

Abstract
Although most experimentally characterized proteins with similar sequences assume the same folds and perform similar functions, an increasing number of exceptions is emerging. One class of exceptions comprises sequence‐similar fold switchers, whose secondary structures shift from α‐helix <‐> β‐sheet through a small number of mutations, a sequence insertion, or a deletion. Predictive methods for identifying sequence‐similar fold switchers are desirable because some are associated with disease and/or can perform different functions in cells. Here, we use homology‐based secondary structure predictions to identify sequence‐similar fold switchers from their amino acid sequences alone. To do this, we predicted the secondary structures of sequence‐similar fold switchers using three different homology‐based secondary structure predictors: PSIPRED, JPred4, and SPIDER3. We found that α‐helix <‐> β‐strand prediction discrepancies from JPred4 discriminated between the different conformations of sequence‐similar fold switchers with high statistical significance (P < 1.8*10−19). Thus, we used these discrepancies as a classifier and found that they can often robustly discriminate between sequence‐similar fold switchers and sequence‐similar proteins that maintain the same folds (Matthews Correlation Coefficient of 0.82). We found that JPred4 is a more robust predictor of sequence‐similar fold switchers because of (a) the curated sequence database it uses to produce multiple sequence alignments and (b) its use of sequence profiles based on Hidden Markov Models. Our results indicate that inconsistencies between JPred4 secondary structure predictions can be used to identify some sequence‐similar fold switchers from their sequences alone. Thus, the negative information from inconsistent secondary structure predictions can potentially be leveraged to identify sequence‐similar fold switchers from the broad base of genomic sequences.
Keywords: bioinformatics, metamorphic proteins, protein fold switching, protein secondary structure prediction, protein structure prediction
1. INTRODUCTION
Most known folded proteins assume one stable structure that performs one specific function. Nevertheless, an increasing number of exceptions to this one‐structure‐one‐function paradigm have been identified.[ 1 , 2 , 3 ] For example, extant (or single‐sequence) fold‐switching proteins remodel their secondary (and tertiary) structures in response to cellular stimuli, leading to changes in function and regulation.[ 4 , 5 ]
Recent evidence suggests that single‐sequence fold‐switching proteins are likely more abundant in nature than currently reflected in the Protein Data Bank (PDB), an online repository with nearly 160 000 experimentally determined protein structures.[ 6 ] To date, there are just under 100 proteins with at least two sequence‐identical PDB entries that have substantially different secondary and tertiary structures[ 5 ] (Figure 1). One explanation for their likely underrepresentation is that the structures of these sequence‐identical fold switchers, because of their multi‐conformational nature, are difficult to characterize experimentally. Indeed, a significant number of these proteins have been discovered since the development of more advanced methodologies for structure determination, such as cryo‐EM and solid‐state NMR.[ 4 , 5 ] Additionally, solution NMR has been used to discover[ 8 , 9 , 10 ] and characterize[ 11 , 12 ] a number of fold switchers, yet NMR structures comprise <8% of all protein structures currently in the PDB. Furthermore, the appropriate stimulus (e.g., pH change, interacting protein) to induce fold switching may also be unknown, leading to only one conformation being found. This lack of functional information could potentially give fold‐switching proteins the false appearance of assuming only one stable fold.
FIGURE 1.
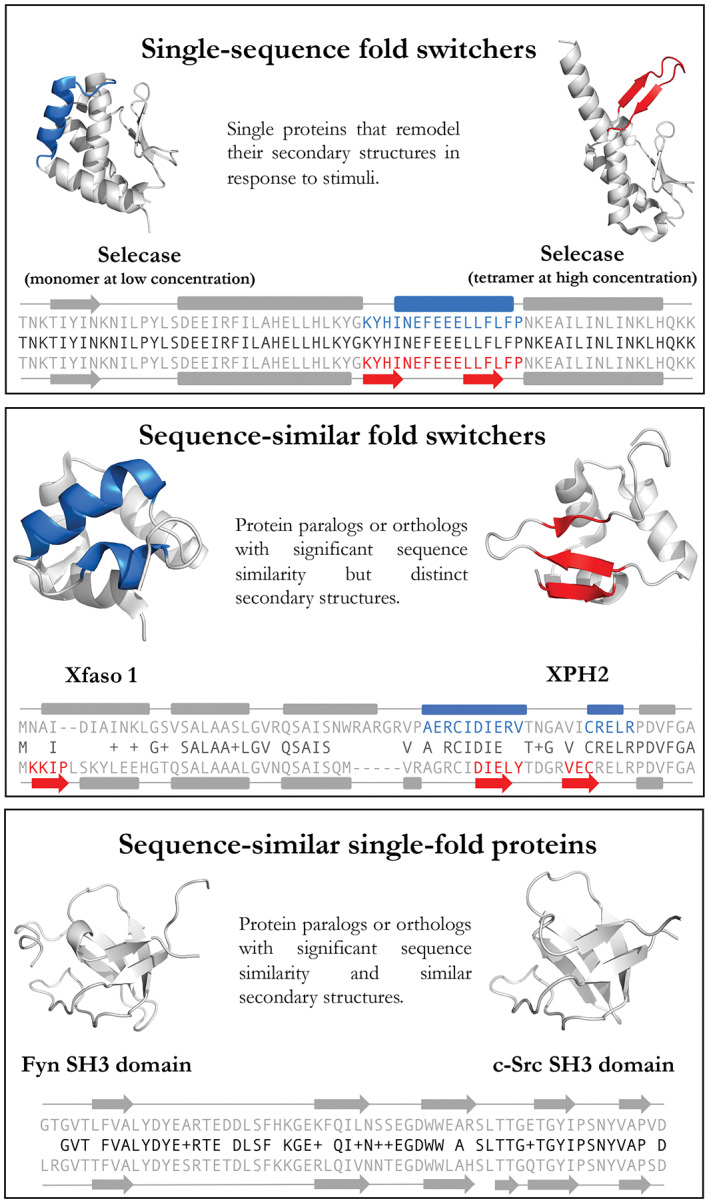
Definitions of single‐sequence fold switchers, sequence‐similar fold switchers, and sequence‐similar single‐fold proteins. Gray regions are structurally unchanged between the two conformations. Upper/lower sequences and secondary structures (rounded rectangle = helix, arrow = strand, line = coil) correspond to protein structures on the left/right, respectively. Middle sequences (black) show amino acid identities (letters) and similarities (+). PDB IDs (Left to right, top to bottom): 4QHF, 4QHH, 3BD1, 5W8Z, 1FYN, 6XVM. Cartoon diagrams were made in PyMOL[ 7 ]
