

Summary
The protocol allows for labeling nascent RNA without isolating nuclei. The cell-permeable uridine analog, 5-ethynyluridine (EU), is added to media to allow in vivo labeling of nascent transcripts. Cells are lysed, total RNA is collected, and biotin is conjugated to EU-labeled RNAs. Custom biotin RNAs are added and biotinylated RNAs are isolated for generation of cDNA libraries. The sequencing data are normalized to controls for quantitative assessment of the nascent transcriptome. The protocol takes 4 days, not including sequencing and analysis.
For complete details on the use and execution of this protocol, please refer to Palozola et al. (2017).
Subject areas: Sequencing, RNAseq, Molecular Biology, Gene Expression
Graphical abstract
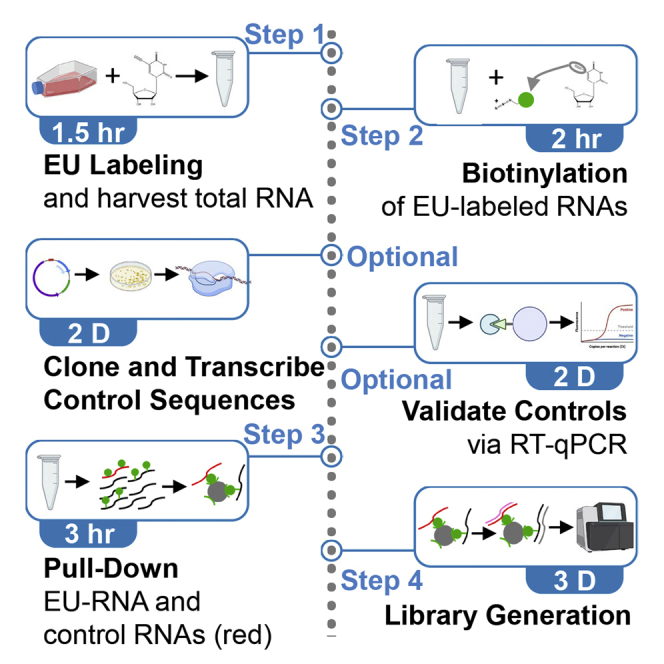
Highlights
-
•
Labeling nascent RNAs with EU does not require nuclear isolation
-
•
EU-RNA-Seq maps nascent transcripts across a large dynamic range
-
•
Control biotin RNAs are customizable
-
•
Custom biotin RNAs allow for global normalization following sequencing
The protocol allows for labeling nascent RNA without isolating nuclei. The cell-permeable uridine analog, 5-ethynyluridine (EU), is added to media to allow in vivo labeling of nascent transcripts. Cells are lysed, total RNA is collected, and biotin is conjugated to EU-labeled RNAs. Custom biotin RNAs are added and biotinylated RNAs are isolated for generation of cDNA libraries. The sequencing data are normalized to controls for quantitative assessment of the nascent transcriptome. The protocol takes 4 days, not including sequencing and analysis.
Before you begin
Commonly used approaches for mapping nascent transcription include Global run-on sequencing (GRO-seq, Core et al., 2008), Precision nuclear run-on (PRO-Seq, Mahat et al., 2016), and native elongating transcript sequencing (NET-seq, Mayer and Churchman, 2016). In GRO-seq, nascent RNAs are metabolically labeled with bromouridine (BrU). However, since BrU is not cell-permeable, this approach requires cell lysis and nuclear isolation prior to the addition of Bru thus making it an in vitro labeling approach. In contrast, both PRO- and NET-seq rely on the detection of RNA Pol II binding rather than nucleotide incorporation. In the NET-seq protocol, nascent transcripts are isolated through their highly stable DNA-RNA-RNA Pol II interaction via antibody pull-down. This methodology relies on the physical isolation of RNA Pol II and thus is not suitable for regions where RNA Pol II levels are low, such as in mitotic chromatin.
Regardless of the state of the nuclear envelope, lysis and nuclear isolation processes may significantly affect the nascent transcriptome and so is not the most suitable choice. To circumvent these issues, others have simply isolated chromatin-associated RNAs and depleted them of poly-adenylated RNAs in an attempt to sequence only nascent RNAs (Liang et al., 2015). This approach, however, does not offer the time resolution of a pulse-label and so the age of the resulting transcripts is not known. TT-seq (Schwalb et al., 2016) circumvents the issue of cell permeability since it uses the cell-permeable 4sU for labeling. However, since 4sU only labels the 3′ end of nascent RNAs, TT-seq requires RNA fragmentation prior to isolation. It is thus suitable for capturing only the most transient of transcripts.
