

Abstract
Advances in understanding of the process of carcinogenesis have undermined the concept of chemicals being classifiable as either carcinogens or non-carcinogens. Elements of carcinogenesis are happening all the time and a proportion of cancers cannot be prevented, the ‘bad luck hypothesis’. Although the proportion that can be prevented is disputed, it is important to continue efforts to reduce it. Factors that increase cancer risk have been grouped into intrinsic factors that cannot be modified, and endogenous and exogenous factors that can be modified. Chemicals are exogenous factors that can be modified by risk management measures. Chemicals can alter three key rates that influence cancer risk: cell division, mutation rate per cell division, transformation rate of mutated cells to cancer. These rates can form the basis of a dynamic cancer risk model, a generic, adverse outcome pathway for carcinogenesis where chemicals are considered for their ability to modify cancer risk rather than simply whether they are classed as carcinogens or non-carcinogens. This allows the development of different strategies for assessing cancer risk that use a range of data sources and are not dependent on using long-term bioassays and epidemiology to identify carcinogens. The framework will also allow difficult questions such as the effect of less than lifetime exposures and the effect of exposures to more than one chemical to be addressed.
Keywords: carcinogenicity, risk assessment, modification of cancer risk, mode of action, dynamic cancer risk model
Introduction: How Much of Cancer Is ‘Bad Luck’?
Cancer remains a major cause of morbidity and mortality. The lifetime probability of being diagnosed with cancer is just under 40% [1] and 22% of deaths in the US in 2018 were cancer-related, making it the second leading cause of death after cardiovascular disease in both men and women [2]. Almost half—46% in 2017—of all people who die from cancer are 70 or older, 41% are between 50 and 69 years old and so 87% of all cancer deaths are in people 50 years or older [3].
The causes of cancer and its treatment have been investigated extensively, but studies of prevention have lagged behind. Hanahan and Weinberg [4] argued that future cancer researchers would be practicing a dramatically different type of science from that applied over the last quarter of the 20th century. They posited that although much of the change would be at the technical level, the more fundamental change would be conceptual. They postulated that the complexities of cancer would become understandable in terms of a small number of underlying principles, and they put forward a set of features that characterize the transformation of normal human cells into cancers, the hallmarks of cancer.
Tomasetti and Vogelstein [5] proposed that the incidence of cancer in tissues correlated with the number of stem cell divisions, and they suggested that over 60% of cancers arose from spontaneous mutations in stem cells; this was dubbed the ‘Bad Luck Hypothesis’ suggesting that opportunity to prevent cancer was therefore limited. This contribution sparked a lively debate and prompted an analysis by Wu and colleagues [6]. They agreed with the concept that a proportion of cancers arose from spontaneous mutations, but they considered that the suggested percentage of non-preventable cancers in this model was too high, although they did not give a precise alternative figure. They considered that there were other factors that could contribute to increasing cancer risk. They put forward a useful framework that classified three types of cancer risk factors:
Intrinsic factors: Random errors in DNA replication (unmodifiable risk)
Non-intrinsic factors: Endogenous—Biologic aging, genetic susceptibility, DNA repair machinery, hormones, growth factors and inflammation (partially modifiable risk)
Non-intrinsic factors: Exogenous—Some forms of radiation, chemical carcinogens, tumour-causing viruses and bad lifestyles such as smoking, lack of exercise and nutrient imbalance (modifiable risk)
The purpose of this paper is to explore the contribution to cancer of one of Wu and colleagues’ extrinsic factors, which they listed as ‘chemical carcinogens’.
Carcinogens and Non-carcinogens
It is impossible to quantify the contribution chemical exposure makes to overall cancer incidence, but it is one of many cancer risk factors cited by Wu et al. [6], and they regarded it as a ‘modifiable’ risk factor. Whether it is a major or a minor factor, if it is modifiable, then steps should be taken to modify it in a downward direction. In order to be modified, the risk factor must be recognized and then steps taken to reduce its impact. Methodology was developed in the 1970s based on the concept that chemicals were either ‘carcinogens’ or ‘non-carcinogens’; elimination and restriction of use for the ‘carcinogens’ would result in lower incidence of cancer. Chemicals were designated as ‘carcinogens’ as a result of epidemiology studies or long-term rodent bioassays. The problems associated with this approach have been extensively explored [7–9]. About half the chemicals that have been tested in long-term rodent bioassays are deemed to have increased the number of tumours. This questions the eliminate/restrict use strategy that was based on an expectation that only a small proportion of chemicals would have carcinogenic potential [7–11]. In addition, rodent bioassays may give different results with the same chemical [12] and only a small percentage of the chemicals with which we may come into contact have been tested in the rodent bioassay because it uses so many animals, takes so long, and is very expensive.
