

Abstract
Alternariol (AOH), a mycotoxin belonging to the genus Alternaria, has been shown to induce cytotoxicity, including apoptosis and cell cycle arrest, in several mammalian cell types. However, its effects on early-stage embryonic development require further investigation. Here, we have shown that AOH exerts embryotoxic effects on mouse blastocyst-stage embryos and long-term adverse effects on immunity in one-day-old newborn mice of the next generation. Significant apoptosis and decrease in total cell number, predominantly through loss of inner cell mass (ICM), and to a minor extent, trophectoderm (TE) cells, were observed in AOH-treated blastocysts. Moreover, AOH exerted detrimental effects on pre- and post-implantation embryo development potential and induced a decrease in fetal weight in in vitro development and embryo transfer assays. Injection of pregnant mice with AOH (1, 3 and 5 mg/kg body weight/day) for 4 days resulted in apoptosis of blastocyst-stage embryos and injurious effects on embryonic development from the zygote to blastocyst stage or embryo degradation and a further decrease in fetal weight. Furthermore, AOH exerted a long-term impact on the next generation, triggering a significant increase in total oxidative stress content and expression of genes encoding antioxidant proteins. Lower expression of CXCL1, IL-1β and IL-8 related to innate immunity was detected in liver tissue extracts obtained from one-day-old newborns of AOH-injected pregnant mice (5 mg/kg body weight/day) relative to their non-treated counterparts. In addition, ROS served as an upstream regulator of AOH-triggered apoptotic processes and impairment of embryonic development. Our collective results highlight the potential of AOH as an embryotoxic and immunotoxic risk factor during embryo and infant development stages in mice.
Keywords: alternariol, apoptosis, oxidative stress, embryonic development, immunotoxicity
Introduction
Alternariol (AOH), the major secondary metabolite produced by the genus Alternaria, is a member of the dibenzo-alfa-pyrone group detected in various fruits, vegetables and grains, including cereal-based infant food and processed fruit products, such as juice and wine [1–4], within a concentration range of .65–54 μg/kg [5, 6]. For analysis of AOH contamination, two tomato producers in Spain were examined, which revealed that 35% organic and 21% non-organic tomato products contained AOH. In addition, 47% concentrates from non-organic tomatoes were contaminated with AOH [7]. Analysis of 45 samples in Italy disclosed contamination with AOH in 56% of the samples. Notably, AOH levels reached 270.7 μg/kg in fresh, 428.4 μg/kg in dried and 1110.8 μg/kg in ground samples [8, 9]. These findings indicate that both humans and livestock are chronically exposed to AOH from their daily food intake [10]. Importantly, the range of AOH concentrations was determined as 1.0–15.2 ng/kg body weight per day in adults with chronic dietary exposure, which is higher than the threshold of toxicological concern (TTC) for potential genotoxic compounds (2.5 ng/kg body weight per day) [1].
AOH is reported to induce cytotoxicity, including apoptosis and cell cycle arrest, in several mammalian cells [11–14], and produce ROS that trigger oxidative stress-mediated hazardous effects [14–19]. Moreover, AOH exerts genotoxic effects that cause unscheduled DNA synthesis, inhibition of DNA synthesis and DNA breaks in mammalian cells [20–23]. Recent investigations on the effects of AOH on porcine female gametes revealed disruption of in vitro maturation of porcine oocytes and deleterious effects on initial embryo development [24]. Regulated apoptosis processes are known to play important roles at various stages of embryogenesis, in particular, early-stage embryo development [25, 26]. However, these processes do not occur from zygote to pre-implantation blastocyst-stage embryos and inappropriate triggering of programmed cell death can exert hazardous effects on embryo implantation, post-implantation and fetal development [27–32]. However, the potential hazardous effects and regulatory mechanisms of AOH on the reproductive system, embryo development and fetal development in vivo require further investigation.
Several studies have highlighted that oxidative stress triggers bidirectional interactions with apoptosis and the immune system [33, 34]. ROS and programmed cell death significantly affect the cytokine responses of lymphocytes and immune cells [35, 36]. Recently, we showed that intravenous injection with either rhein, a glucoside chemical compound derived from rhubarb root, or enniatin B1, a mycotoxin produced by Fusarium genus, into female mice leads to a significant increase in liver oxidative stress content and expression of antioxidant-related genes in next-generation one-day old newborns. Moreover, three innate immunity-related genes (CXCL1, IL-1β and IL-8) were significantly downregulated in next-generation one-day-old newborns [37, 38]. Based on the accumulating evidence and our recent results, the bidirectional interactions among ROS, apoptosis and immunity are believed to be interdependent. However, the bidirectional interactions of apoptosis, cytokine responses and physiological immune responses affected in response to AOH remain unclear.
In this study, mouse blastocyst-stage embryos were used to investigate whether AOH, a ROS producer and apoptosis inducer, has a deleterious impact on pre- and post-embryonic development and immunity of next-generation newborns. Our findings clearly demonstrate that the mycotoxin promotes oxidative stress generation and immunotoxicity during mouse embryonic development.
Materials and Methods
Chemicals and reagents
Alternariol, dimethyl sulfoxide (DMSO), pregnant mare serum gonadotropin (PMSG), bovine serum albumin (BSA), polyvinylpyrrolidone (PVP), propidium iodide (PI), bisbenzimidine, 2′,7′-dichlorofluorescin diacetate (DCFDA), N-acetyl cysteine, human chorionic gonadotropin (hCG) and sodium pyruvate were purchased from Sigma (St. Louis, MO, USA). The TUNEL in situ cell death detection kit (production number: 11684817910) was acquired from Roche (Mannheim, Germany) and CMRL-1066 medium from Thermo Fisher Scientific (Waltham, MA, USA). TRIzol reagent and the reverse transcriptase kit were obtained from Takara Biomedicals (Tokyo, Japan).
Collection of mouse morulas and blastocysts
ICR mice were acquired from the National Laboratory Animal Center (Taiwan, ROC). Animal studies were approved by the Animal Research Ethics Board of Chung Yuan Christian University (Taiwan, ROC). All mice were fed breeder (Harlan Teklad) chow with food and water available ad libitum, and cared for following the guidelines of the Guide to The Care and Use of Experimental Animals (Canadian Council on Animal Care, Ottawa, 1993; ISBN: 0–919 087–18-3). Mice were housed in standard 28 × 16 × 11 cm (height) polypropylene cages with wire-grid tops under a 12 h/12 h night–day regimen. For in vitro and in vivo analyses, 6–8 mice were used in each AOH-treated group. Six- to eight-week-old nulliparous female mice were injected with 5 IU pregnant mare serum gonadotrophin (PMSG) to induce superovulation. After 48 h, PMSG-injected mice were further injected with 5 IU human chorionic gonadotrophin (hCG) and mated overnight with a single fertile male of the same strain. Plug-positive female mice were evaluated the next day, defined as day 0 of gestation, and separated for further embryo collection. Mice were sacrificed via cervical dislocation. Morulas were obtained by flushing uterine tubes on the afternoon of gestation day 3 and blastocysts collected by flushing the uterine horn on day 4. In both cases, the flushing solution for collection consisted of CMRL-1066 medium containing 1 mM glutamine and 1 mM sodium pyruvate. Collected embryos from different females were pooled and randomly allocated into various experimental groups.
Measurement of morula developmental potential in vitro
In vitro assessment morula developmental potential was performed according to our previously reported protocol [38]. In brief, morulas were incubated with culture medium containing AOH (2.5, 5, 10 or 20 μM) or .5% DMSO (vehicle) at 37°C in vitro, transferred to AOH-free culture medium after 24 h, and cultured for an additional 24 h at 37°C. Treatment with 0 μM AOH signifies treatment vehicle (.5% DMSO) only, which was used as the control group in all experiments of this study. Embryos treated with .5% DMSO exhibited no significant differences relative to pure control (PBS- or medium-treated) embryo groups, clearly indicating that .5% DMSO does not exert deleterious effects. Therefore, in our study, embryos treated with .5% DMSO were used as the control (vehicle) group. Blastocyst numbers were counted using phase-contrast microscopy (Olympus BX51, Tokyo, Japan) and developmental percentages calculated.
