

Abstract
Long noncoding RNAs (lncRNAs) have emerged as biomarkers and regulators of cardiovascular disease. However, the expression pattern of circulating extracellular vesicle (EV)-incorporated lncRNAs in patients with coronary artery disease (CAD) is still poorly investigated. A human lncRNA array revealed that certain EV-lncRNAs are significantly dysregulated in CAD patients. Circulating small EVs (sEVs) from patients with (n = 30) or without (n = 30) CAD were used to quantify PUNISHER (also known as AGAP2-antisense RNA 1 [AS1]), GAS5, MALAT1, and H19 RNA levels. PUNISHER (p = 0.002) and GAS5 (p = 0.02) were significantly increased in patients with CAD, compared to non-CAD patients. Fluorescent labeling and quantitative real-time PCR of sEVs demonstrated that functional PUNISHER was transported into the recipient cells. Mechanistically, the RNA-binding protein, heterogeneous nuclear ribonucleoprotein K (hnRNPK), interacts with PUNISHER, regulating its loading into sEVs. Knockdown of PUNISHER abrogated the EV-mediated effects on endothelial cell (EC) migration, proliferation, tube formation, and sprouting. Angiogenesis-related gene profiling showed that the expression of vascular endothelial growth factor A (VEGFA) RNA was significantly increased in EV recipient cells. Protein stability and RNA immunoprecipitation indicated that the PUNISHER-hnRNPK axis regulates the stability and binding of VEGFA mRNA to hnRNPK. Loss of PUNISHER in EVs abolished the EV-mediated promotion of VEGFA gene and protein expression. Intercellular transfer of EV-incorporated PUNISHER promotes a pro-angiogenic phenotype via a VEGFA-dependent mechanism.
Keywords: coronary artery disease, extracellular vesicles, long noncoding RNA, angiogenesis, cardiovascular disease
Graphical abstract
Circulating lncRNAs can be incorporated into circulating extracellular vesicles in patients with coronary artery disease. Shuttling of EV-PUNISHER into target endothelial cells promotes an angiogenic response in a VEGFA-dependent manner. In clinical practice, the EV-PUNISHER might be used as a novel biomarker and therapeutic target in patients.
Introduction
Coronary artery disease (CAD) and its cardiovascular sequelae represent the leading cause of mortality worldwide.1, 2, 3, 4 Emerging data suggest that noncoding RNAs (ncRNAs) are crucial regulators of pathological conditions, such as atherosclerosis. ncRNAs exert their biological effects intracellularly but can also be released into the circulation.5,6 Thus, circulating ncRNAs can provide useful information for the diagnosis of CAD as well as monitoring the progression of the disease,7 which has recently been referred to as a liquid biopsy.8, 9, 10 Although there are abundant ribonucleases in the bloodstream, ncRNAs remain relatively resistant to degradation. The stability of ncRNAs in plasma is mediated by their incorporation into and protection by extracellular vesicles (EVs) or through binding to exogenous proteins or lipids.11, 12, 13
MicroRNAs (miRNAs; ~22 nucleotides in length14) and long ncRNAs (lncRNAs; >200 nucleotides with no potential for translation into proteins15, 16, 17) are the most prominent kinds of ncRNAs.18 We and others have demonstrated that circulating EV-incorporated miRNAs are significantly differentially regulated under pathological conditions and therefore are of high value for the diagnosis of cardiovascular disease.7,19, 20, 21, 22, 23, 24 However, miRNAs are not the most abundant ncRNAs within EVs.25 Recent studies have highlighted that lncRNAs also participate in intercellular communication by way of EV-mediated cell-to-cell transfer. lncRNAs may exert their function by controlling gene expression in the nucleus or regulating other processes, such as RNA stability, in the cytoplasm.26, 27, 28
EVs are usually divided into two main classes according to their diameter: small EVs (sEVs; <150 nm) and large EVs (lEVs; 150−1,000 nm).29 Circulating sEVs and their molecular cargoes have recently emerged as promising diagnostic tools for various diseases including CAD.30 Previous studies have provided evidence that circulating sEV are one of the main sources for lncRNAs in the bloodstream.31, 32, 33, 34 However, whether sEV-incorporated lncRNAs are differentially expressed in CAD and actively involved in disease progression is still poorly understood.
In the present study, we identified the sEV-incorporated lncRNA PUNISHER (also known as AGAP2-antisense RNA 1 [AS1]) as being significantly upregulated in CAD patients. In vitro, we found that PUNISHER can be selectively exported into sEVs via a heterogeneous nuclear ribonucleoprotein K (hnRNPK)-dependent mechanism. sEV-mediated intercellular transfer of PUNISHER increased the expression of vascular endothelial growth factor A (VEGFA) in recipient cells and promoted an angiogenic response. Our mechanistic studies indicate that PUNISHER exerts its function by interacting with the RNA-binding protein (RBP), hnRNPK, to regulate the expression of the proangiogenic protein VEGFA in endothelial cells (ECs).
Results
Isolation and characterization of circulating sEV
A scheme of the workflow for the clinical lncRNA study is shown in Figure 1A. Circulating sEVs were isolated from the plasma of patients using a differential ultracentrifugation method (Figure S1A), as previously described.35 sEVs isolated from plasma samples were characterized prior to the isolation of RNA. The sEVs were characterized according to the current recommendations of the International Society for Extracellular Vesicles (ISEV).35 Immunoblotting analyses revealed that the isolated sEV expressed a distinct set of marker proteins, including CD9, CD81, and Syntenin135,36 (Figure S1B). Nanoparticle tracking analysis (NTA) showed that most of the isolated vesicles were 50–150 nm in diameter (Figure S1C), which is in line with the characteristic size (<150 nm) of sEVs.13 Transmission electron microscopy (TEM) confirmed the size and morphology of the sEVs (Figure S1D).
Figure 1.

lncRNA expression in circulating small extracellular vesicles (sEVs) from patients with coronary artery disease (CAD) or without CAD (non-CAD [NCAD])
(A) Schematic representation of the clinical long noncoding RNA (lncRNA) study. (B) Volcano plot showing differentially regulated human lncRNAs in sEVs derived from patient plasma by using a PCR-based human lncRNA array. Thresholds (dotted lines) of a 2-fold change and p values (false discovery rate [FDR] adjusted) <0.05 were set to distinguish the lncRNAs of interest; n = 3 for NCAD; n = 3 for CAD. (C) Expression of circulating sEV-associated PUNISHER was analyzed in NCAD (n = 30) and stable CAD (n = 30) patients by qRT-PCR. Values were normalized to GAPDH and expressed as 2−[CT(lncRNA) − CT(GAPDH)] log10. Data are presented as the mean ± SD (p = 0.0023, by Student’s t test). (D) List of the four lncRNAs analyzed in plasma sEV from the NCAD (n = 30) and stable CAD (n = 30) groups by qRT-PCR. (E) Top, a schematic representation of the vesicle-RNA degradation assays; bottom, plasma sEVs were treated in parallel using different conditions following RNase A digestion. PUNISHER was quantified by qRT-PCR (∗p < 0.05, compared with the untreated group; ns, not significant, n = 3, by 1-way ANOVA with Bonferroni correction for multiple comparisons test).
Identification of candidate lncRNAs
In order to explore whether sEV-incorporated lncRNAs are regulated differently in patients with or without CAD (non-CAD [NCAD]), a PCR-based human lncRNA array was performed in the screening cohort, which consisted of patients with stable CAD (n = 3) and NCAD (n = 3). The lncRNA array revealed a number of lncRNAs that were differentially expressed in sEVs. When thresholds of a >2-fold difference and a p value <0.05 were applied, the lncRNA PUNISHER was shown to be significantly upregulated in CAD patients, along with several other lncRNAs (ABHD11-AS1, GAS5, APOC1P1, DISC2, etc.) (Figure 1B; Table S5). To validate the lncRNA array results, we prospectively studied another 60 patients, one-half with CAD (n = 30) and one-half NCAD (n = 30). Based on the results of the array and the well-known regulatory roles for certain lncRNAs in atherosclerosis, four lncRNAs (MALAT1,37,38 GAS5,39,40 AGAP2-AS1 [PUNISHER],41 and H1942,43) were selected for single quantitative real-time PCR (qRT (reverse transcriptase)-PCR) analysis in the validation cohort (Table S2).
sEV-encapsulated lncRNA PUNISHER is upregulated in the plasma of CAD patients
Single qRT-PCR results showed that two of the lncRNAs had significantly higher expression levels in patients with stable CAD than patients with NCAD: PUNISHER (p = 0.0023; Figure 1C) and GAS5 (p = 0.0251; Figure S2A). H19 and MALAT1 showed no significant differences between the two groups (Figures S2B and S2C). In summary, PUNISHER showed the most significant difference in expression levels between CAD and NCAD patients among the four lncRNAs studied (Figure 1D).
Baseline characteristics for the validation cohort are presented in Table 1. There was no significant difference with respect to age or gender between the two groups. Stable CAD patients were more frequently on low-dose aspirin and clopidogrel regimens (p = 0.005 and p = 0.001). The NCAD group had higher levels of high-density lipoprotein (HDL) cholesterol (p = 0.016), but other standard laboratory parameters were similar between the two groups (Table 1).
Table 1.
