

Abstract
This review provides a practical guide to myeloperoxidase (MPO) and presents to the reader the diversity of its presence in biology. The review provides a historical background, from peroxidase activity to the discovery of MPO, to its role in disease and drug development. MPO is discussed in terms of its necessity, as specific individuals lack MPO expression. An underlying theme presented throughout brings up the question of the benefit and burden of MPO activity. Enzyme structure is discussed, including accurate masses and glycosylation sites. The catalytic cycle of MPO and its corresponding pathways are presented, with a discussion of the importance of the redox couples of the different states of MPO. Cell lines expressing MPO are discussed and practically summarized for the reader, and locations of MPO (primary and secondary) are provided. Useful methods of MPO detection are discussed, and how these can be used for studying disease processes are implied through the presentation of MPO as a biomarker. The presence of MPO in neutrophil extracellular traps is presented, and the activators of the former are provided. Lastly, the transition from drug metabolism to a target for drug development is where the review concludes.
Keywords: Myeloperoxidase, Neutrophil extracellular traps, Biomarkers, Drug metabolism, Drug development
Graphical abstract

Highlights
-
•
Myeloperoxidase (MPO) is an enzyme with remarkable diversity in health and disease.
-
•
MPO levels usually correlate with disease severity and is a key biomarker.
-
•
MPO has roles in COVID-19, drug oxidation, & neutrophil extracellular traps.
-
•
Clinical MPO inhibitors require further study to elucidate protective activity.
1. Myeloperoxidase – a historical timeline to the present
The initial identification of myeloperoxidase (MPO) occurred by empirical observations of a chemical reaction. The following observations have been summarized from the original article [4,5]. What led up to the discovery of MPO include the observation of a colour change when guaiac tincture (a mixture of guaiacum and alcohols) changed colour in the presence of pus (Klebs, E. 1868; Struve, H., 1872). Others later found that such colour changes occurred in leucocytes and bone marrow (Brandenburg, K., 1900; Meyer, E., 1903). Furthermore, the peroxidase nature of MPO was reflected by the requirement of H2O2 for the oxidation of Nadi reagent (a mixture of N,N-dimethyl-p-phenylenediamine and 1-naphthol, identified by Linossier, G., 1898). Lastly, catalase, which degraded H2O2, also prevented the formation of indophenol blue (Agner, K. 1941). Its green color and peroxidase activity earned this enzyme the name of verdoperoxidase, and its discovery was assigned to Kjell Agner in 1941, from which the above summary and citations were acquired [4]. Nauseef has also provided an excellent historical background, including the fact that the studies by Agner were a part of his medical school doctoral thesis [6]. However, the enzyme name, verdoperoxidase, did not stand the test of time as about nine articles up to 1980 used this nomenclature. This probably avoided future confusion since vascular peroxidase (VPO), was discovered in 2008 [7].
The name myeloperoxidase (MPO) was suggested by Theorell (1943) and was used later in a Methods in Enzymology article dedicated to MPO [8]. The reason for this was to associate the source organ/tissue name with the protein rather than a physical feature. It was initially abbreviated as MyPO (to not confuse it with milk peroxidase, which turns out to be lactoperoxidase). However, in this review, the conventional MPO abbreviation will be adhered to. Soon after its discovery, MPO was studied to “ascertain whether it has a special function in combating infectious diseases” by Agner, where it was observed that the enzyme disappeared from leukocytes during severe infection; mice survived treatment of tetanus toxin only if the toxin was first oxidized with MPO and H2O2 [9]. The idea of such peroxidase activity being bactericidal was also shown earlier by utilizing phenolic oxidation by MPO for bacterial killing [10]. The functional role of MPO in neutrophil-mediated destruction of bacteria has been ascribed to the fundamental studies of Klebanoff [11,12].
1.1. Enzymes related to MPO
MPO belongs to the peroxidase family of enzymes. As this review is focused solely on MPO, only a brief summary of other peroxidases will be discussed.
Thyroid peroxidase has a nucleotide sequence similarity of 58% compared to the functional protein sequence of MPO. The sequence similarity near the haem active site between thyroid peroxidase and MPO is 74%. However, the entire amino acid sequence bears 44% homology with MPO, indicating that these proteins are of the same gene family [13]. Thyroid peroxidase is localized to the thyroid gland and is only involved in thyroid hormone synthesis.
Eosinophil peroxidase is more closely related to MPO in structure and function, even though it is a monomer. This enzyme is located in eosinophils and is considered mainly responsible for generating hypobromous acid to assist in microbial killing. Eosinophil peroxidase shares approximately 70% amino acid sequence homology with MPO [14].
Lactoperoxidase, unlike MPO, has a single chain of 80 kDa containing a single haem. It shares approximately 51% amino acid sequence homology with MPO [15]. Lactoperoxidase is found in milk, tears, saliva, and mucous of the respiratory tract [16].
The most recently discovered enzyme in this family is the somewhat esoteric vascular peroxidase 1 (VPO1) [7]; it is relatively challenging to acquire or isolate. Studies have shown that it is upregulated and is activated in chronic cardiac fibrosis more so than MPO. Fibrosis in a myocardial infarction model was attenuated when VPO1 siRNA was used [17]. A timeline of important discoveries involving MPO is shown in Fig. 1.
Fig. 1.

A timeline of MPO from discovery, function, inhibition, and disease. References indicated have been added for the seminal or foundational publication or first reports. References used to create the timeline: Peroxidase staining [4,5,18], Discovery [19] bactericidal activity [9]; MPO as % dry protein [20]; cells and cellular location [21]; Halogenation as bactericidal mechanism [11,22]; Anti-neutrophil cytoplasmic antibodies are anti-MPO [[23], [24], [25]]; MPO in atherosclerosis [26,27]; cardiotoxicity [[28], [29], [30], [31]]; neurotoxicity [32,33]; drug metabolism [[34], [35], [36]]; requirement for neutrophil extracellular trap formation [37,38]; MPO clinical inhibitor leads [[39], [40], [41]]; MPO levels in COVID-19 and thrombosis events [[42], [43], [44]].
2. MPO: it's in (most of) our genes
MPO was cloned from human acute myelogenous leukemia cells, revealing that a single gene was responsible for its encoding [45]. MPO was determined to be located on chromosome 17 [46,47]. Studies by Morishita et al. reported that the human MPO gene was composed of 12 exons and 11 introns and located at band 17q23.1. In addition, a single mRNA transcription initiation site was identified, which shared homology with sequences found on the 5′-promoter region for the proto-oncogene, c-myc. During differentiation of mouse or human promyelocytic leukemia cells to granulocytes or monocytes, it has been observed that mRNA of MPO and c-myc are both decreased in response to granulocyte-colony stimulating factor [48,49]. Interestingly, other related peroxidase enzymes, including eosinophil peroxidase, lactoperoxidase, and thyroid peroxidase, appear to cluster around the same region, where eosinophil peroxidase is localized 34 kb from the MPO and lactoperoxidase genes [50].
MPO deficiency was first reported about 50 years ago and was highlighted by a patient with a candidiasis infection [51]. This patient appeared to be only deficient in neutrophil MPO, as eosinophil peroxidase (a similar enzyme found in eosinophils) was present. Interestingly, the MPO deficient neutrophils functioned normally otherwise (e.g., phagocytosis was normal) and were able to slowly but eventually kill S. Aureus. This observation suggests that the respiratory burst is more significant in bactericidal activity, but fungal infections require additional support by MPO activity. Early studies have shown that both neutrophils and monocytes lacking MPO have a longer respiratory burst rate, extent and duration [52].
The incidence of MPO deficiency in the general population is 1 in 2000 to 4000 in the United States and Europe [[53], [54], [55]]. The complete deficiency of MPO is characterized by the absence of MPO-related peptides in neutrophils; the partial absence is due to less MPO activity, for a lower overall amount of the protein is visible to the naked eye in neutrophil pellets (Fig. 2) [1,56]. Interestingly, MPO deficiency in Japan appears to be far rarer: approximately 1 in 60 000 for complete deficiency and 1 in 20 000 for partial deficiency [57]. Certain dogs have been noted to have MPO deficiency [58]. The deficiency in MPO is also observed in monocytes. In general, MPO deficiency does not appear to impair patients from fighting infections, except for Candida Albicans (candidiasis) infection, resulting in toxemia [59,60]. It has been suggested that other systems, such as a sustained respiratory burst or increased eosinophil counts (i.e., eosinophil peroxidase) could provide backup mechanisms for the shortfall of chemical oxidants produced by MPO [61].
Fig. 2.
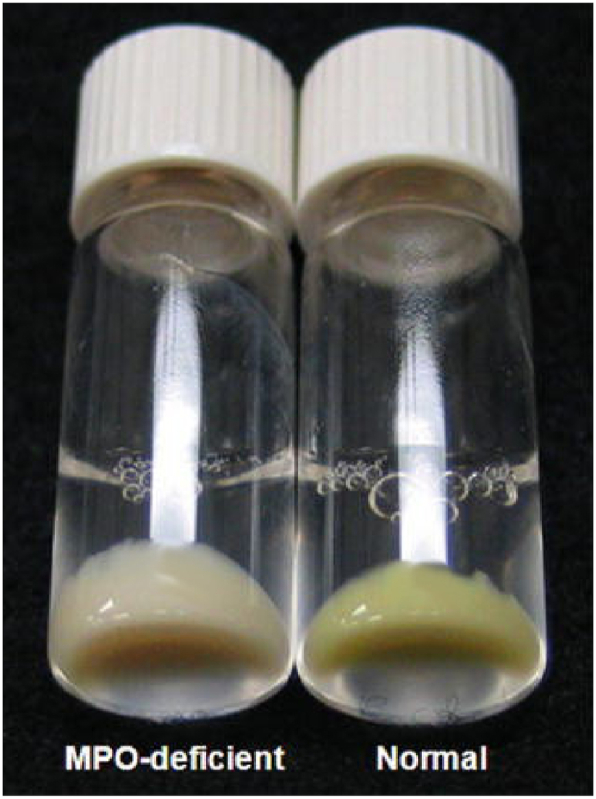
Neutrophils from MPO-normal and MPO-deficient donors demonstrate the haem colour itself as a clear indicator for the presence of MPO (modified from [1], with permission). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
MPO deficiency also reportedly had enhanced phagocytic capability through the absence of ERK1/2 signalling [62]. Lung neutrophil influx induced by LPS in mice was attenuated in MPO knockout mice [63]. Due to the catalytic peroxidase activity of MPO, diagnostic assays for MPO deficiency are readily available [1]. A case study reported a 3-year old boy having recurrent, severe bacterial and fungal infections due to DiGeorge syndrome (chromosome 22q11.2 deletion) combined with MPO deficiency [64]. As such, MPO deficiency may have important effects in susceptibility to infection of particular importance to certain individuals.
3. Do we need MPO?
Despite the apparent importance of MPO in microbial killing, it seems that about 50% of MPO-deficient individuals remain asymptomatic and do not have remarkable health concerns (other than those described above). The MPO deficiency may become more critical if the individual is afflicted with other disease states or mutations. On the other hand, secondary MPO deficiency can result from various disease states or xenobiotic exposure [65]. Indeed, there may be benefits to lower MPO activity. An Alzheimer's disease mouse model (5XFAD) demonstrated improved cognitive, memory, and proinflammatory activity after producing bone marrow deficient MPO [66]. MPO can oxidize high-density lipoprotein, which can lead to its dysfunction and atherosclerotic plaques [67]. However, a study of mice infected with Salmonella proposed that MPO actually mitigated collateral (host) damage in this model [68].