Measurement of apoptotic cells via TUNEL assay
Embryos were treated with various concentrations of AOH as indicated. AOH was dissolved in dimethyl sulfoxide (DMSO) at a final concentration of up to .5% (v/v) for 24 h. Apoptotic cells of embryos were evaluated via the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. To this end, embryos were initially washed in AOH-free medium three times and fixed in 4% paraformaldehyde (PFA) at room temperature for 2 h. Next, embryos were permeabilized and subjected to apoptotic labeling using a TUNEL in situ cell death detection kit according to the manufacturer’s protocol. Briefly, each group of embryos was incubated with 20 μl TUNEL reaction mixture (2 μl enzyme solution and 18 μl labeling solution containing fluorescein-conjugated nucleotides) for 30 min at 37°C and extensively washed with phosphate-buffered saline (PBS) containing .3% (w/v) BSA. Converted POD solution (20 μl) was added to each group of embryos, followed by incubation for 30 min at 37°C. Finally, each group of embryos was incubated with 20 μl 3,3′-diaminobenzidine (DAB) substrate solution for 2 min and observed using fluorescence microscopy. TUNEL-positive (apoptotic) cells, detected as black spots, were counted via microscopy.
Measurement of cell proliferation
Blastocysts were incubated with or without 2.5–10 μM AOH. After 24 h, blastocysts were washed with AOH-free medium and subjected to dual differential staining to facilitate counting of cell numbers in the inner cell mass (ICM) and trophectoderm (TE) [39]. In brief, the zona pellucida of embryos was removed with .4% pronase in M2–BSA medium (M2 containing .1% bovine serum albumin). Denuded blastocysts were incubated with 1 mM trinitrobenzenesulfonic acid (TNBS) in BSA-free M2 medium containing .1% polyvinylpyrrolidone (PVP) at 4°C for 30 min, followed by washing in M2 [40]. After three washes, embryos were incubated with anti-dinitrophenol-BSA complex antibody (30 μg/ml in M2-BSA) at room temperature for 30 min. Following three washes with M2-BSA, embryos were further subjected to complement reaction using whole guinea pig serum as a source of complement. Following incubation with M2 supplemented with 10% whole guinea pig serum along with bisbenzimide (20 μg/ml) and propidium iodide (PI; 10 μg/ml) at room temperature for 30 min, embryonic cells (ICM or TE) were assessed via fluorescence microscopy. Nuclei of ICM cells (which take up bisbenzimidine but exclude PI) were stained with bisbenzimidine (blue fluorescence) and TE cells with both PI and bisbenzimide (orange-red fluorescence). The number of nuclei was considered to represent an accurate measurement of cell number.
Assessment of embryonic development in vitro
Blastocyst development in vitro was measured based on morphological analyses and previous reports [38]. In brief, blastocysts were cultured in fibronectin-coated dishes and incubated with culture medium (1 mM glutamine, 1 mM sodium pyruvate, 50 IU/ml penicillin and 50 mg/ml streptomycin in CMRL-1066 medium) supplemented with 20% heat-inactivated human placental cord serum for 8 days. Embryo development status was measured and recorded with a phase-contrast microscope at a fixed time each day. After 8 days of assessment, morphological scores in each group were evaluated and classified according to previously established methods [38]. Embryo development status was classified attachment only, ICM (+), ICM (++) and ICM (+++). Attachment only status was defined as embryo implantation (binding) with no further development. ICM (+), ICM (++) and ICM (+++) were defined according to shape, ranging from compact and rounded ICM (+++) to a few scattered cells (+) over the trophoblastic layer [38, 41, 42].
Embryo transfer assay
The embryo transfer assay was conducted according our previously published report [38]. Thirty dams (recipient mice) were used for the embryo transfer assay. In brief, pseudopregnant dams were prepared for embryo transfer by mating ICR females (white skin color) with vasectomized males (C57BL/6 J; black skin color). For assessment of AOH-treated blastocyst implantation into uterus of dams and post-implantation developmental status, blastocysts were cultured in medium containing 2.5, 5, or 10 μM AOH or .5% DMSO (control group). After 24 h, 16 embryos were transferred to one pseudopregnant mouse. Eight AOH-treated embryos were transferred into the left uterine horn and eight control embryos into the right uterine horn of dams. All experimental dams were sacrificed on day 18 post-coitus (13 days post-transfer) and embryo developmental status (number of implantation sites and placental and fetus weights) assessed. Implanted embryos that could not further develop into healthy fetuses were subsequently resorbed form as a resorption site in the uterus. Resorption and fetal status were further assessed. The ratio of resorption or survival was calculated as the number of resorbed or surviving fetuses, respectively, per number of implantations. The implantation percentage signifies the number of implantations per number of transferred embryos × 100. The percentage of resorption or survival represents the number of resorbed or surviving fetuses per number of implantations × 100. Weights of surviving fetuses and placenta were measured immediately after dissection.
Intravenous injection of AOH
To determine the effects of AOH on mouse embryonic development in vivo, twenty randomly selected female (42 day-old) mice were intravenously injected with .1 ml PBS containing various doses of AOH (1, 3, 5 mg/kg/day) into the tail vein. The control group included vehicle-injected mice administered an equivalent DMSO dose to the 5 mg/kg/day treatment group. At 24 hours after injection, female mice were mated overnight with a single fertile male of the same strain. AOH (0, 1, 3, 5 mg/kg/day) was continuously intravenously injected for 4 days. Mice were sacrificed on day 4 after mating and blastocysts collected for assessment of apoptosis, proliferation and embryonic development.
ROS measurements
Mouse blastocysts were either pre-incubated with 5 mM N-acetyl cysteine (NAC) or left untreated. After 1 h, embryos were treated with 10 μM AOH for an additional 24 h. DCF-DA (20 μM) dye staining was applied to detect intracellular ROS generation. The intracellular ROS level in each group was measured under a fluorescence microscope and quantitatively analyzed using image J software.
Detection of ROS content in newborn mouse liver
Twenty randomly selected female mice were intravenously injected with AOH (1, 3, 5 mg/kg/day) or vehicle (at an equivalent DMSO dose to the 5 mg/kg/day treatment group). At 24 h after injection, female mice were mated overnight with a single fertile male of the same strain. After mating, mice were continuously intravenously injected with AOH (1, 3, 5 mg/kg/day) or vehicle for 4 days. For detection of ROS content in mouse fetal liver, 10–12 one-day-old mouse newborns were collected for each AOH treatment group. Fetal liver tissue extracted via homogenization with ice-cold phosphate buffered saline (PBS) and lyzed in 1 mL homogenization buffer (20 mM Tris–HCl, pH 7.4, 1 mM ethylene-diaminetetraacetic acid (EDTA), 1 mM ethylene-glycol-bis(b-aminoethylether) N, N, N′, N′-tetraacetic acid (EGTA), 1% Triton X-100, 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride, 50 mM sodium fluoride, 20 mM sodium pyrophosphate and 1 mM sodium orthovanadate). Cell lysates were centrifuged (15 000 × g) for 20 min at 4°C and supernatants collected. ROS were detected using dichlorodihydrofluorescein diacetate (DCFDA) dye. Cell extracts (20 μg) were mixed in 50 μL PBS containing 20 mM DCFDA for 1 h at 37°C and relative ROS levels measured using a fluorescence ELISA reader (excitation at 530 nm, emission at 485 nm).
Analysis of gene expression
Using quantitative real-time PCR, mRNA expression levels of CXCL1, IL-1 beta, IL-8, catalase, glutathione peroxidase, Cu/Zn superoxide dismutase and Mn superoxide dismutase genes were quantified. For extraction of total RNA, 10–12 one-day-old newborn mice were collected for each AOH treatment group and were treated with TRIzol reagent (Life Technologies, CA, USA) and purified with the RNeasy Mini kit (Qiagen) according to the manufacturer’s protocol. Real-time RT-PCR using the ABI 7000 Prism Sequence Detection System (Applied Biosystems, CA, USA) was applied to quantify mRNA levels. β-Actin mRNA was used as the endogenous control for quantitative normalization. The following primers were applied for real-time quantitative RT-PCR: CXCL1 (Forward, 5′-TGA GCT GCG CTG TCA GTG CCT-3′ and Reverse, 5′-AGA AGC CAG CGT TCA CCA GA-3′); IL-1 β (Forward, 5′-AAG GAG AAC CAA GCA ACG ACA AAA-3′ and Reverse, 5′-TGG GGA ACT CTG CAG ACT CAA ACT-3′); IL-8 (Forward, 5’-CAC CTC AAG AAC ATC CAG AGC T-3′ and Reverse, 5’-CAA GCA GAA CTG AAC TAC CAT CG-3′); catalase (Forward, 5’-GCA GAT ACC TGT GAA CTG TC-3′ and Reverse, 5′-GTA GAA TGT CCG CAC CTG AG-3′); glutathione peroxidase (GPX) (Forward, 5’-CCT CAA GTA CGT CCG ACC TG-3′ and Reverse, 5’-CAA TGT CGT TGC GGC ACA CC-3′); Cu/Zn-superoxide dismutase (Cu/Zn-SOD) (Forward, 5′-AAG GCC GTG TGC GTG CTG AA-3′ and Reverse, 5’-CAG GTC TCCA ACA TGC CTC T-3′); Mn-superoxide dismutase (Mn-SOD) (Forward, 5’-GCA CAT TAA CGC GCA GAT CA-3′ and Reverse, 5’-AGC CTC CAG CAA CTC TCC TT-3′); β-Actin (Forward, 5’-CGT ACC ACA GGC ATT GTG ATG-3′ and Reverse, 5’-CTT CTA GGA CTG GCT CGC AC-3′).