Baseline characteristics of the study population in the screening phase
| Characteristic | Total (n = 60) | NCAD (n = 30) | Stable CAD (n = 30) | p value |
|---|---|---|---|---|
| Age (year) | 64.6 ± 10.7 | 63.8 ± 11.1 | 65.3 ± 10.4 | 0.584 |
| Gender (no.; %) | 0.542 | |||
| Female | 14 (23.3%) | 8 (26.7%) | 6 (20.0%) | |
| Male | 46 (76.7%) | 22 (73.3%) | 24 (80.0%) | |
| Cardiovascular risk factors (%) | ||||
| Arterial hypertension | 44 (73.3%) | 23 (76.7%) | 21 (70.0%) | 0.559 |
| Hyperlipoproteinemia | 23 (38.3%) | 11 (36.7%) | 12 (40.0%) | 0.791 |
| Family history | 13 (21.7%) | 9 (30.0%) | 4 (13.3%) | 0.117 |
| Diabetes | 21 (35.0%) | 11 (55.0%) | 10 (33.3%) | 0.524 |
| Smoking | 17 (28.3%) | 7 (23.3%) | 10 (33.3%) | 0.390 |
| Body mass index, kg/m2 | 27.4 ± 3.9 | 27.2 ± 4.6 | 27.7 ± 3.0 | 0.655 |
| Medical history (%) | ||||
| Previous bypass | 0 (0%) | 0 (0%) | 0 (0%) | 1.000 |
| Previous MI (6 months) | 0 (0%) | 0 (0%) | 0 (0%) | 1.000 |
| Previous stroke | 2 (3.3%) | 2 (6.7%) | 0 (0%) | 0.150 |
| Chronic kidney disease | 2 (3.3%) | 1 (3.3%) | 1 (3.3%) | 1.000 |
| PCI | ||||
| Previous | 36 (60%) | 18 (43.3%) | 23 (76.7%) | 0.008 |
| Coronary artery disease (no.; %) | 0.001 | |||
| 1 vessel | 3 (5.0%) | 0 (0%) | 3 (10.0%) | |
| 2 vessels | 12 (20.0%) | 0 (0%) | 12 (40.0%) | |
| 3 vessels | 13 (21.7%) | 0 (0%) | 13 (43.3%) | |
| Medication on administration (no.; %) | ||||
| ACE inhibitors | 42 (70.0%) | 18 (60.0%) | 24 (80.0%) | 0.091 |
| Angiotensin receptor blockers | 11 (18.3%) | 5 (16.7%) | 6 (20.0%) | 0.739 |
| Beta blockers | 51 (85.0%) | 25 (83.3%) | 26 (86.7%) | 0.718 |
| Calcium channel blockers | 10 (16.7%) | 5 (16.7%) | 5 (16.7%) | 1.000 |
| Diuretics | 24 (40.0%) | 13 (43.3%) | 11 (36.7%) | 0.598 |
| Statins | 48 (80.0%) | 20 (66.7%) | 28 (93.3%) | 0.010 |
| Nitrates | 4 (6.7%) | 2 (6.7%) | 2 (6.7%) | 1.000 |
| Aspirin | 47 (78.3%) | 19 (63.3%) | 28 (93.3%) | 0.005 |
| Clopidogrel | 10 (16.7%) | 0 (0%) | 10 (33.3%) | 0.001 |
| Laboratory parameters | ||||
| Glucose | 120.7 ± 60.3 | 116.3 ± 37.2 | 125.2 ± 77.8 | 0.583 |
| Hb A1c (%) | 6.8 ± 3.9 | 6.1 ± 0.7 | 7.5 ± 5.4 | 0.179 |
| Serum creatinine (mg/dL) | 1.0 ± 0.3 | 1.0 ± 0.4 | 1.0 ± 5.4 | 0.637 |
| Glomerular filtration rate (mL/min) | 65.6 ± 10.3 | 65.4 ± 11.1 | 65.7 ± 9.5 | 0.913 |
| Triglycerides (mg/dL) | 131.2 ± 76.3 | 120.5 ± 48.7 | 142.2 ± 96.7 | 0.278 |
| Cholesterol (mg/dL) | 176.6 ± 49.4 | 186.5 ± 54.0 | 166.6 ± 42.9 | 0.121 |
| HDL cholesterol (mg/dL) | 50.5 ± 14.9 | 55.1 ± 16.4 | 40.6 ± 11.8 | 0.016 |
| LDL cholesterol (mg/dL) | 106.0 ± 35.2 | 110.3 ± 39.5 | 101.7 ± 31.3 | 0.349 |
| C-reactive protein (mg/L) | 5.9 ± 10.8 | 5.3 ± 9.4 | 6.5 ± 12.3 | 0.665 |
| Leukocytes (109/L) | 7.3 ± 2.1 | 7.2 ± 1.9 | 7.5 ± 2.3 | 0.600 |
Continuous variables are presented as the mean ± SD, and categorical variables are presented as the percentage of patients. NCAD, non-coronary artery disease; stable CAD, stable coronary artery disease; MI, myocardial infarction; PCI, percutaneous coronary intervention; ACE, angiotensin-converting enzyme; Hb A1c, hemoglobin A1c; LDL, low-density lipoprotein; HDL, high-density lipoprotein. Chronic kidney disease was defined as a glomerular filtration rate <60 mL/min.
Comorbidities or medications have been shown to potentially affect the expression levels of sEV-bound ncRNAs.44 A binary logistic regression analysis showed that higher or lower levels of PUNISHER were not significantly associated with any comorbidities. Furthermore, PUNISHER levels were found to be independent of the use of medications (Table S3).
To explore whether circulating extracellular PUNISHER is encapsulated into sEVs or bound to protein complexes, we performed a vesicle-RNA degradation assay. We found that digestion of proteins only (using proteinase K), before treatment with RNase, did not affect the levels of PUNISHER RNA. In contrast, treatment with Triton X-100, which acts as a detergent to disrupt the phospholipid membrane of vesicles, before treatment with RNase, led to an almost complete degradation of PUNISHER RNA. These findings indicate that the circulating extracellular PUNISHER is primarily incorporated into sEVs, which protect the RNA from resident nucleases (Figure 1E). Additionally, we investigated the stability of the PUNISHER transcript in sEVs isolated from freshly collected plasma versus plasma stored at −80°C for 5 years. There was no significant difference in PUNISHER expression levels between the two groups (Figure S2D), confirming the stability of ncRNAs in frozen plasma over several years, if stored correctly.45
Atherosclerotic stimuli cause endothelial sEV to be enriched in PUNISHER in vitro
Because we found that sEVs containing PUNISHER were significantly upregulated in patients with CAD, and previous studies show that PUNISHER is an endothelial-specific lncRNA,41,46 we next explored whether pro-atherosclerotic conditions might regulate cellular or sEV-incorporated PUNISHER expression levels in vitro. First, EC-derived sEVs were isolated from human coronary artery ECs (HCAECs) through a series of differential ultracentrifugation steps, as previously described (Figure S2A). The resulting sEVs were characterized by immunoblotting for sEV marker expression, NTA for size distribution, and TEM for morphology (Figures S3A−S3C). To confirm that PUNISHER is expressed primarily in the heart, expression profiling of PUNISHER in eight major human tissues was performed by using qRT-PCR on commercially available cDNA from eight different tissues (major organs) of human origin (Human Total RNA Master Panel II; Clontech; #636643) (Figure S3D). The origins of PUNISHER-enriched vesicles were investigated further by the utilization of different vascular cells (vascular smooth muscle cells, HCAECs, and human umbilical vein endothelial cells [HUVECs]), as well as immune cells, such as platelets. Interestingly, we found that the lncRNA PUNISHER is enriched in ECs and their corresponding sEVs (Figure S3E).41 The expression of PUNISHER in HCAECs and the corresponding sEVs was confirmed by qRT-PCR (Figure S4A). Recent investigations indicated that oxidized low-density lipoprotein (oxLDL) or tumor necrosis factor alpha (TNF-α) plays pivotal roles in the development of atherosclerosis and endothelial dysfunction, and they are extensively used in vitro and in vivo as atherosclerotic stimuli.47, 48, 49, 50, 51 Intriguingly, stimulation with oxLDL or TNF-α caused significant upregulation of PUNISHER levels, both in the parental ECs and in EC-derived sEVs, without affecting the overall number of sEVs (Figures 2A-2D; Figure S4B).
Figure 2.

Atherosclerotic stimuli increase PUNISHER expression in endothelial cells (ECs) and corresponding sEVs in vitro
(A and B) PUNISHER was analyzed in ECs upon stimulation with different concentrations of oxidized low-density lipoprotein (oxLDL) or tumor necrosis factor α (TNF-α) by using qRT-PCR (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 4, by 1-way ANOVA with Bonferroni correction for multiple comparisons test). (C and D) PUNISHER was analyzed in sEVs following stimulation with different concentrations of oxLDL or TNF-α by using qRT-PCR. Cycle of threshold (CT) values were normalized to GAPDH, and expression was depicted as fold change (∗p < 0.05, ∗∗p < 0.01, n = 4, by 1-way ANOVA with Bonferroni correction for multiple comparisons test). Data represent the mean ± SD.
Based on the current annotation, AGAP2-AS1 (also known as PUNISHER) is an RNA gene, which has been categorized to the ncRNA class. Unlike protein-coding genes, PUNISHER does not contain any obvious open reading frame (ORF).41
By definition, the lncRNA PUNISHER is not translated into a protein. In silico predictions of the protein-coding potential of PUNISHER indicate that it is a bona fide lncRNA that does not encode proteins. Since the subcellular localization of a given transcript may be considered an important clue about its function, we investigated PUNISHER localization and demonstrated that PUNISHER is localized to both the cytosol and nucleus by fractioning the RNA followed by qRT-PCR-based quantification. Intriguingly, PUNISHER is highly enriched in the cytosolic fraction, compared to the nuclear fraction, of ECs (Figure S5A).
PUNISHER exerts its function via interaction with the RBP hnRNPK
We next aimed to study the mechanism that regulates the enrichment of PUNISHER within sEVs. Recently, RBPs have been shown to be important regulators of the packaging and sorting of ncRNAs into EV.52, 53, 54 However, thus far, there is little known about the mechanisms of packaging lncRNAs into endothelial sEVs. To investigate the molecular mechanism of ncRNA packaging, we re-analyzed our previously published proteomic analysis of EVs.54 We found that a significant portion of the protein candidates that were identified were annotated as RBPs, according to the SwissProt database. In particular, hnRNPs were prominently expressed in isolated EVs, including hnRNPA2B1, hnRNPU, and hnRNPK, as previously published by our group.54
hnRNPs are a large family of RBPs that have been reported to mediate the loading of ncRNAs into EV.53,55 To examine whether the packaging of PUNISHER into EVs is mediated by hnRNPs, we utilized publicly available online tools to predict PUNISHER-hnRNP interaction partners by using the catRAPID omics algorithm.56 In this way, we identified hnRNPK as the top-ranked potential interaction partner for PUNISHER in silico (Table S4). We performed a cross-linked RNA immunoprecipitation (RIP) experiment by using an antibody against hnRNPK in ECs and sEVs to experimentally confirm the binding between PUNISHER and hnRNPK (Figures 3A-3C). Interestingly, qRT-PCR analysis revealed that the interaction of hnRNPK with PUNISHER is specific, and no other lncRNAs (GAS5, MALAT1, and H19) are enriched in either EC or sEVs from the RIP experiments (Figures 3A and 3B). To further investigate whether hnRNPK interacts with other lncRNAs that were upregulated in our clinical samples, we used the RNA Interactome (RNAInter) database to make an in silico prediction of the interaction of hnRNPK with lncRNAs, based on the coverage score. We found that PUNISHER and MALAT1 are predicted to bind to hnRNPK, but not H19 and GAS5, based on the high-interaction scores that they give57 and compared with other databases (Figure 3D; Table S6).58,59
Figure 3.