EU-RNA can be conjugated to biotin fluorophores for visualization of nascent transcriptional events or can be conjugated to biotin and isolated with streptavidin for sequencing. EU-RNA-seq maps across exon and intron sequences, exhibits strand specificity, can be globally normalized to artificial spike-in RNAs, and covers a large dynamic range of nascent transcripts. The approach was developed and has primarily been used to quantify residual transcription in mitotic cells (Palozola et al., 2017; Perea-Resa et al., 2020; Kang et al., 2020), when prior literature had assumed that the vast majority of genes are silent during division. The sensitivity of EU-RNA-seq also allowed the detection of waves of transcription reactivation during mitotic exit (Palozola et al., 2017), a process that has previously been reported as simple on/off transcriptional state change (Hsiung et al., 2016). It will be of interest to compare EU-RNA-seq and methods requiring nuclear isolation in interphase cells to determine the effect of nuclear isolation on the nascent transcriptome. The protocol has been used in cancer cell lines (Yokoyama et al., 2016) to determine the effects of small molecules, as well as to study early transcriptional events in Xenopus embryos (Chen et al., 2019).
Reagent setup
-
1.Growth Media (cell culture specific)
-
a.Can be prepared in advance and stored at 4°C, protected from light.
-
b.Heat to 37°C immediately before use.
-
a.
| Reagent | Final concentration | Volume |
|---|---|---|
| High Glucose DMEM | n/a | 450 mL |
| FBS | 10% | 50 mL |
| Pen/Strep | 1% | 5 mL |
-
2.EU Stock Solution
-
a.Can be prepared in advance and stored at −20°C, protected from light, for several months.
-
b.Mix vigorously by vortexing.
-
c.Thaw on ice immediately prior to use.
-
a.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5-Ethynyluridine | 100 mM | 5 mg |
| H2O | n/a | 186 uL |
Generation of candidate control Biotin-RNAs
Timing: 2 days
Note: The described procedure is for generating the controls used in Palozola et al., 2017. However, experiment-specific considerations should be taken for control choice. Specifically, be sure to choose sequences that will not be transcribed in your samples. In particular, it helps to choose sequences from another species. Once chosen, the control sequences must be tested for their ability to be pulled down from EU-RNA containing samples proportionally and amplified linearly during library generation and qPCR (see relevant Troubleshooting sections below).
| Control | Genomic location | Sequence |
|---|---|---|
| control #1 | hg18 chr7:110045645-110046034 hg19 chr7:110258409-110258798 |
TACCTTCCCTTCTACCTCATCTTTCTTCTG CCTCCATCTTCAACAGCTTCTCTCTAACT TTTCTGTTTTCCTGCTTTGCTTTTAAGG GTTTGTGTGATTACATTAAATATTGCCC CCTCCCCTAGAAACATTCAGGTTCCTT ATCTAATTTAAGGTCAGAATTATTAAA CTTAATTCTGTCTCCAAAGTCCCTTC |
| control #2 | hg18 chr13:105896287-105896586 hg19 chr13:107098286-107098585 |
GTGTATTACAAAAAGCTACAAATGATAAAA ACAAGTAGTATTGTTTTTTAATCAATGAGTT ACATTTTACAGATTTCAGTATACAGAAAAA TTGGGCAGAGTATACAGACAGTCCCATAT GTCACCCCCCATACACATCTCCCTTGTTG TCATCTCACTACATTTGTTACAACCGA TGAATCAATATTGACAGATTATGA |
-
3.Plasmid Linearization
-
a.Digest ~25 ug of each plasmid.
Reagent Final concentration Volume 10× CutSmart Buffer 1× 5 uL HindIII-HF 40 U 2 uL control sequence plasmid∗ 25 ug to 25 ug H2O n/a to 50 uL ∗any sequence that is not expressed in the experimental system -
b.Incubate at 37°C for 4 h.
-
c.Confirm linearization.
-
i.Remove 2 uL (4%) of each reaction, add 8 uL H2O, 2 uL 6× loading buffer, and run on a 0.7% agarose gel at 90 V for 1 h.
-
i.
-
d.Once linearization is confirmed, clean up the remaining reaction with Qiagen PCR purification columns and elute in 20 uL EB.
-
a.
-
4.In Vitro Transcription with Biotin-UTP
-
a.Use MaxiScript kit to make RNA from 1 ug of each linearized control template.