In spite of the technical limitations that likely hamper the discovery of more fold switchers, interest in their structures, functions, and mechanisms has grown for several reasons. Firstly, fold‐switching proteins are associated with a number of diseases, including Alzheimer's,[ 13 ] autoimmune dysfunction,[ 14 ] and bacterial infection.[ 8 ] Secondly, fold switching appears to be a widespread mechanism for biological regulation across all domains of life.[ 4 ] For instance, fold switchers control the expression of bacterial virulence genes,[ 8 ] underlie the periodicity of a cyanobacterial circadian clock,[ 9 ] and regulate Cl− concentrations in human microglia.[ 13 ] Finally, fold switchers, along with intrinsically disordered proteins (IDPs)[ 15 ] and morpheeins,[ 16 ] challenge the paradigm that a protein's amino acid sequence encodes a single secondary, tertiary, and quaternary structure.
It would be ideal to have computational methods that accurately and rapidly identify extant fold‐switching proteins from their sequences alone ‐ both for the basic understanding of protein folding that this would represent, and for the ability to make and test predictions of unknown fold switchers. Although such methods are not yet available, several advances suggest that they could be achieved. Firstly, proteins with identical sequences but different folds have been designed computationally. Specifically, Ambroggio and Kuhlman designed Sw2, a 32‐residue peptide that switches from a trimeric helical bundle into a zinc finger fold in response to changes in pH.[ 17 ] More recently, Wei et al. designed XAA_GVDQ, which forms a homotrimeric helical bundle with long helices in its crystal structure but short helices in its NMR structures.[ 18 ] Secondly, co‐evolutionary methods can correctly identify fold switching and other conformational changes in proteins, such as rotational motions, with reasonable accuracy.[ 19 , 20 ] Thirdly, fold switchers were predicted blindly using a computational method that searches for discrepancies between predicted and experimentally determined secondary structures.[ 5 , 21 ] Although the scope of this latter method is highly constrained because it requires the protein of interest to have at least one experimentally determined structure, it is a step in the right direction.
Our ultimate goal is to expand the predictive scope of algorithms that identify sequence‐identical fold switchers. Specifically, we seek to develop a fast, high‐throughput method to assess fold switchers using their sequences alone (i.e., without using solved protein structures to make predictions). Such a method could be used to identify potential fold‐switching sequences from hundreds of millions of candidates as opposed to the less than 200,000 candidates available in the PDB.
As a step toward this goal, here we present a comparative approach that assesses whether two different proteins with high levels of aligned similarity assume the same fold or different ones (hereafter called sequence‐similar single‐fold proteins and sequence‐similar fold switchers, respectively; Figure 1). In contrast to single‐sequence fold switchers, sequence‐similar fold switchers remodel their secondary structures in response to mutation. High levels of aligned similarity strongly suggest that the secondary structure remodeling of sequence‐similar fold switchers was induced by mutation, as opposed to alternative mechanisms, such as non‐homologous sequence replacement.[ 22 ] For this use, we define high levels of aligned similarity as either (a) >70% similar but less than 100% identical or (b) 100% identical but differing by an insertion or deletion at the N‐ or C‐ terminus. Additionally, we required pairs of sequence‐similar fold switchers to have at least one region of aligned sequence (i.e., sequence: sequence, not sequence: deletion) that assumes an α‐helix in one structure and a β‐strand in the other. These α‐helix <‐> β‐strand discrepancies distinguish sequence‐similar fold switchers from proteins that either shorten/lengthen their pre‐existing secondary structures[ 23 ] or unfold[ 24 ] in response to mutation. Furthermore, since sequences with >70% aligned similarity are expected to assume the same fold,[ 25 ] predicting secondary structure changes in sequences at or above this threshold is a significant result.