Statistical analysis
Experimental data were analyzed using one-way ANOVA followed by Dunnett’s test for multiple comparisons and presented as means ± standard deviation (SD). Different symbols represent significant differences at P < .05.
Results
Embryotoxicity of AOH on mouse blastocysts
For measurement of AOH-induced embryonic toxicity, blastocysts were incubated with 2.5–10 μM AOH or vehicle. After 24 h, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was employed to measure the extent of apoptosis through analysis of DNA fragmentation. DNA fragmentation-positive (apoptotic) cell content and quantitative analysis revealed that 5 and 10 μM AOH-treated groups were significantly increased relative to the vehicle group in blastocysts (Fig. 1A and B).
Figure 1.
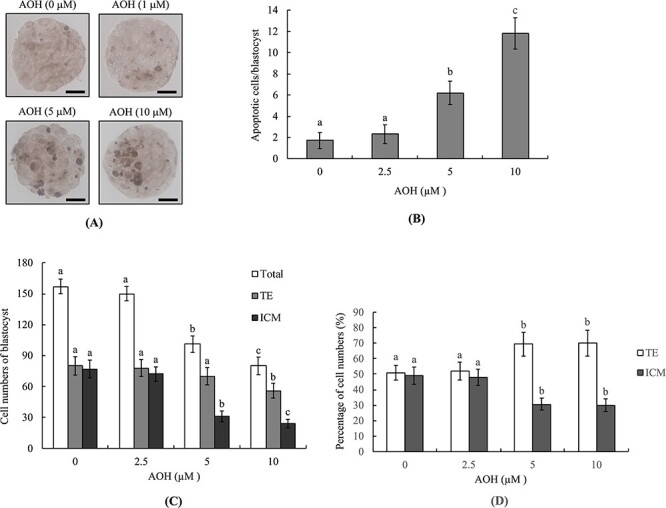
Effects of AOH on mouse blastocysts. Mouse blastocysts were exposed to AOH (2.5, 5 or 10 μM) for 24 h and TUNEL staining employed to evaluate apoptosis. (A) Blastocysts treated with .5% DMSO were used as the control (vehicle) group. TUNEL-positive cells were observed as black spots under light microscopy. (B) Apoptotic (TUNEL-positive) cells were quantitated and apoptotic cells per blastocyst determined. (C) Cell numbers of inner cell mass (ICM) and trophectoderm (TE) per blastocyst were counted via differential staining. Data from each group were analyzed based on 200 blastocysts. Values are presented as means ± SD of eight determinations. Different symbols indicate significant differences at P < .05. The scale bar is 20 μm.
To determine the impact of AOH on cell proliferation, inner cell mass (ICM) and trophectoderm (TE) numbers of blastocysts were assessed via differential staining. Both 5 and 10 μM AOH-incubated groups contained significantly fewer ICM cells while the 10 μM AOH-incubated group contained slightly fewer TE cells in blastocysts compared with the vehicle-treated group (Fig. 1C). The percentage of ICM cells was lower in both AOH than vehicle-treated (control) groups (Fig 1D). In addition, the TE cell percentages in both 5 and 10 μM AOH treatment groups were markedly higher relative to the vehicle-treated (control) group (Fig. 1D). This finding is attributable to the total cell numbers being significantly lower in 5 and 10 μM AOH-incubated groups (Fig. 1C). Our results suggest that 5 and 10 μM AOH-induced deleterious effects are major on ICM cells and minor on TE cells. The significantly lower numbers of ICM and total cell numbers in both treatment groups support the potential of AOH to exert hazardous effects on embryos through inhibition of proliferation and/or induction of apoptosis of ICM (major) and TE (minor) cells in mouse blastocyst-stage embryos.
Impact of AOH on in vitro embryonic development
To further investigate the deleterious impact of AOH on the developmental potential of embryos, the development ratio of AOH-treated morula to blastocyst-stage embryo was assessed. The percentage of 5–10 μM AOH-treated morulas that developed into blastocysts was significantly lower than of the vehicle-treated (control) group (Fig. 2A).
Figure 2.
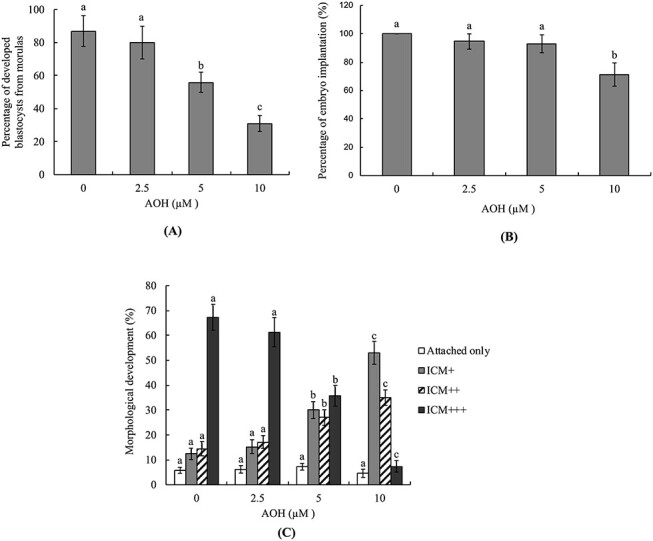
In vitro development of mouse embryos treated with AOH. Mouse morulas were treated with AOH (2.5, 5 or 10 μM) for 24 h and cultured for an additional 24 h. (A) Morulas incubated with .5% DMSO were used as the control (vehicle) group and the blastocyst percentages calculated. (B and C) Mouse blastocysts were treated with or without AOH (2.5, 5 or 10 μM) for 24 h, followed by culture in fibronectin-coated dishes for 7 days. (A) Blastocysts attached to dishes were defined as implantation and the percentages calculated (B). Following culture for 7 days, based on morphological assessment, the status of outgrowth of blastocysts in cultured dishes was classified as attachment only, ICM+, ICM++ and ICM+++, as described in Materials and Methods (C). Values are presented as means ± SD of five determinations. Data from each group were obtained by analysis of 150 blastocyst samples. Different symbols indicate significant differences at P < .05.
For further investigation of the impact of AOH on embryo post-implantation potential in vitro, blastocysts were treated with or without AOH (2.5, 5 or 10 μM) for 24 h, followed by culture in fibronectin-coated dishes for 7 days, and developmental characteristics assessed daily. Embryo implantation potential (based on the percentage of blastocysts attached to fibronectin-coated culture dishes) was significantly lower in 10 μM AOH-treated than vehicle-treated (AOH-free) groups (Fig. 2B). The embryo development morphology index clearly showed that treatment of blastocysts with 5–10 μM AOH led to a lower development score (ICM cluster scoring according to the shape of ICM and trophoblastic layer), indicative of lower post-implantation developmental potential of AOH-incubated embryos (Fig. 2C).
Effect of AOH on embryo development potential in vivo
Embryo development potential in vivo was assessed with the embryo transfer assay. To this end, blastocysts were co-incubated with 2.5–10 μM AOH or vehicle (AOH-free) for 24 h and transferred to the uterus of dams. At day 18 post-coitus (13 days post-transfer), embryo development in the uterus was examined. AOH (5–10 μM)-treated blastocysts clearly showed a lower implantation ratio than the vehicle-treated (control) group (Fig. 3A). In addition, the ratio of embryos that implanted but could not develop further (resorption) was significantly higher in the 5 and 10 μM AOH-treated than control groups (Fig. 3A). Measurement of placental weights revealed that the weight of the 10 μM AOH-treated group was lower than that of the vehicle-treated group while no significant differences were observed between the 2.5 and 5 μM AOH-treated and control groups. (3B). Further measurement of fetal weight at 13 days post-transfer as an indicator of embryo developmental status revealed that the fetal weights of the 5 and 10 μM AOH-treated groups were markedly lower than that of the control group (Fig. 3C). Overall, we observed a significantly lower proportion of fetal weights >600 mg in 5 and 10 μM AOH-treated blastocysts compared to the vehicle-treated (control) group (Fig. 3C).