-
i.Thaw frozen reagents on ice. All reagents should be microfuged briefly before opening to prevent loss.
-
ii.Place the RNA Polymerase on ice.
-
iii.Vortex the 10× Transcription Buffer and ribonucleotide solutions until they are completely in solution.
-
iv.Once they are thawed, store the ribonucleotides on ice, but keep the 10× Transcription Buffer at 20°C–22°C.All reagents should be microfuged briefly before opening to prevent loss.
 CRITICAL: Assemble the transcription reaction at 20°C–22°C.
CRITICAL: Assemble the transcription reaction at 20°C–22°C. -
v.The spermidine in the 10× Transcription Buffer can coprecipitate the template DNA if the reaction is assembled on ice.
-
vi.Add components in order.
Reagent Final concentration Volume H20 n/a to 20 uL linearized plasmid 1 ug to 1 ug 10× Transcription Buffer 1 2 uL 10 mM ATP 1 uL 10 mM CTP 1 uL 10 mM GTP 1 uL 10 mM UTP 0.6 uL bio-11-UTP 0.4 uL T7 enzyme 1× 2 uL -
vii.Mix thoroughly. Gently flick the tube then centrifuge brifly to collect.
-
viii.Incubate 1 h at 37°C.
-
ix.Add 3 uL TURBO DNase, mix well, and incubate at 37°C for 15 min.
-
x.Add 3 uL of 0.5 M EDTA. EDTA blocks the head-induced RNA degradation that can occur in Transcription Buffer.
-
xi.Optional step: Troubleshooting Problem 1.
-
i.
-
b.Removal of free nucleotides
-
i.Add 340.6 uL RNase-free water for a total volume of 400 uL.
-
ii.Add 400 uL phenol chloroform isoamyl alcohol and vortex to mix.
-
iii.Spin max speed 5 min.
-
iv.Transfer upper phase to a fresh tube and repeat steps i to iv.
-
v.Adjust volume to 350 uL with NF water.
-
vi.Add 16 uL 5 M NaCl and 1.5 uL glycogen.
-
vii.Add 375 uL (1 volume) −20°C pre-chilled 100% EtOH.
-
viii.Incubate at −20°C 12–20 h.
-
ix.Spin at 20,000 × g for 10 min at 4°C and discard supernatant.
-
x.Wash with 500 uL −20°C pre-chilled 80% EtOH, spin as above, and remove supernatant.
-
xi.Air dry pellet and resuspend in 20 uL NF water.
-
i.
-
c.Confirm on Gel
-
i.Combine 2 uL sample (10%) and 2 uL gel loading buffer (from MaxiScript kit)
-
ii.Incubate at 95°C for 2 min and immediately transfer to ice.
-
iii.run on a 2% agarose gel at 100 V for 1 h.(1) Suggestion: do not include EtBr in agarose. Instead, stain and destain the gel after running.
-
i.
-
d.Quantify RNA with Nanodrop or Qubit.
-
i.expected concentration: ~500 ng/uL
-
i.
-
a.
Culture cells
Note: Considerations must be taken for the system in use. Here, we refer to the HUH7 cells used for the original development of the protocol in Palozola et al., 2017. As in the original protocol, each replicate consists of one 10-cm plate. Adjustments will need to be made for cells grown in suspension.
-
5.Culture HUH7 cell on 10-cm plates to 80% confluency in 5 mL Growth Media.
-
a.One plate per condition per replicate
-
b.One plate per control per replicate
-
i.Include a NoEU control for each condition to account for background pulldown in Step 26.