Interaction between PUNISHER and heterogeneous nuclear ribonucleoprotein K (hnRNPK) mediates PUNISHER packaging into sEVs
(A) Cross-linked RNA immunoprecipitation (RIP) experiments followed by qRT-PCR quantification was performed to confirm the reciprocal binding of PUNISHER, MALAT1, H19, and GAS5 to hnRNPK in human ECs. From the RIP experiments, strong binding of hnRNPK was observed in comparison to IgG (negative control). Quantification was performed by normalizing the values with the inputs (10%). Data represent the mean ± SD (∗∗∗p < 0.001, n ≥ 4, by Student’s t test). (B) RIP experiments in sEV lysates followed by qRT-PCR quantification were performed to confirm the binding of PUNISHER, MALAT1, H19, and GAS5 in sEVs. Data represent the mean ± SD (∗∗∗p < 0.001, n ≥ 4, by Student’s t test). (C) Immunoprecipitation (IP) of hnRNPK followed by western blotting confirmed the reciprocal binding of hnRNPK to PUNISHER. The respective IgG antibody from the same species was used as an IP negative control. The enrichment was confirmed with input (10%) from the cellular lysates. (D) In silico target prediction of hnRNPK by using publicly available tools, based on the coverage score, by using RNAInter,57 RNACentral,58 and Arena-Idb59 databases. Based on their interaction scores, only MALAT1 and AGAP2-AS1 (PUNISHER) are predicted to bind to hnRNPK. (E) Expression levels of several lncRNAs (PUNIHSER, MALAT1, H19, and GAS5) upon silencing of PUNISHER/hnRNPK in ECs by using siRNAs (∗∗∗p < 0.001, n = 4, by Student’s t test). (F and G) Expression of PUNISHER upon silencing of hnRNPK in ECs and their corresponding sEVs by using siRNA (∗∗∗p < 0.001, n = 4, by Student’s t test). (H) Western blot analysis of hnRNPK protein expression was performed on the hnRNPK siRNA- or control siRNA-transfected ECs. β-actin was used as a marker. hnRNPK protein levels were assessed using ImageJ image analysis software (∗∗p < 0.01, n = 4, by Student’s t test). (I) Prior to oxLDL treatment, ECs were transfected with hnRNPK siRNA or control siRNA. PUNISHER expression was analyzed in the corresponding sEVs by qRT-PCR. GAPDH was used as an endogenous control (∗p < 0.05, n = 3−4, by Student’s t test).
We then evaluated the effect of hnRNPK knockdown on sEV-incorporated PUNISHER levels and other lncRNA expression in ECs (Figure 3E). We found that upon hnRNPK knockdown in EC, the expression of PUNISHER is reduced, whereas other lncRNAs remain unchanged, suggesting that the interaction of hnRNPK to PUNISHER is specific (Figure 3E). Importantly, hnRNPK depletion reduced the levels of PUNISHER in ECs and the corresponding sEVs (Figures 3F-3H), demonstrating that hnRNPK may participate in the loading of PUNISHER into sEVs. Furthermore, in vitro atherosclerotic stimulus, oxLDL-induced PUNISHER upregulation within sEVs, was abrogated upon hnRNPK knockdown, which confirms the importance of hnRNPK-mediated PUNISHER packaging under atherosclerotic conditions (Figure 3H; Figure S5B). Loss of PUNISHER and hnRNPK function leads to a reduction of PUNISHER and hnRNPK mRNA level in the corresponding sEVs, but no significant effects were observed in the case of other lncRNAs, namely, GAS5, H19, and MALAT1, further suggesting that hnRNPK is an interaction partner for PUNISHER and regulates the packaging of ncRNA into sEVs (Figure 3I). In summary, our data clearly indicate that hnRNPK is an interaction partner for PUNISHER, which may regulate the loading of PUNISHER into sEVs.
sEV-incorporated PUNISHER regulates target EC function
Increasing evidence suggests that sEVs act as a vehicle for the intercellular transfer of their contents to modulate the fate of recipient cells.39,53,60,61 Because circulating sEV-containing PUNISHER might exert its main biological effect on target ECs, we first investigated whether endothelial-derived sEVs could deliver PUNISHER into target ECs. By co-incubating sEVs and ECs, we observed a time-dependent internalization of sEVs into the recipient ECs (Figure 4A). The transfer of PUNISHER into target ECs was diminished using PUNISHER-downregulated sEVs (sEVPUNISHER downregulated), which were generated from ECs following transfection with PUNISHER small interfering RNAs (siRNAs) (Figure 4B). The efficiency of PUNISHER downregulation following siRNA transfection was confirmed using qRT-PCR in ECs and the corresponding sEVs (Figure 4C), without affecting other lncRNAs (Figures S6A−S6C). An qRT-PCR-based absolute quantification assay in target ECs confirmed the efficient delivery of PUNISHER by sEV (Figure S6D); furthermore, compared with sEVs, sEVPUNISHER downregulated transferred less PUNISHER into the recipient ECs (Figures 4D-4F). Moreover, to distinguish between endogenous and exogenously delivered PUNISHER, we performed copy number analysis on PUNISHER-downregulated recipient ECs. The results demonstrated that sEV transfer could rescue the reduced level of PUNISHER in PUNISHER-silenced target ECs (Figure 4E; Figure S6D). To confirm whether sEV-mediated PUNISHER transfer exists in vitro, we established a Transwell-based experiment by utilizing PUNISHER-/hnRNPK-silenced donor cells and comparing them with control ECs. We observed that the sEVs have the potential to deliver PUNISHER to the recipient cells. These data indicate that there is reduced transfer of PUNISHER from both PUNISHER- and hnRNPK-silenced donor cells into the corresponding recipient ECs via sEV and suggest that hnRNPK is involved in EV-mediated PUNISHER transfer.
Figure 4.

sEV-incorporated PUNISHER is transferred into recipient ECs
(A) PKH67-labeled sEVs (green) were cultured with recipient ECs for 0, 0.5, 6, and 24 h. The nuclei of the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue), and images were taken by using immunofluorescence microscopy. Scale bar, 20 μm. (B) PUNISHER expression was assessed in target ECs that were treated with sEVPUNISHER downregulated or sEVmock transfected using qRT-PCR. CT values were normalized to GAPDH and were expressed as fold change (∗p < 0.05, n = 3, by Student’s t test). (C) PUNISHER expression was analyzed in PUNISHER-siRNA- or control-siRNA-transfected ECs and the corresponding sEVs by qRT-PCR. GAPDH was used as an endogenous control (∗p < 0.05, ∗∗∗p < 0.001, n = 3−4, by Student’s t test). (D−F) PUNISHER expression was assessed in target ECs that were treated with PBS, sEVs, sEVPUNISHER downregulated, or sEVmock transfected using copy number analysis (∗p < 0.05, ∗∗∗p < 0.001, n = 4, by 1-way ANOVA with Bonferroni multiple comparisons test). (E) Transwell experiments with normal, siScr, siPUNISHER, and sihnRNPK in donor cells and recipient cells. PUNISHER expression was quantified in donor and target ECs that were treated with sEVs from cells transfected with siRNAs. GAPDH was used as a control. (∗∗p < 0.01, ∗∗∗p < 0.001, Student’s t test). HCAECs, human coronary artery ECs.
Next, we investigated the function of sEV-incorporated PUNISHER. Because PUNISHER has been shown to be a major regulator of EC function,41 we investigated whether the PUNISHER RNA found in sEVs can directly affect target EC migration, proliferation, in vitro sprouting, and network formation. Treatment of target ECs with sEVs caused an increase in EC migration (Figure 5A; Figure S7A), proliferation (Figures 5B and 5C), and network formation (Figures 5D and 5E; Figures S7B and S7C), whereas PUNISHER silencing reduces in vitro sprouting of ECs (Figures 5F and 5G). Importantly, sEV-mediated effects on the recipient ECs were abolished when sEVPUNISHER downregulated were used, indicating that the PUNISHER found in the sEVs regulates target EC function and phenotypes by promoting angiogenic responses. Consistent with these findings, the downregulation of PUNISHER in ECs resulted in reduced migration (Figures S8A and S8B), proliferation (Figure S8C), and network formation (Figures S8D−S8G).
Figure 5.

PUNISHER in sEVs regulates target EC function
sEVPUNISHER downregulated and sEVmock transfected were separately derived from parent ECs transfected with PUNISHER siRNA or control siRNA. ECs were co-incubated with sEVs, sEVPUNISHER downregulated, sEVmock transfected, or vehicle. (A) A scratch-migration assay was performed, and representative images of cells migrating into the scratched region after 0, 4, and 8 h are shown. Quantitative analysis of the migration was measured as a percentage of total cell-free area (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 5, by 1-way ANOVA with Bonferroni multiple comparisons test). Scale bars, 100 μm. (B and C) Bromodeoxyuridine (BrdU) incorporation was determined by immunofluorescence (red). Nuclei were stained with DAPI (blue). The percentage of BrdU-positive cells was compared with the total number of cells (∗p < 0.05, ∗∗p < 0.01, n = 6, by 1-way ANOVA with Bonferroni’s multiple comparisons test). Scale bars, 20 μm; 100× magnification. (D and E) Network formation assays of ECs. Capillary tubes were imaged with an immunofluorescence microscope. Total tube length was measured and quantitated by using ImageJ image analysis software (NIH, USA; ∗∗p < 0.01, ∗∗∗p < 0.001, n = 6, by 1-way ANOVA with Bonferroni multiple comparison test). Scale bar, 200 μm. (F and G) In vitro sprouting assay in which ECs are co-incubated with sEVs with or without vascular endothelial growth factor A (VEGFA; 5 μg/mL), sEVPUNISHER downregulated, or sEVmock transfected. Cumulative sprouting length was measured and quantitatively analyzed using digital imaging analysis software (AxioVision Rel. 4.8; Carl Zeiss; ∗∗∗p < 0.001, n = 6, by 1-way ANOVA with Bonferroni multiple comparisons test). Scale bar, 200 μm.
sEV-mediated transfer of PUNISHER regulates VEGFA transcripts in recipient ECs
To explore the underlying mechanism of PUNISHER (delivered via sEVs) for the regulation of angiogenic functions in the recipient ECs, we used two independent experimental approaches: (1) direct downregulation of PUNISHER expression in ECs using siRNAs (PUNISHER siRNA versus control siRNA) and (2) treatment of recipient ECs with sEVs in which PUNISHER has been downregulated (sEVPUNISHER downregulated versus sEVmock transfected). An RT2 Profiler PCR array, which covers 84 key genes that are involved in modulating the biological processes of angiogenesis, was performed on both of the above-mentioned models. In the cellular model of PUNISHER knockdown, analysis of the array results revealed that a series of pro-angiogenic genes, including pro-inflammatory factors (interleukin 1 beta [IL-1β], IL-6, and TNF-α), as well as other cellular factors (chemokines, vascular endothelial factors), were significantly downregulated (Figure 6A). When ECs were treated with sEVPUNISHER downregulated and compared with sEVmock transfected, the array results showed that the anti-angiogenic C-X-C motif chemokine 10 (CXCL10) was upregulated, and four pro-angiogenic genes were downregulated by at least 2-fold, namely jagged1 (JAG1), matrix metalloproteinase-2 (MMP-2), angiopoietin 4 (ANGPT4), and VEGFA (Figure 6B). Interestingly, when the overall angiogenesis gene-expression profile was compared between the two models, VEGFA, a well-known, potent pro-angiogenic factor,62 was found to be decreased upon downregulation of PUNISHER in both arrays (Figures 6A and 6B).
