

Abstract
Breast cancer, a leading cause of death yearly, has been shown to be initiated and propagated by cancer stem cells. CD133, a cell surface antigen, has been shown to be present on cancer stem cells of many solid tumors, including breast cancer. A limitation to targeting CD133 is major histocompatibility complex (MHC)-restricted presentation of epitopes, leading to activation of only one arm of the immune system: either CD4+ helper T cells or CD8+ cytotoxic T cells. Thus, we hypothesized that by creating an MHC-independent vaccination, we would give rise to a sustained immune response against CD133 in triple-negative breast cancer (TNBCs). We transfected CD133 mRNA into dendritic cells and then tested this in animal models of TNBC. We showed in these models the activation of both CD8+ cytotoxic T cells and CD4+ helper T cells by dendritic cell vaccination with modified CD133 mRNA, with subsequent decrease in tumor growth. This study for the first time demonstrates in a syngeneic mouse model of TNBC that targeting CD133, in an MHC-independent manner, is an effective strategy against the cancer stem cell population, leading to tumor abrogation.
Keywords: CD133, breast cancer, dendritic cell vaccine
Graphical abstract
Triple-negative breast cancer stem cells with CD133 expression show increased resistance to current therapies. Tay et al. show that a coordinated cytotoxic and helper T cell response is generated from vaccination with dendritic cells transfected with CD133 mRNA. Mammary tumor growth decreased and survival times increased in a syngeneic breast cancer mouse model.
Introduction
Breast cancer is diagnosed in more than 200,000 patients per year in the United States.1 Worse yet, 15%–50% of all breast cancer cases are triple negative for estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER-2), and this subset of triple-negative breast cancer (TNBC) has worse 5 year outcomes, with survival rates of 14%.2 Emerging evidence points to cancer stem cells (CSCs) as the driver of tumor growth, metastases, and resistance to therapies. Among other cell surface markers of CSCs, CD133 has been shown to be a possible prognostic marker3 and a therapeutic target.4 CD133 has been correlated with larger tumor size, negative PR and ER status, lymph node metastases, higher tumor grade, advanced TNM stage, and poor overall survival in breast cancer.3,5, 6, 7 Notably, there have also been studies showing enrichment of CD133+ CSCs in breast cancer patients after treatment with chemotherapy, suggesting CD133 is associated with increased resistance to therapy.8 Therefore, in these patients with TNBC that are resistant to chemotherapy, immunotherapy targeted against CD133 represents an exciting therapeutic strategy. We hypothesized that the implementation of an active immunotherapy strategy via transducing dendritic cells (DCs) with CD133 mRNA would allow for a vaccine that does not have histocompatibility leukocyte antigen (HLA) restriction, resulting in the treatment of all patients. Furthermore, this method would bypass the major histocompatibility complex (MHC)-dependent presentation of epitopes and thus generate both helper and cytotoxic T cell responses. Here, we test the efficacy of a vaccine comprised of DCs transfected with CD133 mRNA in a syngeneic TNBC mouse model and against human TNBC cell lines.
Results
TNBCs enriched for CD133+ CSCs by microbead isolation using CD133 PE antibody
We chose to use 4T1 cells because once injected into mice models, these cells form a tumor that is known to closely mimic human TNBC. 4T1 cells cultured in SC media resulted in sphere formation (Figure 1A). In order to create a mouse model with TNBCs enriched for CD133+ CSCs, we isolated TNBCs that were CD133+ using a microbead isolation technique. As shown in Figure 1B, after isolation 69.46% of the cells were positive for CD133 versus 7.74% prior to isolation. After these cells were isolated, the mice models were inoculated with these TNBCs in orthotopic breast tissue.
Figure 1.

Sphere Formation and CD133 expression in 4T1 breast cancer stem cells
(A) Sphere formation of 4T1 breast cancer stem cells when cultured in the base medium ATCC-formulated RPMI 1640 medium with 10% fetal bovine serum. (B) CD133 expression on 4T1 breast cancer stem cells before and after microbead isolation as measured by flow cytometry, showing increased number of cells positive for CD133 after isolation.
In vitro testing showed that DCs transfected with CD133 mRNA showed apoptosis in 4T1 cells
We investigated whether DC-based immunotherapy could be an effective approach to treat TNBC by selectively targeting CD133 in cancer stem-like cells in the syngeneic C57BL model. To create a plasmid that would allow for introduction of CD133 into the DCs, we used a signal sorting fragment (SS) and transmembrane-cytoplasmic domain fragment (TM/cyto) and placed them on either side of the mRNA sequence for CD133. This allowed for the CD133 protein to be efficiently transported to both the MHC complex class I (class I) and MHC class II compartment of DCs,9,10 thus allowing for MHC-independent presentation of CD133 epitopes and activation of both the CD4+ and CD8+ pathways.