At the same time, as the long-term bioassay was revealing that a large proportion of chemicals could increase the incidence of tumours in experimental animals, epidemiology was revealing a wide range of factors such as obesity, shift work, alcohol, certain professions and reduced physical activity as being associated with increased incidence of cancer [13]. Taken together, this body of evidence suggests that the concept of trying to identify ‘carcinogens’ is untenable and should be changed [9]. It will never be possible to accurately resolve the issue of how much cancer is due to ‘bad luck’ or intrinsic factors and therefore cannot be prevented, but it is prudent to develop and refine methods that characterize extrinsic factors so that they can be managed.
Problems for Risk Assessors
The role of the risk assessor in cancer prevention is to identify and quantify the factors that can increase the risk of cancer. This information is then used by risk managers who take steps to reduce the exposure to the pertinent factors and thereby reduce the risk. The way this is done for chemicals varies in different organizations and countries but mainly follows the logic exemplified in the decision tree scheme adopted by the UK Committee on Carcinogenicity [14]. The first step is to decide whether the chemical of concern is a carcinogen or not, that is to determine if it shows an increased cancer incidence in epidemiology studies or long-term bioassays. If not, then the chemical is considered to have no impact on cancer, which is a questionable conclusion given the observations of Braakhuis et al. [12] on variation of the results with the same chemical. If yes, then the next question in the decision tree becomes is it genotoxic or not. If it is genotoxic, there is deemed to be no threshold and so exposure should be as low as possible. If it is not genotoxic, there is assumed to be a threshold and an exposure limit is derived from the results of the long-term studies or other studies in which a precursor effect to the cancer has been identified. The exposure limit is set to avoid cancer risk for long-term daily exposure up to and including lifetime.
This provides guidance on the impact of long-term exposure to chemicals, but it is not helpful for the wider range of questions that can be posed to risk assessors, which include the following:
What are the effects of short-term or intermittent exposure?
What are the effects of exposure to more than one chemical either at the same time or at different times?
How can the risk of cancer be assessed if there are no cancer epidemiology studies or long-term bioassays available?
The Concept of Modification of Cancer Risk
These difficult questions are posed against the background that cancer is a probabilistic phenomenon with a high background rate (c40% per lifetime [1]), but safety decisions are required in a world that wants definitive, deterministic assurance of zero risk. How do we then move beyond the idea of carcinogen versus non-carcinogen and the presumption that there is one long-term exposure limit? Perhaps the first step is to revise the terminology. Wu and colleagues [6] stated that ‘exposure to risk factors does not necessitate the development of cancer, nor does absence of exposure to a risk factor provide 100% guarantee to prevent cancer’. They considered that intrinsic and non-intrinsic risk factors interact, and cancer risk can be modified whether or not intrinsic factors play a part.
Applying this logic leads to a different way to consider chemicals and carcinogenesis. Rather than trying to distinguish between carcinogens and non-carcinogens, the aim is to identify and characterize the factors that can modify cancer risk. This would enable the identification and characterization of chemicals that can affect these factors and can thus be called modifiers of cancer risk.
This requires knowledge of cancer pathogenesis, and although there are different types of cancers, a unified theory has emerged [15]. It is a multistage process that starts with mutations in dividing stem cells (or cells with ‘stemness’ properties, though with better understanding of RNA biology and other epigenetic changes even this notion can be questioned). The narrative is that mutations occur randomly, and there is a finite probability of a mutation occurring during each stem cell division. Some mutations lead to the loss of control of cell division. Control of cell division is an adaptation for multicellular organism and when cells lose this control, they behave like single cell organisms. They gain an ‘unfair advantage’ over cells that retain cell division control and thus they thrive and divide.
Once the clone of mutated cells is large enough, it requires some organization to maintain nutrition, and other adaptive or mutational alterations occur, which allow the clone to develop into a tumour, including changes in signalling pathways, all of which create a permissive microenvironment in which tumour growth can occur.
The process of initial mutation and attempts to develop into a tumour happen continuously. Most of these attempts do not result in tumours as there are efficient defence systems including DNA repair, senescence and cell death [16]. As the cumulative total number of stem cell divisions increases, the cumulative total number of initial mutation events increases. Defence systems become less effective as the individual ages, so as time goes by, the probability increases that an initial event will occur and lead to a tumour, and this process results in overall incidence per individual lifetime of c.40%.
Categorizing Modifying Factors
The hallmarks of cancer put forward by Hanahan and Weinberg [4–17] listed characteristics that cancers exhibit, which are listed in Table 1.
Table 1.