Figure 6.

sEV-mediated transfer of PUNISHER RNA regulates the level of VEGFA in recipient ECs
RT2 Profiler PCR array analyses (angiogenesis) were performed on ECs treated with PUNISHER siRNA and control siRNA-transfected ECs (n = 3). The same array was performed on ECs treated with sEVmock transfected or sEVPUNISHER downregulated (n = 3). Ribosomal protein lateral stalk subunit P0 (RPLP0; as PCR array control) was used as an endogenous control in the PCR array. (A) A scatterplot shows the differentially regulated genes between PUNISHER knockdown and control ECs by PCR array. The downregulated genes with a >2-fold change are labeled and listed. (B) A scatterplot shows the differentially regulated genes in target ECs after sEVmock-transfected or sEVPUNISHER-downregulated treatment by PCR array. The five genes with a >2-fold change are labeled and listed. (C) Differentially expressed genes in the PCR array with their respective fold change in donor and recipient ECs. (D) Venn diagram showing common genes in both assays. VEGFA was the only gene shared by the two PCR arrays (fold change = 2.0). (E and F) Individual qRT-PCR validation of VEGFA, which was downregulated in the angiogenesis arrays. The downregulation of the VEGFA gene from the array was confirmed by individual qRT-PCR in ECs. VEGFA RNA expression was analyzed in ECs by qRT-PCR (∗p < 0.5, n = 3, by Student’s t test). (G and H) VEGFA protein levels in donor or recipient ECs were quantified from western blotting.
To confirm the above findings, single qRT-PCR, immunoblotting, and enzyme-linked immunosorbent assay (ELISA) were performed. Notably, PUNISHER downregulation in ECs resulted in a reduction in VEGFA expression on both the mRNA (Figure 6E) and protein levels (Figure 6G; Figure S9A). Besides this, we also analyzed the expression of another dysregulated gene, CXCL10, in donor and recipient cells (Figures S9D and S9E). In line with these findings, ECs treated with sEVPUNISHER downregulated showed reduced expression of VEGFA mRNA (Figure 6F), protein (Figure 6H; Figure S9B), and in the cellular supernatants (Figures 7A and 7B) compared to sEVmock transfected. Furthermore, upon PUNISHER knockdown, although VEGFA mRNA was downregulated in the parent and recipient ECs (Figure 6E; Figure 7B), VEGFA mRNA was not significantly decreased in the corresponding sEVs (sEVPUNISHER downregulate) (Figures S9B and S9C). These data exclude the possibility of VEGFA mRNA transfer via sEVs but instead raise a question: how does PUNISHER regulate mRNA expression or protein synthesis of VEGFA? To explore this mechanistically, we performed hnRNPK immunoprecipitation (IP) and RIP experiments using EC cellular lysates, which confirmed that hnRNPK is an interaction partner for PUNISHER. hnRNPK has been reported to bind to the 3′ UTR (untranslated region) of mRNA, to post-transcriptionally regulate its levels by regulating its stability,63, 64, 65, 66 and is known to regulate the angiogenesis pathway by acting as a transcriptional activator of VEGF.63 To confirm this, we performed RIP followed by qRT-PCR (RIP-qPCR), which indicated a reduction in VEGFA mRNA binding to hnRNPK. Although enrichment of VEGFA mRNA in PUNISHER- or hnRNPK-silenced cells is comparable with scramble control short-interfarying RNA (siScr)-treated cells, a lower level of enrichment was recorded in both cells (Figure 7C). To examine whether such binding has any effect on the transcript stability of VEGFA upon PUNISHER silencing, we treated cells with actinomycin D (5 μg/mL) to inhibit RNA synthesis and measured the VEGFA expression profile via qRT-PCR. Interestingly, we observed that the expression of the VEGFA transcript is downregulated in PUNISHER-silenced cells, in comparison to siScr-treated cells, suggesting that PUNISHER may regulate VEGFA transcription (Figure 7D). Next, to experimentally validate the involvement of hnRNPK in the post-translational regulation of VEGFA, cycloheximide (CHX; an inhibitor of protein synthesis)-mediated protein stability assays were performed upon PUNISHER silencing, and the stability of cellular VEGFA was detected (Figure 7E). In addition, we investigated whether impairment of the angiogenic response in PUNISHER-silenced ECs and recipient ECs can be rescued by exogenous supplementation with the VEGFA protein. To address this point, we performed a recipient EC network formation assay, with or without VEGFA supplementation. Our rescue experiments revealed that the angiogenic activity in EC receiving EVs from PUNISHER-silenced cells can only be rescued by adding a high concentration of extracellular VEGFA (5,000 pg/mL; Figure S10) but not with a lower concentration (500 pg/mL; data not shown). This result excluded that there are different levels of VEGFA mRNA transport by EVs (Figure 7F).
Figure 7.

PUNISHER is crucial for VEGFA regulation
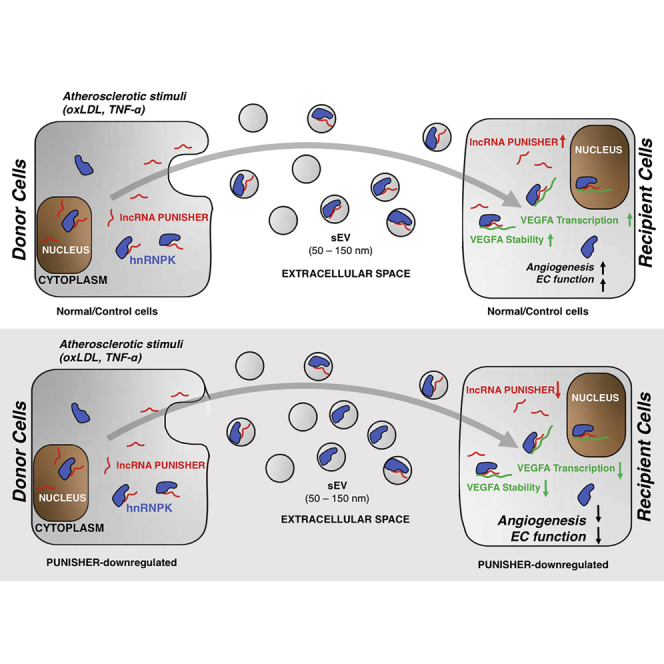
(A and B) ECs were transfected with PUNISHER siRNA or control siRNA. Secreted VEGFA protein levels were quantified via enzyme-linked immunosorbent assay (ELISA) from donor-EC culture media and recipient cells treated with sEVs after siScr and siPUNISHER treatment (∗∗p < 0.01, n = 6 by Student’s t test). (C) Cross-linked RIP experiments followed by qRT-PCR quantification to confirm consequence of PUNISHER or hnRNPK silencing regulate binding of VEGFA mRNA to RBP. This reciprocal binding of PUNISHER to hnRNPK in human ECs is important to control VEGFA protein synthesis. From RIP experiments, binding of hnRNPK was quantified in comparison to IgG (negative control) upon siRNA treatment. Quantification was performed by normalizing the values with the inputs (10%). Data represent the mean ± SD (∗∗∗p < 0.001, n ≥ 4, by Student’s t test). (D) The stability of PUNISHER and VEGFA RNA by using actinomycin D (Ac-D) treatment (5 μg/mL for different time points) upon siRNA knockdown of PUNISHER or a scrambled control. The expression of mRNAs was analyzed by qRT-PCR in donor cells after siRNA treatment (∗p < 0.5, n = 6, by Student’s t test). (E) Stability of VEGFA protein was analyzed at different time points by using cells treated with cycloheximide (CHX) to a final concentration of 25 μg/mL upon siRNA treatment against PUNISHER or a scrambled siRNA control by western blotting. Anti-histone-3 (H3) was used as a loading control. (F) Schematic representation of the working model of sEV-incorporated PUNISHER transport. Atherosclerotic stimuli increase the level of PUNISHER both in donor cells and incorporated into sEVs. hnRNPK regulates the loading of PUNISHER into sEVs from ECs. sEV-mediated transfer of PUNISHER regulates the migration, proliferation, sprouting, and network formation of recipient ECs by controlling VEGFA stability.
Discussion
Circulating ncRNAs are a novel class of biomarkers in cardiovascular disease.7,67 Previous studies have highlighted that circulating EVs are one of the major vehicles for ncRNAs in the bloodstream, providing protection from circulating RNases and promoting stability in the plasma.7,68 Previously, we found that EV-incorporated, but not freely circulating, ncRNAs (miRNA-126 and miRNA-199a) could predict the occurrence of cardiovascular events in patients with stable CAD.69 Thus far, miRNAs and lncRNAs are the most prominent and well-characterized ncRNAs. However, there is evidence that miRNAs are not the most abundant ncRNAs in EVs.70,71 Whereas EV-incorporated miRNAs have already been characterized in a number of studies, the function of EV-incorporated lncRNAs, especially in CAD, is largely unknown. Moreover, in the bloodstream, the quantity of lncRNAs in sEVs is higher than in other types of EVs.72 In this study, we sought to elucidate the role of sEV-incorporated lncRNA PUNISHER in CAD patients and to understand their impact on the regulation of vascular integrity.
We utilized a PCR-based human lncRNA array to investigate differentially regulated lncRNAs in sEVs that were derived from plasma of patients with CAD or NCAD. This array revealed a number of lncRNAs that were highly upregulated in CAD patients. Based on the results of our array as well as the current literature, we investigated the plasma-sEV levels of four atherosclerosis-related lncRNAs: PUNISHER, GAS5, H19, and MALAT1. Among them, the largest difference between NCAD and stable CAD patients was seen for PUNISHER. The majority of lncRNA studies in CAD have focused on whole circulating lncRNAs (in plasma, serum, or whole blood) rather than EV-incorporated circulating lncRNAs.7,73 Notably, a recent study demonstrated that the lncRNA CoroMarker, which is considered to be a novel biomarker for CAD, is primarily found in circulating EVs.74 Our current findings provide further in-depth insights into the regulation of the EV-incorporated lncRNA PUNISHER under the clinical and experimental conditions of vascular diseases.