We tested the activation of both pathways by using an in vitro killing assay comparing co-culture with CD4+ and CD8+ cells versus CD8+ alone. The results showed that antigens were presented to both CD8+ and CD4+ T cells by DCs transfected with modified CD133 mRNA, resulting in a significant activation of both CD8+ and CD4+ T cells as measured by tumor cell death. Using the caspase-3 assay, we measured cell death when DCs transfected with modified CD133 mRNA were co-cultured with CD8+ and CD4+ T cells. When co-cultured with DCs transfected with modified CD133 and both CD8+ and CD4+ T cells, there was 26.36% cell death of 4T1 cells compared with 1.58% cell death found when the 4T1 cells were cultured with DCs transfected with modified CD133 and CD8+ T cells alone (Figure 2). By comparison, we looked at CD8+ and CD4+ T cells co-cultured with non-transfected DCs, which resulted in 15.82% cell death of 4T1 cells versus CD8+ T cells only, which resulted in 2.16% cell death. Without DCs, CD8+ and CD4+ T cells showed 4.84% cell death and CD8+ T cells only showed 3.64% cell death.
Figure 2.

Caspase-3 assay in 4T1 breast cancer stem cells when co-cultured with CD4+ and CD8+ T cells or just CD8+ cells
The first row shows the 4T1 cells cultured with CD8+ T cells only with one of the following, in order from left to right: PBS, non-transfected DCs, or transfected DCs. The second row shows the 4T1 cells cultured with both CD4+ and CD8+ T cells with one of the following, in order from left to right: PBS, non-transfected DCs, or transfected DCs. 4T1 cells co-cultured with CD4+ and CD8+ T cells show significantly more caspase-3 than when the 4T1 cells were co-cultured with CD8+ T cells only across all three groups.
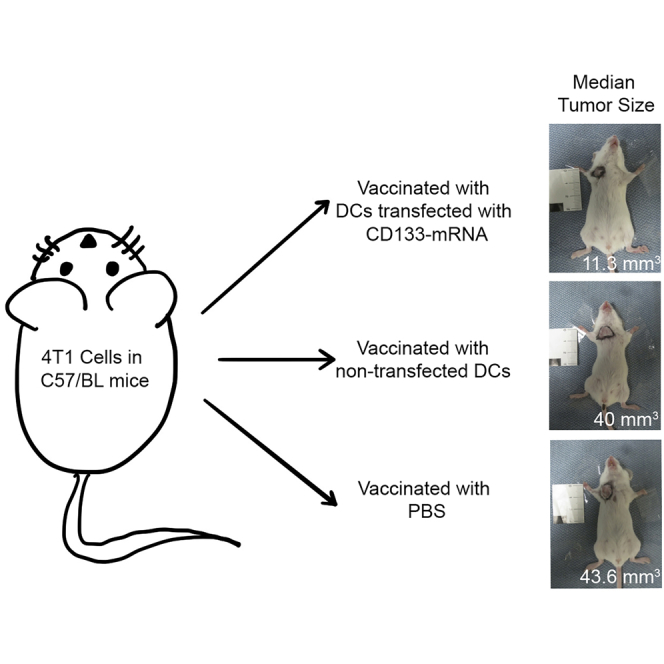
Murine CD133 mRNA-transfected DC vaccination reduced mammary tumor growth
Next, we wanted to see the effect of the DC vaccination on tumor growth in an orthotopic TNBC mouse model (4T1 cells in C57BL mice). We injected the mice with DCs transfected with one of the following: mouse CD133 mRNA or no mRNA. For the control group, we also injected a group of mice with PBS (see Figure 3A for the vaccine schedule). Each group contained 22 mice. After vaccination, these mice were later inoculated with orthotopic TNBCs that were isolated in the earlier sections. Mice vaccinated with DCs transfected with CD133 mRNA had significantly reduced growth of mammary tumors, with a median of 11.3 mm3 at 33 days, whereas the mice vaccinated with non-transfected DCs had a median growth of 40 mm3 and mice vaccinated with PBS had a median growth of 43.6 mm3 (Figure 3B).
Figure 3.