Comparison of the categories of mechanism of carcinogenesis from Wolf et al. [15], the KCs of human carcinogens from Smith et al. [19] and the hallmarks of cancer from Hanahan and Weinberg [4–17]
| Mode of action | Key characteristic of human carcinogens | Precursors of hallmarks of cancer | Key rate affected |
|---|---|---|---|
| Direct interaction with DNA or DNA repair | Is electrophilic or can be metabolically activated Is genotoxic Alters DNA repair or causes genomic instability Induces epigenetic alterations Causes immortalization |
Mutation DNA damage DNA repair |
Rate of mutation per cell division to produce cancer-associated mutation (MR) |
| Receptor-mediated increase in cell division | Modulates receptor-mediated effects Alters cell proliferation, cell death or nutrient supply |
Cell cycle Growth factors Downstream signalling Receptors |
Number of stem cell divisions (CD) Rate of cancer-capable cells progressing to become a cancer (PR) |
| Non-specific increase in cell division | Induces oxidative stress Induces chronic inflammation Alters cell proliferation, cell death or nutrient supply |
Necrosis Autophagy Apoptosis Inflammation Oxidative stress Angiogenic factors |
Number of stem cell divisions (CD) Rate of cancer-capable cells progressing to become a cancer (PR) |
| Modulating the tumour microenvironment | Alters nutrient supply Is immunosuppressive |
Glycolysis/Warburg effect Evading contact inhibition Deregulating checkpoints Immune response Immune suppression Immortalization Senescence |
Rate of cancer-capable cells progressing to become a cancer (PR) |
They also described complex signalling interactions in the tumour microenvironment during the process of tumour progression and metastasis.
Casey et al. [18] postulated mechanisms by which chemicals could affect the tumour microenvironment. However, the range of actions they identified is broad and covers effects that would result in other non-cancer adverse effects such as inflammation and tissue damage. It would be better to rephrase this range of actions as potential mechanisms by which chemicals can modify cancer risk, rather than implying that these are properties that distinguish carcinogenic from non-carcinogenic substances.
Smith et al. [19] were inspired by the concept of the Hallmarks of Cancer to put forward the concept of the key characteristics of carcinogens (KCs) shown in Table 1. They analysed the properties shown by chemicals that have been classified as carcinogens and determined that they show one or more of 10 key characteristics. However, both they and Guyton et al. [20] did not analyse the incidence of one or more KCs in chemicals that did not induce tumours, but inspection of the list shows that some of the so-called key characteristics are shared by chemicals that are not considered to be ‘carcinogens’. For instance, ‘modulates receptor-mediated effects’ would apply to nearly all pharmaceutical compounds; ‘induces chronic inflammation’ is a common result of non-specific cytotoxicity. The characteristics of other adverse outcomes have been postulated in a similar fashion and a number of common themes have emerged. For instance, ‘induces oxidative stress’, ‘is genotoxic’ and ‘induces epigenetic alterations’ appear as characteristics of both male [21] and female [22] reproductive toxicity (as well as characteristics of carcinogenicity).
Wolf et al. [15] postulated a unifying theory of chemical carcinogenesis that outlined three broad mechanisms by which a chemical could modify cancer risk:
Direct action with DNA or DNA repair
Receptor-mediated increase in cell division
Non-specific increase in cell division
These three modes of action are focused on the early stages of carcinogenesis and the key characteristics can be fitted into these broad headings, whereas the hallmarks of cancer also cover the later stages of tumour progression and metastasis. The tumour microenvironment, by definition, focuses on the later stages. A fourth broad mode of action could be added to the Wolf et al. [15] scheme to encompass the later stages: modulating the tumour microenvironment. Table 1 summarizes these concepts.
The question arises whether the four modes of action, the key characteristics and the hallmarks of cancer could or should be used to improve the assessment of cancer risk and to provide answers to the difficult questions posed to risk assessors. Care must be taken to avoid the trap of the ‘carcinogen/non-carcinogen’ mindset. Postulating a mechanism by which cancer risk may be increased and then finding that a chemical activates this mechanism does not make the chemical a ‘carcinogen’. Similarly, postulating a mechanism by which cancer risk may be increased and then citing a chemical that activates this mechanism that is ‘known to be a carcinogen’ does not prove that all ‘carcinogens’ activate this mechanism.
A Proposed Dynamic Cancer Risk Model
The concept of modification of cancer risk shows up the limitations of the simple paradigm of ‘carcinogen yes/no; genotoxin yes/no; avoid all exposure/lifetime exposure limit’, but at first sight, it seems to add nothing but complexity without obvious advantage. There appears to be a long list of potential factors that could influence the process of tumour formation; how could they be addressed in ways that are both scientifically valid and manageable?