In analogy to our clinical findings, the in vitro experiments revealed that PUNISHER levels increased following stimulation with oxLDL and TNF-α in ECs and EC-derived sEVs, which indicates a response to two independent pro-atherogenic stimuli. Recent studies indicated that lncRNAs act as key players in atherosclerosis-related pathological conditions, e.g., EC functional dysregulation and vascularization.75,76 PUNISHER is known to be required for angiogenesis.41
It has been shown previously that ncRNAs are selectively packaged by cells into the corresponding EVs.60 Previous studies have revealed RBPs, including the hnRNP family, which can facilitate selective packaging of ncRNAs into EV (e.g., hnRNPA2B1, Y box binding protein 1 [Y-BOX1], SYNCRIP, and Argonaute protein 2 [AGO2]).52,77 In the present study, we discovered that the multifunctional RBP hnRNPK interacts with PUNISHER and facilitates its loading into sEVs, providing novel mechanistic insights into the cellular sorting and packaging of lncRNAs into sEVs. Finally, in recipient cells, PUNISHER regulates transcription and expression levels of the pro-angiogenic gene VEGFA.
The horizontal transfer of sEV cargoes represents an effective means of biological signaling between parent and recipient cells in diverse settings.78 Within this context, the exosome-transmitted lncRNA ARSR has been shown to ameliorate drug resistance in cancer.53 Furthermore, the cholangiocyte-derived exosomal lncRNA H19 was reported to accelerate cholestatic liver injury.79,80
Although studies have revealed that sEVs deliver lncRNAs in oncologic or other diseases, a role for sEV-mediated lncRNA transfer in the regulation of vascular function has not been previously shown. Our data show, for the first time, that sEV-mediated transfer of a vascular lncRNA, namely PUNISHER, into target ECs promotes an angiogenic response. sEV-incorporated PUNISHER downregulates mRNA levels of the pro-angiogenic protein VEGFA in recipient ECs. The role of angiogenesis in CAD is thought to have a duel effect.41,76,78 In ischemic heart disease (IHD), pro-angiogenetic factors could accelerate neointima formation and promote microvascular network growth, thus contributing to the improvement of myocardial reperfusion.76 In the adult organism, angiogenesis only takes place during the female menstrual cycle and during wound healing. Under pathological conditions, angiogenesis is involved in tumor development, diabetic retinopathy, and IHD, which can be a consequence of atherosclerosis, a sequelae of CAD. In atherosclerosis, angiogenesis facilitates the growth of atherosclerotic lesions; angiogenesis within plaques plays a crucial role in plaque destabilization and rupture.71 In our study, we found that the pro-angiogenic lncRNA PUNISHER is upregulated in the plasma sEVs of stable CAD patients. In vitro, PUNISHER was found to regulate the angiogenic function of recipient ECs via the transport of PUNISHER-incorporated sEVs. This result indicates that the increased level of PUNISHER in sEVs might be triggered by ischemia and can ameliorate myocardial ischemia in CAD patients by promoting neovascularization. However, in patients with stable plaques, the participation of PUNISHER in intraplaque angiogenesis and the potential for a resulting plaque rupture should also be considered. However, these findings are important to understand the mechanism of angiogenesis in CAD as well as other pathological settings (cancer), hinting that future investigations are required to provide deeper explanations for the clinical significance. Our results demonstrate that sEV-incorporated PUNISHER expression is significantly higher in CAD patients when compared with NCAD patients. In vitro, we found that functional PUNISHER can be transferred to target ECs, which promotes an angiogenic response. Pharmacological inhibition of PUNISHER expression is accompanied by an impairment of the angiogenic response and a decrease in cell proliferation.
Mechanistically speaking, the binding partner of PUNISHER, hnRNPK, is a multifunctional RBP that regulates the cytoplasmic fate of mRNAs by regulating their stability or translation, and it is known to bind a number of mRNAs, including VEGFA.63,64 We found that PUNISHER binds to the RBP hnRNPK, which changes the binding of hnRNPK to its cognate mRNA, VEGFA, by binding to its 3′ UTR, as previously reported.63,64 The stability of the VEGFA mRNA transcript is downregulated in PUNISHER-silenced cells in comparison to siScr-treated cells, suggesting that PUNISHER may also regulate the transcription of VEGFA. When we further analyzed the protein stability of VEGFA in these cells, it was confirmed that PUNISHER also regulates the stability of VEGFA protein, thereby controlling the angiogenic function of ECs.
Most importantly, when RIP-qPCR was performed with primer pairs that target the 3′ UTR (which contains the VEGFA-binding regions), significantly less binding of hnRNPK to VEGFA was recorded in PUNISHER- or hnRNPK-silenced cells (Figures 3D-3F). This result suggests that the interaction between PUNISHER and hnRNPK is indispensable for the efficient binding of VEGFA. When hnRNPK is knocked down, it is possible that the expression of VEGFA is reduced, which could be facilitated by the lncRNA PUNISHER, as we observed more binding on normally grown ECs than PUNISHER-silenced ECs and confirmed that PUNISHER is important for EC function. To our knowledge, this is the first time that clinical and experimental atherosclerotic conditions have been shown to promote the packaging of functional PUNISHER into sEVs, thus inducing an angiogenic phenotype in the recipient ECs. Taking all of the above-mentioned data together, we have confirmed that PUNISHER silencing lowers the stability of the basal level of VEGFA, which is regulated via the hnRNPK-PUNISHER interaction, thereby regulating the angiogenic function of ECs.
However, our study has some limitations, namely, the patient samples were all collected at the University of Bonn, Germany; thus, the patient-derived data presented are limited to the Caucasian race. In addition, we generated sEVPUNISHER downregulated through the efficient knockdown of PUNISHER in sEV-producing parent ECs. Although the expression levels of the other four lncRNAs analyzed were not affected in these sEVs, as confirmed by qRT-PCR, it is technically challenging to completely exclude that there was a change to any of the other biological contents of the sEVs.
In conclusion, our findings offer the following insights into the role of lncRNAs as intercellular signaling molecules. (1) We show for the first time that sEV-incorporated lncRNAs are regulated under the clinical and experimental conditions of atherosclerosis. (2) The lncRNA PUNISHER is primarily incorporated within sEVs, and this packaging happens in an hnRNPK-dependent manner. (3) PUNISHER is shuttled between cells via sEVs to increase the angiogenic response by increasing VEGFA expression in the recipient ECs. In summary, our study has revealed that sEV-incorporated PUNISHER has a dual function as both a biomarker and effector of vascular integrity.
Materials and methods
Study subjects
The study enrolled patients presenting in our outpatient and emergency departments between August 2012 and July 2013. All clinical samples were collected with the appropriate informed consent from patients in the department. Ethical approval was granted from the Ethics Committee of the University of Bonn (approval number [no.] 05/12) and followed the principles outlined in the Declaration of Helsinki. Based on the data from the clinical presentation, laboratory parameters, and coronary angiography, patients were classified into two groups: angiographic exclusion of obstructive CAD (<50% stenosis of a major coronary artery, named as NCAD) and stable CAD. Further exclusion criteria for our study were as follows: (1) chronic inflammatory disease, (2) acute kidney or liver disease, (3) malignant disease, and (4) the patients who declined to participate.
Preparation of blood samples
Blood samples were collected under sterile conditions from the cubital vein and buffered by using sodium citrate. In order to generate platelet-deprived plasma samples, blood was first centrifuged at 1,500 × g for 15 min; the supernatant was then centrifuged at 13,000 × g for 20 min. Platelet-deficient plasma samples were stored at −80°C, and repeated freeze-thaw cycles were avoided until the sEVs were isolated.
Cell culture
HCAECs (PromoCell) were cultured in EC growth media with endothelial growth media supplement mix (PromoCell; #C-39225) under standard cell culture conditions (37°C, 5% CO2) in a standard humidified incubator. Cells from passages 5–7 were used. At least three biologically independent batches of cells were studied in each experiment.
For the isolation of sEVs, HCAECs were cultured as previously described.69,81,82 Briefly, confluent cells were first washed 3 times with conditioned medium and then starved in conditioned medium (basal medium without growth media supplements; PromoCell; #C-22020) for 24 h. After starvation, the supernatant was collected for sEV isolation.
sEV generation
A previously described ultracentrifugation method was used for the isolation of sEVs.83 Briefly, 2.5 mL plasma (following 1:4 dilution) or 8 mL conditioned cell culture medium (collected from 8 × 106 donor ECs) was first centrifuged at 3,000 × g, 4°C for 15 min, to deplete the cell debris; lEVs were removed by centrifugation at 20,000 × g, 4°C for 40 min, as previously described.69,81,82 Then the supernatant was centrifuged at 100,000 × g, 4°C for 90 min, in a Beckman Coulter Optima LE-80K ultracentrifuge with a type SW 41Ti rotor (K-factor: 256.6) to pellet the sEVs. The pellet was resuspended in 8 mL sterile ice-cold PBS followed by re-centrifugation at 100,000 × g, 4°C for 90 min, to purify the sEVs.83,84 The sEV pellet was resuspended in sterile 1 × PBS and used immediately.
RNA isolation and qRT-PCR
Total RNA was isolated by using the miRNeasy Mini Kit (QIAGEN; #217004) extraction method according to the instructions of the manufacturer. RNA was diluted in UltraPure DNase/RNase-Free Distilled Water (Invitrogen). RNA concentration and purity (absorbance at 260/280 nm [A260/A280] and A260/A230) were quantified using a Nanodrop spectrophotometer (Table S1). 2 μg RNA (HCAEC) or 300 μg RNA (sEV) was reverse transcribed by using an Omniscript RT kit (QIAGEN; #205113), according to the manufacturer’s protocol. In addition, the purity and integrity of RNA isolated from patient plasma with CAD (n = 6) and NCAD (n = 6) were assessed by using an Agilent Bio-analyzer with the Agilent RNA 6000 Nano Kit (Agilent Technologies; #5067-1511). 5 ng of cDNA template was used for the quantification by using TaqMan assays (Thermo Fisher Scientific) on a 7500 HT Real-Time PCR machine (Applied Biosystems). The intra-/inter-assay variability of PUNISHER detection in plasma sEVs was <5%.
The expression profile of PUNISHER was checked by using human tissue samples, and purified RNA was purchased from commercial vendors as follows: Human Total RNA Master Panel II (Clontech; #636643; lot no. 1202050A); human heart (Amsbio; #R1234122-50; lot no. A804058).