Experimental design and growth of mammary tumors
(A) Experimental design and vaccination schedule of mice. (B) Growth of the mammary tumors in the groups of mice (n = 22 in each group). This shows that the mice in group A (mice vaccinated with DCs transfected with CD133 mRNA) had less tumor growth than both mice in group B (mice vaccinated with DCs that were not transfected) and in group C (mice vaccinated with PBS).
CD133 mRNA DC-vaccinated mice had increased infiltration of T cells in tumor sections
We then examined the mice for T cell infiltration in the tumors using immunohistochemistry techniques. Sections of the tumors were taken on day 33. In Figure 4, the mice that were vaccinated with DCs transfected with CD133 mRNA had increased infiltration of CD3+, CD4+, and CD8+ T lymphocytes compared with the group that was vaccinated with non-transfected DCs and the PBS group. This provides histologic evidence that vaccination with DCs transfected with CD133 mRNA is associated with a robust CD8+ cytotoxic T cell and CD4+ helper T cell infiltration into tumors.
Figure 4.

Immunohistochemistry of tumor sections
Tumor sections showing CD3 (first row), CD4 (second row), and CD8 (third row) infiltration in the various treatment groups (from left to right): PBS group, vaccinated group with DCs that were not transfected, and vaccinated group with DCs transfected with CD133.
Survival benefit in mice vaccinated with DCs transfected with CD133 mRNA
After looking at tumor growth and cell infiltration in vivo, we performed Kaplan Meier analysis for survival benefit on the TNBC mouse model. When mice were vaccinated with DCs transfected with modified CD133 mRNA, there was a significantly prolonged survival compared with the non-transfected DC group and PBS control group, p < 0.05 (Figure 5). Thus, the vaccination with modified CD133 DCs appeared to prolong survival in TNBC-bearing mice, likely from increased activation of CD8+ and CD4+ cells as evidenced in the immunohistochemical analysis.
Figure 5.

Kaplan Meier analysis
Kaplan Meier analysis for the various groups of mice vaccinated with DCs transfected with CD133 mRNA, DCs not transfected, or PBS, showing increased survival in mice vaccinated with DCs transfected with CD133 mRNA. p-value < 0.05.
Human CD133 mRNA-transfected DC vaccination showed increased cytotoxic T cell activity
Next, we looked at whether this DC vaccination would be clinically relevant by testing whether DCs transfected with human CD133 would be able to mount an immune response to human breast cancer cells. We evaluated the antigen surface markers in two known human breast cancer cell lines: MDA-MB-261 and MDA-MB-468. We found that MDA-MB-468 cells had more cells positive for CD133 (Figures 6A and 6B). These data, along with previous evidence showing that ALDHhighCD44+CD133+ cells isolated from MDA-MB-468 cell lines had enhanced malignant behavior in vitro and in vivo,11 led us to choose to study MDA-MB-468 cells. We transfected DCs with human CD133 mRNA and then tested for cytotoxic T cell activity in response to MDA-MB-468 cells. To investigate T cell activation, we determined the interferon (IFN)-gamma production from T cells stimulated with DCs. DCs transfected with CD133 mRNA co-cultured with CD8+ and CD4+ T cells showed high IFN-gamma secretion ability in the presence of MDA-MB-468 cells (Figure 6C) compared with co-culture with CD8+ T cells only.
Figure 6.

Antigen Expression and Inferferon-Gamma Release assays in human breast cancer cells
(A) Expression of antigens in human breast cancer cell MDA-MB-468 was measured by flow cytometry, and percentages are graphically represented. Within the inset are representative data showing the CD133 expression at 86.45%. (B) Expression of antigens in human breast cancer cell MDA-MB-231 was measured by flow cytometry, and percentages are graphically represented. Within the inset are representative data showing the CD133 expression at 1.35%. (C) Interferon-gamma release assay when MDA-MB-468 cells exposed to DCs transfected with human CD133, CD 4+, and CD8+ T cells or DCs transfected with human CD133 and CD8+ T cells or non-transfected DCs with CD4+ and CD8+ T cells or non-transfected DCs with CD8+ T cells. This shows that the group with the largest amount of interferon-gamma production was the culture including MDA-MB-468 cells with DCs transfected with human CD133, CD4+, and CD8+ T cells.