Figure 3 .
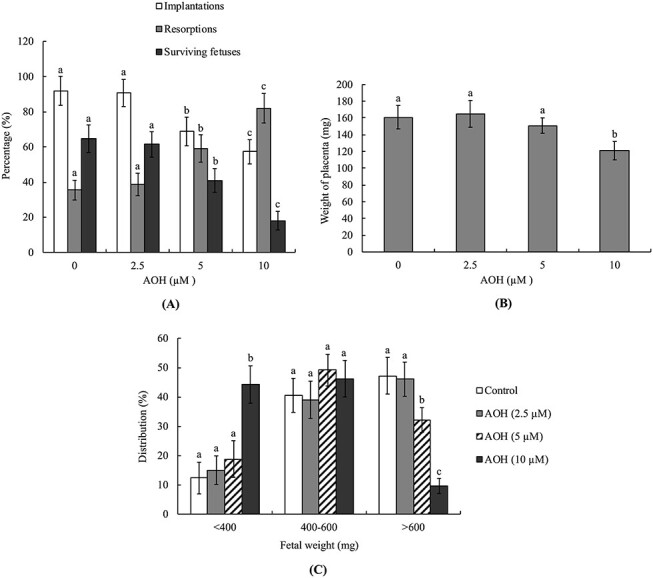
Effects of AOH on in vivo implantation, resorption, fetal survival and fetal weight of mouse blastocysts. (A) Mouse blastocysts were exposed to AOH (2.5, 5 or 10 μM) or .5% DMSO (control group) for 24 h. Embryo implantation, fetal resorption and survival were measured via the embryo transfer assay as described in Materials and Methods. The implantation percentage signifies the number of implantations per number of transferred embryos × 100. The percentage of resorption or survival represents the number of resorbed or surviving fetuses per number of implantations × 100. (B) Average placental weights from 40 dams (recipient mice). (C) Weight distribution of surviving fetuses on day 18 post-coitus (day 13 post-transfer). Surviving fetuses were from embryo transfer of 320 total blastocysts across 40 recipients. Different symbols indicate significant differences at P < .05.
Animal studies via intravenous injection (0, 1, 3, 5 mg/kg/day) were conducted to further investigate the deleterious effects potential of AOH. Notably, AOH induced apoptosis and inhibited cell proliferation in 3 and 5 mg/kg/day injected groups (Fig. 4A and B). Moreover, as evident from the morphological characteristics of collected embryos, AOH induced impairment of embryo development potential from the zygote to blastocyst stage and embryo damage and degradation in 3 and 5 mg/kg/day injected groups (Fig. 4C). The weights of day 18 post-coitus fetuses were lower from 3 and 5 mg/kg/day AOH groups, compared with those of the control group (Fig. 4D). The collective results clearly indicate that AOH exerts hazardous effects that impair implantation and sequent development in vivo.
Figure 4.
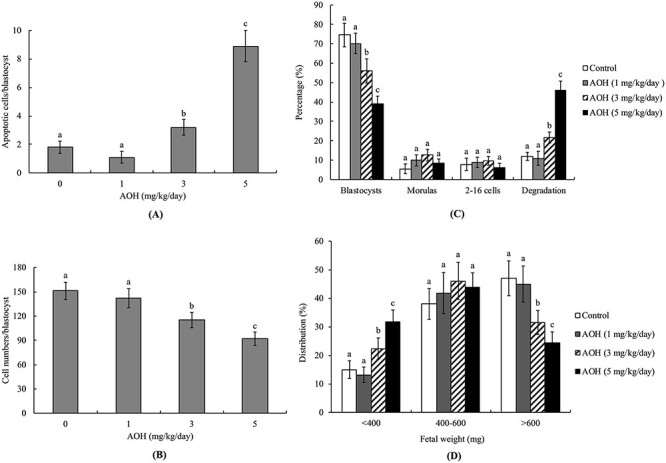
Effects of AOH on apoptosis and blastocyst development following intravenous injection into mice. Twenty randomly selected female mice were intravenously injected with AOH (1, 3, 5 mg/kg/day) or vehicle (.5% DMSO) for 24 h. Female mice were subsequently mated overnight with a single fertile male of the same strain, followed by intravenous injection with AOH (0, 1, 3, 5 mg/kg/day) for 4 days. Blastocysts were collected by flushing the uterine horn on day 4 after mating. (A) Apoptosis of mouse blastocysts was measured via TUNEL staining, followed by light microscopy, and the mean number of TUNEL-positive (apoptotic) cells per blastocyst calculated. (B) Total cell numbers per blastocyst were counted. (C) Developmental stages of embryos were determined by flushing the mouse uterine horn with buffer on day 4. Embryo developmental status was assessed as a percentage of total embryos. (D) Distribution of surviving fetuses measured according to weight on day 13 post-transfer. Experimental data from each group are based on 160 blastocyst samples. Different symbols indicate significant differences at P < .05.
Effects of NAC and caspase inhibitors on AOH-induced impairment of embryonic development
Here, we further examined the possibility that oxidative stress is involved in AOH-triggered apoptosis leading to sequent impairment of embryo development. Intracellular ROS levels were significantly higher in 10 μM AOH-treated blastocysts, compared with the vehicle-treated group. Notably, pretreatment with N-acetylcysteine (NAC), a well-known ROS scavenger, effectively prevented AOH-triggered ROS generation and apoptosis (Fig. 5A–C). In addition, pretreatment with caspase-3 and -9-specific inhibitors significantly and effectively prevented AOH-triggered apoptosis in blastocysts (Fig. 5C) but had no effect on AOH-induced ROS generation (data not shown). In an embryo transfer model, pre-treatment with NAC or specific caspase-9 and caspase-3 inhibitors effectively prevented AOH-promoted decrease in fetal weight at 13 days post-transfer (Fig. 5D). However, pretreatment with the caspase-8-specific inhibitor, IETD, had no preventive effect on AOH-induced apoptosis (Fig. 5C) or decrease in fetal weight (Fig. 5D). Taken together, our results strongly suggest that the regulatory mechanisms triggered by AOH leading to apoptosis in mouse blastocyst cells are ROS-dependent processes involving caspase-9 and -3. Moreover, ROS serve as an upstream regulator of caspase activation and sequential apoptotic cascades.
Figure 5.
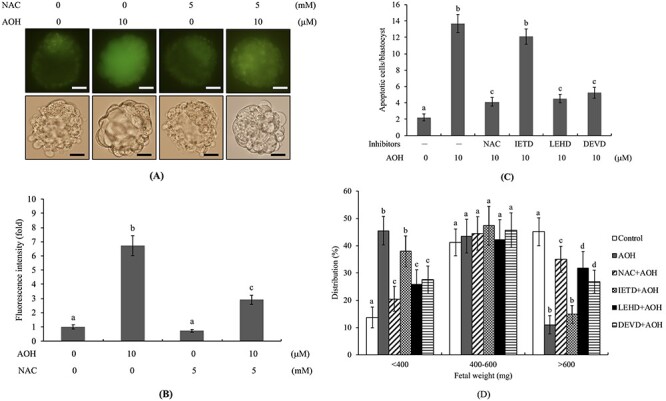
Effects of NAC and caspase inhibitors on developmental status of AOH-treated embryos. Mouse blastocysts were pre-treated with 5 mM N-acetyl cysteine (NAC), 300 μM Z-IETD-FMK (IETD), 300 μM Z-LEHD-FMK (LEHD) or 300 μM Z-DEVD-FMK (DEVD) for 1 h or left untreated, followed by incubation with or without AOH (2.5, 5 or 10 μM) or .5% DMSO (control) for a further 24 h. (A) Detection of ROS generation with DCF-DA fluorescence dye (20 μM). (B) Quantitative analysis of intracellular ROS generation (fluorescence intensity) in each group using image J software. (C) Assessment of apoptosis via TUNEL staining. (D) Mean numbers of apoptotic (TUNEL-positive) cells per blastocyst. (E) Weight distribution of surviving fetuses on day 13 post-transfer via the embryo transfer assay as described in “Materials and Methods” and Figure 3. The percentage of surviving fetuses was analyzed via embryo transfer of AOH-pretreated (a total of 320 blastocysts transferred to 40 recipients) and control blastocysts. Different symbols indicate significant differences at P < .05. The scale bar is 20 μm.
