exposure to irradiation. Taken together, these data suggest that both NF-YB and p300 are vital for irradiation-mediated Cdc25B downregulation by IER5.
In summary, our study investigated the underlying mechanism of IER5 in the cell cycle regulation of irradiation-damaged HeLa cells. The results indicated that the expression of IER5 is upregulated after irradiation, which further causes the release of the coactivator p300 through interaction with NF-YB, leading to the downregulation of Cdc25B expression. However, one limitation might exist in this study. We did not delve into the detailed competitive mechanism between IER5 and NF-YB after irradiation. Further study will be conducted to clarify this issue. Nevertheless, we also hope that these findings will provide an attractive strategy for the treatment of CC.
Conflict of interest statement
The authors declare that they have no competing interests.
Funding
The present work was supported by grants from the National Natural Science Foundation of China (grant nos. 31770907, 31640022) and the Natural Science Foundation of Beijing Municipality (grant no. 7172146).
Supplementary Material
{kind=link}
Contributor Information
Lixin Ding, National Institute for Radiological Protection, Chinese Center for Disease Control and Prevention, Xicheng District, Beijing 100088, P. R. China; Department of Nuclear Medicine, Peking University Cancer Hospital, Haidian District, Beijing 100142, P. R. China.
Xianzhe Zhao, National Institute for Radiological Protection, Chinese Center for Disease Control and Prevention, Xicheng District, Beijing 100088, P. R. China.
Qiang Xiong, National Institute for Radiological Protection, Chinese Center for Disease Control and Prevention, Xicheng District, Beijing 100088, P. R. China.
Xiaoyan Jiang, National Institute for Radiological Protection, Chinese Center for Disease Control and Prevention, Xicheng District, Beijing 100088, P. R. China.
Xiaodan Liu, Beijing Key Laboratory for Radiobiology, Beijing Institute of Radiation Medicine, Haidian District, Beijing 100850, P. R. China.
Kuke Ding, National Institute for Radiological Protection, Chinese Center for Disease Control and Prevention, Xicheng District, Beijing 100088, P. R. China; Office for Public Health Management, Chinese Center for Disease Control and Prevention, Changping District, Beijing 102206, P. R. China.
Pingkun Zhou, Beijing Key Laboratory for Radiobiology, Beijing Institute of Radiation Medicine, Haidian District, Beijing 100850, P. R. China.
References
- 1.Arbyn M, Weiderpass E, Bruni L et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health 2020;8:e191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 3.Subramanya D, Grivas PD. HPV and cervical cancer: updates on an established relationship. Postgrad Med 2008;120:7–13. [DOI] [PubMed] [Google Scholar]
- 4.Gill BS, Lin JF, Krivak TC et al. National Cancer Data Base analysis of radiation therapy consolidation modality for cervical cancer: the impact of new technological advancements. Int J Radiat Oncol Biol Phys 2014;90:1083–90. [DOI] [PubMed] [Google Scholar]
- 5.Pimple S, Mishra G, Shastri S. Global strategies for cervical cancer prevention. Curr Opin Obstet Gynecol 2016;28:4–10. [DOI] [PubMed] [Google Scholar]
- 6.Lin M, Ye M, Zhou J et al. Recent advances on the molecular mechanism of cervical carcinogenesis based on systems biology technologies. Comput Struct Biotechnol J 2019;17:241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kis E, Szatmári T, Keszei M et al. Microarray analysis of radiation response genes in primary human fibroblasts. Int J Radiat Oncol Biol Phys 2006;66:1506–14. [DOI] [PubMed] [Google Scholar]
- 8.Wang HP, Long XH, Sun ZZ et al. Identification of differentially transcribed genes in human lymphoblastoid cells irradiated with 0.5 Gy of gamma-ray and the involvement of low dose radiation inducible CHD6 gene in cell proliferation and radiosensitivity. Int J Radiat Biol 2006;82:181–90. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa Y, Kawabata S, Sakurai H. HSF1 transcriptional activity is modulated by IER5 and PP2A/B55. FEBS Lett 2015;589:1150–5. [DOI] [PubMed] [Google Scholar]
- 10.Williams M, Lyu MS, Yang YL et al. Ier5, a novel member of the slow-kinetics immediate-early genes. Genomics 1999;55:327–34. [DOI] [PubMed] [Google Scholar]
- 11.Asano Y, Kawase T, Okabe A et al. IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci Rep 2016;6:19174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding KK, Shang ZF, Hao C et al. Induced expression of the IER5 gene by gamma-ray irradiation and its involvement in cell cycle checkpoint control and survival. Radiat Environ Biophys 2009;48:205–13. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa Y, Sakurai H. Heat-induced expression of the immediate-early gene IER5 and its involvement in the proliferation of heat-shocked cells. FEBS J 2015;282:332–40. [DOI] [PubMed] [Google Scholar]
- 14.Pan C, Zhu D, Wang Y et al. Human cytomegalovirus miR-UL148D facilitates latent viral infection by targeting host cell immediate early response gene 5. PLoS Pathog 2016;12:e1006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astuti P. Regulation of cdc25B During G2/M Progression: The University of Queensland Diamantina Institute, 2010.