Discussion
There is still debate on whether all TNBCs can be treated with a standard chemotherapy regiment in addition to surgery or should be tailored by subclass of TNBC.12 Clinical trials have addressed treatment for TNBC specific to the various molecular subtypes or based on early or late stage of disease.13,14 Regiments of adjuvant or neoadjuvant therapy include sequential anthracycline and taxane regimens with or without alkylating agents.13 Although immunotherapy has made a great impact on other types of breast cancer (subtypes positive for hormone or ERBB2), it has yet to be incorporated in the standard treatment of TNBC.15 Additionally, while stage 1 TNBC has a 5 year survival of 85%, median survival for metastatic TNBC is approximately 1 year.15 Thus, there is much focus on treatment of metastatic TNBC.
The aggressive pattern of metastatic TNBC has been linked to its ability to mimic patterns of embryonic vasculogenesis, termed vasculogenic mimicry.16,17 The presence of vasculogenic mimicry can be demonstrated by the formation of blood lacunae that are surrounded by tumor cells and indicates poor prognosis.16 This ability appears to be from a subpopulation of cells that are CD133+.18 Furthermore, recent studies have shown that vasculogenic mimicry channels lined with CD133+ CSCs provide the functional blood supply to malignant tumors in human TNBC grafts and thus deliver the necessary molecules to synergize channel formation.19 Other studies have also shown that decreasing the expression of CD133+ CSCs has decreased proliferation and invasion capability20 and that direct targeting of CD133+ cells showed reduction in cancer cell migration and upregulation of downstream tumor suppressor expression,4 with both studies conducted using the TNBC cell line MDA-MB-231. Thus, CD133 stands to be an ideal immunologic target for TNBC. The high expression of CD133 in the MDA-MB-468 line found in our study, but not the MDA-MB-231 line, shows the heterogeneity of CD133 expression in TNBC cell lines derived from metastatic breast tumors, and these data taken together could warrant further study of CD133 in MDA-MB-231 cells.
Unfortunately, the function of CD133 is not well established, although its importance in stemness and therapeutic resistance is clear given that CD133 has been correlated with radio-resistance and chemotherapy resistance.3 CD133 has five transmembrane domains allowing for communication between extracellular environment and intracellular function. It is possible that CD133 may upregulate genes involved in cell survival and DNA repair. In TNBC, not only has it been associated with vasculogenic mimicry as discussed above but also it has been shown to upregulate actin-binding protein tropomyosin4 (Tm4), a protein highly associated with breast cancer metastases,21 and silencing of CD133 led to reduced invasiveness and decreased expression of Tm4.22 Notably, there has been a recent development of using the AC133 epitope of CD133 to develop AC133-specific chimeric antigen receptor (CAR) T cells.23 These AC133-CAR T cells were shown to kill AC133+ glioblastoma SCs both in vitro and in vivo. However, because CAR T cells rely on recognizing the epitope of CD133, theoretically it will not be able to discriminate between CSCs and hematopoetic SCs. Thus, we sought to use a DC vaccination transfected with mRNA CD133, which requires MHC to be presented for activation of the cytotoxic cascade and thus will be able to differentiate CSCs with MHC versus hematopoietic SCs, which do not have MHC on the cell surface. Other current therapies using CD133 include antibody-conjugated nanoparticles24 and radiolabeled AC133 monoclonal antibody (mAb) with iodine-131 to deliver radioactive molecules directly to tumor.25 Given the dismal survival rates of metastatic TNBC, the research findings showing the association between increased invasiveness of metastatic TNBC and CD133, and the mechanism of DC vaccination, we sought to target CD133 through a vaccine with CDs transfected with mRNA of CD133.
This study showed that this vaccination containing DCs transfected with modified mRNA could induce both CD4+ and CD8+ T cell activity against TNBC CSCs both in vitro and in vivo. Importantly, the vaccination also led to decrease tumor growth in our TNBC mouse model. This is in parallel to what we have recently shown in our research (Do et al.26) targeting CD133 cells in glioblastoma mouse model where there was increased survival in mice vaccinated with DCs transfected with CD133 mRNA. Within orthotopic immunocompetent mouse models, targeting of CD133+ cells using DC vaccine transfected with CD133 mRNA resulted in decreased tumor growth. Furthermore, infiltration of the tumor with CD8+ and CD4+ T cells recruited by the DCs was confirmed with immunohistochemical analysis of tumors.
We believe the potent inflammatory response to the tumor was due to the use of CD133 mRNA that allowed for cross-presentation of antigens that was independent of MHC presentation and allowed for stimulation of both CD8+ and CD4+ T cells. We confirmed this robust stimulation in MDA-MB-468 cell lines that showed increased IFN-gamma secretion when DCs were co-cultured with both CD8+ and CD4+ T cells compared with when DCs were co-cultured with CD8+ cells alone. Our data herein, along with our previous study (Do et al.26), strongly support the tumor-antigen-loading method of using modified mRNA transfection of DCs. It also supports CD133 as a therapeutic target in TNBC. The combination of activating CD8+ and CD4+ T cells with mRNA transfection of DCs and using CD133 as a target potentially allows one to specifically target CSCs and can add to the armamentarium of treatment of TNBC.