4. Enzyme structure
The MPO protein is a tetramer, with a total estimated molecular weight of 150 000 kDa. The tetramer is composed of two halves (hemi-MPO), composed of two heavy (~60 kDa) and two light (~15 kDa) components [69]. Previously, a range of molecular weights was reported at 120 000–160 000 kDa [48]. More accurate masses are described later (see below). Human MPO is reported to have 467 amino acids in the large chain and 105 amino acids in its short chain.
It is unique in the peroxidase enzyme family, where MPO has three covalently linked amino acid linkages from the protein chain to the protoporphyrin IX (haem) active site (Fig. 3) [2,70]. The fifth coordination position of the iron is occupied by the imidazole group of histidine, similar to other peroxidases [71]. The three amino acid bonds to the haem have been associated with its green colour, as MPO was originally called verdoperoxidase (Agner, 1940). A curious observation of a protein referred to as “spleen green” protein was identical to MPO [69]. Site-directed mutation studies of MPO revealed that the Met243 → Gln mutation changed the haem absorption peak to 410 nm, approximately 20 nm (blue-shifted) from the native peak at 429 nm, which resembles other peroxidases. The sulfonium linkage by Met243 to the haem produces a distortion of planarity and ligand binding characteristics, which is unique to MPO [72].
Fig. 3.

The MPO active site. The haem prosthetic group is covalently bound to the protein via three amino acid linkages (from [2], with permission).
5. MPO is a glycoprotein
With the advances in glycomics, it is essential to discuss MPO glycosylation and how this affects structure and function. MPO has 38 glycopeptides, and all five expected sites for glycosylation were found to be glycosylated. These sites contain an asparagine residue (Asn323, Asn355, Asn391, Asn483, and Asn729) which is why they are N-linked glycosylation sites [73,74]. Another study was carried out to characterize MPO glycosylation sites [75]. There appear to be specific critical amino acids glycosylated, all of which are Asn residues: Asn322, Asn355, Asn391, Asn483, and Asn729. The glycosylation included phosphorylated N-glycans (Asn323) and paucimannose (Asn483). These in-depth studies have provided a more accurate picture of the mass of MPO, although the glycosylation masses are relatively close to earlier findings and are summarized in Table 1 [73].
Table 1.
Glycosylation sites in MPO from corresponding enzymatic digests.
| Glycosylation site (Asn) | Peptide mass (digestion with PNGase-F) Ref. [73] |
|---|---|
| 323 | 1433 |
| 355 | 1558, 1575, 1958 |
| 391 | 2126 |
| 483 | 1053 |
| 729 | 1965 |
Furthermore, the unusual (truncated) glycosylation pattern on MPO along with the heterogeneity of MPO glycosylation may affect antibody recognition, and in disease states where autoantibodies are generated. Also, antibody recognition may be altered by glycosylation, where a minor decrease in recognition was observed for deglycosylated MPO [74]. Such in-depth analytical studies have produced more accurate calculations regarding the mass of MPO and its corresponding subunits (Table 2).
Table 2.
Mass of MPO and its main components. The data are summarized from [75].
| MPO component | Average MW |
|---|---|
| Holoenzyme | 144170.9 |
| Monomer/hemi-MPO | 72085.5 |
| Haem (prosthetic group) | 616.5 |
| Heavy chain | 53401.5 |
| Light chain | 12375.9 |
6. MPO catalytic cycle
MPO is considered to have two main activities: oxidation of halogens (halogenation cycle) to form hypohalous acids (such as hypochlorous acid) and the peroxidase cycle. As shown in Fig. 3, the resting enzyme is ferric MPO (1). Typically, for the cycle to begin, a source of H2O2 is required to “activate” the enzyme to form Compound I. This form of MPO has an oxidized iron (4+ valence) and a delocalized haem radical (ferryl haem). Compound I can either be reduced to the resting state by oxidizing (pseudo)halogens or go through the peroxidase pathway to Compound II:
| Resting MPO [Fe(III)] + H2O2 → Compound I MPO [Fe(IV O)●+] + H2O | (1) |
| Compound I MPO [Fe(IV O)●+] + halogen → Resting MPO [Fe(III)] + hypohalous acid | (2) |
The halogenation cycle is unique to MPO in terms of its ability to oxidize chloride. Although the latter is a relatively poor substrate for MPO, its abundance in biological fluids (e.g., physiological NaCl levels are ~ 100 mM) allows for its oxidation to occur. It has been proposed, however, that less abundant (pseudo)halogens, like thiocyanate (micromolar amounts in the blood), are more easily oxidized and thus do not require the millimolar quantities of chloride [76]. This notion is supported by the electron donation potential as indicated by the second-order rate constant for Compound I reduction, where thiocyanate (SCN−) is the best reductant (most easily oxidized) for Compound I, followed closely by iodide, bromide, and two orders of magnitude slower for chloride (at neutral pH) [77]. Indeed, the bromination of a phenolic substrate (phenol red commonly used in cell culture) because of the Br− contaminant of laboratory-grade NaCl, has been observed [78].
Although the halogenation cycle is considered the antimicrobial pathway because of the generation of hypohalous acids, there are far many more one-electron substrates for MPO that are metabolized via the peroxidase pathway. Many free radical molecules are produced through this pathway. It is important to emphasize that although these pathways are described as distinct, they can feed into each other. For example, the presence of ascorbic acid and other cellular MPO substrates can ensure that MPO is in its resting state such that it can catalyze chlorination reactions. This is also true of certain xenobiotics and drugs [79]. There is a significant overlap in xenobiotic oxidation of MPO with the cytochrome P450 mixed-function oxidases (P450), which are the predominant drug metabolizing enzymes (and have a similar haem active site). Once Compound I MPO is formed, it is capable of substrate (X) oxidation to produce an initial X free radical (X●):
| Resting MPO [Fe(III)] + H2O2 → Compound I MPO [Fe(IV O)●+] + H2O | (3) |
| Compound I MPO [Fe(IV O)●+] + X → Compound II MPO [Fe(IV O)] + X● | (4) |
Compound II of MPO is less oxidizing than Compound I. If organic substrates (such as drugs or amino acids) have a favourable redox potential, they can reduce Compound II to Resting MPO. It is possible that the X● may undergo additional catalytic oxidation to produce a two-electron oxidized metabolite (X[O]):
| Compound II MPO [Fe(IV O)] + X → Resting MPO [Fe(III)] + X● | (5) |
| Compound II MPO [Fe(IV O)] + X● → Resting MPO [Fe(III)] + X[O] | (6) |
However, a dismutation reaction can also occur that does not require enzymatic catalysis. This will happen at different rates but is usually a thermodynamically favourable reaction:
| X● + X● → X + X[O] | (7) |
Lastly, Compound III MPO is an intermediate that has been considered catalytically inactive, but it may have more value concerning hydroxylation reactions of xenobiotics. To produce Compound III, superoxide is required:
| Resting MPO [Fe(III)] + O2●– → Compound III MPO [Fe(III)–O2●–] |
| ←→ Compound III MPO [Fe(II)–O2] | (8) |
Compound III has been considered inactivated, based on studies in activated neutrophils [80]. Moreover, it has been referred to as a “dead-end” in terms of the more prominent functions of MPO [81], though Compound III has been implicated in oxidizing ascorbic acid [82], acetaminophen [83], salicylic acid [84], dimethyltryptamine [85], lysergic acid diethylamide (LSD, [86], phenol and phenolic drugs [87,88], melatonin [89,90], phenylalanine [91], and possibly phenytoin [92]. An interesting reaction catalyzed by isoniazid and related hydrazines led to the generation of Compound III, where subsequent hydroxylation reactions may occur [93]. This observation was confirmed and likely contributed to identifying 4-aminobenzoic acid as the first potent MPO inhibitor that first converted MPO to Compound III before the loss of activity [94]. A short list of some substrates that MPO oxidizes is shown in Table 3.
Table 3.
Examples of substrates oxidized through the halogenation and peroxidase pathways of MPO.
| MPO Activity or cycle | Reactants | Products |
|---|---|---|
| Halogenation | Halides and pseudohalides | Two-electron oxidized products. |
| Examples | Cl−, Br−, I−, SCN− | HOCl, HOBr, HOI, HOSCN |
| Peroxidase | Aromatic compounds (both endogenous and exogenous); nitric oxide derivatives; small inorganic compounds | One electron oxidation products, most of which are subsequently oxidized. |
| Examples | Ascorbic acid, tyrosine, arylamine drugs, hydrogen sulphide | Dehydroascorbyl radical (a stable radical), Tyrosine phenoxyl radical leading to dityrosine, arylamine radicals leading to multiple products, including oxidizing species that can damage proteins and DNA, hydrogen sulphide radicals |
The catalytic cycle for MPO is shown in Fig. 4. The reduction potentials are also indicated for the relevant redox couple. The higher (more positive) the reduction potential is, the more potent an oxidant is formed. The Compound I/Resting reduction potential (1.16 V) means that MPO can oxidize many compounds. Note that the Compound II/I reduction potential is even higher, which can oxidize numerous xenobiotics and endobiotics. Although these pathways are traditionally regarded as distinct, it is essential to note that the formation of hypochlorous acid, for example, is capable of chlorinating xenobiotics and endobiotics. The latter two may end up appearing to be “oxidized,” although the initial intermediate would be a chlorinated molecule.
Fig. 4.

Different states of MPO and their associated redox potentials. The cycle begins with the “Resting” state, where the redox status of the iron centre of the protoporphyrin IX (haem) active site is indicated. MPO becomes activated in the presence of H2O2, which led to Compound I, which contains the ferryl haem species ([FeIV O]●+) and contains a delocalized free radical. Compound I will transform by either halogen oxidation (“pseudo halogen” is indicated to include SCN−, Cl−, Br−, I−) back to the Resting state, or the peroxidase cycle where xenobiotics and endobiotics (“Substrate”) can be oxidized, resulting in Compound II. In the presence of H2O2 and the absence of substrate, Compound III may form. Compound III can also form by certain substrates, or via reduction of the Resting enzyme to produce Ferrous MPO.
∗ Redox potentials from [95]. These values are useful in predicting if a particular substrate can reduce Compound I/II efficiently. As is apparent, Compound I is capable of oxidizing more substrates than Compound II. ** Indicates the direct conversion of ferrous myeloperoxidase to compound II by hydrogen peroxide [95].
7. Location, location, location
The best known location for MPO is within the primary or azurophilic granules of neutrophils, as this is the cell in which its peroxidase activity was discovered. Neutrophils, however, are end-differentiated and do not synthesize MPO. Monocytes also express MPO, though to a significantly lesser degree than neutrophils. Macrophage expression of MPO has been controversial. Early studies showed that MPO in monocytes granules are lost when differentiated into macrophages in vitro [96,97]. In one study, Kupffer cells, the most prominent resident macrophage population in the body in the human liver, were shown to express MPO [98]. In rats treated with the hepatotoxicant, CCl4, MPO expression could not be replicated [99]. However, a mouse model of atherosclerosis expressing human MPO demonstrated that macrophages in this model express MPO and peroxidase activity [100]. As such, it appears that macrophages cannot be categorically described as expressing MPO, but rather, they may express MPO under specific conditions.
To get a clearer picture of the cellular origins of MPO, one must go to the bone marrow. MPO mRNA synthesis and translation occurs in the late myeloblast or promyelocytes, the precursor to granulocytes [101,102]. A significant subset of bone marrow CD34+ stem cells has been reported to express MPO, although circulating CD34+ cells lacked MPO entirely, suggesting the former would have been committed to granulocyte development [103]. Therefore, MPO synthesis occurs in the bone marrow and not in circulation or within tissues (normally). The steps in processing MPO after translation have been elegantly outlined by Nauseef are summarized here briefly [104]. Readers are directed to that publication for in-depth biosynthesis and processing.
-
•
Step 1 – preMPO is the initial translation product in the endoplasmic reticulum.
