

Abstract
Induced fit‐ (IF) and conformational selection (CS) binding mechanisms have long been regarded to be mutually exclusive. Yet, they are now increasingly considered to produce the final ligand‐target complex alongside within a thermodynamic cycle. This viewpoint benefited from the introduction of binding fluxes as a tool for analyzing the overall behavior of such cycle. This study aims to provide more vivid and applicable insights into this emerging field. In this respect, combining differential equation‐ based simulations and hitherto little explored alternative modes of calculation provide concordant information about the intricate workings of such cycle. In line with previous reports, we observe that the relative contribution of IF increases with the ligand concentration at equilibrium. Yet the baseline contribution may vary from one case to another and simulations as well as calculations show that this parameter is essentially regulated by the dissociation rate of both pathways. Closer attention should be paid to how the contributions of IF and CS compare at physiologically relevant drug/ligand concentrations. To this end, a simple equation discloses how changing a limited set of “microscopic” rate constants can extend the concentration range at which CS contributes most effectively. Finally, it could also be beneficial to extend the utilization of flux‐ based approaches to more physiologically relevant time scales and alternative binding models.
Keywords: binding fluxes, conformational selection, equations, induced fit, rate constants, simulations, thermodynamic cycle
IF always increases with increasing ligand concentration, [L], but minimum may vary from one case to another. A flux‐based approach shows that a limited number of rate constants suffice to describe the curve.

Abbreviations
- CS
conformational selection
- IF
induced‐fit
1. INTRODUCTION
The pharmacokinetic properties as well as the binding kinetics of a drug/ligand are nowadays considered to direct the selectivity and duration of its clinical action.1, 2 In this respect, induced‐fit (IF) binding is often evoked as a means to achieve its long residence time at the target. Traditionally, this mechanism comprises fast binding of the ligand, L, to the free target, T, to yield a transient TL complex, followed by slow transconformation thereof into a more stable T*L species (Figure 1).3, 4, 5, 6 This model represents a compromise between the classical “Occam's razor” principle, which favors a reversible one‐step binding, and increasing experimental evidence for a model in where TL experiences multiple small conformational adjustments.7, 8, 9 Conformational selection (CS) constitutes a well‐known counterpart to IF. Traditionally, this mechanism implies that a slow conformational change between free T and T* precedes a fast but highly selective binding step.5, 6, 10, 11, 12
FIGURE 1.

Schematic representations and equations. (A) Schematic representation of the explored IF‐CS‐ based thermodynamic cycles and parameters for individual pathways. Left side: The “ground state” of the target, T, undergoes a conformational change before the binding of the ligand, L, (to yield T*) according to the conformational selection, CS, model. According to the induced‐fit, IF, model, the binding (to yield TL) precedes the conformational change of the complex. Both pathways give rise to the same final T*L complex. Mid‐ and right side: Schematic representation of individual pathway with focus on the microscopic rate constants (denoted as k1…4) and microscopic fluxes (denoted as F1…4). (B) Top: Relationship between the macroscopic forward and reverse fluxes (i.e., Fon for T to T*L and Foff for T*L to T) and the microscopic fluxes such as presented by Hammes et al..19 Bottom: Relationship between the macroscopic fluxes and the conditional rates.21 (Supporting Information Section S2). Those distinct formulations are mathematically equivalent at equilibrium only (Supporting Information Section S8‐G)
CS‐ binding has often been opposed to IF6, 13, 14 and, in this respect, Copeland15, 16 asserted a decade ago that most of the drugs with high clinical efficacy act via IF. This conclusion was essentially based on the shape of kobs versus [L] plots: i.e., a hyperbolic increase for IF and a hyperbolic decrease for CS.4, 5 However, this strict distinction has been challenged by bio‐mathematical considerations showing that CS may also bring about increasing plots.17, 18 Hence, some previously qualified IF‐ binders could actually bind via CS.14
Such explicit distinction between IF‐ and CS‐binders is now also increasingly regarded to be a false dichotomy.19, 20, 21, 22 Indeed, they are now rather considered to “compete” with one another for producing the final T*L complex within a thermodynamic cycle. This viewpoint benefited from the introduction of binding fluxes, F, as a tool for analyzing the behavior of such cycle.19 Analogous to the rate of product formation in enzymology, a binding flux refers to the rate by which one target species converts into another. As such, it relies on the concentration of a target species as well as on the rate constant for its transformation into the other species. While, a reversible one‐step mechanism only necessitates a “forward” and a “reverse” flux, a two‐step mechanism such as IF and CS already requires four “microscopic” fluxes: i.e., two for each step (Figure 1, and Supporting Information Section S1). The overall conversion of T into T*L and back can also be expressed in terms of “macroscopic” forward‐ and reverse fluxes, Fon and Foff. In essence, they approximate the lowest of the intervening microscopic fluxes (Figure 1B). In this respect, Hammes et al.19 evaluated the relative contribution of IF, Rc‐IF, and CS to T*L within a cycle by comparing their Fon‐ values at equilibrium. This approach is now also adopted by many others.13, 20, 23, 24, 25, 26 The emerging picture is that Rc‐IF invariably increases with [L]. This usually allows CS to dominate at low [L], but not always.19, 24
Prior estimation/knowledge of all the microscopic rate constants is required for calculating Fon. The presently available sets of constants are not only theoretical13, 23, 27 but also based on molecular dynamics simulations and experimental observations.19, 24, 25, 26 In this respect, it is striking that many of the published sets depart from the traditional frame of reference (Section 2.4 and Supporting Information Section S2). Moreover it is still little know that Fon and/or Rc‐IF can also be directly calculated based on the microscopic rate constants without invoking concentration of each target species (e.g., Ref. [21]) and that relevant Rc‐IF‐ estimates can also been obtained by comparing by how much T*L accumulates via each pathway 26, 27 (Supporting Information Section S1).
Binding flux‐ based concepts may appear quite exotic for many pharmacologists who are more acquainted with rate constants. This study aims to partly fill this gap by providing more vivid and applicable insights into this emerging field. Attention is first paid to the link between Fon and the microscopic forward fluxes for IF and CS separately. The impact of the rate of each step of the thermodynamic cycle on the Rc‐IF–[L] relationship is examined next. The role of the most influential microscopic rate constants is finally highlighted.
2. MATERIALS AND METHODS
2.1. Nomenclature
The rate constants that govern the individual steps in two‐step binding models are commonly referred to as “microscopic” are denoted as for IF k1 (in M−1·min−1) and k3 (in min−1) when moving forward from T to T*L and as k2 (in min−1) and k4 (in min−1) when reverting to T. (Figure 1A). The “macroscopic” constants are denoted as kon and koff, respectively.28 To keep the same type of nomenclature, we refer to fluxes that are associated with the individual steps as the “microscopic” F1…F4 and those that account for the “macroscopic” fluxes as Fon and Foff (Figure 1A). To distinguish between the IF and CS pathways of a thermodynamic cycle, those notations are appended by the appellation of each pathway in question. To distinguish between the notations for conventional IF binding (i.e., for which k3 < k2) and bivalent‐ like binding (i.e., for which k3 > k2)29 the latter are appended by “Biv” (Supporting Information Section S2).
2.2. Definitions
For a mono‐ molecular binding step (e.g. from T to T*), F1‐CS amounts to the product of [T] and the associated first‐order rate constant k1‐CS and, for a bimolecular step (e.g. from T* to T*L), F3‐CS amounts to the product of [T*], [L] and the associated second‐order rate constant k3‐CS. Hence, all fluxes can be expressed in % of [Ttotal] × min−1 (Figure 1A). The “competition” between IF and CS in a thermodynamic cycle is presently quantified by the relative contribution (also often denoted as dominance, prevalence...) of IF: i.e., Rc‐IF = Fon‐IF/(Fon‐IF + Fon‐CS).19 When based on how much each pathway has contributed to the accumulation of T*L, Acc, after a given time span (such as proposed by Ordabayev et al.26), Rc‐IF = Acc‐IF/(Acc‐IF + Acc‐CS) (Supporting Information Section S1). Please note that Rc‐CS equals 1−Rc‐IF, that the pathway with the highest Rc value is considered to “dominate” and that both pathways contribute equally when their Rc = 0.5. Finally, it is only at equilibrium that Fon and Foff of each pathway can also be expressed in terms of their conditional rates (Figure 1B).21, 30 While those also act as composite first order rate “constants” (Supporting Information Section S2), they should not be confounded with kobs.