Materials and methods
Cell culture
The 4T1 cells (ATCC, CRL-2539) were cultured in the base medium, ATCC-formulated RPMI 1640 medium (ATCC 30-2001), with 10% fetal bovine serum (FBS). The cells were isolated for CD133+ cells by magnetic-activated cell sorting (MACS) microbead separation protocol provided by the manufacturer using the microbeads conjugated to monoclonal anti-mouse CD133 antibodies (Miltenyi Biotec, Anti-Prominin-1 MicroBeads, mouse). MDA-MB-231 (ATCC, HTB-26) cells were cultured in ATCC-formulated Leibovitz’s L-15 medium (ATCC, 30-2008) with 10% fetal bovine serum, incubated at 37°C without CO2. MDA-MB-468 (ATCC, HTB-132) cells were cultured in ATCC-formulated Leibovitz’s L-15 medium (ATCC, 30-2008) with 10% fetal bovine serum, incubated at 37°C without CO2. Culture and intubation protocols were provided by the manufacturer.
Mouse model
BALB/c mice were obtained from The Jackson Laboratory. All mice weighing between 25 and 35 g were used in this study. Free access to sterilized food and water was provided. This experiment was reviewed by the Institutional Animal Care and Use Committee at the Cedars-Sinai Medical Center.
Modified CD133 mRNA
Plasmid constructs have been described previously.9,10,27 Briefly, for in vitro transcription, the plasmids were cloned with pSP64 vector (Promega). A tyrosinase-related protein-2 (TRP-2) SS and TM/cyto were amplified from TRP-2 cDNA by using PCR (Ex Taq polymerase; Takara Bio). The PCR products were cloned as a Hind III-PstI signal sequence fragment and a BamH I-SmaI TM/cyto into pSP64 to allow in vitro transcription under the control of an SP6 promoter to transport the CD133 protein efficiently to MHC class II compartments for eventual cross-presentation by both classes I and II on DCs in a cognate manner.
Derivation of DCs and nucleofection
Human monocyte enrichment cells were isolated from human peripheral blood mononuclear cells (PBMCs) by EasySep Human Monocyte Enrichment kit (STEMCELL Technologies, BC, Canada) and placed in DC culture medium (CellGenex, New Hampshire, USA) supplied with human granulocyte-macrophage colony stimulating factor (1,000 IU/mL; Genzyme, MA, USA) and human interleukin-4 (IL-4) (1,000 IU/mL; R&D Systems, MN, USA) for up to 7 days. On day 4 of the culture, the same amounts of cytokines were added to the cells. These cells were collected as immature DCs 7 days after culturing with DC medium.
To isolate DCs of mice, the paws, femurs, and tibias of C57BL mice were removed. The marrow was flushed with RPMI 1640 medium by using a syringe with a 26G needle. The marrow was then filtrated through a 70-mm cell strainer. The bone marrow cells were adjusted to 2 × 105 cells/mL in RPMI 1640 medium with 10% of fetal bovine, penicillin/streptomycin, and l-glutamine (all from Life Technologies, NY, USA). They were cultured for up to 7 days in the presence of 1,000 IU/mL of mouse granulocyte-macrophage colony stimulating factor and 500 U/mL of mouse interleukin-4 at 37°C with 5% CO2. On day 4 of the culture, the same amounts of cytokines were added to the cells. These cells also were collected as immature mouse DCs after 7 days of culturing.
Human immature DCs and mouse immature DCs were nucleofected with modified human CD133 mRNA and modified mouse CD133 mRNA, respectively, or were transfected without any mRNA by 4D-nucleofector (Lonza, NJ, USA) (pulse code for human DCs: P3, CB150; pulse code for mouse DCs: P4, DK100). DCs after nucleofection were placed into recovery and maturation medium. Recovery and maturation medium for human DCs was RPMI 1640 with 10% human AB serum, 100 μg/mL streptomycin, 100 U/mL penicillin, 2 mM UltraGlutamine I (Lonza, NJ, USA), 1 mM sodium pyruvate (Life Technologies, NY, USA), and 2.5 μg/mL monophosphoryl lipid A-lipopolysaccharide (MPLA-LPS) (Sigma, MO, USA). Recovery and maturation medium for mouse DCs was RPMI 1640 with 10% fetal bovine serum, 100 μg/mL streptomycin, 100 U/mL penicillin, 2 mM UltraGlutamine I, 1 mM sodium pyruvate, and 2.5 μg/mL MPLA-LPS. The cells were incubated in humidified 37°C/5% CO2 incubator. DCs were used for vaccination therapy after 4 h of incubation or for a CTL assay, a cytokine-releasing assay was used after 24 h of incubation.