-
•
Step 2 – glycosylation of preMPO to form apoproMPO
-
•
Step 3 – acquisition of haem to form proMPO (haemacquisition → catalytically active)
-
•
Step 4 – exit from the endoplasmic reticulum
-
•
Step 5 – proteolytic processing to form hemi-MPO.
-
•
Step 6 – Within the primary/azurophilic granule, the dimerization of the hemi-MPO takes place through a disulphide bond (Cys319) to produce mature MPO.
The most abundant site of MPO synthesis begins in the bone marrow promyelocytes and in specific populations of CD34+ stem cells [103]. In addition, some studies suggest that MPO expression occurs in peripheral blood lymphocytes of mice and humans [105,106]. These reports indicated that lymphocytes had a higher mRNA level than neutrophils or monocytes but had significantly lower protein expression. Interestingly, in chronic HIV cases, there appears to be a statistically significant higher expression of MPO in CD4 and CD8 T lymphocytes. The functional significance of MPO in lymphocytes is yet unknown and more studies are required to understand how HIV infection, which causes immunosuppression, relates to MPO.
Other cells that express MPO include cells in the CNS, such as astrocytes, microglia, and neurons. Studies from human autopsies or animal models have revealed that MPO from these cells is associated with Alzheimer's disease, multiple sclerosis, stroke or cerebral ischemia, Parkinson's disease, depression or bipolar disorder, bacterial meningitis, neuroinflammation, and epilepsy (reviewed in [107]).
8. Cell lines containing MPO
Commercially available cell lines that highly express MPO are expectedly cell lines isolated from leukemia patients. However, not all leukemia cell lines express MPO or express MPO at different levels. Specific examples of cell lines that highly express MPO include HL-60, NB4, Kasumi-1, and MOLM13. Examples of low MPO-expressing cell lines include K562, U937, KG1, and OCI-AML3 [108]. A cell line that has been used as a good model for human macrophage precursors is THP-1 cells. The reason is that they can be differentiated into macrophages and demonstrate inflammasome function. Although THP-1 cells are leukemia cells, they are acute monocytic leukemia. No MPO activity has been reported in this cell line, and our lab has not detected any MPO expression (unpublished data). One study investigated the role of MPO inhibition for chemotherapy using cytarabine (AraC) in vitro and in vivo using the MOLM13 cell line. MPO inhibition in cells highly expressing MPO resulted in sensitization to AraC, which appeared to result from mitochondrial redox changes resulting in oxidative stress [109]. For reference, the reader is directed to Table 4 that lists related acute myeloid leukemia cell lines. As can be observed, myelocytic cell lines typically express MPO, whereas monocytic cell lines do not.
Table 4.
Related leukemia cell lines and their relative expression of MPO.
| Cell line | Disease | MPO relative expression |
|---|---|---|
| HL-60 | Acute Myeloblastic Leukemia, (myelocytic) | ↑↑↑ |
| NB-4 | Acute Myeloblastic Leukemia, (myelocytic) | ↑↑↑ |
| Kasumi-1 | Acute Myeloblastic Leukemia, (myelocytic) | ↑↑↑ |
| MOLM-13 | Acute Myeloid Leukemia, (monocytic) | ↑ |
| K-562 | Chronic Myelogenous Leukemia | Not detected |
| KG-1 | Acute Myeloid Leukemia, (myelocytic) | Not detected |
| OCI-AML3 | Acute Myeloid Leukemia, (myelocytic) | Not detected |
| U937 | Acute Myeloid Leukemia, (monocytic) | Not detected |
| THP-1 | Acute Myeloid Leukemia, (monocytic) | Not detected |
8.1. Secondary sites of MPO localization
It should be noted that neutrophils, through chemotaxis (e.g., IL-8, TNFα), can infiltrate organs and catalyze inflammatory reactions, including the release and activation of MPO. This is clear in specific instances of liver injury. What may complicate the matter in this case is the activation of liver-resident macrophages (Kupffer cells) which may express MPO during M1 polarization during inflammation (discussed above). Otherwise, Kupffer cells or macrophages do generally not express MPO. Another important site of MPO is in the atherosclerotic lesions in the blood vessel endothelium. It has been shown that MPO plays an important role in the formation of the plaques (discussed below). Other locations for MPO include the heart and the CNS.
Neurodegenerative conditions appear to have a significant association with the expression of MPO. Although cells will show a basal level of MPO, as demonstrated from human brain homogenate, disease states have the propensity to induce expression of MPO similar to phenomena in liver Kupffer cells described above. In Alzheimer's disease patient brain autopsies, the presence of MPO in the frontal cortex is increased (detected by Western blot). Importantly, biochemical markers of MPO activity were also found, indicating not only the presence but activity also were increased in Alzheimer's disease patient brain autopsies. From a disease pathology point of view, MPO was associated with amyloid β deposits [111]. MPO has been found human saliva, though peroxidase activity can also be derived from lactoperoxidase also found in tears, saliva, and milk [112]. The presence of MPO in healthy and disease states is summarized in Fig. 5.
Fig. 5.

The presence of MPO in tissues, and association with various disease states. Please refer to the text description of locations for greater detail. MPO usefulness as a biomarker in disease states.
9. MPO usefulness as a biomarker in disease states
9.1. Detection strategy
In order to be helpful as a biomarker, MPO must be effectively detected in the specimen of interest. MPO can be detected through several methods, which are either direct or indirect (but specific). Direct methods involve using commercially available antibodies for immunoassays, including ELISA, Western blot, or microscopy. Cytochemical staining, however, is usually accomplished by using MPO substrates (e.g., 3,3′-diaminobenzidine, o-dianisidine), which undergo metabolism upon adding H2O2 to form coloured products (usually red-brown). The consumption of H2O2 through the use of a H2O2 electrode is also possible, although competing reactions with catalase must be considered.
Indirect detection focuses on products of MPO, chiefly hypochlorous acid (HOCl), considered to be the main physiological product of MPO. HOCl can oxidize amino acids such as tyrosine to form 3-chloro-tyrosine [113,114]. 3,5-Dichlorotyrosine is also a suitable and stable detection product, though not used as often [115]. It would be ideal to detect 3-chloro-tyrosine via antibodies, but there do not appear to be any successful ones that have widespread use; thus, LC-MS is the detection method commonly employed. Other intermediates such as lipid chlorohydrins, chlorinated nucleosides, protein carbonyls, glutathione sulfonamide are also potential biomarkers [116]. 2-Chlorofatty acids have been demonstrated to be detected in models of sepsis and sepsis-associated acute respiratory distress syndrome [[117], [118], [119]]. Fluorescent detection is usually a desirable strategy in cell studies due to providing a simple means for cell and tissue imaging. There are some challenges, however, since fluorescent yield, solubility, and permeability need to be considered in the useful implementation of such probes. A recent study demonstrated that a large lipophilic, charged compound (TPY) could detect micromolar HOCl ratiometrically. This compound lost green fluorescence upon reacting with HOCl in HepG2 cells [120]. One study used a dopamine scaffold to generate a fluorescent product that demonstrated specificity for hypochlorite and responded to exogenous HOCl (NaOCl) addition to cells [121,173]. An acute model of mouse liver ischemia showed fluorescent detection of a probe, though this required the exposure of the organs (i.e., reflection of the abdominal wall) for visualization. An interesting colorimetric probe based on the reaction with HOCl and glutathione could provide a snapshot of the balance between the oxidant (HOCl) and antioxidant (glutathione) status of the cell or tissue [122]. Other approaches may be better suited for in vivo approaches, whereas in vitro approaches are more diverse [123]. Further information on such probes was recently described in [124].
Methods have employed MPO substrates conjugated with fluorescent or paramagnetic probes for fluorescent microscopy and magnetic resonance imaging, respectively [125]. As shown in Fig. 6, a substrate molecule (serotonin shown here) is conjugated with the desired probe initially via chemical synthesis [126], which then can be used in the test specimen for active MPO detection. A key consideration here is that MPO activity is needed, which indicates the need for an inflammatory/immunological process for the successful detection of active MPO localization.
Fig. 6.

Fluorescent and magnetic resonance detection methodology. MPO peroxidase activity catalyzes the one-electron oxidation of the substrate, which converts it to a free radical metabolite. Although the free radical metabolite was proposed to bind to protein [126], there is more evidence to suggest that a quinoneimine or o-quinone serotonin metabolite is the proximate electrophile that will bind to protein [127,128]. (Modified from [126]).
9.2. Leukemia
MPO serves as a valuable marker for acute myelogenous leukemia, as alluded to above in describing cell lines (see “Cell lines containing MPO”). Acute myelogenous leukemia is characterized by a rapid increase in the myeloid cells associated with an arrest in their maturation. Its diagnosis was initially based solely on the pathological and morphological examination of the bone marrow and the blood. However, with growing knowledge about the heterogeneity of acute myelogenous leukemia, subcategorization into nine subtypes has been established by the French-American-British group, depending on the particular myeloid lineage, morphologic appearance and the reactivity with three histochemical stains, including MPO, Sudan black, and the nonspecific esterase α-naphthylacetate and naphthylbutyrate. Six (M1 to M4, M4EO and M6) out of the nine acute myelogenous leukemia subtypes are MPO-positive [129,130]. MPO expression in this disease is associated with a favourable outcomes, in addition to its aforementioned role as a biomarker.
9.3. Cardiovascular diseases
MPO has been considered an important biomarker in cardiovascular health for about two decades. Cardiovascular diseases in which increased MPO levels have been detected include acute coronary syndrome, heart failure, pulmonary arterial hypertension, hypertension, heart transplant and graft rejection, diabetes mellitus, atrial fibrillation, and atherosclerosis (reviewed in [131]). MPO serum level are a predictor for future cardiovascular events and adverse outcomes in patients with acute coronary [31,132]. It has been known for some time that macrophage and neutrophil activity in the plaques of coronary blood vessels leads to oxidation and destabilization of those lesions [133]. Unstable plaques in thrombi have been found to contain MPO-positive monocytes and neutrophils [134].
Concerning MPO, it is well established to be the catalyst for oxidation of LDL and HDL, where oxidation of the latter is the primary concern. Tyrosine residues in this molecule serve as facile targets of oxidation by MPO and its products [135]. In a study evaluating Chinese coronary artery disease patients, there was a difference in comparison to US patients; there was no increase in MPO mediated oxidation of HDL observed, though several limitations were discussed, such as ambient air quality [136,137]. Prediabetic patients affected by adverse cardiovascular outcomes also demonstrated that MPO levels were correlated with cardiovascular risk factors. As such metabolic diseases are multifactorial, it should be noted that these subjects also had increased blood pressure, body-mass index, waist-to-hip ratio and dyslipidemia, which sets the stage for atherosclerosis [138].
The pathogenic role for MPO in cardiovascular diseases is reasonably established. The use of MPO as a prognostic indicator of cardiovascular disease status will likely continue to gain momentum. It remains to be seen if MPO will be used solely as a biomarker of cardiovascular disease status, or become a therapeutic target.