2.3. Simulations
Microscopic rate constants are provided for each investigated case in Supporting Information Section S3. Accumulation of T*L at time t’ via a given pathway is obtained by integrating the fluxes of the last microscopic step thereof,26 such as shown Supporting Information Section S1. The sum of both yields the total [T*L]. In general, numerical solutions for the differential equations are achieved by consecutively solving all their segments in parallel over very small time intervals.31 All simulations take account of the pre‐existing equilibrium between [T] and [T*]. Simulated data are analyzed by non‐linear regression analysis with GraphPad Prism® (GraphPad Software Inc.).
2.4. Paradigms
Simulations are also based on the widely adopted paradigm according to which [L] vastly exceeds [Ttot] so that [L] remains constant throughout. This is often the case for drug‐ binding.17, 19, 21, 23, 32, 33, 34 The transconformation step should significantly contribute to the ligand's affinity for genuine IF binding, i.e., k3‐IF > k4‐IF (so that [T*L] exceeds [TL] at equilibrium). Also, [T*] should also only represent a small fraction of the initial unbound targets, i.e., k2‐CS > k1‐CS.13, 18 The microscopic rate constants should comply with the classical “rapid equilibrium” paradigm according to which the binding proceeds faster than the conformational change for both pathways, i.e., k2‐IF > k3‐IF 29 and k4‐CS > k1‐CS.5, 13, 17, 18, 19 Yet, exceptions to those rules are nowadays tolerated, and those are here also taken into consideration. Finally, ratios between microscopic fluxes are only equivalent to the ratios between their constituent rate constants when they “originate” from the same target species and when they do not, or equally depend on [L]. This only concerns the F2‐IF/F3‐IF and F4‐IF/F4‐CS ratios.
Since the difference in Gibbs free energy between T and T*L needs to be rigorously the same for the two pathways of a thermodynamic cycle, their thermodynamic KD’s (i.e., the k2·k4/k1·k3‐ ratios) need to be equal as well. This constraint has been referred to as the “detailed balance rule“.21 When only approximate rate constants are provided in the literature, one of them needs to be adjusted to conform to this rule. Also, when starting from a compliant situation, it is only permitted to change two (or more) of them in parallel, such as when changing the forward and reverse rate constants of a single step equally (Figures 5 and 6). On the other hand, the two forward (or reverse) rate constants of a single pathway have to changed oppositely and so on.
FIGURE 5.
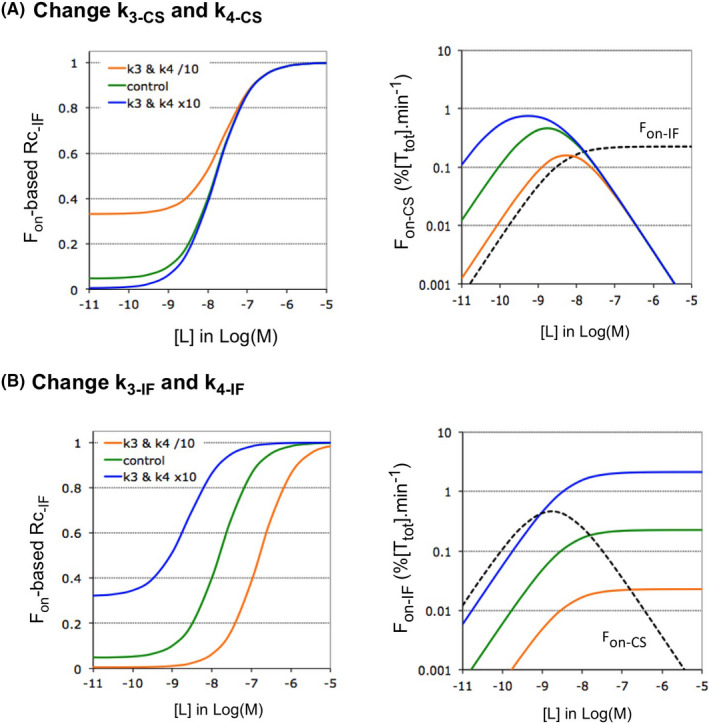
Impact of the rate of the second step of each pathway on Rc‐IF at equilibrium. Microscopic rate constants k3 and k4 are changed 10‐fold in tandem (to keep the KD unchanged) for a single pathway. The impact thereof on the Rc‐IF versus [L] plots is here shown for Case A (as control). Similar outcomes are also observed for Case B (Supporting Information Section S5). While Case A allows a better appreciation of the horizontal shifts, Case B allows a better appreciation of the vertical ones. (A) Left side: Decreasing or increasing the value of the k3‐CS–k4‐CS tandem does not affect the ascending portion of the Rc‐IF versus [L] plots but shifts Rc‐IF at low [L] in the opposite way. Right side: This shift can be related to the ability of changing k3‐CS to produce a similar change of F3‐CS at low [L] (not shown) and thus also of Fon‐CS. Hence, the Fon‐IF/Fon‐CS ratio changes in the opposite way. (B) Left side: Decreasing or increasing the value of the k3‐IF–k4‐IF tandem shifts the ascending portion of the Rc‐IF versus [L] plots in the opposite way (i.e., to higher and lower [L], respectively) but shifts Rc‐IF at low [L] in a similar way. Right side: This latter shift can be related to the ability of changing k3‐IF to produce an alike change of Fon‐IF (via F3‐IF) and thus also of the Fon‐IF/Fon‐CS ratio at low [L]. The more complex link between k3‐IF and the horizontal shifts is documented in Supporting Information Section S6
FIGURE 6.
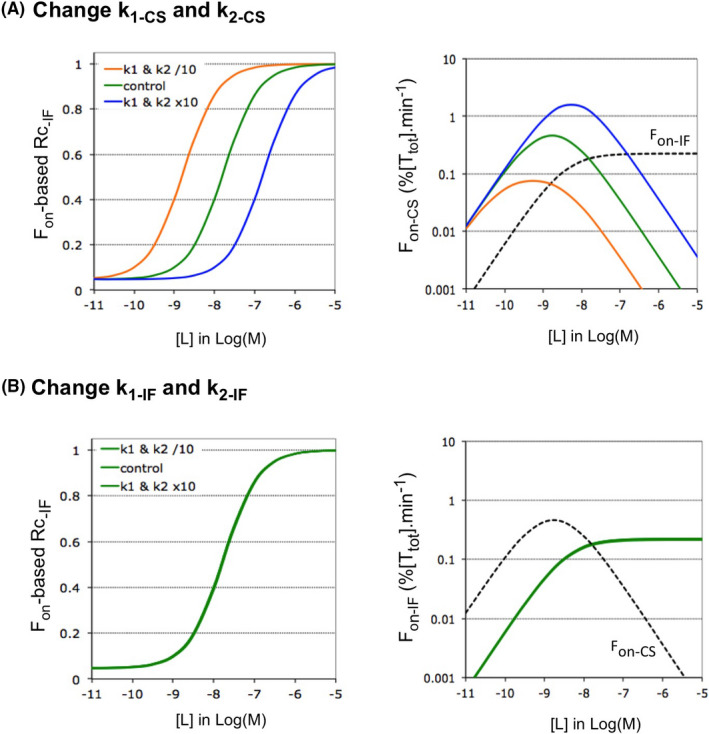
Impact of the rate of the first step of each pathway on Rc‐IF at equilibrium. Microscopic rate constants k1 and k2 are changed 10‐fold in tandem for a single pathway of Case A. The impact thereof on the Rc‐IF is also shown for Case B in Supporting Information Section S5. (A) Left side: Decreasing or increasing the value of the k1CS–k2‐CS tandem shifts the ascending portion of the Rc‐IF versus [L] plots in the same way (i.e., to lower and higher [L], respectively) without affecting Rc‐IF at low [L]. Right side: The horizontal shifts can be related to the ability of Fon‐CS to start descending at lower and higher [L], respectively. The more complex link between k1CS and the shifts is further documented in Supporting Information Section S6. (B) Left side: Decreasing or increasing the value of the k1IF–k2‐IF tandem does not affect the Rc‐IF versus [L] plots. Left side: the Fon‐IF versus [L] plots are not affected either
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,35 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.36, 37
3. RESULTS
3.1. Bivalent‐like‐ (Biv) versus genuine IF binding
It is of note that some authors have also classified as ‘IF’ cases that deviate from the classical “rapid equilibrium binding”‐ premise (i.e., with k3 < k2) by allowing the transconformation to proceed equally fast (i.e., k3 = k2)19 or even faster than the dissociation (i.e., k3 > k2).14 Kinetically speaking, the latter act like genuine bivalent ligands (Supporting Information Section S2) and are therefore abbreviated as ‘biv’. Here we show that this k3/k2 ratio also determines the microscopic step that mostly impacts Fon.