Human lncRNA PCR array
The expression of 372 lncRNAs with known biological functions or disease associations as well as well-characterized functional lncRNAs was quantified via an Arraystar Human Functional lncRNA Array (Arraystar, Rockville, MD, USA). The samples were prepared based on the manufacturer’s standard protocols, with only minor modification. Briefly, total RNAs from sEVs isolated from the plasma of NCAD and CAD patients were isolated by using a miRNeasy kit (QIAGEN, Hilden, Germany), and 1.5 μg RNA was reverse transcribed by using an rt-STAR First-strand cDNA synthesis kit (Arraystar; catalog [cat.] #AS-FS-001), according to the manufacturer’s protocol. Six human housekeeping genes, ACTB, B2M, Gusb, Hsp90ab1, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and 18S rRNA, were included as the internal qPCR normalization references.
In-plate genomic DNA controls (GDCs) were used on the array plate to confirm the quality of the cDNA template for qRT-PCR analysis. Quantification of lncRNAs was performed by using the 7900HT Fast Real-Time PCR System. Data were normalized by using an in-plate housekeeping gene, GAPDH. Data analysis was performed with SDS 2.4 data analysis software (Applied Biosystems, USA), and further analyses were conducted with an RQ Manager 1.2.1 data analysis tool.
Atherosclerotic stimuli treatment in vitro
Confluent HCAECs were treated with 25 μg/mL or 50 μg/mL oxLDL (Alfa Aesar; #J65591) for 24 h. Similarly, HCAECs were treated by using different TNF-α (R&D Systems; #210-TA-020) concentrations (10 ng/mL, 20 ng/mL, or 50 ng/mL). Then the cells were subjected to basal media for 24 h to generate the corresponding sEVs.
Endothelial sEV internalization into recipient EC
HCAECs were stained with PKH67 (Sigma-Aldrich; #MIDI67), according to the manufacturer’s instructions. PKH67-labeled sEVs were washed twice with PBS. 2 × 105 recipient ECs were co-incubated with PKH67-labeled sEVs (1.5 μg sEV, equivalent to sEVs collected from 8 × 106 donor ECs) for 0, 0.5, 6, or 24 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories; #H-1200). A Zeiss Axiovert 200M microscope and ZEN 2.3 Pro software was used to visualize the uptake of sEVs into the recipient HCAECs.
Co-incubation of endothelial sEVs and recipient ECs
2 × 105 recipient ECs were co-incubated with 1.5 μg sEV (equivalent to sEV collected from 8 × 106 donor ECs) or PBS for 24 h. Then the ECs were washed three times with PBS. Total RNA was isolated from all EC samples for qRT-PCR analysis.
Knockdown experiments in ECs
Transient siRNA transfection (10 nM final concentrations) of semi-confluent HCAECs (50%–60% confluent) was carried out for 48 h using the HiPerFect reagent (QIAGEN), according to the manufacturer’s protocol.
RIP
A Magna RIP Kit (Millipore; #17-700) was used according to the manufacturer’s protocol. For each RIP reaction, 100 μL of the cellular pellet was fixed with 1% formaldehyde in PBS at room temperature for 10 min. For one RIP reaction (i.e., one IP using one antibody), 100 μL of cell lysate or ~5.0 × 106 cells were used. The cross-linking reaction was stopped by adding 590 μL of 2.5 M glycine. Fixed cells were subsequently harvested and resuspended in RIP lysis buffer supplemented with protease/RNase inhibitors. The lysates were obtained using a Dounce homogenizer on ice (10 passes to release the nuclei) followed by incubation on ice for 15 min. An equal volume of RIP lysis buffer was added to the cellular pellet. From the solution, 10 μL (10%) of the lysates was removed and stored as an “input,” as recommended by the manufacturer. For each RIP reaction, 100 μL of lysate was mixed with 5 μg of mouse anti-immunoglobulin G (IgG; negative control provided with the kit) or anti-hnRNPK antibody (Abcam) that had been previously conjugated with protein A/G magnetic beads (provided with the kit). After incubating overnight at 4°C, the RNA-protein precipitate was extensively washed with RIP Wash Buffer (provided with the kit). The cross-linking was reversed by incubation with proteinase K. The RIP was purified through phenol/chloroform/isoamyl alcohol (125:24:25) isolation. The purified RIP was treated with DNase I (Thermo Fisher Scientific; #AM2239) and reverse transcribed using SuperScript VILO Master Mix (Thermo Fisher Scientific; #11755050).
hnRNPK IP
IP was performed using the Dynabeads Protein G Immunoprecipitation Kit (Invitrogen). For one sample, with respective controls, 7.2 × 107 HCAECs were lysed in Nonidet P-40 (NP-40) cell lysis buffer (Invitrogen) and centrifuged at 13,000 × g for 20 min at 4°C. The lysate was pre-cleared with 50 μL Dynabeads per 1,000 μL lysate for 60 min at 4°C. After coupling of 10 μg of monoclonal mouse anti-hnRNPK antibody (Abcam) or mouse IgG1 kappa monoclonal isotype control antibody (Abcam) to 50 μL Protein G Dynabeads for 1 h at room temperature, 1,000 μL of lysate was incubated with the antibody-coupled beads for 16 h at 4°C. Samples were washed extensively, and the RNA was eluted twice with 20 μL of the provided elution buffer. Protein samples were isolated according to the recommended protocol of the manufacturer.
Western blotting
As a control for the sEVs isolated from plasma in immunoblotting, whole plasma was used, whereas for endothelial sEVs, cell lysate and conditioned media (growth media without growth media supplements; PromoCell; #C-22020) were used as the control. Cells were homogenized in radioimmunoprecipitation assay buffer (RIPA) buffer (Sigma-Aldrich; #R0278) including a protease inhibitor cocktail (Roche; #11 873 580 001) on ice. The protein concentration of the corresponding samples was measured by a Lowry protein assay (BioRad; #500-0116). An equal amount of protein (20 μg) for sEVs, plasma, or cells was loaded onto 4%–15% precast polyacrylamide gels (BioRad; #456-1084). Then the gel was transferred to a polyvinylidene fluoride (PVDF) membrane (Thermo Fisher Scientific; #88585), followed by blocking in 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TBST). The blots were incubated with the appropriate primary antibodies, and detection was performed by using the appropriate secondary antibody. The membranes were imaged using chemiluminescence via an Enhanced Chemiluminescence (ECL) Detection Kit (GE Healthcare; #RPN2232).
Electron microscopy
For TEM, sEVs from plasma or from ECs were pelleted by centrifugation, fixed in 1.25% glutaraldehyde in 0.1 M cacodylate buffer overnight, dehydrated with ethanol and propylene oxide, and embedded in Epon 812 (Serva), also overnight. After double-contrast staining with uranyl acetate and an aqueous lead solution, images were taken with a CM 10 electron microscope (Philips).
Vesicle-RNA degradation assay
For the degradation assay, 45 μL proteinase K (Thermo Fisher Scientific; #25530049) was used for protein digestion, with or without the presence of 45 μL Triton X-100 (Sigma-Aldrich; #T8787) for 30 min at 37°C. The samples were then treated with 5 μL RNase A (Thermo Fisher Scientific; #AM2271) for 20 min at 37°C. The untreated group was used as a normal control. Total RNA was isolated from all sEV samples for qRT-PCR analysis.
Manipulated sEV generation and recipient EC treatment
To generate sEVPUNISHER downregulated and sEVmock transfected, HCAECs were transfected with PUNISHER siRNA or control siRNA using HiPerFect for 48 h and exposed to conditioned media for 24 h to generate the modified sEVs. 2 × 105 recipient HCAECs were co-incubated with the same amount (1.5 μg) of sEVs, sEVPUNISHER downregulated, or sEVmock transfected and PBS for 24 h. ECs were washed three times with PBS. The ECs from different groups were then used for the following functional assays.
mRNA and protein stability
A total of 20,000 HUVECs were plated into a 6-well plate (Greiner bio-one; cat. #657160) prior to incubation for 24 h under optimal cell-growth conditions (5% CO2 and 37°C in an incubator). Afterward, the growth medium was replenished, followed by two times more after incubation. Cells were induced with 5 μg/mL actinomycin D (Sigma-Aldrich; product no. A9415) added to the culture medium, followed by a time-wise collection of RNA with TRIzol (Invitrogen) at 1, 2, 4, 6, 12, and 24 h. For CHX, 500 mg CHX powder was dissolved and homogenized in 5 mL sterile DMSO to prepare 10 mg/mL stock solution. A similar number of cells were treated with CHX to a final concentration of 25 μg/mL. Protein and RNA from untreated cells at 0 h were also collected as a control. For cDNA preparation, 1 μg RNA was treated with DNase I and reverse transcribed by using SuperScript VILO Master Mix (Invitrogen; cat. no. 11755050) prior to cDNA preparation, which was finally diluted to 1 ng/μL for qRT-PCR analysis.
In vitro network formation assay
After thawing overnight at 4°C, 250 μL Growth Factor Reduced (GFR) Matrigel (Thermo Fisher Scientific; #A1413202) was placed into each well of cold 24-well plates using cold pipette tips, and the plates were then placed at room temperature for 30 min. 4 × 104 HCAECs were placed in each of the Matrigel-coated wells and incubated for 24 h under standard cell culture conditions (37°C, 5% CO2). Wells containing PBS were used as controls. To analyze the dose-dependent rescue, three different exogenous VEGFA concentrations: 0 pg/mL, 500 pg/mL, and 5,000 pg/mL were used. Network formation was quantified by measuring the number of branches, number of loops, and total length of the tube. Digital images of microtiter well sections were obtained by using a Zeiss Axiovert 200M microscope and ZEN 2.3 Pro software. Data were analyzed with ImageJ image analysis software (NIH, USA).
Human VEGFA ELISAs
Protein levels of the human VEGFA (Abcam; #ab119566) were monitored in the supernatant of EC cultures by using commercial ELISAs, following the manufacturer’s instructions. Each sample was assayed with two replicates (duplicates), and the absorbance was measured by a spectrophotometer (TECAN; Infinite M200 Microplate reader) at 450 nm as the primary wavelength. Curve fitting and data analysis were accomplished by CurveExpert Pro software (Hyams Development, USA).
RT2 Profiler angiogenesis PCR array gene expression
Total RNA was isolated out of HCAECs by using an RNeasy Mini Kit (QIAGEN; #74104), according to the manufacturer’s instructions. Then, 1 μg of the total RNA was reverse transcribed by using an RT2 First Strand Kit (QIAGEN), according to the manufacturer’s protocol. RT2 Profiler PCR Array Analysis (QIAGEN; #PAHS-024Z) was performed to measure the expression of 84 key genes involved in modulating the biological processes of angiogenesis. PCR was carried out on an Applied Biosystems 7500 HT Real-Time PCR machine. Detailed data analysis was performed by and exported from the QIAGEN GeneGlobe Data Analysis Center.
Data availability
The raw proteomic data were recently published by our group.54 The Human Functional lncRNA Array and in silico target prediction data have been provided in Tables S5 and S6. All further data that support the findings of this manuscript are available on request from the corresponding authors.