Vaccine schedule
C57BL mice were vaccinated subcutaneously on days 0, 4, 8, and 12 with different tumor-antigen-pulsed DC vaccines: control (PBS alone, n = 22; DC unpulsed, n = 22; DC pulsed with CD133 mRNA, n = 22). Mice were inoculated with 4T1 cells in the intramammary fat pad with tumor on day 16.
Measurement of mammary tumors
The tumor was measured in two perpendicular directions, and the volume was calculated according to the following equation: where L is the longest diameter and W is the perpendicular diameter to L.
Immunocytochemistry
The perfusion-fixed tumor tissue were cut into 40-μm sections were treated with 10% donkey serum (Sigma) for 30 min at room temperature and then stained with primary antibodies for anti-CD4 (rat mAb, 1:50; BD Biosciences, CA, USA), anti-CD8 (rat mAb, 1:100; BD Biosciences), anti-CD3 (rat mAb, 1:50; BD Biosciences), and isotype control antibodies. The primary antibodies were detected Vector Elite ABC kit (Vector Laboratories, Burlingame, CA) and developed with diaminobenzidine (Sigma) and counterstained with hematoxylin before mounting the sections. Experiments were done once.
In vivo anti-tumor effect
DCs were obtained from mice and were then used for introduction of murine CD133 mRNA that then served as a vaccine against experimentally generated mice bearing mammary TNBC tumors with appropriate controls (i.e., PBS, no transfection).
Mouse DCs were derived from mouse bone marrow by previously described methods.26 24 h after incubation, supernatant was recovered and maturation medium was collected for interleukin-12 ELISA assay (BD Biosciences). At the same time, T cells (CD8+ cells and CD4+ cells) or CD8+ T cells were isolated from mouse spleen cells by Dynabeads T Cell Isolation kit or Dynabeads CD8 T Cell Isolation kit (Invitrogen, Carlsbad, CA). Murine DCs (1 × 104) transfected with murine CD133 mRNA were then cultured in the presence of 1 × 105 T cells (CD8+ cells and CD4+ cells) or in the presence of 1 × 105 CD8+ cells alone or with non-T cells in a 96-well plate. Similarly, 1 × 104 murine DCs that were transfected without any mRNA were cultured in the presence of 1 × 105 T cells (CD8+ cells and CD4+ cells) or in the presence of 1 × 105 CD8+ cells alone or with non-T cells in a 96-well plate.
As a further control, PBS was used to replace murine DCs and exposed to 1 × 105 T cells (CD8+ cells and CD4+ cells) or 1 × 105 CD8+ cells alone in a 96-well plate. The same amount of T cells or CD8+ T cells was added every week for a total of 4 times. After 4 weeks of co-culture, T cells or CD8+ T cells were collected as effector cells for CTL analysis, and supernatant was also collected for IFN-gamma ELISA (BD Biosciences). LT-1 mammosphere cells were stained with DDAO-SE (Life Technologies) and were prepared as target cells. Target cells and effector cells were co-cultured overnight (E:T ratio = 20:1). After overnight co-culture, supernatants were collected for an IFN-gamma ELISA assay (BD Biosciences) and CTL assay by caspase-3 apoptosis kit PE (BD Biosciences). Experiments were done once.
Tumor growth and evaluation of IFN-gamma were done to confirm the efficacy and efficiency of DC vaccination on TNBC-bearing mice. This was determined through ELISA for IFN-gamma release and immunohistochemical analysis and caspase killing assay. Studies on the mice were carried out in accordance with protocols approved by Cedars-Sinai Medical Center Institutional Animal Care and Use Committee.