One approach is to adapt the concept of the adverse outcome pathway or AOP. An AOP is defined by the OECD [23] as an analytical construct that describes a sequential chain of causally linked events at different levels of biological organization that lead to an adverse health or ecotoxicological effect. The process of cancer formation can be considered as an AOP.
The process starts with stem cell division:
Each stem cell division has a probability of a mutation occurring.
A cell needs to accumulate more than one mutation in the same or in subsequent cell divisions to start the process of losing cell division control.
DNA repair mechanisms act to correct a proportion of these mutations.
Some mutated cells are not viable and die.
Other mutated cells are recognized as abnormal and are destroyed.
The portion of mutated cells that survive, then proliferate and enter the transformation process, which can lead ultimately to a cancer.
Most of these will fail to become a clinically detectable neoplasm but, in spite of the high attrition rate at each stage, in c.40% of humans, at least one mutated cell clone will survive all the stages to become a cancer [1]. This dynamic risk model is outlined in Fig. 1.
Figure 1.

The basic AOP based Dynamic Cancer Risk model showing stages in the process where chemicals could act to modify cancer risk.
The model highlights the points in the process that could be affected by chemicals to modify the cancer risk. First, chemicals can increase the number of stem cell divisions thereby increasing the number of divisions with a mutation even if the mutation rate remains constant. Chemicals can interact directly with DNA to increase the probability of a mutation during a stem cell division, thus increasing the mutation rate. They can inhibit DNA repair mechanisms thus increasing the probability that a mutation will survive. They can also inhibit mechanisms that cause the death of abnormal cells and increase the probability of their survival. They can cause increased rate of cell division thereby increasing the number of mutated daughter cells with the potential to transform into precancerous cells. They can interfere with the complex signalling in the cell microenvironment and increase the probability that the altered cells become clinically evident neoplasms.
It is important to bear in mind that the process is continuous, not just when a chemical of interest is present. There is a probability of the initial mutation, and a probability for each subsequent stage and probabilities at each stage accumulate. The model can become the basis of a framework for assessing the effect of chemicals upon the process, and thus for assessing the modification of cancer risk.
It may be helpful to think of the process as a series of cohorts as shown in Fig. 2. Increasing the number of cells undergoing division and/or exposing those cells to agents that can promote mutations will result in a larger cohort of mutated cells. The cohort of cells then enters the multistage attrition process. Altering the survival rate at any stage in the process will modify the overall risk of cancer.
Figure 2.

Diagrammatic representation of cohorts of number of cells with the potential to become cancer declining with a half-life of 10 years.
Some insight into the length of the attritional process might be deduced by looking at the decline in the risk of cancer from a factor that is present for a time period and is then withdrawn. This will give an indication of how long it takes for the effect of the modification of risk to cease. Lung cancer risk after cessation of smoking has been extensively studied [24] and the risk declines exponentially with a half-life of 10 years. This is illustrated in Fig. 2, which shows the number of cells with cancer potential building up and then declining with a 10-year half-life in subsequent periods. This assumption would not apply if the material modifying the cancer risk itself had a long half-life and so can exert its effect for a long period, for instance asbestos fibres that are not eliminated [25].
Using the data from Tomasetti and Vogelstein [6] who correlated the number of stem cell divisions with the incidence of cancer in 21 organs, there is estimated be a total of 1013 stem cell divisions in a human lifetime. Jackson and Loeb [26] estimate that the spontaneous mutation rate for somatic cells is 2 × 10−7 per gene per cell division. They state there are 5 × 104 genes in the genome and that for 100 of them, mutations are recognized as possible driver mutations. Furthermore, two to eight of these genes must be mutated in the same cell for the cell to have the potential to transform into a cancer [27].
It is therefore possible to speculate about the number of cells produced per lifetime with the potential to become a cancer. The probability that there will be at least one gene associated with cancer mutating for each cell division is:
Mutation rate per gene per cell x number of cancer genes = 2 × 10−7 × 102 = 2 × 10−5.
Which when multiplied by the assumed number of stem cell divisions in a lifetime gives the number of cells with one ‘cancer gene’ mutation as: 2 × 10−5 × 1013 = 2 × 108.
Cells need more than one relevant mutation to have the potential to transform, the additional mutations occurring during subsequent cell divisions [28]. Only in c40% of lifetimes [1] will one of these 2 × 108 cells lead to a cancer.
There are three key rates involved in the process that can be modified as follows:
Number of stem cell divisions per time period (CD).
Rate of mutation per cell division to produce cancer-associated mutation (MR).
Rate of cancer-capable cells progressing to become a cancer (PR).