Statistical analysis
Normally distributed continuous variables were presented as the mean ± SD. Continuous variables were tested for normal distribution with the use of the Kolmogorov-Smirnov test. Categorical variables are given as frequencies and percentages. For continuous variables, a Student’s t test or Mann-Whitney U test was used for the comparison between two groups. For the comparison of >2 groups, the one-way ANOVA with Bonferroni correction for multiple comparisons test was used. The chi-square test was used for categorical data that resulted from classifying objects. Binary logistic regression was applied to identify factors that were independently associated with PUNISHER. All tests were two sided, and statistical significance was assumed when the null hypothesis could be rejected at p < 0.05. Statistical analysis was performed with IBM SPSS Statistics version 20 (IBM, USA) and GraphPad Prism 7 (GraphPad, USA).
An extended Supplemental materials and methods can be found in the online Supplemental information.
Acknowledgments
We thank Ms. Anna Flender, Ms. Paula Levermann, and Ms. Sarah Arahouan for their excellent technical assistance. We also thank Dr. Meghan Lucas for her critical reading of the manuscript. We thank the Core Facility Mass Spectrometry at the Institute of Biochemistry and Molecular Biology, Medical Faculty, University of Bonn, for performing mass spectrometry analysis. The authors also acknowledge cooperation with the MPI, Dortmund, for support with electron microscopy. M.R.H. is funded by the German Society of Cardiology (DGK). This study was supported by the German Research Foundation (DFG) under grants WE 4139/8-1 and JA 2351/2-1. The study was further supported by a translational grant from the Corona Foundation and Deutsche Forschungsgemeinschaft (German Research Foundation) project ID 397484323-TRR 259 (project B04).
Author contributions
M.R.H. and F.J. supervised and conceptualized the study and prepared the manuscript. Q.L., M.R.H., Y.Y., A.Z., K.M., and P.G. performed the in vitro/in vivo experiments. S.U., E.L., N.W., G.N., and F.J. contributed intellectually to the development and funding of the project. All authors have read and approved the final manuscript.
Declaration of interests
The authors have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.05.023.
Contributor Information
Mohammed Rabiul Hosen, Email: hosenmr@uni-bonn.de.
Felix Jansen, Email: felix.jansen@ukbonn.de.
Supplemental information
References
- 1.Boulanger C.M., Loyer X., Rautou P.-E., Amabile N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017;14:259–272. doi: 10.1038/nrcardio.2017.7. [DOI] [PubMed] [Google Scholar]
- 2.Ray D.M., Spinelli S.L., Pollock S.J., Murant T.I., O’Brien J.J., Blumberg N., Francis C.W., Taubman M.B., Phipps R.P. Peroxisome proliferator-activated receptor γ and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thromb. Haemost. 2008;99:86–95. doi: 10.1160/TH07-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brill A., Dashevsky O., Rivo J., Gozal Y., Varon D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc. Res. 2005;67:30–38. doi: 10.1016/j.cardiores.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Owens A.P., 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y., Cai Y., Wu G., Chen X., Liu Y., Wang X., Yu J., Li C., Chen X., Jose P.A. Plasma long non-coding RNA, CoroMarker, a novel biomarker for diagnosis of coronary artery disease. Clin. Sci. (Lond.) 2015;129:675–685. doi: 10.1042/CS20150121. [DOI] [PubMed] [Google Scholar]
- 6.Fiedler J., Baker A.H., Dimmeler S., Heymans S., Mayr M., Thum T. Non-coding RNAs in vascular disease - from basic science to clinical applications: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018;114:1281–1286. doi: 10.1093/cvr/cvy121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viereck J., Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ. Res. 2017;120:381–399. doi: 10.1161/CIRCRESAHA.116.308434. [DOI] [PubMed] [Google Scholar]
- 8.Helsmoortel H., Everaert C., Lumen N., Ost P., Vandesompele J. Detecting long non-coding RNA biomarkers in prostate cancer liquid biopsies: Hype or hope? Noncoding RNA Res. 2018;3:64–74. doi: 10.1016/j.ncrna.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Born L.J., Harmon J.W., Jay S.M. Therapeutic potential of extracellular vesicle-associated long noncoding RNA. Bioeng. Transl. Med. 2020;5:e10172. doi: 10.1002/btm2.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Zhang L., Wang Y., Ding H., Xue S., Qi H., Li P. MicroRNAs or long noncoding RNAs in diagnosis and prognosis of coronary artery disease. Aging Dis. 2019;10:353–366. doi: 10.14336/AD.2018.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arroyo J.D., Chevillet J.R., Kroh E.M., Ruf I.K., Pritchard C.C., Gibson D.F., Mitchell P.S., Bennett C.F., Pogosova-Agadjanyan E.L., Stirewalt D.L. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabet F., Vickers K.C., Cuesta Torres L.F., Wiese C.B., Shoucri B.M., Lambert G., Catherinet C., Prado-Lourenco L., Levin M.G., Thacker S. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014;5:3292. doi: 10.1038/ncomms4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tkach M., Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 14.He L., Hannon G.J. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 15.Kung J.T., Colognori D., Lee J.T. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 17.Wilusz J.E., Sunwoo H., Spector D.L. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayoumi A.S., Sayed A., Broskova Z., Teoh J.-P., Wilson J., Su H., Tang Y.-L., Kim I.M. Crosstalk between long noncoding RNAs and microRNAs in health and disease. Int. J. Mol. Sci. 2016;17:356. doi: 10.3390/ijms17030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Li Q., Hosen M.R., Zietzer A., Flender A., Levermann P., Schmitz T., Frühwald D., Goody P., Nickenig G. Atherosclerotic conditions promote the packaging of functional MicroRNA-92a-3p into endothelial microvesicles. Circ. Res. 2019;124:575–587. doi: 10.1161/CIRCRESAHA.118.314010. [DOI] [PubMed] [Google Scholar]
- 20.Jansen F., Yang X., Hoelscher M., Cattelan A., Schmitz T., Proebsting S., Wenzel D., Vosen S., Franklin B.S., Fleischmann B.K. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013;128:2026–2038. doi: 10.1161/CIRCULATIONAHA.113.001720. [DOI] [PubMed] [Google Scholar]
- 21.Loyer X., Vion A.-C., Tedgui A., Boulanger C.M. Microvesicles as cell-cell messengers in cardiovascular diseases. Circ. Res. 2014;114:345–353. doi: 10.1161/CIRCRESAHA.113.300858. [DOI] [PubMed] [Google Scholar]
- 22.De Rosa S., Curcio A., Indolfi C. Emerging role of microRNAs in cardiovascular diseases. Circ. J. 2014;78:567–575. doi: 10.1253/circj.cj-14-0086. [DOI] [PubMed] [Google Scholar]
- 23.Thum T., Mayr M. Review focus on the role of microRNA in cardiovascular biology and disease. Cardiovasc. Res. 2012;93:543–544. doi: 10.1093/cvr/cvs085. [DOI] [PubMed] [Google Scholar]
- 24.Fichtlscherer S., De Rosa S., Fox H., Schwietz T., Fischer A., Liebetrau C., Weber M., Hamm C.W., Röxe T., Müller-Ardogan M. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 25.Hinger S.A., Cha D.J., Franklin J.L., Higginbotham J.N., Dou Y., Ping J., Shu L., Prasad N., Levy S., Zhang B. Diverse long RNAs are differentially sorted into extracellular vesicles secreted by colorectal cancer cells. Cell Rep. 2018;25:715–725.e4. doi: 10.1016/j.celrep.2018.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumarswamy R., Bauters C., Volkmann I., Maury F., Fetisch J., Holzmann A., Lemesle G., de Groote P., Pinet F., Thum T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014;114:1569–1575. doi: 10.1161/CIRCRESAHA.114.303915. [DOI] [PubMed] [Google Scholar]
- 27.Uchida S., Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015;116:737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 28.Skroblin P., Mayr M. “Going long”: long non-coding RNAs as biomarkers. Circ. Res. 2014;115:607–609. doi: 10.1161/CIRCRESAHA.114.304839. [DOI] [PubMed] [Google Scholar]
- 29.Jimenez L., Yu H., McKenzie A.J., Franklin J.L., Patton J.G., Liu Q., Weaver A.M. Quantitative Proteomic Analysis of Small and Large Extracellular Vesicles (EVs) Reveals Enrichment of Adhesion Proteins in Small EVs. J. Proteome Res. 2019;18:947–959. doi: 10.1021/acs.jproteome.8b00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen F., Li Q., Pfeifer A., Werner N. Endothelial- and Immune Cell-Derived Extracellular Vesicles in the Regulation of Cardiovascular Health and Disease. JACC Basic Transl. Sci. 2017;2:790–807. doi: 10.1016/j.jacbts.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q., Shao Y., Zhang X., Zheng T., Miao M., Qin L., Wang B., Ye G., Xiao B., Guo J. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumour Biol. 2015;36:2007–2012. doi: 10.1007/s13277-014-2807-y. [DOI] [PubMed] [Google Scholar]
- 32.Naderi-Meshkin H., Lai X., Amirkhah R., Vera J., Rasko J.E.J., Schmitz U. Exosomal lncRNAs and cancer: connecting the missing links. Bioinformatics. 2019;35:352–360. doi: 10.1093/bioinformatics/bty527. [DOI] [PubMed] [Google Scholar]
- 33.Dragomir M., Chen B., Calin G.A. Exosomal lncRNAs as new players in cell-to-cell communication. Transl. Cancer Res. 2018;7(Suppl 2):S243–S252. doi: 10.21037/tcr.2017.10.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi P., Zhou X.Y., Du X. Circulating long non-coding RNAs in cancer: current status and future perspectives. Mol. Cancer. 2016;15:39. doi: 10.1186/s12943-016-0524-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Théry C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michalik K.M., You X., Manavski Y., Doddaballapur A., Zörnig M., Braun T., John D., Ponomareva Y., Chen W., Uchida S. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 38.Vausort M., Wagner D.R., Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ. Res. 2014;115:668–677. doi: 10.1161/CIRCRESAHA.115.303836. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y.-N.-Z., Shan K., Yao M.-D., Yao J., Wang J.-J., Li X., Liu B., Zhang Y.-Y., Ji Y., Jiang Q., Yan B. Long noncoding RNA-GAS5: a novel regulator of hypertension-induced vascular remodeling. Hypertension. 2016;68:736–748. doi: 10.1161/HYPERTENSIONAHA.116.07259. [DOI] [PubMed] [Google Scholar]
- 40.Yin Q., Wu A., Liu M. Plasma Long Non-Coding RNA (lncRNA) GAS5 is a New Biomarker for Coronary Artery Disease. Med. Sci. Monit. 2017;23:6042–6048. doi: 10.12659/MSM.907118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurian L., Aguirre A., Sancho-Martinez I., Benner C., Hishida T., Nguyen T.B., Reddy P., Nivet E., Krause M.N., Nelles D.A. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015;131:1278–1290. doi: 10.1161/CIRCULATIONAHA.114.013303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan J.X. LncRNA H19 promotes atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017;21:322–328. [PubMed] [Google Scholar]