9.4. Neurodegenerative diseases
MPO has been associated with the neuropathology observed in Alzheimer's disease, multiple sclerosis, stroke, Parkinson's disease, depression, and traumatic brain injury and epilepsy. As described above, the peripheral blood MPO concentration appears to correlate with disease progress or intensity. In Alzheimer's disease patients, MPO peripheral blood concentration was significantly higher in these patients compared to control [139]. Furthermore, this biochemical measurement is considered reflective of neutrophil activity in this disease [140]. (See “Secondary sites of MPO localization” section above for more details on MPO and Alzheimer's disease). Similarly, inhibition of MPO in experimental autoimmune encephalitis – which shares similarity to multiple sclerosis as a neuroinflammatory autoimmune disease – reduced disease severity [141]. Both the levels of MPO and its activity reflected by 3-chlorotyrosine were reduced by an experimental MPO inhibitor, which correlated with disease severity, including demyelination, oligodendrocyte regeneration, and neurogenesis [142]. In Parkinson's disease, elevated MPO has been detected in functionally relevant brain regions compared to healthy controls, including the putamen, caudate nucleus, and substantia nigra [143]. In a transgenic mouse model expressing human MPO, significant damage to α-synuclein was observed characteristic of MPO-catalyzed oxidants. In human Parkinson's disease patients, post-mortem analysis also revealed elevated levels of MPO present in the substantia nigra [144]. A mouse model of Parkinson's disease (using MPTP) showed that treatment with a plant extract (Tomentosin) reduced proinflammatory cytokines and MPO levels [145]. Numerous other compounds of natural origins have been proposed to be potentially therapeutic, and some target MPO. However, it remains to be seen if MPO inhibition by novel inhibitors will be utilized as an additional treatment regimen for Parkinson's disease patients.
In patients admitted to the emergency department presenting with acute ischemic stroke, the levels of MPO were shown to rise [146]. Interestingly, in a murine stroke model, the use of an MPO inhibitor in wild-type mice or homozygous MPO knockout mice showed less cell loss, degeneration, better behavioural outcomes and decreased damage overall [147]. A damaging role of MPO has also been proposed in traumatic brain injury [148]. Studies in mice demonstrated elevated peroxidase activity in the cerebrum of mice that appeared to last for a month. SPECT/CT imaging confirmed that MPO activity was inhibited. Also, MPO was present in the temporal lobe of an epileptic patient, which was absent in controls [149]. Studies on the role of MPO in brain injury are essential, but so is the specific type of injury. A study evaluating biomarkers in sports-related concussion in males and females reported that in female athletes only, symptom severity (based on the Sport Concussion Assessment Tool) was negatively related to proinflammatory biomarkers, including MPO [150]. While interesting, it also raises questions on sex-specific responses to MPO induction and the temporal relationship between brain injury and MPO.
9.5. Autoimmune disease – idiopathic and drug-induced systemic lupus erythematosus (SLE)
SLE is an autoimmune disease that can be expressed with systemic symptoms or limited to one specific organ, e.g., skin, kidney, blood vessels. The serum profile is characterized by two main types of autoantibodies: one against a cell's nuclear components, including single-stranded or double-stranded DNA, histones, and histone-DNA complexes, referred to as anti-nuclear antibodies (ANAs). The other class of antibodies is directed towards cytoplasmic lysosomal components of neutrophils and monocytes, including the enzymes such as proteinase-3 and MPO. These are collectively termed anti-neutrophil cytoplasmic antibodies (ANCAs). Within this subset, specific antibodies towards a single protein, i.e., anti-MPO = p-ANCA and anti-proteinase-3 = c-ANCA, have been identified in patients with specific variants of SLE. The presence of these antibodies is used to diagnose certain rare forms of vasculitis, which include three different forms of polyangiitis, renal nephritis, and drug-induced vasculitis [151]. Anti-MPO antibodies in particular, will be discussed.
One study showed that high titers of anti-MPO antibodies are observed with SLE or a drug-induced SLE-like disease, drug-induced vasculitis or nephritis, and idiopathic vasculitis [152]. In these instances, i.e., where anti-MPO antibodies are observed, ANA is usually absent. It has been reported that anti-MPO antibodies have been reported in an inconsistent manner in SLE patients, which has been ascribed to detection methodology. The same study, however, reported that anti-MPO is deposited in the kidney glomerular basement membrane, the first study to do so [153].
Drugs can produce a rare, unpredictable adverse drug reaction (idiosyncratic drug reaction), which can produce vasculitis characterized by the presence of anti-MPO. Although rare, approximately one hundred drugs have been associated with drug-induced SLE. Some notable drugs which have been definitively associated with this condition includes hydralazine, isoniazid, procainamide, methyldopa (withdrawn), quinidine, minocycline, and chlorpromazine [154,155]. In a study of seven patients taking hydralazine, six were positive for ANA. Five of the seven showed anti-MPO antibodies. Interestingly, anti-MPO was normalized after six months after drug withdrawal. Leukopenia was also observed in these patients [156]. Hydralazine appears to be the drug most recently and often reported that report both anti-MPO and ANAs [[157], [158], [159]]. The role of anti-MPO antibodies in either idiopathic SLE or drug-induced lupus is considered to involve mechanisms that activate neutrophils leading to the NET formation and tissue damage. Interestingly, a study reported statistically higher MPO plasma levels in SLE patients compared to healthy patients, although this did not correlate with disease severity [160].
10. Neutrophil extracellular traps (NETs) at the crossroads of defence and disease
The foundational publication regarding NETs was reported by the Zylchinsky group [161]. The NETs are composed of a web of chromatin (DNA) and proteins that form a matrix that can trap invading pathogens. NET formation has been synonymously referred to as NETosis, which implies a form of cell death occurs. Although debatable, suicidal NETosis has been found to occur with certain stimuli (e.g., phorbol 12-myristate 13-acetate), which is characterized by NET release accompanied by cell lysis. On the other hand, vital NETosis involves a more regulated but rapid release of NETs induced by appropriate stimuli (bacterial lipopolysaccharide, for example), through toll-like receptor 2/4 activation [162]. A third NETosis pathway involved mitochondrial DNA release and is dependent on reactive oxygen species and granulocyte-macrophage colony stimulating factor [163]. NET formation involves three significant steps: 1) activation to produce superoxide (and subsequently H2O2) via NADPH oxidase; 2) nuclear and granule membrane destabilization and release of respective contents into the cytosol; 3) mixing of nuclear and granule contents and complete loss of their membranes; 4) cell contraction and release of NETs [164]. What results in this process is termed NETosis (initially coined in [165]). Thus, the NET can provide defence against pathogens, such as bacteria and fungi [166]. Significant to this review is the involvement of MPO in the chromatin (DNA) strands of NETs. MPO is considered to exert its oxidative capacity to cause damage to invading microbes, or host tissue, within the scope of the NET. Importantly, individuals lacking MPO appear not to be capable of forming neutrophil extracellular traps (NETs) [38]. In addition, the superoxide (and subsequently H2O2) generated by the respiratory burst (NADPH oxidase) is also required to produce NETs together with the fundamental role of MPO [167]. The re-addition of H2O2 to chronic granulomatous disease (patients lacking NADPH oxidase activity) neutrophils reactivated their capacity to form neutrophil extracellular traps [168]. This may partially explain the reason behind the inability to combat C. Albicans infection.
Linking the previous topic of ANCA (anti-MPO antibodies), there is a relationship between ANCAs and NETosis. Studies on this topic first were indicated over a decade ago when examining vasculitis, where the NET formation was proposed to trigger vasculitis and anti-MPO formation [170]. A recent study reported a role for the TIM-3 surface receptor on T-cells as a critical contributor to NET maintenance in MPO-ANCA-associated vasculitis [171]. This is an essential connection between the adaptive and innate immune systems via NETs. The NET formation, where MPO can have an important role, has appeared in numerous reports of diseases and morbidities. NET formation and complement (C3, C5) were observed in COVID-19 patients and were associated with disease severity. MPO-DNA levels were significantly higher in severe vs. mild COVID-19 cases [43]. A study investigating thrombosis mechanisms in COVID-19 patients reported increased circulating NET formation in these patients, including increased MPO-DNA complexes (MPO associated with NETs) [44]. As a catalyst for thrombotic events in COVID-19, NETs were reported to be an exciting interplay between platelets and neutrophils in this disease [42]. As previously discussed, circulating MPO levels are an independent risk factor associated with cardiovascular disease. NETs, as a more complex structure (that also includes MPO), are involved in cardiac inflammation, myocardial infarction, and atrial fibrillation, though it is unclear if they are the cause or the consequence of these morbidities [172,173].
Furthermore, higher NET levels were detected in the plasma of Brazilian Alzheimer's disease patients compared to healthy controls, which may be associated to complement system activity [174]. Importantly, damage-associated molecular patterns (such as mitochondrial DNA, fibronectin extra domain A, and galectin-3) released during tissue damage may activate NETs through interaction with neutrophil surface proteins [172]. In addition, NETs can be triggered by cytokines [175], platelets [176], nitric oxide and peroxynitrite [177], and numerous microcrystals (of urate, calcium, cholesterol, and silica) [178], and numerous microbes (bacteria, viruses, yeast, parasites) [179]. The activation step for NETs as well as inducers of NETs is shown in Fig. 7.
Fig. 7.

Major NET formation steps and summary of inducers of NETosis. For illustrative purposes, the small green spheres in the NET represent MPO, which were housed in the primary granules (large green spheres). ANCA-induced activation is suggested to require neutrophil priming in advance. TIM-3 is a surface protein on T-cells that is proposed to activate NETosis. DAMPs (damage associated molecular patterns) have been described in the text, but include mitochondrial DNA, galectin-3, for example. Uric acid microcrystals, and other microcrystals can activate NETosis, and so could the combination of the cytokines IL-6 and TNFα. Concepts and findings were applied in this figure from [42,164,169]. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Overall, the involvement in NETs in many disease processes is continually evolving. As NETs are a heterogeneous mixture of components, including different enzymes, singling out the specific contribution of MPO in this context may be challenging. On the other hand, the fact that MPO is required for NET formation indicates a vital need for MPO activity in both protective and deleterious NET–associated processes.
11. Drug metabolism and MPO inhibition
Drug metabolism reactions catalyzed by MPO have been known for some time. The fact that MPO is a haem enzyme, similar to the P450 enzyme family, also suggests its drug metabolism capacity. Neutrophil metabolism of drugs, specifically via the concerted action of NADPH oxidase and MPO, has been proposed to have a role in their side effects and adverse drug reactions [35,36]. MPO is unique in that it has at least four mechanisms to catalyze reactions mimicking P450-like drug metabolism: 1) its peroxidase activity [84]; 2) oxidizing equivalents via HOCl/–OCl generation, and 3) superoxide radical catalyzed hydroxylation [84].
Simplified Hydroxylation reactions of MPO:
Peroxidase reactions
| (9) |
| (10) |
An example of this reaction is the phenol parahydroxylation mechanism to form hydroquinone and eventually benzoquinone. This pathway proposes a localized carbocation reaction with H2O to produce a hydroxylated product [180], although a superoxide mechanism was initially suggested [181].
| (11) |
The example of amine-containing drugs is used, however, this can apply to other compounds such as the amine of nucleosides. Indeed, a primary radical is observed (an aminyl radical in this instance) [182,183], though the question of the products may vary. There will be an aldehyde product in the degradation pathway of these chloramines [184,185]. The latter may undergo reduction to an alcohol to produce a putative hydroxylated product. However, if the aldehyde (in this case, nitroso intermediate) is highly reactive, free products will likely not be observed in a biological matrix, as observed with sulfamethoxazole and procainamide [186,187].
| (12) |
The superoxide-dependent reaction involves an initial MPO Compound III formed between superoxide and resting MPO (in the ferric state). This is followed by a subsequent reduction by superoxide molecule of the MPO Compound III to produce a hydroxylated enzyme similar to P450, where superoxide is bound to ferrous MPO to interact with the substrate [84]. It is important to note that unlike P450, where a radical pair (both the enzyme and substrate) are caged, so the free radical (drug) is not diffusible to the medium, MPO substrates usually are not caged. For example, aromatic carbon oxidation reactions of MPO, where molecular oxygen favourably adds to the radical, forming a peroxyl radical (as seen with lipid peroxidation):
| (13) |
Numerous compounds can be oxidized by MPO through one of these processes, including olefin oxidation (epoxidation) [188], drug oxidation such as carbon oxidation [189] and arylamine oxidation [[190], [191], [192], [193]]. Recently, the oxidation of edaravone, a pyrazolone antioxidant drug, was detected as a carbon radical [79]. A relevant functional group with respect to this review that undergoes oxidation by MPO is that of the hydrazines and hydrazides. In particular, this scaffold has provided research tools that have paved the road to new drugs, which turn out to be MPO inhibitors.