-
i.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| High glucose DMEM | Thermo Fisher | 10566-016 |
| FBS | HyClone | 7195 |
| Pen/Strep | Thermo Fisher | 15140122 |
| PBS | Invitrogen | 1113 |
| TRIzol Reagent | Ambion | 15596026 |
| miRNAeasy Kit | QIAGEN | 217004 |
| Glycogen | Sigma-Aldrich | 10901393001 |
| Ammonium acetate | Sigma-Aldrich | 631-61-8 |
| Nuclease-free water | Invitrogen | 10977-015 |
| 10× CutSmart Buffer | New England Biolabs | B7204 |
| HindIII-HF | New England Biolabs | R3104 |
| PCR Purification Kit | QIAGEN | n/a |
| Critical commercial assays | ||
| miRNeasy Kit | QIAGEN | |
| Click-iT Nascent RNA Capture Kit | Invitrogen | C10365 |
| Biotin-UTP | Thermo Fisher | AM8450 |
| MAXIscript T7 In Vitro Transcription Kit | Thermo Fisher | AM1320 |
| RNaseOUT | Thermo Fisher | 10777019 |
| iScript | Bio-Rad | 1708890 |
| Agencourt RNAClean XP beads | Beckman Coulter | A66514 |
| Ovation Human FFPE RNA-Seq Kit | NuGEN | 0340-32 |
| Experimental models: cell lines | ||
| HUH7 cells | n/a | n/a |
| Oligonucleotides | ||
| oligo 5078: TTA GCT GAA GAC CCC TGC TC | IDT | n/a |
| oligo 5079: TGG AAA CCA GTG ATG GGA CT | IDT | n/a |
| oligo 5080: CCT GGG ACA GCT TCA GCT AC | IDT | n/a |
| oligo 5081: GGG AGG TCA ACT GCA TGA AT | IDT | n/a |
| oligo 5082: AGA CCC CTG CTC TTT CAA CA | IDT | n/a |
| oligo 5083: TGA GCA TGG ATT TTG TGA GAG AA | IDT | n/a |
| oligo 5084: ACC CTA CGC AGC ACA GTT TT | IDT | n/a |
| oligo 5089: TGC ACA TCC TTC TGT CTC TCT | IDT | n/a |
| Recombinant DNA | ||
| Plasmid 1408: control #1 | Zaret Lab | n/a |
| Plasmid 1409: control #2 | Zaret Lab | n/a |
| Other | ||
| 1.5 mL Nuclease-free microcentrifuge tubes | USA Scientific | 1615-5510 |
| 10 cm Plates | Greiner Bio-One | 664061 |
| 18 cm Cell lifters | GeneMate | T-2443-4 |
| Microcentrifuge | n/a | n/a |
| Vortex mixer | n/a | n/a |
| Magnetic stand for 1.5 mL tubes | n/a | n/a |
| Qubit/NanoDrop | n/a | n/a |
Step-by-step method details
EU labeling
EU-labeling allows for the incorporation of 5-ethynyluridine (EU) into nascent transcripts as they are being generated. For most cell lines, it takes approximately 30–40 min for EU to cross both the cell and nuclear membranes and begin to be incorporated at detectable rates.
-
1.Prepare fresh Labeling Media
-
a.Prepare 1 mL per 10-cm plate of cells.
-
b.Use pre-warmed Growth Media.
-
c.Invert the solution several times to thoroughly resuspend the EU.
-
a.
| Reagent | Final concentration | Volume |
|---|---|---|
| 5-Ethynyluridine (100 mM) | 3 mM | 30 uL |
| Growth Media | n/a | 970 uL |
-
2.Add 1 mL of pre-warmed Labeling Media to the 5 mL of Growth Media in which you’ve cultured your cells on a 10 cm plate to 80% confluency.
-
a.Swirl the plate gently to mix for a final concentration of 0.5 mM EU.
-
b.Incubate at 37°C for 40 min.
-
a.
Note: The incubation time in labeling media should be optimized for the cell line in use and should occur under normal culturing conditions. See Troubleshooting Problem 2.
Harvest total RNA, including EU-RNA
Total RNA is harvested from the cells. This includes all cellular RNA, including that which was labeled with EU during the 40-min labeling step.
-
3.Gently aspirate the Labeling Media from the cells.
-
a.Add 1 mL PBS to the side of the plate and gently swirl around to rinse the remaining media off of the cells.
-
b.Gently aspirate the PBS from the cells.
-
a.
-
4.Add 700 uL TRIzol reagent directly to the cells and use a cell lifter to spread around the TRIzol and dislodge the cells from the plate.
-
a.Transfer the cells in TRIzol to a 1.5 mL microcentrifuge tube
-
b.Vortex on maximum speed for 1 min.
-
a.
-
5.
Snap freeze in liquid nitrogen.
Pause point: Samples can be stored at −80°C.
-
6.
Purify RNA with the miRNeasy Kit, as per manufacturer’s protocol.
Note: Other RNA isolation kits may also be used, but we chose the miRNeasy kit to capture as many forms of nascent RNAs as possible.
-
7.
Quantify the total RNA recovered.
Biotinylation of cellular EU-RNA
This step conjugates biotin-azide to the EU in any EU-labeled RNAs present in the total RNA harvested from the cells. Be sure to perform this step on an unlabeled, “NoEU” control as well.
-
8.
Prepare 2.3 ug of total RNA in a final volume of 15.75 uL water.
-
9.
Perform the click reaction as per manufacturer’s protocol in 50 uL total volume.
-
10.