The simulated plots in Figure 2 gradually move from Biv‐ (left side) to classical IF binding (right side) by lowering k3 and k4. (i.e., to keep the KD constant and because the slow dissociation of IF binders is linked to a low k4 5). Alike previous observations,29 Figure 2A shows that binding equilibrium is reached faster for Biv‐ than for IF binding at equal [L]. Figure 2B compares how the microscopic forward fluxes for each step—F1 and F3—evolve with time. Although both change considerably early on, they eventually reach a plateau when equilibrium is approximated and this takes again less time for Biv‐ than for IF binding. This difference also applies to the macroscopic Fon (Figure 2C).
FIGURE 2.
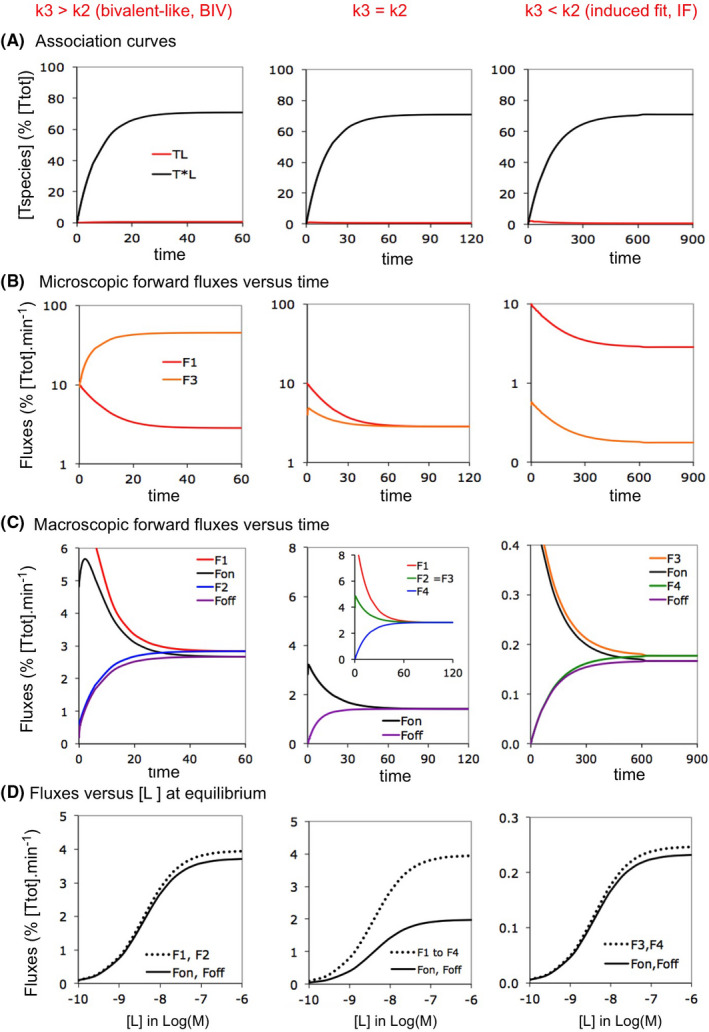
Fluxes for Biv‐ versus IF binding: implication of the k3/k2 ratio. Bivalent‐like, “Biv” refers to IF binding in where the conformational change proceeds faster than the binding, i.e., with k3 > k2 compared to k3 < k2 for classical IF. It is only from the kinetic viewpoint that such Biv‐ binding behaves similarly as genuine bivalent ligand binding (More information in Supporting Information Section S2). Simulations refer to isolated pathways. From left to right: the binding shifts from a Biv‐ to an IF pattern when the k3/k2 ratio decreases. Data for t = 0 not presented. (A) Simulated association/”accumulation” plots of the bound target species (i.e., TL for the intermediate complex and T*L for the final complex) as a function of the incubation time. Microscopic rate constants are provided in Supporting Information Section S3 and comply with the classical frame of reference for each model. [L] = 2.5 × KD for each example. Please note that [TL] still remains minimal (i.e., <1% of [Ttot]) at all times. It is only at higher [L] that transient rises in [TL] become significant for IF (not shown). (B) Evolution of the microscopic forward fluxes with time. Note that, at equilibrium, F1 < F3 for Biv and F1 > F3 for IF. (C) Evolution of the macroscopic Fon and Foff with time. Note that at equilibrium, all forward and reverse fluxes cancel out, that Fon is dictated by the smallest microscopic forward flux for Biv and IF and that Fon equals half of the microscopic forward fluxes for the intermediate situation in where F1 = F3. (D) The above considerations apply to all [L] at equilibrium
At equilibrium, F3 predominates for Biv binding, F1 and F3 are equal for the intermediate case (i.e., with k2 = k3) and F1 predominates for IF binding. As expected, Fon is largely determined by the smallest of the microscopic fluxes (i.e., F1 for Biv and F3 for IF) (Figure 2C). Similar relationships apply to the reverse fluxes. Figure 2C,D show that Foff always equals Fon at equilibrium and that this rule also applies to the microscopic fluxes (Figure 1A). Finally, the intermediate case (Figure 2C‐mid) represents a special situation: the microscopic fluxes are all equal at equilibrium and Fon only equals 50% thereof. Figure 2D further shows that, at equilibrium, Fon increases with [L] until a plateau value for all three cases. Interestingly, the proportionality between Fon and its most influential microscopic flux (or fluxes) remains constant throughout. This invariance owes to fact that the F2/F3 ratio always equals the k2/k3 ratio (see Section 2.4).
3.2. CS binding
Hyperbolically decreasing kobs versus [L] plots were regarded to be a hallmark for CS binding.4, 5 Yet, this shape only applies when k4 > k1 (Supporting Information Section S4). Otherwise (i.e., when k4 < k1), the plots rather increase hyperbolically.17, 32 In spite of this, all the forward (and reverse) fluxes still respond in the same way to increasing [L] for those two cases.
Figure 3 depicts the time‐wise evolution of the forward fluxes for both cases at low‐ (left side) as well as at high [L] (mid). Here also, they tend to reach a standstill when equilibrium is approximated. All the connected forward and reverse fluxes then become equal as well (not shown). However, it is noteworthy that F1 predominates at low [L] while F3 predominates at high [L]. This shift is gradual such as shown by the bell‐shaped profile of the Fon versus [L] plots (right side). This shape reflects the fact that Fon is governed by the lowest of the microscopic fluxes (i.e., from F3 to F1 when [L] increases). From the strict viewpoint of fluxes, increasing [L] allows CS‐ binding to shift from an IF‐ like‐ profile to a Biv‐ like profile.