11.1. The evolution of MPO-catalyzed drug metabolism towards MPO inhibitors
The seminal study by Kettle and Winterbourn on benzoic acid hydrazides led to 4-aminobenzoic acid hydrazide as a tool to study the effect of MPO inhibition [94,194]. Almost 700 publications have cited the use of 4-aminobenzoic acid hydrazide to date. A mechanistic study of inhibition revealed that 4-aminobenzoic acid hydrazide catalyzed ester bond cleavage between the haem and amino acid protein chain [195]. Although quite valuable for enzymatic, cellular, and short-term in vivo animal studies, 4-aminobenzoic acid hydrazide is not ideal as a drug candidate as hydrazides have undesirable side effects, which must be outweighed by their benefit to be clinically used (such as the anti-mycobacterial drug, isoniazid).
A detailed review outlining patents that have been filed since 2003 reported 15 MPO inhibitor patents [3]. Studies have come quite a long way since hydrazides, and there is considerable chemical heterogeneity for MPO inhibitors. Computational and experimental screening efforts directed towards identifying reversible and irreversible MPO inhibitors have led to diverse scaffolds [[196], [197], [198], [199]]. These chemical scaffolds include triazolopyridine macrocyclics, peptides, ferulic acids, indole alkylamines, thioxanthines, thioxodihydroquinazolinones, triazolopyridines, and thiopyrimidinones [3]. Despite this chemical diversity, the most favourable scaffolds thus far are based on 2-thioxanthine and 2-thiopyrimidinone. A 2-thiopyrimidinone (PF06282999, Fig. 8) produced by Pfizer did not progress after Phase I clinical trials, in spite of desirable potency and pharmacokinetics parameters [3]. Work by Kettle and scientists at AstraZeneca demonstrated the potent MPO inhibition of 2-thioxanthines, which act as mechanism-based inhibitors. The initial radical product forms a haem-adduct before it is able to leave the MPO active site [39]. In this sense, the radical of the inhibitor is effectively caged, resulting in haem inactivation. The 2-thioxanthines demonstrated sub-micromolar IC50 values. Studies on aromatic hydroxamates revealed novel reversible inhibition [200]; however, it appears the utility of MPO inhibitors in disease settings favours irreversible inhibition. Even though 2-thioxanthines and a 2-thiopyrimidone advanced to clinical trials, none have yet passed phase II. There is currently a clinical trial for an AstraZeneca MPO inhibitor (AZD4831, Fig. 8) being run at the Mayo Clinic, which is currently in phase II (Clinical trial: NCT03611153) [201]. The study aims to determine if a single dose of this compound can affect hemodynamics in heart failure patients. It appears as though MPO inhibition is currently aimed at ameliorating cardiovascular disease, although work should continue to attenuate oxidative stress which is a key component in many of the diseases discussed in this review.
Fig. 8.

Chemical structure of AZD4831 (AstraZeneca), currently in phase II clinical trials, and PF06282999 (Pfizer) did not proceed from Phase I clinical trials [3].
12. Concluding comments
MPO has remarkable diversity in the spectrum of its functions in health and disease. After reading this review, and considering clinical MPO inhibitors are being evaluated, one may be tempted to suppose that we may be better off without this enzyme. However, this is over simplistic, since the healthy milieu intérieur must be achieved with the normal presence and activity of MPO. Indeed, reflecting on the fact that most individuals express MPO indicates a fundamental (protective) function, and perhaps regulation of over active MPO is more desirable. When disease and pathobiology comes into play, MPO inhibition becomes a viable target, though clinical trials will be telling exactly how well this is achieved.
Funding source
Select parts of this review, where relevant articles were cited, were funded by the Natural Sciences and Engineering Research Council (NSERC) grant number RGPIN-2014-04878 and RGPIN-2020-06305.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102109.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Nauseef W.M. In: Neutrophil Methods and Protocols. Quinn M.T., DeLeo F.R., Bokoch G.M., editors. Humana Press; Totowa, NJ: 2007. Diagnostic assays for myeloperoxidase deficiency; pp. 525–530. [Google Scholar]
- 2.Battistuzzi G. Influence of the covalent heme–protein bonds on the redox thermodynamics of human myeloperoxidase. Biochemistry. 2011;50(37):7987–7994. doi: 10.1021/bi2008432. [DOI] [PubMed] [Google Scholar]
- 3.Soubhye J., Van Antwerpen P., Dufrasne F. A patent review of myeloperoxidase inhibitors for treating chronic inflammatory syndromes (focus on cardiovascular diseases, 2013-2019) Expert Opin. Ther. Pat. 2020;30(8):595–608. doi: 10.1080/13543776.2020.1780210. [DOI] [PubMed] [Google Scholar]
- 4.Agner K., Verdoperoxidase . In: In Advances in Enzymology and Related Areas of Molecular Biology. Werkman F.F.N.a.C., editor. 2006. pp. 137–148. [Google Scholar]
- 5.Agner K., Verdoperoxidase . 1943. In Advances in Enzymology and Related Areas of Molecular Biology; pp. 137–148. [Google Scholar]
- 6.Nauseef W.M. Myeloperoxidase in human neutrophil host defence. Cell Microbiol. 2014;16(8):1146–1155. doi: 10.1111/cmi.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng G. Identification and characterization of VPO1, a new animal heme-containing peroxidase. Free Radic. Biol. Med. 2008;45(12):1682–1694. doi: 10.1016/j.freeradbiomed.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maehly A.C. MYELOPEROXIDASE. Methods in Enzymology. 1955;2:794–801. [Google Scholar]
- 9.Agner K. Detoxicating effect of verdoperoxidase on toxins. Nature. 1947;159(4034):271. doi: 10.1038/159271a0. [DOI] [PubMed] [Google Scholar]
- 10.Kojima S. Studies ON peroxidase: II. The effect of peroxidase on the bactericidal action of phenols. J. Biochem. 1931;14(1):95–109. [Google Scholar]
- 11.Klebanoff S.J. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 1968;95(6):2131–2138. doi: 10.1128/jb.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klebanoff S.J. Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science. 1970;169(3950):1095–1097. doi: 10.1126/science.169.3950.1095. [DOI] [PubMed] [Google Scholar]
- 13.Kimura S., Ikeda-Saito M. Human myeloperoxidase and thyroid peroxidase, two enzymes with separate and distinct physiological functions, are evolutionarily related members of the same gene family. Proteins: Structure, Function, and Bioinformatics. 1988;3(2):113–120. doi: 10.1002/prot.340030206. [DOI] [PubMed] [Google Scholar]
- 14.Sakamaki K. Molecular cloning and characterization of a chromosomal gene for human eosinophil peroxidase. J. Biol. Chem. 1989;264(28):16828–16836. [PubMed] [Google Scholar]
- 15.Ueda T. Molecular cloning and characterization of the chromosomal gene for human lactoperoxidase. Eur. J. Biochem. 1997;243(1‐2):32–41. doi: 10.1111/j.1432-1033.1997.0032a.x. [DOI] [PubMed] [Google Scholar]
- 16.Magacz M. The significance of lactoperoxidase system in oral health: application and efficacy in oral hygiene products. Int. J. Mol. Sci. 2019;20(6):1443. doi: 10.3390/ijms20061443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z. Vascular peroxidase 1 is a novel regulator of cardiac fibrosis after myocardial infarction. Redox Biology. 2019;22:101151. doi: 10.1016/j.redox.2019.101151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischel R. Der mikrochemische Nachweis der Peroxydase und Pseudoperoxydase in tierischen Geweben. Archiv für mikroskopische Anatomie. 1913;83(1):A130–A175. [Google Scholar]
- 19.Agner K. Verdoperoxidase: a ferment isolated from leukocytes. Acta Physiol. Scand. 1941;2:1–62. [Google Scholar]
- 20.Schultz J., Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 1962;96(3):465–467. doi: 10.1016/0003-9861(62)90321-1. [DOI] [PubMed] [Google Scholar]
- 21.Dunn W.B., Hardin J.H., Spicer S.S. Brief report: ultrastructural localization of myeloperoxidase in human neutrophil and rabbit heterophil and eosinophil leukocytes. Blood. 1968;32(6):935–944. [PubMed] [Google Scholar]
- 22.Klebanoff S.J. Iodination of bacteria: a bactericidal mechanism. J. Exp. Med. 1967;126(6):1063–1078. doi: 10.1084/jem.126.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falk R.J., Jennette J.C. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N. Engl. J. Med. 1988;318(25):1651–1657. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 24.Bosch X., Guilabert A., Font J. Antineutrophil cytoplasmic antibodies. Lancet. 2006;368(9533):404–418. doi: 10.1016/S0140-6736(06)69114-9. [DOI] [PubMed] [Google Scholar]
- 25.Kallenberg C.G.M., Leontine Mulder A.H., Cohen Tervaert J.W. Antineutrophil cytoplasmic antibodies: a still-growing class of autoantibodies in inflammatory disorders. Am. J. Med. 1992;93(6):675–682. doi: 10.1016/0002-9343(92)90202-m. [DOI] [PubMed] [Google Scholar]
- 26.Daugherty A. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 1994;94(1):437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hazen S.L., Heinecke J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Invest. 1997;99(9):2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yagmurca M. Erdosteine prevents doxorubicin-induced cardiotoxicity in rats. Pharmacol. Res. 2003;48(4):377–382. doi: 10.1016/s1043-6618(03)00185-3. [DOI] [PubMed] [Google Scholar]
- 29.Ky B. Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. J. Am. Coll. Cardiol. 2014;63(8):809–816. doi: 10.1016/j.jacc.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan M.L. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003;349(17):1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 31.Baldus S. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108(12):1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 32.Pennathur S. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o'-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J. Biol. Chem. 1999;274(49):34621–34628. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds W.F. MPO and APOEε4 polymorphisms interact to increase risk for AD in Finnish males. Neurology. 2000;55(9):1284–1290. doi: 10.1212/wnl.55.9.1284. [DOI] [PubMed] [Google Scholar]
- 34.Mason R.P. Free-radical metabolite formation by mammalian peroxidases. Life Sci. Res. Rep. 1987;37:67–83. Mech. Cell Inj.: Implic. Hum. Health. [Google Scholar]
- 35.Uetrecht J.P. Idiosyncratic drug reactions: possible role of reactive metabolites generated by leukocytes. Pharmaceut. Res. 1989;6(4):265–273. doi: 10.1023/a:1015934104984. [DOI] [PubMed] [Google Scholar]
- 36.Tafazoli S., O'Brien P.J. Peroxidases: a role in the metabolism and side effects of drugs. Drug Discov. Today. 2005;10(9):617–625. doi: 10.1016/S1359-6446(05)03394-5. [DOI] [PubMed] [Google Scholar]
- 37.Papayannopoulos V. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. JCB (J. Cell Biol.) 2010;191(3):677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metzler K.D. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117(3):953–959. doi: 10.1182/blood-2010-06-290171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiden A.-K. 2-Thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J. Biol. Chem. 2011;286(43):37578–37589. doi: 10.1074/jbc.M111.266981. S37578/1-S37578/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanson S., Nordvall G., Tiden A.-K. AstraZeneca AB; Swed.: 2003. Preparation of Xanthinethione Derivatives as Myeloperoxidase Inhibitors; p. 55. [Google Scholar]
- 41.Carpino, P.A., et al., 2-Thiopyrimidineones as Myeloperoxidase Inhibitors and Their Preparation. 2013, Pfizer Inc., USA . p. 198pp.