Cytotoxicity against human MB-MDA-468 cells
Human DCs were derived from human PBMCs by previously described methods.26 At the same time, T cells (CD8+ cells and CD4+ cells) or CD8+ T cells were isolated from human PBMCs by Dynabeads T Cell Isolation kit or Dynabeads CD8 T Cell Isolation kit (Invitrogen). Human DCs (1 × 104) transfected with human CD133 mRNA were then cultured in the presence of 1 × 105 T cells (CD8+ cells and CD4+ cells) or in the presence of 1 × 105 CD8+ cells alone or with non-T cells in a 96-well plate. Similarly, 1 × 104 human DCs that were transfected without any mRNA were cultured in the presence of 1 × 105 T cells (CD8+ cells and CD4+ cells) or in the presence of 1 × 105 CD8+ cells alone or with non-T cells in a 96-well plate. The same amount of T cells or CD8+ T cells was added every week for a total of 4 times. After 4 weeks of co-culture, T cells or CD8+ T cells were collected as effector cells for IFN-gamma ELISA (BD Biosciences). MB-MDA-468 cells were used as target cells. Target cells and effector cells were co-cultured overnight (E:T ratio = 20:1). After overnight co-culture, supernatants were collected for an IFN-gamma ELISA assay (BD Biosciences). Experiments were done once.
Statistical analysis
Based on preliminary data, we assume a less than 10% survival rate in control animals at approximately 40 days. Therefore, sample sizes of 22 in each study group will have 80% power to detect a difference of 36% at the 0.05 significance level in an analysis of survival times with a log rank test.
Acknowledgments
This work is supported by a grant from FasterCures and the Lowell Milken Foundation (to J.S.Y.) and a grant from the National Institute for Neurological Disorders and Stroke (NIH) (2R01# NS 048959 to J.S.Y.).
Author contributions
T.A. initiated experiments. T.A. and J.S.Y. designed experiments, and A.S.T., T.A., L.A.E., and J.S.Y. wrote the manuscript. T.A. performed experiments. T.A. and L.A.E. performed data analysis. T.A. provided experimental materials.
Declaration interests
The authors declare no competing interests.
References
- 1.DeSantis C., Ma J., Bryan L., Jemal A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 2.Bauer K.R., Brown M., Cress R.D., Parise C.A., Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109:1721–1728. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 3.Brugnoli F., Grassilli S., Al-Qassab Y., Capitani S., Bertagnolo V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019;2019:7512632. doi: 10.1155/2019/7512632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin H., Xiong G., Guo S., Xu C., Xu R., Guo P., Shu D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019;27:1252–1261. doi: 10.1016/j.ymthe.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Q., Li J.G., Zheng X.Y., Jin F., Dong H.T. Expression of CD133, PAX2, ESA, and GPR30 in invasive ductal breast carcinomas. Chin. Med. J. (Engl.) 2009;122:2763–2769. [PubMed] [Google Scholar]
- 6.Currie M.J., Beardsley B.E., Harris G.C., Gunningham S.P., Dachs G.U., Dijkstra B., Morrin H.R., Wells J.E., Robinson B.A. Immunohistochemical analysis of cancer stem cell markers in invasive breast carcinoma and associated ductal carcinoma in situ: relationships with markers of tumor hypoxia and microvascularity. Hum. Pathol. 2013;44:402–411. doi: 10.1016/j.humpath.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Xia P. CD133 mRNA may be a suitable prognostic marker for human breast cancer. Stem Cell Investig. 2017;4:87. doi: 10.21037/sci.2017.10.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nadal R., Ortega F.G., Salido M., Lorente J.A., Rodríguez-Rivera M., Delgado-Rodríguez M., Macià M., Fernández A., Corominas J.M., García-Puche J.L. CD133 expression in circulating tumor cells from breast cancer patients: potential role in resistance to chemotherapy. Int. J. Cancer. 2013;133:2398–2407. doi: 10.1002/ijc.28263. [DOI] [PubMed] [Google Scholar]
- 9.Fukui M., Ueno K., Suehiro Y., Hamanaka Y., Imai K., Hinoda Y. Anti-tumor activity of dendritic cells transfected with mRNA for receptor for hyaluronan-mediated motility is mediated by CD4+ T cells. Cancer Immunol. Immunother. 2006;55:538–546. doi: 10.1007/s00262-005-0027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amano T., Kajiwara K., Yoshikawa K., Morioka J., Nomura S., Fujisawa H., Kato S., Fujii M., Fukui M., Hinoda Y., Suzuki M. Antitumor effects of vaccination with dendritic cells transfected with modified receptor for hyaluronan-mediated motility mRNA in a mouse glioma model. J. Neurosurg. 2007;106:638–645. doi: 10.3171/jns.2007.106.4.638. [DOI] [PubMed] [Google Scholar]