FIGURE 3.
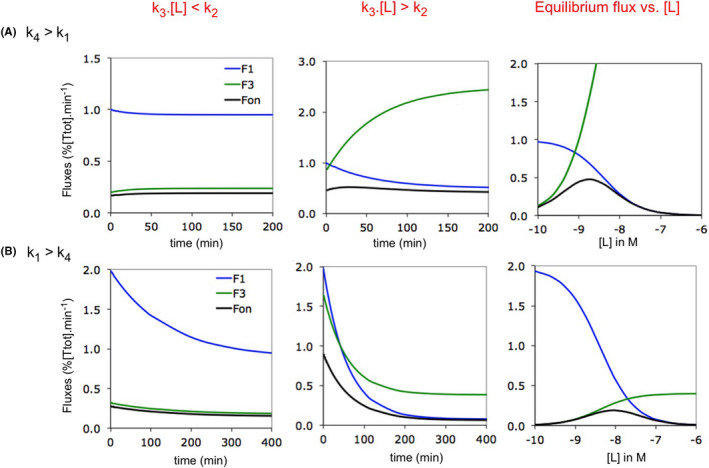
Forward fluxes for CS binding: implication of the ([L]·k3)/k2 and k4/k1 ratios. Simulated plots are for isolated CS binding. Microscopic rate constants are provided in Supporting Information Section S3. Data for t = 0 not presented. (A) The model complies with the traditional “rapid equilibrium binding” paradigm (i.e., with k4 > k1). Left side: forward fluxes versus time at low [L] = 0.2 nM. Such as for IF (Figure 2C), Fon is essentially dictated by F3 at all times. Mid: forward fluxes versus time at higher [L] = 4 nM. Fon becomes essentially dictated by F1 (i.e., Biv‐like, Figure 2C) as time goes by. Right side: forward fluxes versus [L] at equilibrium. The Fon versus [L] plot adopts a bell‐shaped pattern with a F3‐ dictated ascending portion and a F1‐ dictated descending portion. (B) Plots such as in (A) for the alternative CS binding model14, 17, 32 in where the conformational change proceeds faster (i.e., with k4 < k1). Low [L] = 5 nM and high [L] = 100 nM. The observations are quite similar as for the traditional model in (A). This contrasts with the hyperbolically decreasing kobs versus [L] plot for the traditional model and hyperbolically decreasing plot for the alternative model (Supporting Information Section S4). Finally, Fon is not maximal when the F1‐ and F3 versus [L] plots intersect because of their different curvature (right side)
3.3. Thermodynamic cycles: from macroscopic to microscopic fluxes
Next, we evaluate the contribution of the IF‐ and CS‐ pathways to T*L within a thermodynamic cycle. To this end, Figure 4 focuses on three cases with a distinct relative contribution of IF, Rc‐IF, at low [L] and at equilibrium. Although this difference received little attention until now, it offers a key to a better understanding of how this relative contribution is related to some of the microscopic rate constants.
FIGURE 4.
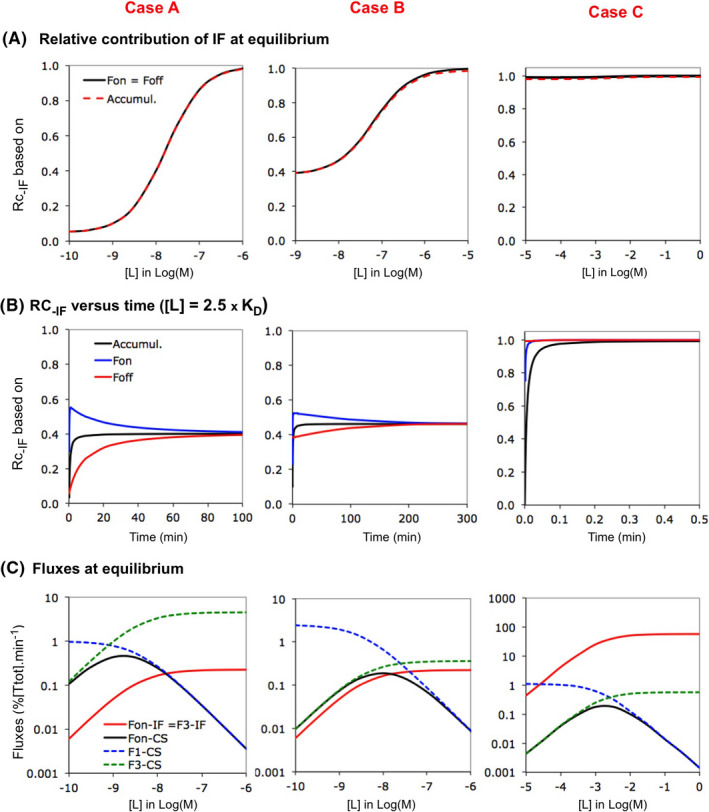
Competition between pathways of a thermodynamic cycle: IF versus CS. Competition between IF and CS is evaluated for three Cases. Both pathways comply with the classical frame of reference for Case A (see “Definition”). The two other cases diverge. The conformational change proceeds faster than the binding for CS For Case B (such as in Figure 3B) and Case C is based on kinetic data provided by Zhou et al.24 The latter values are 106‐ fold lower for the sake of comparison. Please see Supporting Information Section S3 for microscopic rate constants and important ratios thereof. (A) The relative contribution of IF to T*L, “Rc‐IF” increases with [L] for the three Cases at equilibrium. Rc‐IF can be expressed in terms of Fon (i.e., Rc‐IF = Fon‐IF/(Fon‐IF + Fon‐CS)), Foff and the accumulation of T*L (same type of equations). These three modes of calculation yield nearly‐ overlapping plots for each Case (hence only one color. Rc‐IF at very low [L] differs among the Cases: i.e., from <0.5 (CS‐ dominant) for Case A to nearly 1 (outspokenly IF‐ dominant) for Case C.24 (B) The evolution of Rc‐IF with time is shown for the three cases for [L] = 2.5 × KD. Note that, while the three approaches provide different Rc‐IF‐ values early on for each given case, they become closely the same at equilibrium. (C) Evolution of microscopic and macroscopic fluxes at equilibrium as a function of [L]. Such as for the individual pathways in Figures 2 and 3, Fon is dominated by F3 for both IF and CS at low [L]. Hence, the different Rc‐IF values low [L] relates to F3‐IF << F3‐CS for Case A, F3‐IF ≈ F3‐CS for Case B and F3‐IF >> F3‐CS for Case C. The bell‐shaped Fon versus [L] profile for CS is preserved when moving from the isolated CS pathway (Figure 3) to the cycle and the drop of Fon‐CS at higher [L] permits Rc‐IF to increase
Figure 4A compares their Rc‐IF versus [L] plots. The microscopic rate constants for each case and their compliance with the classical frame of reference are related in Supporting Information Section S3. Case A represents the conventional model. For this and several other cases in the literature (e.g., Refs. [23, 26, 27]), CS largely dominates at low [L] and Rc‐IF increases gradually with [L] until full IF‐ dominance. Such as for the two examples by Hammes et al.,19 Rc‐IF already resides well above baseline at low [L] for Case B. Case C relies on molecular dynamics simulation‐ based rate constants for MDM2 binding to the intrinsically disordered transactivation domain of the tumor suppressor p53 protein.24 This case represents an extreme situation in where IF always overwhelmingly dominates at low [L]. Interestingly, the Fon‐, the Foff‐ and the accumulation‐ based approaches26 yield nearly alike plots for each of the three cases (Figure 4A). Yet, Rc‐IF‐ values may differ from one another for each case early on (Figure 4B). We will only deal with equilibrium binding below.