- 43.Bitarafan S., Yari M., Broumand M.A., Ghaderian S.M.H., Rahimi M., Mirfakhraie R., Azizi F., Omrani M.D. Association of Increased Levels of lncRNA H19 in PBMCs with Risk of Coronary Artery Disease. Cell J. 2019;20:564–568. doi: 10.22074/cellj.2019.5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bei Y., Das S., Rodosthenous R.S., Holvoet P., Vanhaverbeke M., Monteiro M.C., Monteiro V.V.S., Radosinska J., Bartekova M., Jansen F. Extracellular Vesicles in Cardiovascular Theranostics. Theranostics. 2017;7:4168–4182. doi: 10.7150/thno.21274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge Q., Zhou Y., Lu J., Bai Y., Xie X., Lu Z. miRNA in plasma exosome is stable under different storage conditions. Molecules. 2014;19:1568–1575. doi: 10.3390/molecules19021568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juni R.P., Abreu R.C., da Costa Martins P.A. Regulation of microvascularization in heart failure - an endothelial cell, non-coding RNAs and exosome liaison. Noncoding RNA Res. 2017;2:45–55. doi: 10.1016/j.ncrna.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehta J.L., Saldeen T.G., Rand K. Interactive role of infection, inflammation and traditional risk factors in atherosclerosis and coronary artery disease. J. Am. Coll. Cardiol. 1998;31:1217–1225. doi: 10.1016/s0735-1097(98)00093-x. [DOI] [PubMed] [Google Scholar]
- 48.Zhang C., Xu X., Potter B.J., Wang W., Kuo L., Michael L., Bagby G.J., Chilian W.M. TNF-α contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2006;26:475–480. doi: 10.1161/01.ATV.0000201932.32678.7e. [DOI] [PubMed] [Google Scholar]
- 49.Pirillo A., Norata G.D., Catapano A.L. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013;2013:152786. doi: 10.1155/2013/152786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D., Mehta J.L. Oxidized LDL, a critical factor in atherogenesis. Cardiovasc. Res. 2005;68:353–354. doi: 10.1016/j.cardiores.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Sitia S., Tomasoni L., Atzeni F., Ambrosio G., Cordiano C., Catapano A., Tramontana S., Perticone F., Naccarato P., Camici P. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010;9:830–834. doi: 10.1016/j.autrev.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 52.Villarroya-Beltri C., Gutiérrez-Vázquez C., Sánchez-Cabo F., Pérez-Hernández D., Vázquez J., Martin-Cofreces N., Martinez-Herrera D.J., Pascual-Montano A., Mittelbrunn M., Sánchez-Madrid F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qu L., Ding J., Chen C., Wu Z.J., Liu B., Gao Y., Chen W., Liu F., Sun W., Li X.F. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell. 2016;29:653–668. doi: 10.1016/j.ccell.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Zietzer A., Hosen M.R., Wang H., Goody P.R., Sylvester M., Latz E., Nickenig G., Werner N., Jansen F. The RNA-binding protein hnRNPU regulates the sorting of microRNA-30c-5p into large extracellular vesicles. J. Extracell. Vesicles. 2020;9:1786967. doi: 10.1080/20013078.2020.1786967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janas T., Janas M.M., Sapoń K., Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015;589:1391–1398. doi: 10.1016/j.febslet.2015.04.036. [DOI] [PubMed] [Google Scholar]
- 56.Agostini F., Zanzoni A., Klus P., Marchese D., Cirillo D., Tartaglia G.G. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 2013;29:2928–2930. doi: 10.1093/bioinformatics/btt495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Y., Liu T., Cui T., Wang Z., Zhang Y., Tan P., Huang Y., Yu J., Wang D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020;48(D1):D189–D197. doi: 10.1093/nar/gkz804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bateman A., Agrawal S., Birney E., Bruford E.A., Bujnicki J.M., Cochrane G., Cole J.R., Dinger M.E., Enright A.J., Gardner P.P. RNAcentral: A vision for an international database of RNA sequences. RNA. 2011;17:1941–1946. doi: 10.1261/rna.2750811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonnici V., Caro G., Constantino G., Liuni S., D’Elia D., Bombieri N., Licciulli F., Giugno R. Arena-Idb: a platform to build human non-coding RNA interaction networks. BMC Bioinformatics. 2018;19(Suppl 10):350. doi: 10.1186/s12859-018-2298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J., Li C., Zhang L., Wang X. Extracellular Vesicles as Carriers of Non-coding RNAs in Liver Diseases. Front. Pharmacol. 2018;9:415. doi: 10.3389/fphar.2018.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hobor F., Dallmann A., Ball N.J., Cicchini C., Battistelli C., Ogrodowicz R.W., Christodoulou E., Martin S.R., Castello A., Tripodi M. A cryptic RNA-binding domain mediates Syncrip recognition and exosomal partitioning of miRNA targets. Nat. Commun. 2018;9:831. doi: 10.1038/s41467-018-03182-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu Y., Wu W., Han Q., Wang Y., Li C., Zhang P., Xu H. Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 2019;9:180239. doi: 10.1098/rsob.180239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z., Qiu H., He J., Liu L., Xue W., Fox A., Tickner J., Xu J. The emerging roles of hnRNPK. J. Cell. Physiol. 2020;235:1995–2008. doi: 10.1002/jcp.29186. [DOI] [PubMed] [Google Scholar]
- 65.Leal G., Comprido D., de Luca P., Morais E., Rodrigues L., Mele M., Santos A.R., Costa R.O., Pinto M.J., Patil S. The RNA-binding protein hnRNP K mediates the effect of BDNF on dendritic mRNA metabolism and regulates synaptic NMDA receptors in hippocampal neurons. eNeuro. 2017;4 doi: 10.1523/ENEURO.0268-17.2017. ENEURO.0268-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoon Y., McKenna M.C., Rollins D.A., Song M., Nuriel T., Gross S.S., Xu G., Glatt C.E. Anxiety-associated alternative polyadenylation of the serotonin transporter mRNA confers translational regulation by hnRNPK. Proc. Natl. Acad. Sci. USA. 2013;110:11624–11629. doi: 10.1073/pnas.1301485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Roosbroeck K., Pollet J., Calin G.A. miRNAs and long noncoding RNAs as biomarkers in human diseases. Expert Rev. Mol. Diagn. 2013;13:183–204. doi: 10.1586/erm.12.134. [DOI] [PubMed] [Google Scholar]
- 68.Kim K.M., Abdelmohsen K., Mustapic M., Kapogiannis D., Gorospe M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA. 2017;8:e1413. doi: 10.1002/wrna.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jansen F., Yang X., Proebsting S., Hoelscher M., Przybilla D., Baumann K., Schmitz T., Dolf A., Endl E., Franklin B.S. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J. Am. Heart Assoc. 2014;3:e001249. doi: 10.1161/JAHA.114.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fritz J.V., Heintz-Buschart A., Ghosal A., Wampach L., Etheridge A., Galas D., Wilmes P. Sources and Functions of Extracellular Small RNAs in Human Circulation. Annu. Rev. Nutr. 2016;36:301–336. doi: 10.1146/annurev-nutr-071715-050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mateescu B., Kowal E.J., van Balkom B.W., Bartel S., Bhattacharyya S.N., Buzás E.I., Buck A.H., de Candia P., Chow F.W., Das S. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell. Vesicles. 2017;6:1286095. doi: 10.1080/20013078.2017.1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dong L., Lin W., Qi P., Xu M.D., Wu X., Ni S., Huang D., Weng W.W., Tan C., Sheng W. Circulating long RNAs in serum extracellular vesicles: their characterization and potential application as biomarkers for diagnosis of colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 2016;25:1158–1166. doi: 10.1158/1055-9965.EPI-16-0006. [DOI] [PubMed] [Google Scholar]
- 73.Busch A., Eken S.M., Maegdefessel L. Prospective and therapeutic screening value of non-coding RNA as biomarkers in cardiovascular disease. Ann. Transl. Med. 2016;4:236. doi: 10.21037/atm.2016.06.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cai Y., Yang Y., Chen X., Wu G., Zhang X., Liu Y., Yu J., Wang X., Fu J., Li C. Circulating ‘lncRNA OTTHUMT00000387022’ from monocytes as a novel biomarker for coronary artery disease. Cardiovasc. Res. 2016;112:714–724. doi: 10.1093/cvr/cvw022. [DOI] [PubMed] [Google Scholar]
- 75.Yu B., Wang S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics. 2018;8:3654–3675. doi: 10.7150/thno.26024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aryal B., Rotllan N., Fernández-Hernando C. Noncoding RNAs and atherosclerosis. Curr. Atheroscler. Rep. 2014;16:407. doi: 10.1007/s11883-014-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guduric-Fuchs J., O’Connor A., Camp B., O’Neill C.L., Medina R.J., Simpson D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics. 2012;13:357. doi: 10.1186/1471-2164-13-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mathieu M., Martin-Jaular L., Lavieu G., Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019;21:9–17. doi: 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- 79.Li X., Liu R., Huang Z., Gurley E.C., Wang X., Wang J., He H., Yang H., Lai G., Zhang L. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology. 2018;68:599–615. doi: 10.1002/hep.29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Suppl 2):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jansen F., Yang X., Hoelscher M., Cattelan A., Schmitz T., Proebsting S., Wenzel D., Vosen S., Franklin B.S., Fleischmann B.K. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013;128:2026–2038. doi: 10.1161/CIRCULATIONAHA.113.001720. [DOI] [PubMed] [Google Scholar]
- 82.Jansen F., Zietzer A., Stumpf T., Flender A., Schmitz T., Nickenig G., Werner N. Endothelial microparticle-promoted inhibition of vascular remodeling is abrogated under hyperglycaemic conditions. J. Mol. Cell. Cardiol. 2017;112:91–94. doi: 10.1016/j.yjmcc.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 83.Théry C., Amigorena S., Raposo G., Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 84.Tkach M., Kowal J., Zucchetti A.E., Enserink L., Jouve M., Lankar D., Saitakis M., Martin-Jaular L., Théry C. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. 2017;36:3012–3028. doi: 10.15252/embj.201696003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw proteomic data were recently published by our group.54 The Human Functional lncRNA Array and in silico target prediction data have been provided in Tables S5 and S6. All further data that support the findings of this manuscript are available on request from the corresponding authors.