- 42.Middleton E.A. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y. Carboxypeptidase B blocks ex vivo activation of the anaphylatoxin-neutrophil extracellular trap axis in neutrophils from COVID-19 patients. Crit. Care. 2021;25(1) doi: 10.1186/s13054-021-03482-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuo Y. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis. 2021;51(2):446–453. doi: 10.1007/s11239-020-02324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang K.S. Human myeloperoxidase gene: molecular cloning and expression in leukemic cells. Blood. 1986;68(6):1411–1414. [PubMed] [Google Scholar]
- 46.Kudoh J. Assignment of the myeloperoxidase gene MPO to human chromosome 17 using somatic cell hybrids and flow-sorted chromosomes. Jinrui Idengaku Zasshi. 1988;33(3):315–324. doi: 10.1007/BF02032861. [DOI] [PubMed] [Google Scholar]
- 47.Van Tuinen P. Localization of myeloperoxidase to the long arm of human chromosome 17: relationship to the 15;17 translocation of acute promyelocytic leukemia. Oncogene. 1987;1(3):319–322. [PubMed] [Google Scholar]
- 48.Morishita K. Chromosomal gene structure of human myeloperoxidase and regulation of its expression by granulocyte colony-stimulating factor. J. Biol. Chem. 1987;262(31):15208–15213. [PubMed] [Google Scholar]
- 49.Inazawa J. Assignment of the human myeloperoxidase gene (MPO) to bands q21.3→q23 of chromosome 17. Cytogenet. Genome Res. 1989;50(2–3):135–136. doi: 10.1159/000132742. [DOI] [PubMed] [Google Scholar]
- 50.Sakamaki K. The eosinophil peroxidase gene forms a cluster with the genes for myeloperoxidase and lactoperoxidase on human chromosome 17. Cytogenet. Genome Res. 2000;88(3–4):246–248. doi: 10.1159/000015529. [DOI] [PubMed] [Google Scholar]
- 51.Salmon S.E. Myeloperoxidase deficiency. N. Engl. J. Med. 1970;282(5):250–253. doi: 10.1056/NEJM197001292820505. [DOI] [PubMed] [Google Scholar]
- 52.Locksley R., Wilson C., Klebanoff S. Increased respiratory burst in myeloperoxidase-deficient monocytes. Blood. 1983;62(4):902–909. [PubMed] [Google Scholar]
- 53.Bos A.J. Characterization of hereditary partial myeloperoxidase deficiency. J. Lab. Clin. Med. 1982;99(4):589–600. [PubMed] [Google Scholar]
- 54.Kutter D. Prevalence of myeloperoxidase deficiency: population studies using Bayer-Technicon automated hematology. J. Mol. Med. 1998;76(10):669–675. doi: 10.1007/s001090050266. [DOI] [PubMed] [Google Scholar]
- 55.Parry M.F. Myeloperoxidase deficiency: prevalence and clinical significance. Ann. Intern. Med. 1981;95(3):293–301. doi: 10.7326/0003-4819-95-3-293. [DOI] [PubMed] [Google Scholar]
- 56.Nauseef W.M., Root R.K., Malech H.L. Biochemical and immunologic analysis of hereditary myeloperoxidase deficiency. J. Clin. Invest. 1983;71(5):1297–1307. doi: 10.1172/JCI110880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nunoi H. Prevalence of inherited myeloperoxidase deficiency in Japan. Microbiol. Immunol. 2003;47(7):527–531. doi: 10.1111/j.1348-0421.2003.tb03414.x. [DOI] [PubMed] [Google Scholar]
- 58.Gentilini F. A nonsense mutation in the myeloperoxidase gene is responsible for hereditary myeloperoxidase deficiency in an Italian hound dog. Anim. Genet. 2016;47(5):632–633. doi: 10.1111/age.12463. [DOI] [PubMed] [Google Scholar]
- 59.Lehrer R.I., Cline M.J. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J. Clin. Invest. 1969;48(8):1478–1488. doi: 10.1172/JCI106114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalinski T. Lethal candida sepsis associated with myeloperoxidase deficiency and pre-eclampsia: case report. APMIS. 2007;115(7):875–880. doi: 10.1111/j.1600-0463.2007.apm_600.x. [DOI] [PubMed] [Google Scholar]
- 61.Lanza F. Clinical manifestation of myeloperoxidase deficiency. Journal of Molecular Medicine-Jmm. 1998;76(10):676–681. doi: 10.1007/s001090050267. [DOI] [PubMed] [Google Scholar]
- 62.Fujimoto K. Myeloperoxidase deficiency enhances zymosan phagocytosis associated with up-regulation of surface expression of CD11b in mouse neutrophils. Free Radic. Res. 2016;50(12):1340–1349. doi: 10.1080/10715762.2016.1244821. [DOI] [PubMed] [Google Scholar]
- 63.Haegens A. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 2009;182(12):7990–7996. doi: 10.4049/jimmunol.0800377. [DOI] [PubMed] [Google Scholar]
- 64.Abraitytė S. Unexpected combination: DiGeorge syndrome and myeloperoxidase deficiency. BMJ Case Rep. 2020;13(2) doi: 10.1136/bcr-2019-232741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanza F. Clinical manifestation of myeloperoxidase deficiency. J. Mol. Med. 1998;76(10):676–681. doi: 10.1007/s001090050267. [DOI] [PubMed] [Google Scholar]
- 66.Volkman R. Myeloperoxidase deficiency inhibits cognitive decline in the 5XFAD mouse model of Alzheimer's disease. Front. Neurosci. 2019;13(990) doi: 10.3389/fnins.2019.00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin Z. Myeloperoxidase targets apolipoprotein A-I for site-specific tyrosine chlorination in atherosclerotic lesions and generates dysfunctional high-density lipoprotein. Chem. Res. Toxicol. 2021 doi: 10.1021/acs.chemrestox.1c00086. [DOI] [PubMed] [Google Scholar]
- 68.Schürmann N. Myeloperoxidase targets oxidative host attacks to Salmonella and prevents collateral tissue damage. Nature Microbiology. 2017;2(4):16268. doi: 10.1038/nmicrobiol.2016.268. [DOI] [PubMed] [Google Scholar]
- 69.Ikeda-Saito M. On the analogy in the structure of the spleen green heme protein and granulocyte myeloperoxidase. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1986;202(2):245–250. doi: 10.1016/0014-5793(86)80695-0. [DOI] [PubMed] [Google Scholar]
- 70.Fiedler T.J., Davey C.A., Fenna R.E. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution. J. Biol. Chem. 2000;275(16):11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 71.La Mar G.N., de Ropp J.S. Assignment of exchangeable proximal histidine resonances in high-spin ferric hemoproteins: substrate binding in horseradish peroxidase. Biochem. Biophys. Res. Commun. 1979;90(1):36–41. doi: 10.1016/0006-291x(79)91586-9. [DOI] [PubMed] [Google Scholar]
- 72.Kooter I.M. The sulfonium ion linkage in myeloperoxidase: direct spectroscopic detection BY isotopic labeling and effect OF mutation. J. Biol. Chem. 1999;274(38):26794–26802. doi: 10.1074/jbc.274.38.26794. [DOI] [PubMed] [Google Scholar]
- 73.Van Antwerpen P. Glycosylation pattern of mature dimeric leukocyte and recombinant monomeric myeloperoxidase: glycosylation is required for optimal enzymatic activity. J. Biol. Chem. 2010;285(21):16351–16359. doi: 10.1074/jbc.M109.089748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ravnsborg T., Houen G., Højrup P. The glycosylation of myeloperoxidase. Biochim. Biophys. Acta Protein Proteonomics. 2010;1804(10):2046–2053. doi: 10.1016/j.bbapap.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 75.Reiding K.R. Neutrophil myeloperoxidase harbors distinct site-specific peculiarities in its glycosylation. J. Biol. Chem. 2019;294(52):20233–20245. doi: 10.1074/jbc.RA119.011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Dalen C.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997;327(2):487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Furtmüller P.G., Burner U., Obinger C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 1998;37(51):17923–17930. doi: 10.1021/bi9818772. [DOI] [PubMed] [Google Scholar]
- 78.Morgan A. Caution for the routine use of phenol red – it is more than just a pH indicator. Chem. Biol. Interact. 2019;310:108739. doi: 10.1016/j.cbi.2019.108739. [DOI] [PubMed] [Google Scholar]
- 79.Suh L.Y.K. Myeloperoxidase-mediated oxidation of edaravone produces an apparent non-toxic free radical metabolite and modulates hydrogen peroxide-mediated cytotoxicity in HL-60 cells. Free Radic. Biol. Med. 2019;143:422–432. doi: 10.1016/j.freeradbiomed.2019.08.021. [DOI] [PubMed] [Google Scholar]
- 80.King C.C., Jefferson M.M., Thomas E.L. Secretion and inactivation of myeloperoxidase by isolated neutrophils. J. Leukoc. Biol. 1997;61(3):293–302. doi: 10.1002/jlb.61.3.293. [DOI] [PubMed] [Google Scholar]
- 81.Davies M.J. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2011;48(1):8–19. doi: 10.3164/jcbn.11-006FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marquez L.A., Dunford H.B. Reaction of compound III of myeloperoxidase with ascorbic acid. J. Biol. Chem. 1990;265(11):6074–6078. [PubMed] [Google Scholar]
- 83.Marquez L.A., Dunford H.B. Interaction of acetaminophen with myeloperoxidase intermediates: optimum stimulation of enzyme activity. Arch. Biochem. Biophys. 1993;305(2):414–420. doi: 10.1006/abbi.1993.1440. [DOI] [PubMed] [Google Scholar]
- 84.Kettle A.J., Winterbourn C.C. Superoxide-dependent hydroxylation by myeloperoxidase. J. Biol. Chem. 1994;269(25):17146–17151. [PubMed] [Google Scholar]
- 85.Gomes M.M. Biosynthesis of N,N-dimethyltryptamine (DMT) in a melanoma cell line and its metabolization by peroxidases. Biochem. Pharmacol. 2014;88(3):393–401. doi: 10.1016/j.bcp.2014.01.035. [DOI] [PubMed] [Google Scholar]
- 86.Gomes M.M. Oxidation of lysergic acid diethylamide (LSD) by peroxidases: a new metabolic pathway. Forensic Toxicol. 2012;30(2):87–97. [Google Scholar]
- 87.Subrahmanyam V.V., Kolachana P., Smith M.T. Hydroxylation of phenol to hydroquinone catalyzed by A human myeloperoxidase-superoxide complex: possible implications in benzene-induced myelotoxicity. Free Radic. Res. Commun. 1991;15(5):285–296. doi: 10.3109/10715769109105224. [DOI] [PubMed] [Google Scholar]
- 88.Reszka K.J., McGraw D.W., Britigan B.E. Peroxidative metabolism of β2-agonists salbutamol and fenoterol and their analogues. Chem. Res. Toxicol. 2009;22(6):1137–1150. doi: 10.1021/tx900071f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ximenes V.F. Superoxide-dependent oxidation of melatonin by myeloperoxidase. J. Biol. Chem. 2005;280(46):38160–38169. doi: 10.1074/jbc.M506384200. [DOI] [PubMed] [Google Scholar]
- 90.Ximenes V.F., Catalani L.H., Campa A. Oxidation of melatonin and tryptophan by an HRP cycle involving compound III. Biochem. Biophys. Res. Commun. 2001;287(1):130–134. doi: 10.1006/bbrc.2001.5557. [DOI] [PubMed] [Google Scholar]
- 91.Fujimoto S. Hydroxylation of phenylalanine by myeloperoxidase-hydrogen peroxide system. Chem. Pharm. Bull. 1991;39(6):1598–1600. [Google Scholar]
- 92.Mays D.C. Metabolism of phenytoin and covalent binding of reactive intermediates in activated human neutrophils. Biochem. Pharmacol. 1995;50(3):367–380. doi: 10.1016/0006-2952(95)00151-o. [DOI] [PubMed] [Google Scholar]
- 93.van der Walt B.J., van Zyl J.M., Kriegler A. Aromatic hydroxylation during the myeloperoxidase-oxidase oxidation of hydrazines. Biochem. Pharmacol. 1994;47(6):1039–1046. doi: 10.1016/0006-2952(94)90415-4. [DOI] [PubMed] [Google Scholar]
- 94.Kettle A.J., Gedye C.A., Winterbourn C.C. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem. J. 1997;321(2):503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arnhold J., Furtmüller P.G., Obinger C. Redox properties of myeloperoxidase. Redox Rep. 2003;8(4):179–186. doi: 10.1179/135100003225002664. [DOI] [PubMed] [Google Scholar]
- 96.Nakagawara A., Nathan C.F., Cohn Z.A. Hydrogen peroxide metabolism in human monocytes during differentiation in vitro. J. Clin. Invest. 1981;68(5):1243–1252. doi: 10.1172/JCI110370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Locksley R.M. Loss of granule myeloperoxidase during in vitro culture of human monocytes correlates with decay in antiprotozoa activity. Am. J. Trop. Med. Hyg. 1987;36(3):541–548. doi: 10.4269/ajtmh.1987.36.541. [DOI] [PubMed] [Google Scholar]