Effects of AOH on innate immune and ROS-related gene expression
To establish the effects of AOH on genes related to the immune system, mRNA levels of CXCL1, IL-1β and IL-8 in one-day-old infants born from female mice intravenously injected with AOH were examined (0, 1, 3, 5 mg/kg/day). CXCL1, IL-1β and IL-8 mRNA levels of newborns from female mice injected with 5 mg/kg/day AOH were significantly lower than those in newborns from mice injected with vehicle (Fig. 6A). Notably, higher intracellular ROS content of liver cell extracts was detected in infants from the 5 mg/kg/day ENN B1-injected than the vehicle-injected (control) group (Fig. 6B). We further analyzed the transcript levels of antioxidant-related enzyme genes in one-day-old infants from AOH (0, 1, 3, 5 mg/kg/day)-injected female mice. Our results clearly revealed increased relative transcript levels of catalase, glutathione peroxidase and Cu/Zn superoxide dismutase in infant liver from female mice injected with 5 mg/kg/day AOH than that from vehicle-injected groups. In addition, expression of Mn superoxide dismutase in 1 and 3 mg/kg/day AOH-injected groups was higher than that in the vehicle-injected group (Fig. 6C).
Figure 6.

Effects of AOH injection into female mice on expression of genes related to innate immunity in liver of newborn mice. Twenty randomly selected female mice were intravenously injected with AOH (1, 3, 5 mg/kg/day) or DMSO (using the same dosage as the 5 mg/kg/day AOH-injected group). After 24 h, female mice were mated overnight with a single fertile male of the same strain and intravenously injected with AOH (0, 1, 3, 5 mg/kg/day) for 4 days. One-day after birth, newborn mice were sacrificed and liver tissue extracts collected and prepared. (A) Gene expression levels of CXCL1, IL-1β and IL-8 were analyzed by quantitative real-time RT-PCR. (B) ROS levels were detected with 2′,7′-dichlorofluorescin diacetate (DCF-DA) using a fluorescence ELISA reader (excitation 530 nm, emission 485 nm). (C) Expression levels of catalase, glutathione peroxidase, Cu/Zn superoxide dismutase and Mn superoxide dismutase genes were analyzed from six independent determinations and presented as means ± SD. Experimental results are from at least 35 samples for each group. Different symbols indicate significant differences at P < .05.
Discussion
Embryogenesis is a complex and precisely orchestrated event. External chemical, physical and environmental factors have the potential to exert deleterious effects on normal embryo development, even leading to miscarriage. Therefore, it is important to determine the potential embryotoxic effects of various external agents, in particular, common food contaminants such as mycotoxins. Alternariol (AOH) produced by alternaria fungi, a natural compound in foodstuffs, is a frequent contaminant of fruit and grain products. Several studies have shown that AOH induces cytotoxicity, including apoptosis and cell cycle arrest, in multiple mammalian cell types, such as human colon carcinoma, murine hepatoma and human Caco-2, as well as the murine macrophage cell line RAW 264.7 [11–14]. These results clearly indicate that AOH is potentially harmful for human health, exerting adverse effects on the reproductive system. Another recent investigation showed that AOH disrupts in vitro maturation of porcine oocytes and exerts deleterious effects on initial embryo development [24]. However, the potential harmful effects and regulatory mechanisms of action of AOH on pre- and post-implantation embryo development are unclear at present. Earlier investigations on various models have demonstrated potential hazardous effects of several mycotoxins and their secondary metabolites on embryo development. For instance, ochratoxin A (OTA), T-2 toxin, deoxynivalenol (DON) and zearalenone (ZEN), and its metabolites, alpha-zearalenol (α-ZOL) and beta-zearalenol (β-ZOL) were recently reported to exert deleterious effects on zebrafish larva development [43]. In addition, exposure to OTA and ZEN and its metabolites triggered negative behavioral effects [43]. Recent experiments by our group have consistently shown that mycotoxins, such as citrinin (CTN), OTA, enniatin B and enniatin B1 have embryotoxic effects on mouse oocyte maturation, fertilization and pre- and post-implantation mouse embryo development [29, 38, 44–47]. Moreover, the mycotoxin beauvericin (BEA) is reported to trigger oocyte mitochondrial dysfunction and affect embryo development in juvenile sheep [48]. Another recent study showed that AOH (10–20 μM) exerts deleterious effects on oocyte in vitro maturation and preimplantation development of porcine female gametes [24]. The collective results highlight potential teratogenic effects of mycotoxins and their secondary metabolites on embryonic development in vertebrate animal models. Here, we further validated the teratogenic effects of AOH on early-stage mouse embryonic development, both in vitro and in vivo. In our experiments, preincubation of mouse blastocysts with 5–10 μM AOH caused impairment of early-stage embryonic development and post-implantation development status (Figs. 1–4).
Several reports have shown that AOH exerts toxicity in vitro. For instance, treatment with 10 μM AOH causes genotoxic effects in the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene locus in V79 cells [22]. AOH at a treatment concentration of 30 μM induces DNA damage and modifies macrophage phenotype and inflammatory responses [49]. Importantly, AOH triggers various types of DNA damage or breaks, typically within a concentration range of 15 to 60 μM, in vitro [14, 21, 50]. Moreover, these cytotoxic effects of AOH are exerted through blocking the cell cycle, induction of mitochondrial damage, suppressing cell proliferation and increasing apoptosis/necrosis in Caco-2 cells [13]. Recently, AOH (10–20 μM) was reported to exert deleterious effects on oocyte in vitro maturation and preimplantation development of porcine female gametes [24]. Based on the collective previous findings, we investigated the effects of 2.5–10 μM AOH on early-stage embryonic development in this study. Our results demonstrated that 5 and 10 μM AOH induce ROS generation and subsequent activation of caspase-9 and caspase-3, in turn, triggering apoptotic processes that exert harmful effects on sequent embryonic development in mouse blastocysts in vitro. The in vitro treatment dosage of AOH in our study is thus consistent with biological concentrations. Limited data are available on the toxicity of AOH in vivo but acute toxicity of AOH is considered to be low [51]. Earlier reports have documented AOH contamination of 31% feed samples and agricultural commodities in Europe, with concentrations ranging from 6.3 to 1840 mg/kg [51]. The estimated human dietary exposure of AOH is ~1.9–39 ng/kg/bw/day. However, this dosage in humans exceeds the threshold of toxicological concern for potential genotoxic compounds (2.5 ng/kg/bw/day) [51]. In addition, AOH exposure is a long-term issue, since we consume contaminated foods on a daily basis. The degree of accumulation of AOH in human tissues and organs is unclear. For our in vivo model, pregnant mice were exposed to AOH (1, 3 and 5 mg/kg body weight/day) for a short period of 4 days to investigate effects on pre- and post-implantation embryonic development. However, serum concentrations upon intake of AOH-contaminated foods are unclear.
A previous study analyzed the toxicokinetics of radiolabeled AOH administered orally as a single dose of 200 or 1000 mg/kg in mice [52]. The authors noted that AOH exhibited low systemic absorption, with about 90% of the total dose excreted through feces and up to 9% via urine. Importantly, the blood level did not exceed .06% of the administered dose during the first 24 h after intake of AOH [52]. A more recent study found that AOH formed stable complexes with human serum albumin (HSA), as assessed by spectroscopic measurement [53]. The same authors tested the binding affinity of AOH with bovine, porcine and rat serum albumin and found that AOH bound rat albumin with considerably higher affinity the other tested albumins [53]. Based on these previous findings, we speculate that the interaction of AOH with serum albumins is important for the toxicokinetics of AOH in vivo and that, in our animal model, the vast majority of the injected-AOH is excreted through urine, carried in blood as AOH-albumin complexes, or metabolized into 4-hydroxy alternariol and 4-hydroxy alternariol monomethyl ether secondary metabolites by cytochrome P450 enzymes [54]. The potential embryotoxic effects of AOH-albumin complexes and/or secondary metabolites of AOH should be investigated in the future. However, these experimentally determined outcomes for AOH administration may help explain why a high injected dosage of AOH (3 and 5 mg/kg body weight) was needed in our in vivo model to cause ROS generation and injurious effects on embryonic development. We acknowledge that dietary intake of AOH cannot achieve such a high level of AOH in blood. However, AOH and its secondary metabolites have high potential to be developed as effective anti-cancer drugs, given their ability to trigger cytotoxicity or genotoxicity in several cancer cell lines [55]. For anti-cancer drug development, it might be necessary to develop a high-dosage formulation of AOH and/or its second metabolites, such as by using an optimized biomaterial coating to increase its absorption rate and improve delivery efficiency via oral intake, with the goal of reaching an effective (high) dosage level in blood. In this context, our study results could provide important insights for the dosage safety parameters of AOH or its derivatives as anticancer drugs in clinical therapy for pregnant women.