Together, they determine the probability of an individual being diagnosed with cancer in his or her lifetime. It is the composite changes to these rates that modify the risk of clinically detectable cancer.
Very broadly these rates can be estimated for humans.
Dividing the total number of stem cell divisions in a lifetime by the number of years in a lifetime suggests a rate of number of stem cell divisions per time period (CD) = 1011/year.
Taking into account the mutation rate per gene per division and the number of genes associated with cancer suggests a rate of the mutation rate per cell division to produce cancer-associated mutation (MR) = 1 in 105.
These rates can be used to calculate the total number of capable cells progressing to become a cancer and that progression to cancer occurs in only 40% of lifetimes, the progression rate of cancer-capable cells (PR) = 1 in 108.
Potential of Chemicals to Modify Cancer Risk
Chemicals have the potential to modify the number of stem cell divisions, rate of mutation and the balance of cell death and survival. The greater the potency of a chemical to change these rates, the greater will be its impact on the modification of cancer risk. Long-term rodent bioassays can be considered to be an assessment of the potential of the chemical to modify the overall rate, but it is not possible to know which of the rates has been modified. However, understanding the mode of action, even in the broad categories proposed by Wolf et al. [15], can allow sensible deductions to be made as to which rate or rates are being modified.
This model provides ways to address the difficult risk assessor’s dilemma, which was posed earlier.
What are the effects of short-term or intermittent exposure?
Figure 3 shows a representation of different time periods of exposure. The first diagram (A) shows the time period in a human life of 70 years, which is equivalent to the duration of exposure in a rat’s life for a long-term rodent bioassay. It shows that a chemical dosed over this time period will have the opportunity to modify the key rates for a large number of cohorts of cells going through the process. The other diagrams show one (B), two (C) or three (C) shorter time periods and indicate that the number of cohorts of cells they can modify is reduced. This will lead to a reduction in the probability of a cancer developing.
Figure 3.
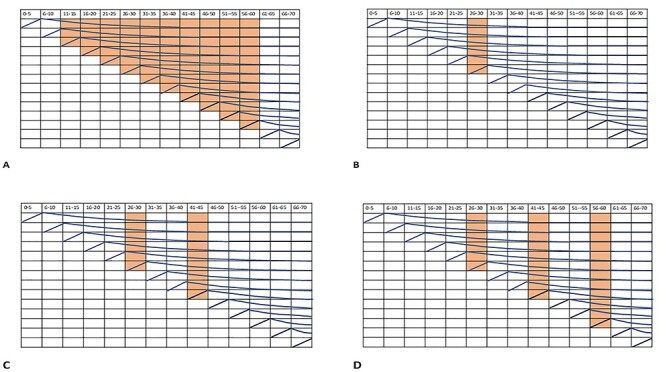
Diagrammatic representation of different exposure time periods and the cohorts of cancer potential cells they can influence. A. shows the equivalent in a human lifetime of the duration of exposure of a long term rodent bioassay. B. C. and D. show the reduced cohorts of cells affected by one, two or three periods of exposures.
The modification of the cancer risk will be proportional to the duration of the exposure; the longer the exposure, the larger the number of cohorts of cells going through the process towards the formation of a cancer. The process depends on accumulating probabilities; the longer the effect lasts, the greater the accumulated probability. The exact relationship between duration of exposure and the modification of cancer risk will be difficult to determine, but a Haber’s rule approach can be considered. Simply put Haber’s rule states that for any given effect dose x time will be constant; if the dose is doubled and the time halved, the result will be the same. This assumes a linear dose response curve, and the rule can be modified to be ‘effective dose’ x time will be constant; if the dose is increased until the magnitude of the effect is doubled and the time halved, the result will be the same. This provides further support for the Haber’s rules–based framework proposed [29] to assess the carcinogenic risk of less than lifetime exposures. Kinetic factors need to be considered when applying Haber’s rule, with long half-life extending the time period over which a modification to cancer risk would take place.
What are the effects of exposure to more than one chemical either at the same time or at different times?
Two different chemicals may act on the same part of the carcinogenic pathway and modify the same key rate, or they may act on different parts of the pathway and modify different key rates. Exposure to two chemicals at the same time may cause the key rates to be modified at the same time to impact on the same cohort of cells. The overall effect on cancer risk modification would be additive, and it would have the same effect as an increased dose of one chemical. The effect of exposure to two chemicals at different times depends on whether the same cohort of cells is going through the pathway when the second modifying factor is present. If the two exposures are far enough apart in time so that no cells from the previous modified cohort are still vulnerable, then the effects of the two exposures will be independent.