- 16.Kohama Y, Saito M, Yada M et al. Regulation of the stability and activity of CDC25A and CDC25B by protein phosphatase PP2A and 14-3-3 binding. Cell Signal 2019;54:10–6. [DOI] [PubMed] [Google Scholar]
- 17.Miyata H, Doki Y, Shiozaki H et al. CDC25B and p53 are independently implicated in radiation sensitivity for human esophageal cancers. Clin Cancer Res 2000;6:4859–65. [PubMed] [Google Scholar]
- 18.Körner K, Jerôme V, Schmidt T et al. Cell cycle regulation of the murine cdc25B promoter: essential role for nuclear factor-Y and a proximal repressor element. J Biol Chem 2001;276:9662–9. [DOI] [PubMed] [Google Scholar]
- 19.Biggin MD. To bind or not to bind. Nat Genet 2001;28:303–4. [DOI] [PubMed] [Google Scholar]
- 20.Yu X-P, Wu Y-M, Liu Y et al. IER5 is involved in DNA double-strand breaks repair in association with PAPR1 in hela cells. Int J Med Sci 2017;14:1292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalvai M, Mondesert O, Bourdon JC et al. Cdc25B is negatively regulated by p53 through Sp1 and NF-Y transcription factors. Oncogene 2011;30:2282–8. [DOI] [PubMed] [Google Scholar]
- 22.Zehavi Y, Kedmi A, Ideses D et al. TRF2: TRansForming the view of general transcription factors. Transcription 2015;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani R. The molecular biology of the CCAAT-binding factor NF-Y. Gene 1999;239:15–27. [DOI] [PubMed] [Google Scholar]
- 24.Klyuchko KO, Gargin VV. Influence of neoadjuvant chemoradiotherapy for locally advanced cervical cancer. Pol Merkur Lekarski 2020;48(288):406–409. [PubMed] [Google Scholar]
- 25.Yang SS, Yu DY, Du YT et al. Inhibition of Delta-like Ligand 4 enhances the radiosensitivity and inhibits migration in cervical cancer via the reversion of epithelial-mesenchymal transition. Cancer Cell Int 2020;20(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni M, Li J, Zhao H et al. BRD4 inhibition sensitizes cervical cancer to radiotherapy by attenuating DNA repair. Oncogene 2021;40(15):1–14. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Pang ST, Sahlin L et al. Gene expression profiling of the effects of castration and estrogen treatment in the rat uterus. Biol Reprod 2003;69:1308–17. [DOI] [PubMed] [Google Scholar]
- 28.Tavakoli H, Manoochehri M, Modarres MS et al. Dose-dependent and gender-related radiation-induced transcription alterations of Gadd45a and Ier5 inhuman lymphocytes exposed to gamma ray emitted by 60Co. Radiat Prot Dosim 2013;154:37–44. [DOI] [PubMed] [Google Scholar]
- 29.Pan L, Lemieux ME, Thomas T et al. IER5, a DNA damage response gene, is required for Notch-mediated induction of squamous cell differentiation. elife 2020;9:e58081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding KK, Yang F, Jiang HQ et al. Overexpression of the immediate early response 5 gene increases the radiosensitivity of HeLa cells. Oncol Lett 2019;18:2704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindqvist A, Källström H, Lundgren A et al. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol 2005;171:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabrielli BG, Clark JM, McCormack AK et al. Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J Biol Chem 1997;272:28607–14. [DOI] [PubMed] [Google Scholar]
- 33.Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem 2016;416:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalvai M, Mondesert O, Bugler B et al. Doxorubicin promotes transcriptional upregulation of Cdc25B in cancer cells by releasing Sp1 from the promoter. Oncogene 2013;32:5123–8. [DOI] [PubMed] [Google Scholar]
- 35.Husmann M, Dragneva Y, Romahn E et al. Nuclear receptors modulate the interaction of Sp1 and GC-rich DNA via ternary complex formation. Biochem J 2000;352 Pt 3:763–72. [PMC free article] [PubMed] [Google Scholar]
- 36.Romier C, Cocchiarella F, Mantovani R et al. The NF-YB/NF-YC structure gives insight into DNA binding and transcription regulation by CCAAT factor NF-Y. J Biol Chem 2003;278:1336–45. [DOI] [PubMed] [Google Scholar]
- 37.Calaf GM, Hei TK. Ionizing radiation induces alterations in cellular proliferation and c-myc, c-jun and c-fos protein expression in breast epithelial cells. Int J Oncol 2004;25:1859–66. [DOI] [PubMed] [Google Scholar]
- 38.Elshawi OE, Nabeel AI. Modulatory effect of a new benzopyran derivative via COX-2 blocking and down regulation of NF-κB against γ-radiation induced- intestinal inflammation. J Photochem Photobiol B 2019;192:90–6. [DOI] [PubMed] [Google Scholar]
- 39.Wang W, Xiong Y, Ding X et al. Cathepsin L activated by mutant p53 and Egr-1 promotes ionizing radiation-induced EMT in human NSCLC. J Exp Clin Cancer Res 2019;38:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurtner A, Manni I, Piaggio G. NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim Biophys Acta Gene Regul Mech 2017;1860:604–16. [DOI] [PubMed] [Google Scholar]
- 41.Ly LL, Yoshida H, Yamaguchi M. Nuclear transcription factor Y and its roles in cellular processes related to human disease. Am J Cancer Res 2013;3:339–46. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.