Comparing how Fon is related to its constituent microscopic fluxes offers better insight into why Rc‐IF may be so different at low [L] (Figure 4C). Although both pathways now act in unison (so that they can potentially influence one another), the relationships that applied to the separate pathways (Figures 2 and 3) remain largely preserved. In particular, Fon‐CS is limited by F3‐CS at low [L] only while Fon‐IF is always limited by F3‐IF. The differences between the three cases can thus be explained as follows: IF is marginal for Case A because F3‐CS exceeds F3‐IF, both pathways contribute almost alike for Case B because F3‐IF and F3‐CS are nearly equal and IF dominates for Case C because F3‐CS exceeds F3‐IF (Figure 4C).
3.4. Link between Rc‐IF and the rate of individual steps
Ideally, it should be of interest to examine the impact of each rate constant on the Rc‐IF versus [L] plots separately. Unfortunately, the “detailed balance rule“ (Section 2.4) requires two (or more) rate constants to be changed in parallel. We here compare how changing the two rate constants of each step affect those plots.
Figure 5 focuses on the k3‐ and k4 values for Case A. Decreasing or increasing those values for CS prompts an inverse change of Rc‐IF at low [L] (Figure 5A). This is to be expected since changing k3‐CS produces an identical change of F3‐CS and Fon‐CS (right side) and thus an opposite change of the Fon‐IF/Fon‐CS ratio. Hence, decreasing k3‐CS favors IF while increasing this constant favors CS. This effect is even more explicitly illustrated for Case B (Supporting Information Section S5). In contrast, changing the k3‐IF‐ k4‐IF tandem produces an alike change of the Fon‐IF/Fon‐CS ratio and thus also of Rc‐IF at low [L] (Figure 5B). However, changing this tandem also prompts an appreciable horizontal shift of the Rc‐IF versus [L] plot, i.e., a decrease thereof allows CS to remain efficacious until higher [L] whereas an increase narrows this concentration range. As shown at the right side, those horizontal shifts reflect the ability of k3‐IF to change the amplitude of the Fon‐IF versus [L] plot (see also Supporting Information Section S6).
Figure 6 focuses on the k1‐ and k2 values for Case A. Here, decreasing or increasing this tandem for CS produces a (respectively) left‐ and rightward shift of the Rc‐IF versus [L] plot (Figure 6A). In essence, these shifts reflect the ability of k1‐CS to change the overall shape of the biphasic Fon‐CS versus [L] plot (right side). Please see also Supporting Information Section S6 for more detailed information. Finally, Figure 6B shows that increasing or decreasing the k1‐IF–k2‐IF tandem does not affect those plots.
Taken together, the present observations disclose the diversity of outcomes that that can be obtained by changing the overall rate of each step. In particular, they shed light on the ability of both transconformation steps to modulate the ligand's concentration range within which CS remains most efficacious (Figures 5B and 6A) and also on the ability of both k3–k4 tandems to modulate Rc‐IF at low [L] (Figure 5A,B). Comparable outcomes are also observed for Case B (Supporting Information Section S5) and the above principles can also be applied for transforming the IF‐ dominated situation for Case C (Figure 4C) into a situation in where CS remains dominant for an extended range of ligand concentrations (Supporting Information Section S7).
3.5. What do equations tell?
In line with the common practice, the previous figures essentially referred to forward fluxes. Yet, it is of note that substituting the forward fluxes by their reverse counterparts yields equivalent outcomes at equilibrium. Moreover, this special condition also allows Fon and Foff to be expressed by mathematically equivalent conditional rate‐ based equations (e.g., Refs. [21, 30]) (Figure 1B and Supporting Information Section S8‐G). As a major advantage, these allow the Rc‐IF–[L] relationship to be expressed in terms of the eight microscopic rate constants and [L] as only input (Figure 7A). This relationship can also be resolved in terms of minRc, i.e., the minimal value of Rc‐IF (for [L] = 0, Figure 7B); [L]0.5, the ligand concentration at which IF and CS contribute equally21 (Figure 7C) and [L]med, the median/midpoint of the sigmoidal plots (Figure 7D). Elaboration of all the equations is shown in Supporting Information Section S8. Taken together, those equations shed light on two major aspects.
FIGURE 7.
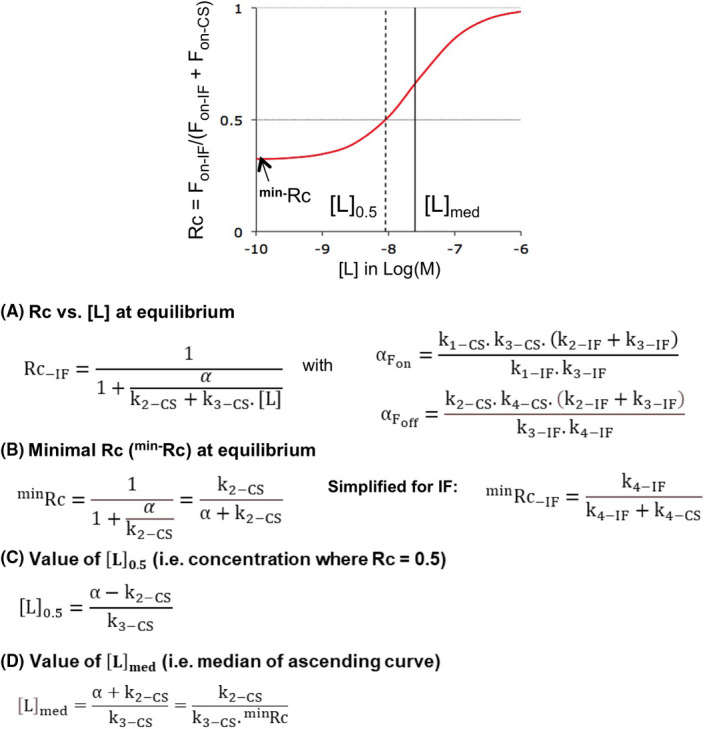
All equations refer to equilibrium binding and, unless explicitly specified, Rc applies to IF as well as to Biv (Supporting Information Section S2). The graph at the top shows the parameters that define a Rc versus [L] plot: minRc for the minimal value of Rc (i.e., when [L] reaches 0), [L]med for [L] at which the ascending portion of the plot is half‐maximal and [L]0.5 for [L] at which IF and CS contribute equally (i.e., when Rc = 0.5). Please see Supporting Information Section S8 for the elaboration of the equations. They are all based on the conditional rate‐ based equations for Fon and Foff (Figure 1B).21 (A) Equations for Rc at all [L]: The Fon‐ and Foff‐ based approaches yield the same value for the recurrent parameter, α. (B) minRc: The conditional rate‐ based equations can be simplified when [L] = 0 (see also Supporting Information Section S2). Simplification of the Foff‐ based equations discloses the implication of k4‐IF and k4‐CS for IF‐CS‐ based cycles. (C) IF and CS contribute equally at [L]0.5. The presented equation is mathematically equivalent to the one in.21 Those do obviously not apply when minRc exceeds 0.5. (D) [L]med refers to “Median” value of [L], i.e., at which the Rc versus [L] plot is half‐maximal between minRc and 1. Please see Table 1 for numerical values
First, it is only for a genuine IF–CS combination that the equation for minRc draws attention to a major impact of the two k4 values (Figure 7B and Supporting Information Sections S2 and S8‐C). Indeed, the calculations and simulations also reveal that the role of k4‐IF is taken over by the “k1‐Biv.KD ” product for a Biv–CS combination (Supporting Information Sections S2, S8‐D and S9). This shift corroborates with the fact that Fon‐Biv is largely dictated by F1‐Biv instead of F3‐Biv at low [L] (Figure 2).