Blastocyst-stage embryos are composed of TE and ICM cells, both of which play critical roles in embryo implantation and sequent development. During early embryo development, TE and inner lineage cells of the uterus develop to form the placenta [56]. Loss and/or impairment of the TE cell lineage has been shown to exert deleterious effects on embryo implantation, promoting miscarriage [39, 57–59]. In addition, loss and/or impairment of ICM cells represents the major cause of impairment of post-implantation fetal development [28, 56, 60, 61]. In our experiments, 5–10 μM AOH-triggered embryonic cell death predominantly occurred in the ICM and minor impairment of TE cells was evident in the 10 μM AOH-treated group (Figs. 1C). These findings reflect major impairment of post-implantation embryonic and fetal development and minor effects of AOH on embryo attachment and growth in vitro, implantation in vivo and placenta formation (Figs 2B, C and3A-C). However, the precise mechanisms by which AOH triggers programmed cell death and/or affects proliferation in ICM while exerting no concomitant effects on TE remain to be established. Moreover, intravenous injection of mice with 5 mg/kg/day AOH exerted deleterious effects on pre-implantation embryo development, including loss of cell number and apoptosis in blastocysts, and post-implantation embryonic and fetal development (Fig. 4). These findings clearly indicate that AOH is a potential embryotoxic agent that exerts significant injurious effects on early mouse embryo development.
The innate immune system plays a critical role in defense and prevention of infections during the newborn and early life stages of infants [62, 63]. Several environmental and endocrine-disrupting chemicals are known to interfere with synthesis or transcription of chemokines and cytokines, even affecting the physiological function of the immune system [64–66]. Two recent studies by our group disclosed that rhein, a glucoside chemical extracted from rhubarb root and enniatin B1, a mycotoxin and secondary metabolite produced by species of the Fusarium genus, downregulate the transcription of innate-immunity related genes in newborns and infants of dams [37, 38]. Rhein and enniatin B1 suppressed the expression of CXCL1, IL-1 β and IL-8 in newborn mouse liver, supporting their potential to disrupt immune system development and induce embryonic immunotoxicity during mouse embryo development [37, 38]. In the current study, we further investigated whether AOH affects expression of innate immune-related gene in next-generation infants. Our data showed significant downregulation of mRNA levels of CXCL-1, IL-1β and IL-8, three innate immune-related genes, in one-day-old newborns from 5 mg/kg/day AOH-injected female mice (Fig. 6A). These chemokines attract and activate leukocytes to trigger inflammation responses for protecting against or reducing infection. Moreover, both neutrophils and macrophages are activated by IL-1β in infected tissues [67]. In our experiments, CXCL1, IL-1β and IL-8 mRNA levels in one-day-old newborns from female mice injected with 5 mg/kg/day AOH were reduced, leading to increased number of infections at the infant and early life stages (Fig. 6A). Our results highlight the potential of AOH to cause damage to the immune system.
Ongoing evidence has shown that intracellular oxidative stress plays critical roles in regulating a series of signaling cascades and/or activation or expression of transcription factors to determine cell fate [68]. Under conditions of exposure to external or internal risk factors or intracellular ROS generation, the level may exceed the capacity of the physiological redox buffering system to scavenge oxidative stress to the normal physiological state. Several critical biomolecules are damaged under conditions of oxidative stress, including DNA, protein, lipid and cell membrane [68]. In normal physiological conditions, expression levels and activities of anti-oxidative enzymes, including catalase, glutathione peroxidase and superoxide dismutase, are critical for scavenging intracellular free radicals or oxidative stress in order to facilitate return to normal ROS levels and avoid ROS-induced harmful effects. Thus, the anti-oxidative enzyme activities and/or amounts provide an important evaluation index for determining the potential of oxidative stress-generating compounds to cause injury [69, 70]. AOH is reported to induce ROS in various mammalian cell lines, including Caco-2, HT29, HCT116 [16, 18, 19] and RAW264.7 mouse macrophages [14]. Earlier results also support the hypothesis that the source of intracellular ROS in AOH-treated cells is AOH metabolism [14, 16]. Experiments from the current investigation demonstrated an increase in intracellular ROS in AOH-treated blastocysts. Excess ROS triggered sequential apoptotic signaling cascades resulting in impairment of early-stage mouse embryonic development. Moreover, pre-treatment of blastocysts with ROS scavengers as well as caspase-9 and caspase-3-specific inhibitors effectively blocked or prevented AOH-triggered apoptotic processes (Fig. 5A-D). Further animal studies revealed a significant increase in ROS content in liver cell extracts of one-day-old newborn mice during exposure of pregnant mice to AOH (Fig. 6B). Importantly, mRNA levels of several important anti-oxidative enzymes for physiological detoxification, including catalase, glutathione peroxidase, Cu/Zn superoxide dismutase and Mn superoxide dismutase, were markedly increased in the animal assay model (Fig. 6C). The collective findings clearly highlight that AOH promotes oxidative stress and further induces gene expression of antioxidant enzyme to facilitate removal of excess ROS.
Several types of DNA damage are triggered by AOH, including single-stranded DNA breaks (SSB), double-stranded breaks (DSB) and oxidative DNA damage [3]. Two possible regulatory mechanisms of AOH-mediated DNA damage are AOH-induced ROS and interactions with DNA topoisomerase. Previous investigations have demonstrated that AOH is an inhibitor of DNA topoisomerase I and II enzymes with selectivity for the IIα isoform [18, 21]. In addition to inhibiting DNA topoisomerase enzyme activity, AOH could stabilize covalent topoisomerase–DNA intermediates as a topoisomerase poison via interactions with topoisomerase IIα isoforms [71]. Collision of the stabilized DNA complex with DNA replication forks or transcriptional machinery is reported to generate DSB, providing a feasible regulatory mechanism for the DNA clastogenic effects of AOH. Importantly, overexpression of human tyrosyl-DNA phosphodiesterase I (TDP1), a central enzyme in the repair of covalent DNA topoisomerase adducts, led to significant suppression of DNA breaks compared to cells expressing an inactive form of TDP1 in AOH-treated groups [71]. Using RAW264.7 cells as the assay model, several investigations have confirmed that activity as a topoisomerase poison is a key mechanism underlying AOH-induced DSB [14, 49, 72, 73]. In our experiments, pretreatment with N-acetylcysteine (NAC), a well-known ROS scavenger, effectively prevented AOH-triggered ROS generation and apoptosis (Fig. 5). Interesting, preliminary results from our studies also showed that pretreatment with N-acetylcysteine (NAC) could not fully prevent AOH-induced impairment of embryo development (data not shown). The results imply that AOH-triggered deleterious effects on embryo development are exerted not only through ROS-mediated apoptosis but also topoisomerase poison activity. However, the regulatory mechanisms triggered by AOH leading to apoptosis in mouse blastocyst cells are clearly ROS-dependent processes involving caspase-9 and -3 (Fig. 5). The specific effects of the topoisomerase poison property of AOH on embryonic development require further investigation.
Conclusion
Our collective results have demonstrated for the first time that AOH triggers apoptosis predominantly in the ICM and to a minor extent in TE of mouse blastocysts, leading to impairment of cell viability and embryonic development, both in vitro and in vivo. We propose that AOH induces ROS generation and subsequent activation of caspase-9 and caspase-3 that, in turn, trigger apoptotic processes exerting harmful effects on sequent embryonic development in mouse blastocysts. In conclusion, our collective results demonstrate the potential of AOH as an embryotoxic and immunotoxic risk factor during embryo and infant development stages in mice and AOH should be classified as an embryotoxic agent.
Supplementary Material
Contributor Information
Chien-Hsun Huang, Department of Obstetrics and Gynecology, Taoyuan General Hospital, Ministry of Health & Welfare, Taoyuan City 33004, Taiwan.
Fu-Ting Wang, Rehabilitation and Technical Aid Center, Taipei Veterans General Hospital, Taipei City 11217, Taiwan.
Wen-Hsiung Chan, Department of Bioscience Technology and Center for Nanotechnology, Chung Yuan Christian University, Chung Li District, Taoyuan City 32023, Taiwan.
Funding
This work was supported by grants from the Ministry of Science and Technology, Taiwan, ROC (MOST 107-2221-E-033-022-MY2 and MOST 109-2311-B-033-001).