- 98.Brown K.E., Brunt E.M., Heinecke J.W. Immunohistochemical detection of myeloperoxidase and its oxidation products in Kupffer cells of human liver. Am. J. Pathol. 2001;159(6):2081–2088. doi: 10.1016/S0002-9440(10)63059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amanzada A. Myeloperoxidase and elastase are only expressed by neutrophils in normal and in inflamed liver. Histochem. Cell Biol. 2011;135(3):305–315. doi: 10.1007/s00418-011-0787-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McMillen T.S., Heinecke J.W., LeBoeuf R.C. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111(21):2798–2804. doi: 10.1161/CIRCULATIONAHA.104.516278. [DOI] [PubMed] [Google Scholar]
- 101.Austin G.E. Control of myeloperoxidase gene expression in developing myeloid cells. Leuk. Res. 1996;20(10):817–820. doi: 10.1016/s0145-2126(96)00032-x. [DOI] [PubMed] [Google Scholar]
- 102.Lin K.M., Austin G.E. Functional activity of three distinct myeloperoxidase (MPO) promoters in human myeloid cells. Leukemia. 2002;16(6):1143–1153. doi: 10.1038/sj.leu.2402514. [DOI] [PubMed] [Google Scholar]
- 103.Strobl H. Myeloperoxidase expression in CD34+ normal human hematopoietic cells. Blood. 1993;82(7):2069–2078. [PubMed] [Google Scholar]
- 104.Nauseef W.M. Biosynthesis of human myeloperoxidase. Arch. Biochem. Biophys. 2018;642:1–9. doi: 10.1016/j.abb.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Henrique de Araujo T. Intracellular localization of myeloperoxidase in murine peritoneal B-lymphocytes and macrophages. Cell. Immunol. 2013;281(1):27–30. doi: 10.1016/j.cellimm.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 106.Okada S.S. Myeloperoxidase in human peripheral blood lymphocytes: production and subcellular localization. Cell. Immunol. 2016;300:18–25. doi: 10.1016/j.cellimm.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 107.Ray R.S., Katyal A. Myeloperoxidase: bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 2016;68:611–620. doi: 10.1016/j.neubiorev.2016.06.031. [DOI] [PubMed] [Google Scholar]
- 108.Kim Y.R. Myeloperoxidase expression as a potential determinant of parthenolide-induced apoptosis in leukemia bulk and leukemia stem cells. J. Pharmacol. Exp. Therapeut. 2010;335(2):389–400. doi: 10.1124/jpet.110.169367. [DOI] [PubMed] [Google Scholar]
- 109.Hosseini M. Targeting myeloperoxidase disrupts mitochondrial redox balance and overcomes cytarabine resistance in human acute myeloid leukemia. Canc. Res. 2019;79(20):5191–5203. doi: 10.1158/0008-5472.CAN-19-0515. [DOI] [PubMed] [Google Scholar]
- 110.Quentmeier H. The LL-100 panel: 100 cell lines for blood cancer studies. Sci. Rep. 2019;9(1):8218. doi: 10.1038/s41598-019-44491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Green P.S. Neuronal expression of myeloperoxidase is increased in Alzheimer's disease. J. Neurochem. 2004;90(3):724–733. doi: 10.1111/j.1471-4159.2004.02527.x. [DOI] [PubMed] [Google Scholar]
- 112.Thomas E.L. Leukocyte myeloperoxidase and salivary lactoperoxidase: identification and quantitation in human mixed saliva. J. Dent. Res. 1994;73(2):544–555. doi: 10.1177/00220345940730021001. [DOI] [PubMed] [Google Scholar]
- 113.Kettle A.J. Neutrophils convert tyrosyl residues in albumin to chlorotyrosine. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1996;379(1):103–106. doi: 10.1016/0014-5793(95)01494-2. [DOI] [PubMed] [Google Scholar]
- 114.Hazen S.L. Human neutrophils employ chlorine gas as an oxidant during phagocytosis. J. Clin. Invest. 1996;98(6):1283–1289. doi: 10.1172/JCI118914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chapman A.L.P. Comparison of mono- and dichlorinated tyrosines with carbonyls for detection of hypochlorous acid modified proteins. Arch. Biochem. Biophys. 2000;377(1):95–100. doi: 10.1006/abbi.2000.1744. [DOI] [PubMed] [Google Scholar]
- 116.Winterbourn C.C., Kettle A.J. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic. Biol. Med. 2000;29(5):403–409. doi: 10.1016/s0891-5849(00)00204-5. [DOI] [PubMed] [Google Scholar]
- 117.Amunugama K., Pike D.P., Ford D.A. The lipid biology of sepsis. J. Lipid Res. 2021;62 doi: 10.1016/j.jlr.2021.100090. 100090-100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meyer N.J. Myeloperoxidase-derived 2-chlorofatty acids contribute to human sepsis mortality via acute respiratory distress syndrome. JCI Insight. 2017;2(23) doi: 10.1172/jci.insight.96432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pike D.P. 2-Chlorofatty acids are biomarkers of sepsis mortality and mediators of barrier dysfunction in rats. J. Lipid Res. 2020;61(7):1115–1127. doi: 10.1194/jlr.RA120000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yuan Y. Ratiometric fluorescent detection of hypochlorite in aqueous solution and living cells using an ionic probe with aggregation-induced emission feature. Sensor. Actuator. B Chem. 2021;330:129324. [Google Scholar]
- 121.Jiang Y. A fast-response fluorescent probe for hypochlorous acid detection and its application in exogenous and endogenous HOCl imaging of living cells. Chem. Commun. 2017;53(91):12349–12352. doi: 10.1039/c7cc07373a. [DOI] [PubMed] [Google Scholar]
- 122.Han Q. A redox-switchable colorimetric probe for “naked-eye” detection of hypochlorous acid and glutathione. Molecules. 2019;24(13):2455. doi: 10.3390/molecules24132455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang Y. Detection of hypochlorous acid fluctuation via a selective near-infrared fluorescent probe in living cells and in vivo under hypoxic stress. J. Mater. Chem. B. 2019;7(15):2557–2564. doi: 10.1039/c9tb00079h. [DOI] [PubMed] [Google Scholar]
- 124.Ashoka A.H. Recent advances in fluorescent probes for detection of HOCl and HNO. ACS Omega. 2020;5(4):1730–1742. doi: 10.1021/acsomega.9b03420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wadghiri Y.Z. High-resolution imaging of myeloperoxidase activity sensors in human cerebrovascular disease. Sci. Rep. 2018;8(1):7687. doi: 10.1038/s41598-018-25804-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bogdanov A. Synthesis and testing of a binary catalytic system for imaging of signal amplification in vivo. Bioconjugate Chem. 2007;18(4):1123–1130. doi: 10.1021/bc060392k. [DOI] [PubMed] [Google Scholar]
- 127.Kato Y. Myeloperoxidase catalyzes the conjugation of serotonin to thiols via free radicals and tryptamine-4,5-dione. Chem. Res. Toxicol. 2012;25(11):2322–2332. doi: 10.1021/tx300218f. [DOI] [PubMed] [Google Scholar]
- 128.Wrona M.Z., Dryhurst G. Oxidation chemistry of 5-hydroxytryptamine. 1. Mechanism and products formed at micromolar concentrations. J. Org. Chem. 1987;52(13):2817–2825. [Google Scholar]
- 129.Löwenberg B., Downing J.R., Burnett A. Acute myeloid leukemia. N. Engl. J. Med. 1999;341(14):1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 130.Wakui M. Diagnosis of acute myeloid leukemia according to the WHO classification in the Japan Adult Leukemia Study Group AML-97 protocol. Int. J. Hematol. 2008;87(2):144–151. doi: 10.1007/s12185-008-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramachandra C.J.A. Myeloperoxidase as a multifaceted target for cardiovascular protection. Antioxidants Redox Signal. 2019;32(15):1135–1149. doi: 10.1089/ars.2019.7971. [DOI] [PubMed] [Google Scholar]
- 132.Meuwese M.C. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-norfolk prospective population study. J. Am. Coll. Cardiol. 2007;50(2):159–165. doi: 10.1016/j.jacc.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 133.Kameda T. Effects of myeloperoxidase-induced oxidation on antiatherogenic functions of high-density lipoprotein. Journal of Lipids. 2015;2015:592594. doi: 10.1155/2015/592594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tavora F.R. Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc. Disord. 2009;9(1):27. doi: 10.1186/1471-2261-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zheng L. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Invest. 2004;114(4):529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang G. Myeloperoxidase mediated HDL oxidation and HDL proteome changes do not contribute to dysfunctional HDL in Chinese subjects with coronary artery disease. PloS One. 2018;13(3) doi: 10.1371/journal.pone.0193782. e0193782-e0193782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li J. Ambient air pollution is associated with HDL (High-Density lipoprotein) dysfunction in healthy adults. Arterioscler. Thromb. Vasc. Biol. 2019;39(3):513–522. doi: 10.1161/ATVBAHA.118.311749. [DOI] [PubMed] [Google Scholar]
- 138.Mahat R.K., Singh N., Rathore V. Association of myeloperoxidase with cardiovascular disease risk factors in prediabetic subjects. Diabetes & Metabolic Syndrome: Clin. Res. Rev. 2019;13(1):396–400. doi: 10.1016/j.dsx.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 139.Wu C.-Y. Neutrophil activation in Alzheimer's disease and mild cognitive impairment: a systematic review and meta-analysis of protein markers in blood and cerebrospinal fluid. Ageing Res. Rev. 2020;62:101130. doi: 10.1016/j.arr.2020.101130. [DOI] [PubMed] [Google Scholar]
- 140.Bawa K.K. A peripheral neutrophil-related inflammatory factor predicts a decline in executive function in mild Alzheimer's disease. J. Neuroinflammation. 2020;17(1) doi: 10.1186/s12974-020-01750-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang H. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016;136(4):826–836. doi: 10.1111/jnc.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu G., Zheng S., Zhang H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport. 2018;29(3):208–213. doi: 10.1097/WNR.0000000000000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gellhaar S. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson's and Alzheimer's disease. Cell Tissue Res. 2017;369(3):445–454. doi: 10.1007/s00441-017-2626-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maki R.A. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson's disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019;141:115–140. doi: 10.1016/j.freeradbiomed.2019.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fan Y.H. Tomentosin reduces behavior deficits and neuroinflammatory response in MPTP-induced Parkinson's disease in mice. J. Environ. Pathol. Toxicol. Oncol. 2021;40(1):75–84. doi: 10.1615/JEnvironPatholToxicolOncol.v40.i1.70. [DOI] [PubMed] [Google Scholar]
- 146.Tay A. Serum myeloperoxidase levels in predicting the severity of stroke and mortality in acute ischemic stroke patients. Eur. Rev. Med. Pharmacol. Sci. 2015;19(11):1983–1988. [PubMed] [Google Scholar]
- 147.Kim H.J. Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke. J. Cerebr. Blood Flow Metabol. 2019;39(9):1864–1877. doi: 10.1177/0271678X18771978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eastman C.L., D'Ambrosio R., Ganesh T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology. 2020;172:107907. doi: 10.1016/j.neuropharm.2019.107907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Y. Myeloperoxidase nuclear imaging for epileptogenesis. Radiology. 2016;278(3):822–830. doi: 10.1148/radiol.2015141922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Di Battista A.P. The relationship between symptom burden and systemic inflammation differs between male and female athletes following concussion. BMC Immunol. 2020;21(1):11. doi: 10.1186/s12865-020-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qasim A, P.J. ANCA Positive Vasculitis. StatPearls [Internet] 2020 2021 Jan 5; Available from: https://www.ncbi.nlm.nih.gov/books/NBK554372/.