Figure 4 illustrates this concept with a second exposure occurring directly after the first exposure or the second exposure occurring after an interval of 15 years. The number of cells affected by the first exposure is reduced.
Figure 4.

Diagrammatic representation of exposure to two different chemicals. A. shows the second exposure occurring directly after the first exposure. B. shows the second exposure after an interval of 15 years. The number of cells affected by the first exposure is reduced.
However, given the assumed half-life of 10 years, there will be potential neoplastic cells in the pathway from cohorts that were exposed to the first chemical for many years so that true independence is unlikely but the probability will decline as time goes by as shown in Table 2.
Table 2.
Percentage of cancer-potential cells remaining after increasing delays between exposures assuming 10-year half-life
| Delay between exposures (years) | Assumed % of cancer-potential cells remaining |
|---|---|
| 10 | 50% |
| 20 | 25% |
| 30 | 12.5% |
| 40 | 6.25% |
| 50 | 3.125% |
These considerations provide a framework in which to place the concept of ‘initiators’ and ‘promoters’: the initiator acts on a cohort of cells to increase the number of cells with cancer potential and the promoter acts on that cohort later in its pathway to decrease the rate of attrition and so increase the cancer risk.
How can the risk of cancer be assessed if there are no cancer epidemiology studies or long-term bioassays available?
Current cancer risk assessment starts with a consideration of whether a chemical is a carcinogen or not with the decision being taken on the basis of human epidemiology or long-term rodent bioassay. This limits the number of chemicals that can be considered. Information from in vivo, ex vivo and in vitro studies can indicate effects on processes that can modify cancer risk and these can be taken into account when considering the overall impact of the chemical on human health.
The dynamic cancer risk model provides the basis for a framework in which this information can be considered in a structured way built around a series of questions such as the following:
Is there evidence of an effect of the chemical that could modify one or more of the critical rates determining cancer risk?
Is there information about dose response, either direct experimental evidence or implied from mode of action evidence?
What is the dose and duration of the exposure to the chemical (or chemicals) in the situation being assessed?
Are there other factors that could modify the key rates that need to be considered?
The information can then be used to derive a health-based guidance value (HBGV). This can be done by estimating a point of departure (PoD) for the modifying factor, using in vitro to in vivo extrapolation (IVIVE) pharmacokinetic modelling if the evidence is from in vitro studies. The PoD can then be divided by uncertainty factors to derive an HBGV. Much cancer risk modification results from events such as inflammation or hormonal activity, which themselves can lead to adverse effects other than cancer, but which may also lead to changes in the rate of cell division or cell signalling that could affect the rate of progression of potentially cancerous cells. The HBGV would be set to provide protection against all the adverse effects, both cancer and non-cancer, and it could be considered to be an HBGV for ill health including cancer rather than a cancer-specific HBGV.
Age-related cancer sensitivity
The incidence of cancer increases with age. The incidence per 100 000 of cancer diagnosis in 5-year age periods is shown in Fig. 5 (data from English cancer statistics [30]).
Figure 5.

Diagnosis of cancer per 100,000 by 5-year age ranges in UK data from English cancer statistics, ONS 2016.
There are several possible explanations for this age-related increase in the incidence rate. Clearly, the development of a cancer takes time and many cancers diagnosed within a five-year period would likely have been initiated in an earlier period; thus, the incidence of cancer will inevitably increase as time passes. However, it would be expected that the rate of diagnosis would plateau after 5 half-lives [31], which would be 50 years based on the half-life derived from the cessation of smoking studies [24], but diagnostic rates continue to increase up to the late 1980s so there are likely to be other factors in play. From the model we propose, the key rates are number of stem cell divisions, the mutation and mutation repair rate, and the survival rate of mutated cells to become cancers. The number of stem cell divisions will decrease once adulthood is reached so that is unlikely to be a major factor, but the other key rates are known to increase as part of the ageing process. Lopez Otin et al. [32] have identified what they called the ‘key hallmarks of ageing’: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication. These are remarkably similar to the hallmarks of cancer and will impact the key rates of mutation, mutation repair and mutated cell survival to become a cancer and offer an explanation for the increase in cancer diagnosis rate with age. This leaves open the likelihood that the sensitivity to cancer risk modifiers increases with age.