Proceed immediately to RNA precipitation.
RNA precipitation
This part of the protocol is largely from the Invitrogen Click-iT Nascent RNA Capture kit, with some small modifications. The purpose is to clean up the samples following the click reaction.
-
11.
Add 1 uL of glycogen, 50 uL of 7.5 M ammonium acetate, and 700 uL of chilled 100% EtOH, invert the tube to mix and incubate at −70°C for 12–20 h.
-
12.
Spin at 13,000 × g for 20 min at 4°C.
-
13.
Remove the supernatant from the tube without disturbing the pellet and add 700 uL of chilled 75% EtOH.
-
14.
Vortex briefly to mix and spin at 13,000 × g for 10 min at 4°C.
-
15.
Repeat the 75% EtOH wash.
-
16.
Let the pellet air dry at 20°C–22°C.
-
17.
Thoroughly resuspend the pellet in 10 uL water.
-
18.
Quantify RNA by Qubit.
Biotin-EU-RNA and biotin-spike-in pull-down
-
19.
Thaw sample RNA and biotin-control RNAs on ice.
-
20.
Add 5e-5 ug spike-in control #1 and 5e-4 spike-in control #2 to 1.7 ug of sample RNA in a total volume of 5 uL.
-
21.
Dispense 5 uL beads per sample into 1.5 mL microcentrifuge tubes.
-
22.Per sample:
-
a.Wash the beads 3× with 9 uL Wash Buffer 2.
-
b.Resuspend beads in 5 uL Wash Buffer 2.
-
c.Dilute RNaseOUT 1:10 then prepare Master Mix as follows per reaction.Master Mix
Reagent Volume Click-iT Buffer (G) 12.5 uL RNaseOUT 0.2 uL H20 2.3 uL -
d.Add 15 uL of master mix to each 10 uL sample.
-
a.
-
23.
Incubate on heat block at 69°C for 5 min.
-
24.Add 5 uL pre-washed beads to each reaction.
-
a.Pipet up and down to mix.
-
a.
-
25.Incubate at 24°C for 30 min with gentle agitation.
-
a.Suggestion: tap tubes every few minutes to keep beads in suspension.
-
a.
-
26.Place samples on a magnetic rack and wash the beads, as per manufacturer’s protocol.
-
a.5× with 50 uL wash buffer 1
-
b.5× with 50 uL wash buffer 2
-
c.Resuspend beads in 5 uL Wash Buffer 2.
-
a.
-
27.
Optional, but recommended: proceed to Troubleshooting Problems 3 and 4.
cDNA library generation
This section of the protocol is largely based on the manufacturer’s protocol, with some small modifications to account for using beads as the starting template.
-
28.Perform the library generation with NuGen’s Ovation Human FFPE RNA-Seq Kit, as per the manufacturer’s protocol, with the following adjustments:
-
a.Begin with the 5 uL of resuspended beads from step 26.
-
b.Briefly remove the sample from the PCR machine every 5 min during PCR programs 2 and 3 and tap/flick the side of the tube to keep the beads from settling at the bottom.
-
c.After adding the Second Strand Stop Buffer (B3), transfer the tube to a magnetic rack, let stand for 2 min to clear the solution, and carefully remove the supernatant from the beads with a pipette while the tube is still on the rack.
-
a.
-
29.
Optional, but recommended: proceed to Troubleshooting Problem 5.
Sequencing
Paired-end sequencing to 75 bp was performed on the Illumina NextSeq500 High Output Kit v2.5 75 Cycles. Reads were trimmed to 35 nucleotides and aligned to human genome build hg19, as described below.
Background subtraction and spike-in normalization
Sequencing data must be aligned to the genome and processed to produce per-gene tag counts adjusted for library size, interphase contamination, and spike-in proportion. Data may be visualized by the use of bigWig format track files on UCSC Genome Browser or IGV.
-
30.Align reads to the genome using bowtie2 / tophat with command-line parameter --very-sensitive in end-to-end mode. Sort aligned tags by tag ID using samtools sort -n, then remove PCR duplicates using PICARD MarkDuplicates with command-line parameters REMOVE_DUPLICATES=True and ASSUME_SORT_ORDER=queryname.