Conflict of interest
The authors have no conflict of interest to declare.
References
- 1.EFSA DA, Eskola M, Gomez Ruiz JA. Scientific report on the dietary exposure assessment to Alternaria toxins in the European population. EFSA J 2016;14:4654–86. [Google Scholar]
- 2.Ackermann Y, Curtui V, Dietrich R, et al. Widespread occurrence of low levels of alternariol in apple and tomato products, as determined by comparative immunochemical assessment using monoclonal and polyclonal antibodies. J Agric Food Chem 2011;59:6360–8. [DOI] [PubMed] [Google Scholar]
- 3.Solhaug A, Eriksen GS, Holme JA. Mechanisms of Action and Toxicity of the Mycotoxin Alternariol: A Review. Basic Clin Pharmacol Toxicol 2016;119:533–9. [DOI] [PubMed] [Google Scholar]
- 4.López P, Venema D, de Rijk T, et al. Occurrence of Alternaria toxins in food products in The Netherlands. Food Control 2016;60:196–204. [Google Scholar]
- 5.Hickert S, Bergmann M, Ersen S, et al. Survey of Alternaria toxin contamination in food from the German market, using a rapid HPLC-MS/MS approach. Mycotoxin Res 2016;32:7–18. [DOI] [PubMed] [Google Scholar]
- 6.Gotthardt M, Asam S, Gunkel K, et al. Quantitation of six alternaria toxins in infant foods applying stable isotope labeled standards. Front Microbiol 2019;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Estiarte N, Crespo-Sempere A, Marin S, et al. Occurrence of Alternaria mycotoxins and quantification of viable Alternaria spp. during the food processing of tomato products in Spain. World Mycotoxin J 2018;11:625–33. [Google Scholar]
- 8.Gambacorta L, Magista D, Perrone G, et al. Co-occurrence of toxigenic moulds, aflatoxins, ochratoxin A, Fusarium and Alternaria mycotoxins in fresh sweet peppers (Capsicum annuum) and their processed products. World Mycotoxin J 2018;11:159–73. [Google Scholar]
- 9.Puntscher H, Kutt ML, Skrinjar P, et al. Tracking emerging mycotoxins in food: development of an LC-MS/MS method for free and modified Alternaria toxins. Anal Bioanal Chem 2018;410:4481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escriva L, Oueslati S, Font G, Manyes L. Alternaria mycotoxins in food and feed: an overview. J Food Qual 2017;2017:1569748. [Google Scholar]
- 11.Schreck I, Deigendesch U, Burkhardt B, et al. The Alternaria mycotoxins alternariol and alternariol methyl ether induce cytochrome P450 1A1 and apoptosis in murine hepatoma cells dependent on the aryl hydrocarbon receptor. Arch Toxicol 2012;86:625–32. [DOI] [PubMed] [Google Scholar]
- 12.Bensassi F, Gallerne C, el Dein OS, et al. Mechanism of Alternariol monomethyl ether-induced mitochondrial apoptosis in human colon carcinoma cells. Toxicology 2011;290:230–40. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Blanco C, Juan-Garcia A, Juan C, et al. Alternariol induce toxicity via cell death and mitochondrial damage on Caco-2 cells. Food Chem Toxicol 2016;88:32–9. [DOI] [PubMed] [Google Scholar]
- 14.Solhaug A, Vines LL, Ivanova L, et al. Mechanisms involved in alternariol-induced cell cycle arrest. Mutat Res 2012;738-739:1–11. [DOI] [PubMed] [Google Scholar]
- 15.Wollenhaupt K, Schneider F, Tiemann U. Influence of alternariol (AOH) on regulator proteins of cap-dependent translation in porcine endometrial cells. Toxicol Lett 2008;182:57–62. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Blanco C, Font G, Ruiz MJ. Oxidative stress of alternariol in Caco-2 cells. Toxicol Lett 2014;229:458–64. [DOI] [PubMed] [Google Scholar]
- 17.Vila-Donat P, Fernandez-Blanco C, Sagratini G, et al. Effects of soyasaponin I and soyasaponins-rich extract on the alternariol-induced cytotoxicity on Caco-2 cells. Food Chem Toxicol 2015;77:44–9. [DOI] [PubMed] [Google Scholar]
- 18.Tiessen C, Fehr M, Schwarz C, et al. Modulation of the cellular redox status by the Alternaria toxins alternariol and alternariol monomethyl ether. Toxicol Lett 2013;216:23–30. [DOI] [PubMed] [Google Scholar]
- 19.Bensassi F, Gallerne C, Sharaf El Dein O, et al. Cell death induced by the Alternaria mycotoxin Alternariol. Toxicol In Vitro 2012;26:915–23. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann L, Wagner J, Metzler M. Estrogenic and clastogenic potential of the mycotoxin alternariol in cultured mammalian cells. Food Chem Toxicol 2006;44:398–408. [DOI] [PubMed] [Google Scholar]
- 21.Fehr M, Pahlke G, Fritz J, et al. Alternariol acts as a topoisomerase poison, preferentially affecting the IIalpha isoform. Mol Nutr Food Res 2009;53:441–51. [DOI] [PubMed] [Google Scholar]
- 22.Brugger EM, Wagner J, Schumacher DM, et al. Mutagenicity of the mycotoxin alternariol in cultured mammalian cells. Toxicol Lett 2006;164:221–30. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Blanco C, Font G, Ruiz MJ. Oxidative DNA damage and disturbance of antioxidant capacity by alternariol in Caco-2 cells. Toxicol Lett 2015;235:61–6. [DOI] [PubMed] [Google Scholar]
- 24.Schoevers EJ, Santos RR, Roelen BAJ. Alternariol disturbs oocyte maturation and preimplantation development. Mycotoxin Res 2020;36:93–101. [DOI] [PubMed] [Google Scholar]
- 25.Hardy K. Cell death in the mammalian blastocyst. Mol Hum Reprod 1997;3:919–25. [DOI] [PubMed] [Google Scholar]
- 26.Hardy K, Stark J, Winston RM. Maintenance of the inner cell mass in human blastocysts from fragmented embryos. Biol Reprod 2003;68:1165–9. [DOI] [PubMed] [Google Scholar]
- 27.Byrne AT, Southgate J, Brison DR, Leese HJ. Analysis of apoptosis in the preimplantation bovine embryo using TUNEL. J Reprod Fertil 1999;117:97–105. [DOI] [PubMed] [Google Scholar]
- 28.Chan WH. Ginkgolide B induces apoptosis and developmental injury in mouse embryonic stem cells and blastocysts. Hum Reprod 2006;21:2985–95. [DOI] [PubMed] [Google Scholar]
- 29.Chan WH. Effects of citrinin on maturation of mouse oocytes, fertilization, and fetal development in vitro and in vivo. Toxicol Lett 2008;180:28–32. [DOI] [PubMed] [Google Scholar]
- 30.Chan WH. Cytotoxic effects of 2-bromopropane on embryonic development in mouse blastocysts. Int J Mol Sci 2010;11:731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratno Budiarto B, Chan WH. Oxidative stresses-mediated apoptotic effects of ginsenoside Rb1 on pre- and post-implantation mouse embryos in vitro and in vivo. Environ Toxicol 2017;32:1990–2003. [DOI] [PubMed] [Google Scholar]
- 32.Anggelia MR, Chan WH. Impairment of preimplantation and postimplantation embryonic development through intrinsic apoptotic processes by ginsenoside Rg1 in vitro and in vivo. Environ Toxicol 2017;32:1937–51. [DOI] [PubMed] [Google Scholar]
- 33.Jiang J, Wu S, Liu X, et al. Effect of acetochlor on transcription of genes associated with oxidative stress, apoptosis, immunotoxicity and endocrine disruption in the early life stage of zebrafish. Environ Toxicol Pharmacol 2015;40:516–23. [DOI] [PubMed] [Google Scholar]
- 34.Tu W, Niu L, Liu W, Xu C. Embryonic exposure to butachlor in zebrafish (Danio rerio): endocrine disruption, developmental toxicity and immunotoxicity. Ecotoxicol Environ Saf 2013;89:189–95. [DOI] [PubMed] [Google Scholar]
- 35.Livingstone DR. Contaminant-stimulated reactive oxygen species production and oxidative damage in aquatic organisms. Mar Pollut Bull 2001;42:656–66. [DOI] [PubMed] [Google Scholar]
- 36.Murugesan P, Kanagaraj P, Yuvaraj S, et al. The inhibitory effects of polychlorinated biphenyl Aroclor 1254 on Leydig cell LH receptors, steroidogenic enzymes and antioxidant enzymes in adult rats. Reprod Toxicol 2005;20:117–26. [DOI] [PubMed] [Google Scholar]