Conclusions
The development of the hallmarks of cancer by Hanahan and Weinberg [4–17] stimulated thinking about the influence of chemicals on the process of carcinogenesis, which has questioned the established concept of chemicals being either carcinogens or non-carcinogens. Tomasetti and Vogelstein [5] put forward the hypothesis that 60% of cancer was caused by spontaneous mutations or as they put it ‘bad luck’ and as such would not be preventable, with 40% being preventable. Wu et al. [6] accepted the principle but questioned the proportion that would be preventable, suggesting that it should be higher. They also segregated out the factors that could modify cancer risk:
Intrinsic factors: Random errors in DNA replication (unmodifiable)
Non-intrinsic factors: Endogenous—Biologic aging, genetic susceptibility, DNA repair machinery, hormones, growth factors and inflammation (partially modifiable)
Non-intrinsic factors: Exogenous—Radiation, chemical carcinogens, tumour-causing viruses and bad lifestyles such as smoking, lack of exercise and nutrient imbalance (modifiable)
Chemicals would be considered to be exogenous non-intrinsic factors using the Wu et al. [6] scheme. Wolf et al. [15] developed the Tomasetti and Vogelstein concept to postulate three modes of action by which chemicals could modify cancer risk:
Direct action with DNA or DNA repair
Receptor-mediated increase in cell division
Non-specific increase in cell division
Smith et al. [19] adapted the Hallmarks of Cancer to try to determine the key characteristics of carcinogens, which seem to cover a wide range of characteristics including those indicative of general toxicity. Casey et al. [18] postulated that influencing the tumour microenvironment will have an impact on whether cells bearing mutations transform and develop into clinically detectable neoplasms.
In this paper, we have put these concepts together and suggested a dynamic cancer risk model (Fig. 1) that describes how mutations arising from stem cell divisions survive, transform and cause development of neoplasms. The process is happening continually and results in a clinically detectable cancer in almost half of all human lifetimes. The model identifies the key points in the process where chemicals can modify cancer risk and there are three key rates that control the process:
Number of stem cell divisions
Rate of mutation per cell division to produce cancer-associated mutation
Rate at which abnormal cells progress to detectable neoplasms
Together, they determine the probability of an individual being diagnosed with cancer in his or her lifetime.
Chemicals can modify cancer risk if they can change any of the key rates and there are many ways in which these rates can be modified including increasing cell division, mutagenicity and/or interaction with cell signalling. The range of potential modification points is consistent with the observation of so many chemicals and other factors such as lifestyle changing the incidence of cancer.
This concept will allow the development of strategies for protecting human health, which do not rely solely upon the identification of ‘carcinogens’ from cancer epidemiology and long-term rodent bioassays. The model can provide a framework within which strands of evidence such as non-cancer epidemiology, general toxicology, in vivo and in vitro laboratory studies can be put together to assess the modification of cancer risk. It should be possible to develop models that can quantify the modification of cancer risk, but this will require more detailed, data-rich studies that relate the changes in the key rates to cancer outcomes as reported by Greenfield et al. [33]. However, in the meantime, it should be possible to develop semi-quantitative estimates of level of concern based on margin of exposure from effects that would modify one or more key rates.
Conflicts of Interest
D.J.H. and J.E.D. are members of the UK Government Committee on Carcinogenicity, but the opinions expressed are their own.
Contributor Information
David J Harrison, School of Medicine, University of St Andrews, North Haugh, St Andrews KY16 9TF, UK.
John E Doe, School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Byrom Street, Liverpool L3 3AF, UK.
References
- 1.Sasieni PD, Shelton J, Ormiston-Smith N et al. What is the lifetime risk of developing cancer? The effect of adjusting for multiple primaries. Br J Cancer 2011;105:460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control (CDC) , Mortality in the United States, 2018. NCHS Data Brief No 355 January 2020
- 3.Roser M and Ritchie H (2019) Cancer: Our World in Data Published online at OurWorldInData.org. Retrieved from: https://ourworldindata.org/cancer
- 4.Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 5.Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015;347:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu S, Zhu W, Thompson P, Hannun YA. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat Commun 2018;9:3490. 10.1038/s41467-018-05467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ames BN, Gold LS. Chemical carcinogenesis: too many rodent carcinogens. Proc Natl Acad Sci 1990b;87:7772–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boobis AR, Cohen SM, Dellarco VL et al. Classification schemes for carcinogenicity based on hazard-identification have become outmoded and serve neither science nor society. Regul Toxicol Pharmacol 2016;82:158–66. 10.1016/j.yrtph.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Doe JE, Boobis AR, Dellarco V et al. Chemical carcinogenicity revisited 2: modern knowledge of carcinogenesis shows that carcinogen or non-carcinogen categorization is not scientifically credible. Regul Toxicol Pharmacol 2019;103:124–9. 10.1016/j.yrtph.2019.01.024. [DOI] [PubMed] [Google Scholar]
- 10.Crump KS, Krewski D, Van Landingham C. Estimates of the proportion of chemicals that were carcinogenic or anticarcinogenic in bioassays conducted by the national toxicology program. Environ Health Perspect 1999;107:83–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaylor D. Are tumor incidence rates from chronic bioassays telling us what we need to know about carcinogens? Regul Toxicol Pharmacol 2005;41:128–33. [DOI] [PubMed] [Google Scholar]
- 12.Braakhuis H, Slob W, Olthof E et al. Is current risk assessment of non-genotoxic carcinogens protective? Crit Rev Toxicol 2018;48:500–11. 10.1080/10408444.2018.1458818. [DOI] [PubMed] [Google Scholar]
- 13.IARC (2020) International Agency for Research on Cancer list of classifications. Lyon, France: World Health Organisation, https://monographs.iarc.fr/list-of-classifications.