-
a.To quantify tag levels at transcript models, first download transcript models in BED format from an online database such as UCSC Genome Browser. Critically, the file should have the chromosome / contig, TSS, and TES of each transcript model in columns 1, 2, and 3, respectively. If spike-in controls were used, include the coordinates of the relevant loci in this file. Use deepTools multiBamSummary in BED-file mode and write counts for all samples to a file with option --outRawCounts. Calculate the RPM for each sample as 1,000,000 / number of aligned tags and then adjust each count as itself multiplied by the RPM, divided by the number of kilobases in the transcript model, defined as (max(TSS,TES)-min(TSS,TES))/1000. Subtract NoEU samples from experimental samples, then zero out negative values. Where spike-ins were added, sum the tags aligned to all spike-in loci and adjust for RPM, then divide all counts by this scalar.
-
b.To make bigWig track files, use deepTools bamCoverage to obtain bigWigs with command-line parameter --normalizeUsing CPM. Do this for experimental and NoEU control samples, then subtract NoEU using deepTools bigWigCompare --operation subtract. If a spike-in was used, control the spike-in ratio using the --scaleFactors parameter and the scalar calculated above.
-
a.
Expected outcomes
The expected yield of total RNA isolated after labeling (Step 7) is approximately 0.005 ng/cell for a mitotic population and 0.015 ng/cell for an asynchronous population. There should be very little, if any, loss of RNA following biotinylation (Step 18).
Since EU labels nascent transcripts, it is expected that mapped reads will span both exon and introns (Figures 1A and 1B). This should be visible on the genome browser (Figure 1A). We recommend visualizing low count genes to empirically determine how sensitive the method can be in a given experimental system.
Figure 1.

Expected outcomes from EU-RNA-seq
(A) Browser view shows mapped reads span both introns and exons.
(B) Genomic quantification of read coverage over exons, introns, and intergenic regions. p < 0.001.
(C) Log2-based read density of all transcripts in an asynchronous population.
Due to the 40-min incubation period, a portion of the nascent transcripts may have been spliced by the time they are captured, thus signal may be stronger over exons than over introns. This will usually be visible at the level of the genome browser, but should also be quantified to determine the proportion of transcripts in your data set that follow this pattern (Figure 1B).
EU-RNA-seq labels transcripts over a large dynamic range and in our hands results in a bimodal distribution of expression in an asynchronous population. When viewing transcripts on the genome browser that fall in either mode, we found that the modes separate background signal from true transcriptional events.
Limitations
The main limitation to our approach is that it takes approximately 40 min for 5-ethynyluridine to permeate the cell membrane, nuclear membrane, and be significantly incorporated (Caravaca et al., 2013). This is, however, based on the HUH7 cells that we used. We encourage you to use click tagging of Alexafluorophore to determine the timing needed in each cell type or system to determine the incubation period needed to achieve fluorophore signal of the labeling event. For instance, see Chen et al, 2019 where they performed labeling in embryos.
Troubleshooting
These subprotocols provide detailed overviews of the optimization of EU-labeling and the design and validation of the custom, biotin-RNA controls sequences used as spike-ins for global normalization in Palozola et al., 2017. The goal is to sufficiently label nascent transcripts for sequencing and to control for global differences in transcription level between two populations (Figure 2) and the resulting over-amplification of sequences in low-abundance populations. Known quantities of control sequences are added to each EU-labeled RNA sample prior to pull down and cDNA library generation (Figure 3).
Figure 2.

Different basal levels of transcription affect library generation
(A) An unknown fraction of the total RNA harvested is labeled with EU.
(B) Biotin-azide is conjugated to EU via a “click” reaction.
(C) All biotinylated RNAs are pulled down on streptavidin-coated magnetic beads.
(D and E) (D) cDNA libraries are generated directly off the beads and the (E) relative gene expression between populations is misrepresented after library amplification.
Figure 3.

Spike-in controls globally normalize for differences in basal transcriptional levels between samples
(A) An unknown fraction of the total RNA harvested is labeled with EU.
(B) Biotin-azide is conjugated to EU via a “click” reaction.
(C) Custom, biotin-RNA sequences are spiked-in at known levels.
(D) All biotinylated RNAs are pulled down on streptavidin-coated magnetic beads.
(E) cDNA libraries are generated directly off the beads.
Problem 1
Confirm the successful in vitro transcription of the control sequences using biotin-UTP.
Potential solution
We recommend running the reaction product on a gel to confirm transcription has occurred. Combine 6.6 μL of 2X loading buffer and 6.6 μL of each reaction, and at 95°C for 2 minutes, and immediately return to ice. Run on a 2% agarose gel at 100V for 1 hr. To improve visualization of the transcription product, we suggest pouring the gel as thin as possible and excluding EtBr. Instead, stain and destain the gel after running. To stain, submerge the gel in 1X TAE containing EtBr and rock at 20-22°C for 5 minutes. Then remove the excess EtBr by washing the gel 3X 5 minutes in 1X TAE before imaging.