Second, the surprisingly simple expression of [L]med as k2‐CS/(k3‐CS. minRc) implies that only a limited set of—largely CS related—microscopic rate constants suffices to quantify the concentration range of the ligand for which CS remains most efficacious. As shown in Table 1, the so‐ calculated values of [L]med correspond to those that are based on the simulations in Figures 5 and 6. Among others, this equation also provides a straightforward explanation for why [L]med is unaffected by varying the rate of ligand association for IF (Figure 6B). It also links the only minimal effect of the association rate for CS (Figure 5A) to the opposite contributions of minRc and k3‐CS.
TABLE 1.
[L]med and related parameters for Case A
| Parameters | minRc‐IF (approximated) | k3‐CS/k2‐CS in nM | [L]med in nM (calculated) | [L]med in nM (simulated)a |
|---|---|---|---|---|
| 0.0476 | 0.8 | 16.8 | 16.9 | |
| Fold‐ shift in Figures 5 and 6 by changing | ||||
| k3‐CS and k4‐CS | ||||
| ×0.1 | ×7.0 | ×10 | ×1.43 | ×1.43 |
| ×10 | ×0.104 | ×0.1 | ×0.96 | ×0.96 |
| k3‐IF and k4‐IF | ||||
| ×0.1 | ×0.104 | ×1.0 | ×9.57 | ×9.52 |
| ×10 | ×7.0 | ×1.0 | ×0.143 | ×0.148 |
| k1‐CS and k2‐CS | ||||
| ×0.1 | ×1.0 | ×0.1 | ×0.1 | ×0.1 |
| ×10 | ×1.0 | ×10 | ×10 | ×10 |
| k1‐IF and k2‐IF | ||||
| ×0.1 | ×1.0 | ×1.0 | ×1.0 | ×1.05 |
| ×10 | ×1.0 | ×1.0 | ×1.0 | ×1.0 |
The ascending portion of the sigmoidal Rc‐IF versus Log[L] plot is half‐maximal at [L]med. This median/midpoint is calculated via equation 25 in Supporting Information Section S8 (also presented in Figure 7D) based on the k2‐CS/k3‐CS ratio and the simplified expression of the baseline value of the relative contribution of IF (i.e., minRc‐IF as k4‐IF/(k4‐IF + k4‐CS)) for Case A and variations thereof such as shown in Figures 5 and 6. Those calculated [L]med values are already in excellent agreement with those that are obtained by non‐linear regression analysis with GraphPad Prism® (Graphpad Software Inc.) of the Fon‐ based Rc‐IF versus Log[L] plots (in where Fon‐ values are based on the microscopic rate constants and simulated concentrations of the different target species at equilibrium, such as related).19
Finally, please note that the present differential equation‐ and conditional rate‐ based calculations provide closely the same Rc‐IF versus [L] plots as the reported ones for cases whose input data were based on molecular dynamics simulations and experimental observations19, 24, 26 (not shown). Conclusions about the impact of microscopic rate constants on those plots (see Discussion) remain pertinent as well.
4. DISCUSSION
IF and CS pathways are increasingly considered to operate alongside within a thermodynamic cycle. This allows both of them to contribute to the final T*L complex instead of being mutually exclusive. The notion by Hammes et al.19 that binding fluxes are more appropriate than rate constants for evaluating this contribution is also gaining ground. The present article aims to provide better insight into the basic aspects of this new approach, with special focus on the connection between such fluxes and their constituent rate constants (Figure 1).
Although the concept of binding‐ or “reactive” fluxes is already pretty ancient,38 the equations that were presented by Hammes et al.19 have the merit to express the “macroscopic” fluxes of each pathway (Fon for T to T*L) in terms of the “microscopic” fluxes of the intervening steps (e.g., F1‐IF for T to TL) under equilibrium‐ as well as under non‐equilibrium conditions. The present simulations show such relationships as a function of time, the ligand concentration and of the kinetic properties of each pathway. While they confirm that Fon is chiefly dictated by the lowest of the microscopic fluxes (Figures 2 and 3), they also call attention to differences between IF and CS at equilibrium. Fon‐IF is governed by the flux of the transconformation step (F3‐IF) at all [L] whereas Fon‐CS is successively controlled by the binding step (F3‐CS) and the transconformation step (F1‐CS) when [L] increases. This shift is gradual and confers a bell‐shaped profile to the Fon‐CS versus [L] plots.
The relative contribution of both pathways to T*L within a cycle is now also customary evaluated by comparing their Fon‐ values at equilibrium (here referred to as Rc‐IF = Fon‐IF/(Fon‐IF + Fon‐CS). Like all previous reports, Figure 4A shows that Rc‐IF increases with [L] until IF fully dominates for three distinct Cases. However, matching profiles can also be obtained by the accumulation‐ based approach26 (Supporting Information Section S1). This correspondence is especially striking because both approaches embody different perceptions of the term “contribution”. Indeed, the former refers to a macroscopic flux at a given time point which, by definition, has to originate from T. Alternatively, the accumulation‐ based approach plays the role of an “archivist” by adding up all the net contributions each pathway to T*L during a given time frame.
Figure 4 also clearly shows that the positive relationship between Rc‐IF and [L] does not imply that CS necessarily dominates at low [L]. It is therefore of interest to find out how this parameter is governed by microscopic fluxes and even rate constants. To this end, comparing the Cases in Figure 4C, changing the k3–k4 tandems for each pathway (Figure 5) as well as simplifying the conditional rate‐ based equations (Figure 7B and Supporting Information Sections S2 and S8‐D) shed light on a major impact of the two k4‘s on the baseline value of Rc‐IF (minRc‐IF) for a genuine IF–CS‐ composed cycle. Taken together, minRc‐IF can be approximated as F3‐IF/(F3‐IF + F3‐CS), such as already hinted at in Figure 4C, but also as F4‐IF/(F4‐IF + F4‐CS) and even as k4‐IF/(k4‐IF + k4‐CS). This owes to the equivalence between the forward‐ and reverse fluxes for each step at equilibrium as well as to the equivalence between the F4‐IF/F4‐CS‐ and k4‐IF/k4‐CS ratios. In this respect, it is of note that the ratios between the F3‐ and the more elemental k3‐ values are not equivalent (see Section 2.4).
Because of the limiting role of the k4’s with respect to the dissociation of T*L via both IF and CS,5 it can be inferred that Rc‐IF at low [L] is essentially controlled by the rates of the bidirectional transit between T and T*L. In other words, it implies that the fastest pathway dominates. Such conclusion was previously also reached for heterobivalent ligand binding39 and it is also central to a model in where TL to T*L transition goes along with the formation of a ‘lid’ that occludes bound L from the aqueous surrounding.15, 40, 41 This shielding is considered to greatly impair the spontaneous escape of the ligand so that its dissociation is most prone to proceed via a retrograde IF mechanism.
The most effective contribution of CS can also be perceptibly extended to higher [L] by changing the transconformation rates of IF and CS (Figures 5B and 6A). Although this issue has only been scantly addressed in previous studies 42 it also merits particular attention since it relates to how much each pathway is able to contribute to T*L within a physiological range of [L]. While horizontal shifts of the Rc‐IF versus [L] plots (left side) are quite arduous to interpret in terms of microscopic fluxes (right side), this is not the case when turning to the conditional rate‐ based equations. Indeed, these allow the Rc‐IF–[L] relationship to be expressed in terms of the microscopic rate constants and [L] only (Figure 7A, see also13) and they also allow the median/midpoint of such plots, [L]med, to be calculated. This equation is even surprisingly simple since it only embraces minRc‐IF and the k2‐CS/k3‐CS ratio (Figure 7D). Those parameters fully account for the shifts of Lmed in Figures 5 and 6 (Table 1) and, since F1‐CS and F2‐CS are equal at equilibrium, it can be inferred that k2‐CS/k3‐CS ratio corresponds to [L] at which the F1‐CS‐ and F3‐CS plots do intersect in Figure 3.