- 11.Croker A.K., Goodale D., Chu J., Postenka C., Hedley B.D., Hess D.A., Allan A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009;13(8B):2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jitariu A.A., Cîmpean A.M., Ribatti D., Raica M. Triple negative breast cancer: the kiss of death. Oncotarget. 2017;8:46652–46662. doi: 10.18632/oncotarget.16938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergin A.R.T., Loi S. Triple-negative breast cancer: recent treatment advances. F1000Res. 2019;8:1342. doi: 10.12688/f1000research.18888.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mancini P., Angeloni A., Risi E., Orsi E., Mezi S. Standard of care and promising new agents for triple negative metastatic breast cancer. Cancers (Basel) 2014;6:2187–2223. doi: 10.3390/cancers6042187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waks A.G., Winer E.P. Breast Cancer Treatment: A Review. JAMA. 2019;321:288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 16.Plantamura I., Casalini P., Dugnani E., Sasso M., D’Ippolito E., Tortoreto M., Cacciatore M., Guarnotta C., Ghirelli C., Barajon I. PDGFRβ and FGFR2 mediate endothelial cell differentiation capability of triple negative breast carcinoma cells. Mol. Oncol. 2014;8:968–981. doi: 10.1016/j.molonc.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luan Y.Y., Liu Z.M., Zhong J.Y., Yao R.Y., Yu H.S. Effect of grape seed proanthocyanidins on tumor vasculogenic mimicry in human triple-negative breast cancer cells. Asian Pac. J. Cancer Prev. 2015;16:531–535. doi: 10.7314/apjcp.2015.16.2.531. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D., Sun B., Zhao X., Ma Y., Ji R., Gu Q., Dong X., Li J., Liu F., Jia X. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer. 2014;13:207. doi: 10.1186/1476-4598-13-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H., Yao N., Cheng S., Li L., Liu S., Yang Z., Shang G., Zhang D., Yao Z. Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer. Cancer Biol. Med. 2019;16:299–311. doi: 10.20892/j.issn.2095-3941.2018.0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brugnoli F., Grassilli S., Lanuti P., Marchisio M., Al-Qassab Y., Vezzali F., Capitani S., Bertagnolo V. Up-modulation of PLC-β2 reduces the number and malignancy of triple-negative breast tumor cells with a CD133+/EpCAM+ phenotype: a promising target for preventing progression of TNBC. BMC Cancer. 2017;17:617. doi: 10.1186/s12885-017-3592-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Li F., Qian Z.M., Sun H. Structure and topology of the transmembrane domain 4 of the divalent metal transporter in membrane-mimetic environments. Eur. J. Biochem. 2004;271:1938–1951. doi: 10.1111/j.1432-1033.2004.04104.x. [DOI] [PubMed] [Google Scholar]
- 22.Brugnoli F., Grassilli S., Piazzi M., Palomba M., Nika E., Bavelloni A., Capitani S., Bertagnolo V. In triple negative breast tumor cells, PLC-β2 promotes the conversion of CD133high to CD133low phenotype and reduces the CD133-related invasiveness. Mol. Cancer. 2013;12:165. doi: 10.1186/1476-4598-12-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X., Prasad S., Gaedicke S., Hettich M., Firat E., Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6:171–184. doi: 10.18632/oncotarget.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaminathan S.K., Roger E., Toti U., Niu L., Ohlfest J.R., Panyam J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release. 2013;171:280–287. doi: 10.1016/j.jconrel.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Weng D., Jin X., Qin S., Lan X., Chen C., Sun X., She X., Dong C., An R. Radioimmunotherapy for CD133(+) colonic cancer stem cells inhibits tumor development in nude mice. Oncotarget. 2017;8:44004–44014. doi: 10.18632/oncotarget.16868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do A.S.S., Amano T., Edwards L.A., Zhang L., De Peralta-Venturina M., Yu J.S. CD133 mRNA-Loaded Dendritic Cell Vaccination Abrogates Glioma Stem Cell Propagation in Humanized Glioblastoma Mouse Model. Mol. Ther. Oncolytics. 2020;18:295–303. doi: 10.1016/j.omto.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saka M., Amano T., Kajiwara K., Yoshikawa K., Ideguchi M., Nomura S., Fujisawa H., Kato S., Fujii M., Ueno K. Vaccine therapy with dendritic cells transfected with Il13ra2 mRNA for glioma in mice. J. Neurosurg. 2010;113:270–279. doi: 10.3171/2009.9.JNS09708. [DOI] [PubMed] [Google Scholar]