Problem 2
The amount of labeling time necessary to sufficiently label nascent transcripts for detection by sequencing will vary depending on the biological system in use. We recommend using a labeling time that allows for sufficient incorporation to visualize the nascent transcripts when the EU is conjugated to a fluorophore.
Potential solution
Label cells as above for a minimum of 10 minutes. For adherent cells cultured in a monolayer, such as the HUH7 cells used in Palozola et al, we recommend a minimum labeling of 30 minutes. Follow manufacturer's protocol to complete the click reaction with fluorophore and visualize with a fluorescent microscope to ensure sufficient incorporation of the EU.
Problem 3
Once the control sequences are generated as described above, the next step is to determine whether the control sequences are pulled down proportionally on streptavidin-coated magnetic beads in the presence of cellular RNA containing an unknown amount of biotin-EU-RNA. This is important to ensure that the in vivo labeled transcripts do not out-compete the synthetic sequences for binding to the beads, thus affecting their pull-down.
Potential solution
Pull down the spike-in controls in the presence of cellular biotin-EU-RNA, in excess of what will be used in the final assay. Quantify the resulting control sequences by qPCR. We recommend pulse labeling HUH7 cells with 0.5 uM EU for 40 minutes, harvesting total RNA, and performing the click reaction as in steps 1-18. Next, add control sequences to 1.5 ug total RNA and pull down with streptavidin-coated magnetic beads as described in steps 19-26. Combine 4 uL of 5X iScript reaction mix and 1 uL of iScript reverse transcriptase with the 15 uL click-it reaction. Incubate reaction mix on heat blocks with gentle shaking: 5 minutes at 25°C, 30 minutes at 42°C (Use a pipet to keep the beads suspended during this longer incubation.), and 5 minutes at 85°C. Use a magnetic rack to remove the beads and adjust the total volume to 50 uL with NF-water. Precipitate as in steps 11-18 and quantify controls by qPCR (Figure 4A). All controls should pulled down proportionally in the presence of biotin-EU-RNA (Figure 4B-C).
Figure 4.

The custom biotin-RNA control sequences retain their proportionality when pulled down and converted to cDNA
(A–C) (A) Experimental approach to validating controls. Ct values following RT-qPCR of (B) control #1 or (C) control #2, when added at the indicated levels to 1.5 ug of asynchronous biotin-EU-RNA and pulled down with streptavidin-coated magnetic beads. Expected values are based on linear pull down and cDNA conversion.
Problem 4
Once you have shown that control sequences are pulled down proportionally, the next step is to determine whether the amount of cellular biotin-EU-RNA affects pull down of the control biotin-RNA sequences. This is important since lower basal levels of transcription in one sample could result in the enrichment of the spike-in upon pull down.
Potential solution
Pull down the spike-in controls in the presence of various amounts of cellular biotin-EU-RNA. Quantify the resulting control sequences by qPCR. We recommend pulse labeling HUH7 cells with 0.5 uM EU for 40 minutes, harvesting total RNA, and performing the click reaction as in steps 1-18. Next, add control sequences 1 and 2 to 0.5, 1.0, or 1.5 ug total RNA and pull down with streptavidin-coated magnetic beads as described in steps 19-26. Generate first and second strand cDNA, remove beads, and quantify controls by qPCR. (Figure 5A). The amount of input cellular EU-RNA should not predictably affect pull down of the control sequences (Figure 5B-C).
Figure 5.

The custom biotin-RNA control sequences retain their proportionality independent of cellular EU-RNA level
(A–C) (A) Experimental approach to validating controls. Ct values following RT-qPCR of (B) control #1 or (C) control #2, when added at the indicated levels to 0.5, 1.0, or 1.5 ug of asynchronous biotin-EU-RNA and pulled down with streptavidin-coated magnetic beads. Expected values are based on linear pull down and cDNA conversion. Error bars represent standard error of the mean (SEM).
Problem 5
Once you have shown that control sequences are pulled down proportionally in the presence of various amounts of cellular biotin-EU-RNA, the next step is to determine whether the process of generating cDNA libraries affects the relative proportion of the control sequences. This is important since it is the amount of the spike-in sequence present in the final libraries that will be used to scale the transcriptomic data after sequencing.