- 37.Huang CH, Chan WH. Rhein induces oxidative stress and apoptosis in mouse blastocysts and has immunotoxic effects during embryonic development. Int J Mol Sci 2017;18:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang CH, Wang FT, Chan WH. Enniatin B1 exerts embryotoxic effects on mouse blastocysts and induces oxidative stress and immunotoxicity during embryo development. Environ Toxicol 2019;34:48–59. [DOI] [PubMed] [Google Scholar]
- 39.Pampfer S, de Hertogh R, Vanderheyden I, et al. Decreased inner cell mass proportion in blastocysts from diabetic rats. Diabetes 1990;39:471–6. [DOI] [PubMed] [Google Scholar]
- 40.Hardy K, Handyside AH, Winston RM. The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development 1989;107:597–604. [DOI] [PubMed] [Google Scholar]
- 41.Pampfer S, Wuu YD, Vanderheyden I, De Hertogh R. In vitro study of the carry-over effect associated with early diabetic embryopathy in the rat. Diabetologia 1994;37:855–62. [DOI] [PubMed] [Google Scholar]
- 42.Huang LH, Shiao NH, Hsuuw YD, Chan WH. Protective effects of resveratrol on ethanol-induced apoptosis in embryonic stem cells and disruption of embryonic development in mouse blastocysts. Toxicology 2007;242:109–22. [DOI] [PubMed] [Google Scholar]
- 43.Khezri A, Herranz-Jusdado JG, Ropstad E, Fraser TW. Mycotoxins induce developmental toxicity and behavioural aberrations in zebrafish larvae. Environ Pollut 2018;242:500–6. [DOI] [PubMed] [Google Scholar]
- 44.Chan WH. Citrinin induces apoptosis via a mitochondria-dependent pathway and inhibition of survival signals in embryonic stem cells, and causes developmental injury in blastocysts. Biochem J 2007;404:317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang FJ, Chan WH. Effects of ochratoxin a on mouse oocyte maturation and fertilization, and apoptosis during fetal development. Environ Toxicol 2016;31:724–35. [DOI] [PubMed] [Google Scholar]
- 46.Hsuuw YD, Chan WH, Yu JS. Ochratoxin a inhibits mouse embryonic development by activating a mitochondrion-dependent apoptotic signaling pathway. Int J Mol Sci 2013;14:935–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang CH, Wang FT, Chan WH. Enniatin B induces dosage-related apoptosis or necrosis in mouse blastocysts leading to deleterious effects on embryo development. Drug Chem Toxicol 2021. (In Press). [DOI] [PubMed] [Google Scholar]
- 48.Mastrorocco A, Martino NA, Marzano G, et al. The mycotoxin beauvericin induces oocyte mitochondrial dysfunction and affects embryo development in the juvenile sheep. Mol Reprod Dev 2019;86:1430–43. [DOI] [PubMed] [Google Scholar]
- 49.Solhaug A, Wisbech C, Christoffersen TE, et al. The mycotoxin alternariol induces DNA damage and modify macrophage phenotype and inflammatory responses. Toxicol Lett 2015;239:9–21. [DOI] [PubMed] [Google Scholar]
- 50.Pfeiffer E, Eschbach S, Metzler M. Alternaria toxins: DNA strand-breaking activity in mammalian cellsin vitro. Mycotoxin Res 2007;23:152–7. [DOI] [PubMed] [Google Scholar]
- 51.EFSA, European Food Safety Authority . Scientific opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J 2011;9:2407. [Google Scholar]
- 52.Schuchardt S, Christina Ziemann C, Hansen T. Combined toxicokinetic and in vivo genotoxicity study on Alternaria toxins. European Food Safety Authority 2014;EN-679:130. [Google Scholar]
- 53.Fliszar-Nyul E, Lemli B, Kunsagi-Mate S, et al. Interaction of Mycotoxin Alternariol with Serum Albumin. Int J Mol Sci 2019;20:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiessen C, Ellmer D, Mikula H, et al. Impact of phase I metabolism on uptake, oxidative stress and genotoxicity of the emerging mycotoxin alternariol and its monomethyl ether in esophageal cells. Arch Toxicol 2017;91:1213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaferanloo B, Pepper SA, Coulthard SA, et al. Metabolites of endophytic fungi from Australian native plants as potential anticancer agents. FEMS Microbiol Lett 2018;365. [DOI] [PubMed] [Google Scholar]
- 56.Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science 1994;266:1508–18. [DOI] [PubMed] [Google Scholar]
- 57.Kelly SM, Robaire B, Hales BF. Paternal cyclophosphamide treatment causes postimplantation loss via inner cell mass-specific cell death. Teratology 1992;45:313–8: 313–318. [DOI] [PubMed] [Google Scholar]
- 58.Chan WH. Ginkgolides induce apoptosis and decrease cell numbers in mouse blastocysts. Biochem Biophys Res Commun 2005;338:1263–7. [DOI] [PubMed] [Google Scholar]
- 59.Chan WH, Shiao NH. Effect of citrinin on mouse embryonic development in vitro and in vivo. Reprod Toxicol 2007;24:120–5. [DOI] [PubMed] [Google Scholar]
- 60.Lane M, Gardner DK. Differential regulation of mouse embryo development and viability by amino acids. J Reprod Fertil 1997;109:153–64. [DOI] [PubMed] [Google Scholar]
- 61.Chan WH. Embryonic toxicity of sanguinarine through apoptotic processes in mouse blastocysts. Toxicol Lett 2011;205:285–92. [DOI] [PubMed] [Google Scholar]
- 62.Lam SH, Chua HL, Gong Z, et al. Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev Comp Immunol 2004;28:9–28. [DOI] [PubMed] [Google Scholar]
- 63.Trede NS, Langenau DM, Traver D, et al. The use of zebrafish to understand immunity. Immunity 2004;20:367–79. [DOI] [PubMed] [Google Scholar]
- 64.Eder KJ, Clifford MA, Hedrick RP, et al. Expression of immune-regulatory genes in juvenile Chinook salmon following exposure to pesticides and infectious hematopoietic necrosis virus (IHNV). Fish Shellfish Immunol 2008;25:508–16. [DOI] [PubMed] [Google Scholar]
- 65.Jiang J, Wu S, Wu C, et al. Embryonic exposure to carbendazim induces the transcription of genes related to apoptosis, immunotoxicity and endocrine disruption in zebrafish (Danio rerio). Fish Shellfish Immunol 2014;41:493–500. [DOI] [PubMed] [Google Scholar]
- 66.Jin Y, Zheng S, Fu Z. Embryonic exposure to cypermethrin induces apoptosis and immunotoxicity in zebrafish (Danio rerio). Fish Shellfish Immunol 2011;30:1049–54. [DOI] [PubMed] [Google Scholar]
- 67.Baggiolini M, Moser B, Clark-Lewis I. Interleukin-8 and related chemotactic cytokines. The Giles Filley Lecture, Chest 1994;105:95S–8S. [DOI] [PubMed] [Google Scholar]
- 68.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 2007;39:443–55. [DOI] [PubMed] [Google Scholar]
- 69.Risso-de Faverney C, Orsini N, de Sousa G, Rahmani R. Cadmium-induced apoptosis through the mitochondrial pathway in rainbow trout hepatocytes: involvement of oxidative stress. Aquat Toxicol 2004;69:247–58. [DOI] [PubMed] [Google Scholar]
- 70.Valavanidis A, Vlahogianni T, Dassenakis M, Scoullos M. Molecular biomarkers of oxidative stress in aquatic organisms in relation to toxic environmental pollutants. Ecotoxicol Environ Saf 2006;64:178–89. [DOI] [PubMed] [Google Scholar]
- 71.Fehr M, Baechler S, Kropat C, et al. Repair of DNA damage induced by the mycotoxin alternariol involves tyrosyl-DNA phosphodiesterase 1. Mycotoxin Res 2010;26:247–56. [DOI] [PubMed] [Google Scholar]
- 72.Solhaug A, Torgersen ML, Holme JA, et al. Autophagy and senescence, stress responses induced by the DNA-damaging mycotoxin alternariol. Toxicology 2014;326:119–29. [DOI] [PubMed] [Google Scholar]
- 73.Solhaug A, Holme JA, Haglund K, et al. Alternariol induces abnormal nuclear morphology and cell cycle arrest in murine RAW 264.7 macrophages. Toxicol Lett 2013;219:8–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.