The present binding flux‐ based approaches provide better insight into how the microscopic rate constants of IF and CS pathways affect their relative contribution to T*L at equilibrium. Relying on this special condition is now common practice since it facilitates comparative studies by getting rid of time‐ related variations. Also, it is only at equilibrium that Rc‐IF can be expressed in terms of simple equations. On the backside, it is obvious that equilibrium binding is only rarely met in real‐ life situations.41 This important consideration pleads for extending the use of the present—as well as additional flux‐related approaches to more physiologically relevant time scales.27, 43
Ligand‐target interactions are still often classified as either IF or CS based on the shape of their kobs versus [L] plots (e.g., Ref. [44]). Yet, while the already many reported hyperbolically decreasing plots do still point at CS, increasing plots are nowadays considered as inconclusive.17, 45 Interestingly, the thermodynamic cycle model allows them to be biphasic (i.e., with an initial decrease and subsequent increase)23, 27 and such pattern has also been observed experimentally (e.g., Refs. [46, 47, 48, 49, 50]). Structural biology provides complementary information. Refinements in molecular dynamics simulations and in experimental techniques like nuclear magnetic resonance‐ and fluorescence spectroscopy already provided the kinetic parameters for some of the reported relative contributions of IF and CS.19, 24, 26 They also greatly improved our insight about the conformational plasticity of many targets. In this respect, it is now considered that CS‐ binding can take place when the unbound target is already able to adopt a “binding‐competent” conformation51 and especially when ligand binding increases the population thereof. On the other hand, IF is to be preferred when new conformations are produced. Based on a mixed profile, Sušac et al.52 recently concluded that agonist‐mediated adenosine A2A receptor activation might utilize both mechanisms. The constitutive activity of many wild‐type and mutant G protein‐ coupled receptors, the existence of inverse agonists12 and the results from above‐ mentioned simulations and measurements53, 54, 55 point to the ability of such receptors to perform highly dynamic sampling between “inactive” and “active” conformations. Such CS‐ prone sampling has also been observed for other target proteins. The Hsp90 heat shock protein offers a striking example. Based on the ascending profile of its kobs versus [L] plot, it was initially assumed that the antagonist geldanamycin binds to this protein via IF.15, 16, 56 However, this profile has meanwhile been related to its conversion into a more potent species in the assay medium.57, 58 Instead, simulations and measurements now indicate that the involved N‐terminal domain can sample a wide range of conformations in between the fully open‐ and the ATP‐ bound closed ones59, 60, 61, 62, 63 and recent kinetic considerations further suggest that the binding of related antagonists implicate both IF and CS.45
In this respect, there is also increasing awareness that conformational changes can take place within a large range of timescales and that the energy landscape of a binding process may be funnel‐like with rapidly interconverting conformations at each bottom.9, 16, 64, 65, 66 While some authors opted to neglect such fast transconformations,32, 33 others rather opted to extend the CS model to an increasingly popular one in where a fast CS‐ binding precedes a slower IF‐ like conformational change of the complex.67, 68, 69 Since this hybrid model is thought to allocate high selectivity (via CS) and high affinity (via IF) to drug binding,22 it could be of particular interest to pay more attention to this model in terms of fluxes.
DISCLOSURES
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AUTHOR CONTRIBUTIONS
G. Vauquelin: conception and design of this work, generation of data and writing of this manuscript. D Maes: design of this work, generation of mathematical data and writing of this manuscript.
ETHICS APPROVAL STATEMENT
N/A.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We are very much indebted to Dr Denis Michel (Univ. Rennes‐1) for helpful comments.
Vauquelin G, Maes D. Induced fit versus conformational selection: From rate constants to fluxes… and back to rate constants. Pharmacol Res Perspect. 2021;9:e00847. 10.1002/prp2.847
DATA AVAILABILITY STATEMENT
No data have been shared.
REFERENCES
- 1.Swinney DC. Biochemical mechanisms of drug action, what does it take for success? Nat Rev Drug Discov. 2004;3:801‐808. [DOI] [PubMed] [Google Scholar]
- 2.Copeland RA, Pompliano DL, Meek TD. Drug‐target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730‐739. [DOI] [PubMed] [Google Scholar]
- 3.Koshland DE. (1958) Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strickland S, Palmer G, Masset V. Determination of dissociation constants and specific rate constants of enzyme‐ substrate (or protein‐ligand) interactions from rapid reaction kinetic data. J Biol Chem. 1975;250:4048‐4052. [PubMed] [Google Scholar]
- 5.Tummino PJ, Copeland RA. Residence time of receptor‐ ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481‐5492. [DOI] [PubMed] [Google Scholar]
- 6.Du X, Li YI, Xia Y‐L, et al. Insights into protein–ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 2016;17:144‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garvey EP. Structural mechanism of slow‐onset, two‐step enzyme inhibition. Curr Chem Biol. 2010;4:64‐73. [Google Scholar]
- 8.Dror RO, Pan AC, Arlow DH, et al. Pathway and mechanism of drug binding to G‐protein‐coupled receptors. Proc Natl Acad Sci USA. 2011;108:13118‐13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weis WI, Kobilka BK. The molecular basis of G protein‐coupled receptor activation. Annu Rev Biochem. 2018;87:897‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88‐118. [DOI] [PubMed] [Google Scholar]
- 11.Burgen AS. Conformational changes and drug action. Fed Proc. 1981;40:2723‐2728. [PubMed] [Google Scholar]
- 12.Changeux JP, Edelstein S. Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol Rep. 2011;3:19. 10.3410/B3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greives N, Zhou H‐X. Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc Natl Acad Sci USA. 2014;11:10197‐10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty P, Di Cera P. Induced fit is a special case of conformational selection. Biochemistry. 2017;56(22):2853‐2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copeland RA. The dynamics of drug‐target interactions: drug‐ target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5:305‐310. [DOI] [PubMed] [Google Scholar]
- 16.Copeland RA. Conformational adaptation in drug–target interactions and residence time. Future Med Chem. 2011;3:1491‐1501. [DOI] [PubMed] [Google Scholar]
- 17.Vogt AD, Di Cera E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry. 2012;51:5894‐5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gianni S, Dogan J, Jemth P. Distinguishing induced fit from conformational selection. Biophys Chem. 2014;189:33‐39. [DOI] [PubMed] [Google Scholar]
- 19.Hammes GG, Chang YC, Oas TG. Conformational selection or induced fit: a flux description of reaction mechanism. Proc Natl Acad Sci USA. 2009;106:13737‐13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daniels KG, Tonthat NK, McClure DR, et al. Ligand concentration regulates the pathways of coupled protein folding and binding. J Am Chem Soc. 2014;136:822‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michel D. Conformational selection or induced fit? New insights from old principles. Biochimie. 2016;128–129:48‐54. [DOI] [PubMed] [Google Scholar]
- 22.Meyer‐Almes FJ. Discrimination between conformational selection and induced fit protein‐ligand binding using Integrated Global Fit analysis. Eur Biophys J. 2016;45:245‐257. [DOI] [PubMed] [Google Scholar]
- 23.Galburt EA, Rammohan JA. Kinetic signature for parallel pathways: conformational selection and induced fit. Links and disconnects between observed relaxation rates and fractional equilibrium flux under pseudo‐first‐order conditions. Biochemistry. 2016;55:7014‐7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou G, Pantelopoulos GA, Mukherrjee S, Voelz VA. Bridging microscopic and macroscopic mechanisms of p53‐MDM2 binding with kinetic network models. Biophys J. 2017;113:785‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sekhar A, Velyvis A, Zoltsman G, Rosenzweig G, Bouvignies G, Kay LE. Conserved conformational selection mechanism of HP70 chaperone‐substrate interactions. Elife. 2018;7:e32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ordabayev YA, Nguyen B, Kozlov AG, Jia H, Lohman TM. UvrD helicase activation by MutL involves rotation of its 2B sub‐domain. Proc Natl Acad Sci USA. 2019;116:16320‐16325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vauquelin G, Maes D, Swinney DC. Fluxes for unraveling complex binding mechanisms. Trends Pharmacol Sci. 2020;41:923‐932. [DOI] [PubMed] [Google Scholar]