Potential solution
Pull down the spike-in controls in the presence of various amounts of cellular biotin-EU-RNA. Generate cDNA libraries as above, and quantify the resulting control sequences by qPCR. Pulse-label asynchronous HUH7 cell with 0.5 uM EU for 40 minutes, harvest total RNA, and perform click reaction to conjugate biotin to the unknown fraction of that contains EU. Add control sequences 1 and 2 to 2 ug total RNA, pull down with streptavidin-coated magnetic beads, generate cDNA libraries (step 27) and quantify controls by qPCR (Figure 6A). The proportionality of the control sequences should be retained after library generation (Figure 6B-C).
Figure 6.

The custom biotin-RNA control sequences retain their proportionality when converted to cDNA libraries
(A–C) (A) Experimental approach to validating controls. Ct values following RT-qPCR of (B) control #1 or (C) control #2, when added at the indicated levels to 1.5 ug of asynchronous biotin-EU-RNA and pulled down with streptavidin-coated magnetic beads and converted to cDNA libraries. Expected values are based on linear pull down and cDNA conversion.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ken Zaret (zaret@pennmedicine.upenn.edu).
Materials availability
Spike-in control sequences generated for this study are available upon request.
Data and code availability
Custom scripts are available upon request.
Acknowledgments
We thank training grant T32GM00812 to K.C.P. and NIH grant GM36477 to K.S.Z.
Author contributions
K.C.P. developed the spike-ins, pull-down, and library generation methods, with guidance from K.S.Z. G.D. developed the method of background normalization and NoEU subtraction.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Katherine C. Palozola, Email: palozola@pennmedicine.upenn.edu.
Kenneth S. Zaret, Email: zaret@pennmedicine.upenn.edu.
References
- Caravaca J.M., Donahue G., Becker J.S., He X., Vinson C., Zaret K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes & Development. 2013;27 doi: 10.1101/gad.206458.112. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Einstein L.C., Little S.C., Good M.C. Spatiotemporal patterning of zygotic genome activation in a model vertebrate embryo. Dev. Cell. 2019;49:852–866. doi: 10.1016/j.devcel.2019.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L.J., Waterfall J.J., Lis J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung C.C.S., Bartman C.R., Huang P., Ginart P., Stonestrom A.J., Keller C.A., Face C., Jahn K.S., Evans P., Sankaranarayanan L. A hyperactive transcriptional state marks genome reactivation at the mitosis-G1 transition. Genes Dev. 2016;30:1423–1439. doi: 10.1101/gad.280859.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K., Woodfin A.R., Slaughter B.D., Unruh J.R., Box A.C., Rickels R.A., Gao X., Haug J.S., Jaspersen S.L., Shilatifard A. Mitotic transcriptional activation: clearance of actively engaged Pol II via transcriptional elongation control in mitosis. Mol. Cell. 2015;60:1–34. doi: 10.1016/j.molcel.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H., Shokhirev M.N., Xu Z., Chandran S., Dixon J.R., Hetzer M.W. Dynamic regulation of histone modifications and long-range chromosomal interactions during postmitotic transcriptional reactivation. Genes Dev. 2020;34:913–930. doi: 10.1101/gad.335794.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat D.B., Kwak H., Booth G.T., Jonker I.H., Danko C.G., Patel R.K., Waters C.T., Munson K., Core L.J., Lis J.T. Base-pair resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq) Nat. Protoc. 2016;11:1455–1476. doi: 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A., Churchman L.S. Genome-wide profiling of RNA polymerase transcription at nucleotide resolution in human cells with native elongating transcript sequencing. Nat. Protoc. 2016;11:813. doi: 10.1038/nprot.2016.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola K.C., Donahue G., Liu H., Grant G.R., Becker J.S., Cote A., Yu H., Raj A., Zaret K.S. Mitotic transcription and waves of gene reactivation during mitotic exit. Science. 2017;358:119–122. doi: 10.1126/science.aal4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea-Resa C., Bury L., Cheeseman I.M., Blower M.D. Cohesin removal reprograms gene expression upon mitotic entry. Mol. Cell. 2020;78:127–140. doi: 10.1016/j.molcel.2020.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B., Michel Zacher M.B., Fruhauf K., Demel C., Tresch A., Gagneur J., Cramer P. TT-seq maps the human transient transcriptome. Science. 2016;352:1225–1228. doi: 10.1126/science.aad9841. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y., Zhu H., Lee J.H., Kossenkov A.V., Wu S.Y., Wickramasinghe J.M., Yin X., Palozola K.C., Gardini A., Showe L.C. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016;76:6320–6330. doi: 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Custom scripts are available upon request.