- 14.CoC . A guidance statement from the committee on carcinogenicity of chemicals in food, consumer products and the environment (COC)—a strategy for the risk assessment of chemical carcinogens COC/G1. . England, UK: Public Health, 2012, Version 4 (2012).
- 15.Wolf DC, Cohen SM, Boobis AR et al. Chemical carcinogenicity revisited 1: A unified theory of carcinogenicity based on modern knowledge. Regul Toxicol Pharmacol 2019;103:86–92. 10.1016/j.yrtph.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2007;481:287–94. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 18.Casey SC, Vaccari M, Al-Mulla F et al. The effect of environmental chemicals on the tumour microenvironment. Carcinogenesis 2015;36:S160–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MT, Guyton KZ, Gibbons CF et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ Health Perspect 2016;124:713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guyton K, Rusyn I, Chiu W et al. Application of the key characteristics of carcinogens in cancer hazard identification. Carcinogenesis 2018;39:614–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arzuaga X, Smith MT, Gibbons CF et al. Proposed key characteristics of male reproductive toxicants as an approach for organizing and evaluating mechanistic evidence in human health hazard assessments. Environ Health Perspect 2019;127:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luderer U, Eskenazi B, Hauser R et al. Proposed key characteristics of female reproductive toxicants as an approach for organizing and evaluating mechanistic data in hazard assessment. Environ Health Perspect 2019;127:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.OECD (2016) Environment, Health and Safety Publications Series on Testing and Assessment No. 233 USERS’ HANDBOOK SUPPLEMENT TO THE GUIDANCE DOCUMENT FOR DEVELOPING AND ASSESSING AOPs. Paris, France: Organisation for Economic Co-operation and Development, https://www.oecd-ilibrary.org/docserver/5jlv1m9d1g32-en.pdf?expires=1605799693&id=id&accname=guest&checksum=7F0F98C3AF19EB34F0A07A87D63F0076.
- 24.Fry J, Lee P, Forey B, Coombs J. How rapidly does the excess risk of lung cancer decline following quitting smoking? A quantitative review using the negative exponential model. Regul Toxicol Pharmacol 2013;67:13–26. [DOI] [PubMed] [Google Scholar]
- 25.Feder I, Tischoff I, Theile A et al. The asbestos fibre burden in human lungs: new insights into the chrysotile debate. Eur Respir J, 2017;49:1602534. 10.1183/13993003.02534-2016. [DOI] [PubMed] [Google Scholar]
- 26.Jackson AL, Loeb LA. The mutation rate and cancer. Genetics 1998;148:1483–90 PMID: 9560368; PMCID: PMC1460096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anandakrishnan R, Varghese RT, Kinney NA et al. Estimating the number of genetic mutations (hits) required for carcinogenesis based on the distribution of somatic mutations. PLoS Comput Biol 2019;15:e1006881. 10.1371/journal.pcbi.1006881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomasetti C, Marchionni L, Nowak M et al. Only three driver gene mutations are required for the development of lung and colorectal cancers. PNAS 2015;112:118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felter S, Conolly R, Bercu J et al. A proposed framework for assessing risk from less-than lifetime exposures to carcinogens. Crit Rev Toxicol 2011, 2011;41:507–44. [DOI] [PubMed] [Google Scholar]
- 30.ONS Cancer Registration Statistics, England , 2017. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/datasets/cancerregistrationstatisticscancerregistrationstatisticsengland
- 31.Gupta PK. Fundamentals of Toxicology: Essential Concepts and Applications. Amsterdam, NL: Academic Press, 2016, ISBN 978-0-12-805426-0. [Google Scholar]
- 32.López-Otín C, Blasco MA, Partridge L et al. The hallmarks of aging. Cell 2013;153:1194–217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenfield R, Ellwein LD, Cohen S. A general probabilistic model of carcinogenesis: analysis of experimental urinary bladder cancer. Carcinogensis 1984;5:437–45. [DOI] [PubMed] [Google Scholar]