- 152.Cambridge G. Autoantibodies to myeloperoxidase IN idiopathic and drug-induced systemic lupus-erythematosus and vasculitis. Br. J. Rheumatol. 1994;33(2):109–114. doi: 10.1093/rheumatology/33.2.109. [DOI] [PubMed] [Google Scholar]
- 153.Mannik M., Merrill C.E., Wener M.H. Antibodies to human myeloperoxidase in glomerular immune deposits of systemic lupus erythematosus. Lupus. 2000;9(8):607–613. doi: 10.1191/096120300678828758. [DOI] [PubMed] [Google Scholar]
- 154.Vasoo S. Drug-induced lupus: an update. Lupus. 2006;15(11):757–761. doi: 10.1177/0961203306070000. [DOI] [PubMed] [Google Scholar]
- 155.West S., Kolfenbach J. Elsevier Health Sciences; 2019. Rheumatology Secrets E-Book. [Google Scholar]
- 156.Timlin H. Clinical characteristics of hydralazine-induced. Lupus. Cureus. 2019;11(6) doi: 10.7759/cureus.4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rubin R.L., Haluptzok R.F., Davila L.M. Severe hydralazine-induced lupus presenting as systemic lupus erythematosus. Lupus. 2020;29(5):509–513. doi: 10.1177/0961203320906265. [DOI] [PubMed] [Google Scholar]
- 158.Kumar B. Hydralazine-associated vasculitis: overlapping features of drug-induced lupus and vasculitis. Semin. Arthritis Rheum. 2018;48(2):283–287. doi: 10.1016/j.semarthrit.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 159.Skaljic M. A hydralazine-induced triumvirate: lupus, cutaneous vasculitis, and cryptococcoid Sweet syndrome. JAAD case reports. 2019;5(11):1006–1009. doi: 10.1016/j.jdcr.2019.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Telles R.W. Increased plasma myeloperoxidase levels in systemic lupus erythematosus. Rheumatol. Int. 2010;30(6):779–784. doi: 10.1007/s00296-009-1067-4. [DOI] [PubMed] [Google Scholar]
- 161.Brinkmann V. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 162.Yipp B.G., Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 163.Delgado-Rizo V. Neutrophil extracellular traps and its implications in inflammation: an overview. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.00081. 81-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Brinkmann V., Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007;5(8):577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 165.Steinberg B.E., Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci. STKE. 2007;2007(379):pe11. doi: 10.1126/stke.3792007pe11. [DOI] [PubMed] [Google Scholar]
- 166.Urban C.F. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5(10) doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Parker H. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012;92(4):841–849. doi: 10.1189/jlb.1211601. [DOI] [PubMed] [Google Scholar]
- 168.Fuchs T.A. Novel cell death program leads to neutrophil extracellular traps. The Journal of cell biology. 2007;176(2):231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heeringa P., Rutgers A., Kallenberg C.G.M. The net effect of ANCA on neutrophil extracellular trap formation. Kidney Int. 2018;94(1):14–16. doi: 10.1016/j.kint.2018.03.010. [DOI] [PubMed] [Google Scholar]
- 170.Kessenbrock K. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009;15(6):623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Su B. TIM-3 regulates the NETs-mediated dendritic cell activation in myeloperoxidase-ANCA-associated vasculitis. Clin. Exp. Rheumatol. 2021;39(2):13–20. doi: 10.55563/clinexprheumatol/6y0bjb. [DOI] [PubMed] [Google Scholar]
- 172.Ling S., Xu J.-W. NETosis as a pathogenic factor for heart failure. Oxidative Medicine and Cellular Longevity. 2021;2021:6687096. doi: 10.1155/2021/6687096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Döring Y., Libby P., Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases. Circ. Res. 2020;126(9):1228–1241. doi: 10.1161/CIRCRESAHA.120.315931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kretzschmar G.C. Neutrophil extracellular traps: a perspective of neuroinflammation and complement activation in Alzheimer's disease. Frontiers in Molecular Biosciences. 2021;8(143) doi: 10.3389/fmolb.2021.630869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Du J. Neutrophil extracellular traps induced by pro-inflammatory cytokines enhance procoagulant activity in NASH patients. Clinics and Research in Hepatology and Gastroenterology. 2021:101697. doi: 10.1016/j.clinre.2021.101697. [DOI] [PubMed] [Google Scholar]
- 176.Clark S.R. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13(4):463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 177.Manda-Handzlik A. Nitric oxide and peroxynitrite trigger and enhance release of neutrophil extracellular traps. Cell. Mol. Life Sci. 2020;77(15):3059–3075. doi: 10.1007/s00018-019-03331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rada B. Neutrophil extracellular traps and microcrystals. Journal of immunology research. 2017 doi: 10.1155/2017/2896380. 2896380-2896380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Delgado-Rizo V. Neutrophil extracellular traps and its implications in inflammation: an overview. Front. Immunol. 2017;8(81) doi: 10.3389/fimmu.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ullrich R., Hofrichter M. Enzymatic hydroxylation of aromatic compounds. Cell. Mol. Life Sci. 2007;64(3):271–293. doi: 10.1007/s00018-007-6362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Subrahmanyam V.V., Kolachana P., Smith M.T. Hydroxylation of phenol to hydroquinone catalyzed by a human myeloperoxidase-superoxide complex: possible implications in benzene-induced myelotoxicity. Free Radic. Res. Commun. 1991;15(5):285–296. doi: 10.3109/10715769109105224. [DOI] [PubMed] [Google Scholar]
- 182.Hawkins C.L., Davies M.J. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2002;15(1):83–92. doi: 10.1021/tx015548d. [DOI] [PubMed] [Google Scholar]
- 183.Zheng L. Aminyl radical generation via tandem norrish type I photocleavage, β-fragmentation: independent generation and reactivity of the 2′-deoxyadenosin- N6-yl radical. J. Org. Chem. 2017;82(7):3571–3580. doi: 10.1021/acs.joc.7b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Davies M.J., Hawkins C.L. The role of myeloperoxidase in biomolecule modification, chronic inflammation, and disease. Antioxidants Redox Signal. 2020;32(13):957–981. doi: 10.1089/ars.2020.8030. [DOI] [PubMed] [Google Scholar]
- 185.Szabó M. Decomposition of N-chloroglycine in alkaline aqueous solution: kinetics and mechanism. Chem. Res. Toxicol. 2015;28(6):1282–1291. doi: 10.1021/acs.chemrestox.5b00084. [DOI] [PubMed] [Google Scholar]
- 186.Uetrecht J.P., Zahid N. N-Chlorination and oxidation of procainamide by myeloperoxidase: toxicological implications. Chem. Res. Toxicol. 1991;4(2):218–222. doi: 10.1021/tx00020a015. [DOI] [PubMed] [Google Scholar]
- 187.Uetrecht J.P., Shear N.H., Zahid N. N-chlorination of sulfamethoxazole and dapsone by the myeloperoxidase system. Drug Metab. Dispos. 1993;21(5):830–834. [PubMed] [Google Scholar]
- 188.Zhang X.Y., Elfarra A.A. Advances in Molecular Toxicology. 2018. Myeloperoxidase-mediated bioactivation of olefins; pp. 123–150. [Google Scholar]
- 189.Aljuhani N. Phenylbutazone oxidation via Cu,Zn-sod peroxidase activity: an EPR study. Chem. Res. Toxicol. 2015;28(7):1476–1483. doi: 10.1021/acs.chemrestox.5b00152. [DOI] [PubMed] [Google Scholar]
- 190.Siraki A.G. Advances in Molecular Toxicology. 2013. Free radical metabolites in arylamine toxicity; pp. 39–82. [Google Scholar]
- 191.Siraki A.G. N-oxidation of aromatic amines by intracellular oxidases. Drug Metab. Rev. 2002;34(3):549–564. doi: 10.1081/dmr-120005657. [DOI] [PubMed] [Google Scholar]
- 192.Siraki A.G., Jiang J., Mason R.P. Investigating the mechanisms of aromatic amine-induced protein free radical formation by quantitative Structure−Activity relationships: implications for drug-induced agranulocytosis. Chem. Res. Toxicol. 2010;23(5):880–887. doi: 10.1021/tx900432d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Khan S.R. Proteomic profile of aminoglutethimide-induced apoptosis in HL-60 cells: role of myeloperoxidase and arylamine free radicals. Chem. Biol. Interact. 2015;239:129–138. doi: 10.1016/j.cbi.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 194.Kettle A.J. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem. J. 1995;308:559–563. doi: 10.1042/bj3080559. Pt 2)(Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Huang J., Smith F., Panizzi P. Ordered cleavage of myeloperoxidase ester bonds releases active site heme leading to inactivation of myeloperoxidase by benzoic acid hydrazide analogs. Arch. Biochem. Biophys. 2014;548:74–85. doi: 10.1016/j.abb.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Soubhye J. Discovery of novel potent reversible and irreversible myeloperoxidase inhibitors using virtual screening procedure. J. Med. Chem. 2017;60(15):6563–6586. doi: 10.1021/acs.jmedchem.7b00285. [DOI] [PubMed] [Google Scholar]
- 197.Soubhye J. From dynamic combinatorial chemistry to in vivo evaluation of reversible and irreversible myeloperoxidase inhibitors. ACS Med. Chem. Lett. 2017;8(2):206–210. doi: 10.1021/acsmedchemlett.6b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Aldib I. Evaluation of new scaffolds of myeloperoxidase inhibitors by rational design combined with high-throughput virtual screening. J. Med. Chem. 2012;55(16):7208–7218. doi: 10.1021/jm3007245. [DOI] [PubMed] [Google Scholar]
- 199.Soubhye J. Design, synthesis, and structure–activity relationship studies of novel 3-alkylindole derivatives as selective and highly potent myeloperoxidase inhibitors. J. Med. Chem. 2013;56(10):3943–3958. doi: 10.1021/jm4001538. [DOI] [PubMed] [Google Scholar]
- 200.Forbes L.V. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J. Biol. Chem. 2013;288(51):36636–36647. doi: 10.1074/jbc.M113.507756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gan L.-M. Safety, tolerability, pharmacokinetics and effect on serum uric acid of the myeloperoxidase inhibitor AZD4831 in a randomized, placebo-controlled, phase I study in healthy volunteers. Br. J. Clin. Pharmacol. 2019;85(4):762–770. doi: 10.1111/bcp.13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.