- 28.Neubig R, Spedding M, Kenakin T, Christopoulos A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev. 2003;55:597‐606. [DOI] [PubMed] [Google Scholar]
- 29.Vauquelin G. Distinct in vivo target occupancy by bivalent‐ and induced‐fit‐like binding drugs. Br J Pharmacol. 2017;173:1268‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malygin EG, Hattman S. A probabilistic approach to compact steady‐state kinetic equations for enzymic reactions. J Theor Biol. 2006;242:627‐633. [DOI] [PubMed] [Google Scholar]
- 31.Vauquelin G, Morsing P, Fierens FLP, De Backer JP, Vanderheyden PML. A two‐state receptor model for the interaction between angiotensin II AT1 receptors and their non‐peptide antagonists. Biochem Pharmacol. 2001;61:277‐284. [DOI] [PubMed] [Google Scholar]
- 32.Vogt AD, Pozzi N, Chen Z, Di Cera E. Essential role of conformational selection in ligand binding. Biophys Chem. 2014;186:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pozzi N, Vogt AD, Gohara DW, Di Cera E. Conformational selection in trypsin‐like proteases. Curr Op Struct Biol. 2012;22:421‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul F, Weikl TR. How to distinguish conformational selection and induced fit based on chemical relaxation rates. PLoS Comput Biol. 2016;12(9):e1005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2019: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexander SPH, Christopoulos A, Davenport AP, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Gprotein‐coupled receptors. Br J Pharmacol. 2019;176(Suppl 1):S20‐S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexander SPH, Fabbro D, Kelly E, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(Suppl 1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong P, Cara JEF, Tager HS. Importance of receptor occupancy, concentration differences, and ligand exchange in the insulin‐like growth factor I receptor system. Proc Natl Acad Sci USA. 1993;90:11451‐11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vauquelin G. Simplified models for heterobivalent ligand binding: when are they applicable and which are the factors that affect their target residence time. Naunyn‐Schmiedeberg’s Arch Pharmacol. 2013;386:949‐962. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan SM, Holyoak T. Enzymes with lid‐gated active sites must operate by an induced fit mechanism instead of conformational selection. Proc Natl Acad Sci USA. 2008;105:3829‐13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Copeland RA. The drug‐target residence time model: a 10‐year retrospective. Nat Rev Dug Discov. 2016;15:87‐95. [DOI] [PubMed] [Google Scholar]
- 42.Li D, Liu Ming S, Ji B. Mapping the dynamics landscape of conformational transitions in enzyme: the adenylate kinase case. Biophys J. 2015;109:647‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffmann C, Castro M, Rinken A, Leurs R, Hill SJ, Vischer HF. Ligand residence time at G‐protein–coupled receptors ‐ why we should take our time to study it. Mol Pharmacol. 2015;88:552‐560. [DOI] [PubMed] [Google Scholar]
- 44.Guengerich FP, Wilkey CJ, Phan TTN. Human cytochrome P450 enzymes bind drugs and other substrates mainly through conformational‐ selection modes. J Biol Chem. 2019;294:10928‐10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amaral M, Kokh DB, Bomke J, et al. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat Commun. 2007;8:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burton RL, Chen S, Xu XL, Grant GA. Transient kinetic analysis of the interaction of l‐serine with Escherichia colid‐3‐phosphoglycerate dehydrogenase reveals the mechanism of V‐type regulation and the order of effector binding. Biochemistry. 2009;48:12242‐12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faleev NG, Zakomirdina LN, Vorob'ev MM, et al. A straightforward kinetic evidence for coexistence of ′′induced fit′′ and “selected fit” in the reaction mechanism of a mutant tryptophan indole lyase Y72F from Proteus vulgaris. Biochim Biophys Acta, Proteins Proteomics. 2014;1844:1860‐1867. [DOI] [PubMed] [Google Scholar]
- 48.Rammohan J, Ruiz‐Manzano A, Garner AL, Stallings CL, Galburt EA. CarD stabilizes mycobacterial open complexes via a two‐tiered kinetic mechanism. Nucleic Acids Res. 2015;43:3272‐3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakrabarti K, Agafonov R, Pontiggia F, et al. Conformational selection in a protein‐protein interaction revealed by dynamic pathway analysis. Cell Rep. 2016;14:32‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sreemantee S, Udgaonkar JB. Binding‐induced folding under unfolding conditions: switching between induced fit and conformational selection mechanisms. J Biol Chem. 2019;294:16942‐16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuxreiter M. Classifying the binding modes of disordered proteins. Int J Mol Sci. 2020;21:8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sušac L, Eddy MT, Didenko T, Stevens RC, Wüthrich K. A2A adenosine receptor functional states characterized by 19F‐NMR. Proc Natl Acad Sci USA. 2018;115:12733‐12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS. Activation of the A2A adenosine G‐protein‐coupled receptor by conformational selection. Nature. 2016;533:265‐270. [DOI] [PubMed] [Google Scholar]
- 54.Shimada I, Ueda T, Kofuku Y, Eddy MT, Wüthrich K. GPCR drug discovery: integrating solution NMR data with crystal and cryo‐EM structures. Nat Rev Drug Discov. 2019;18:59‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu F‐J, Williams LM, Abdul‐Ridha A, et al. Probing the correlation between ligand efficacy and conformational diversity at the α1A‐adrenoreceptor reveals allosteric coupling of its microswitches. J Biol Chem. 2020;295:7404‐7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gooljarsingh LT, Fernandes C, Yan K, et al. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc Natl Acad Sci USA. 2006;103:7625‐7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maroney AC, Marugan JJ, Mezzasaima TM, et al. Dihydroquinone ansamycins: toward resolving the conflict between low in vitro affinity and high cellular potency of geldanamycin derivatives. Biochemistry. 2006;45:5678‐5685. [DOI] [PubMed] [Google Scholar]
- 58.Onuoha SC, Mukund SR, Coulstock ET, et al. Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J Mol Biol. 2007;372:287‐297. [DOI] [PubMed] [Google Scholar]
- 59.Colombo G, Morra G, Meli M, Verkhivker G. Understanding ligand‐based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc Natl Acad Sci USA. 2008;105:7976‐7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krukenberg KA, Förster F, Rice LM, Sali A, Agard DA. Multiple conformations of E. coli Hsp90 in solution: insights into the conformational dynamics of Hsp90. Structure. 2008;16:755‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Q Rev Biophys. 2011;44:229‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morra G, Verkhivker G, Colombo G. Modeling signal propagation mechanisms and ligand‐ based conformational dynamics of the Hsp90 molecular chaperone full‐length dimer. PLoS Comput Biol. 2009;5:e1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H, Zhou C, Chen W, et al. A Dynamic View of ATP‐coupled functioning cycle of Hsp90 N‐terminal domain. Sci Rep. 2015;5:9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598‐1603. [DOI] [PubMed] [Google Scholar]
- 65.Henzler‐Wildman KA, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964‐972. [DOI] [PubMed] [Google Scholar]
- 66.Okazaki K, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: induced‐fit versus population‐shift mechanisms. Proc Natl Acad Sci USA. 2008;105:11182‐11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agafonov RV, Wilson C, Otten R, Buosi V, Kern D. Energetic dissection of Gleevec’s selectivity toward human tyrosine kinases. Nat Struct Mol Biol. 2014;21:848‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Paula VS, Jude KM, Nerli S, Glassman CR, Garcia KC, Sgourakis NG. Interleukin‐2 druggability is modulated by global conformational transitions controlled by a helical capping switch. Proc Natl Acad Sci USA. 2020;117:7183‐7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim E, Lee S, Jeon A, et al. A single‐molecule dissection of ligand binding to a protein with intrinsic dynamics. Nat Chem Biol. 2013;9:313‐318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
No data have been shared.