With the wealth of non‐redundant protein sequences currently available,[ 26 ] we reasoned that even sequences with subtle differences (i.e., one amino acid change) might align to alternative sequences in a large database, providing potentially discriminatory structural information. In other words, large sequence repositories could be “crowdsourced” by search methods such as PSI‐BLAST[ 27 ] to associate very similar query sequences with different sequences found by the searches. These differential hits could then be leveraged to predict structural differences between sequence‐similar fold switchers.
We benchmarked our method on 19 experimentally validated proteins from four different fold families with significant α‐helix <‐> β‐strand discrepancies, the most robust predictor of fold switchers found previously.[ 21 ] Specifically, we quantified α‐helix <‐> β‐strand discrepancies derived from three high‐performing secondary structure predictors with reasonably high accuracies: PSIPRED, 84%[ 28 ]; JPred4, 82%[ 29 ]; SPIDER3, 73%.[ 30 ] All three of these predictors query large sequence databases to suggest the elements of secondary structure assumed by a given amino acid sequence. JPred4 predictions showed significant α‐helix <‐> β‐strand discrepancies in 14/19 cases, which we predicted to switch folds. By comparison, only 1/207 sequence‐similar single‐fold proteins (Figure 1) showed appreciable α‐helix <‐> β‐strand discrepancies. Thus, as a classifier this method yielded a Matthews Correlation Coefficient of 0.82, demonstrating its robustness. We then addressed the question of why JPred4 predicted sequence‐similar fold switchers much better than PSIPRED and SPIDER3. We found that JPred4 had two prominent features that distinguished it from the other two algorithms. The first was its curated library used for PSI‐BLAST[ 27 ] searches. Rerunning PSIPRED and SPIDER3 using JPred4's library improved PSIPRED's predictions considerably (2/4 correct as opposed to 0/4) and SPIDER3's predictions slightly (1/4 correct as opposed to 0/4). JPred4's second distinguishing feature was its use of HMMer[ 31 , 32 ] to generate sequence profiles. HMMer is a software suite that generates profiles of multiple sequence alignments (MSAs) using Hidden Markov Models (HMMs), statistical models with many biological applications.[ 33 ] Finally, we traced JPred4's different secondary structure predictions back to unique sequences hit by PSI‐BLAST searches of one sequence‐similar fold‐switched sequence, but not another. Even sequences differing by just one amino acid yielded unique PSI‐BLAST hits.
2. METHODS
2.1. Identification of sequence‐similar fold switchers
As previously,[ 5 ] we ran a BLAST[ 27 ] search of all PDBs from a culled set (PISCES[ 34 ] list from 4/4/20 including all NMR structures and X‐ray/cryo‐EM structures with a minimum resolution of 3.0 Å and a maximum R‐value of 0.3; all sequences had ≤99% identity to one another) against all other PDBs within the set. Secondary structure annotations of each PDB, by DSSP,[ 35 ] were aligned in register with their corresponding BLAST alignments and compared one‐by‐one, residue‐by‐residue. A potential hit was required to have a continuous region of at least 15 residues in which at least 50% of the residues showed α‐helix <‐> β‐sheet differences.
We classified the resulting protein pairs as sequence‐similar fold switchers if they satisfied the following criteria:
Visual inspection confirmed significant differences in regular secondary structure. Specifically, the protein pair had to have at least one aligned region where one protein segment folded into an α‐helix while the other folded into a β‐strand. This requirement was based on our previous work[ 21 ] showing that α<‐>β discrepancies appear to be the strongest predictor of fold switching when using homology‐based secondary structure predictors.
Both structures were reported in the published literature.
2.2. Secondary structure predictions of sequence‐similar fold switchers
All amino acid sequences from 19 sequence‐similar fold switchers were downloaded from the Protein Data Bank[ 6 ] (PDB) and saved as individual FASTA[ 36 ] files. Separate secondary structure predictions were run on each file using JPred4, PSIPRED, and SPIDER3. JPred4 predictions were run remotely using a publicly downloadable scheduler available on the JPred4 website (http://compbio.dundee.ac.uk/jpred/); jnetpred predictions were used for all calculations unless specified otherwise (IE JNET_HMM and JNET_PSSM predictions specified in JPred4's robustness results from its sequence database and HMMer profiles). SPIDER3 calculations were performed on the Spark's Lab webserver (https://sparks-lab.org/server/spider3). PSIPRED calculations were run on their webserver (http://bioinf.cs.ucl.ac.uk/psipred/). Secondary structure predictions from .jnetpred (JPred), .horiz files (PSIPRED), and .spd33 files (SPIDER3) were converted into FASTA format. Each residue was assigned one of three secondary structures: “H” for helix, “E” for extended β‐strand, and “C” for coil. Chain breaks were annotated “‐.” PDB IDs from each family of sequence‐similar fold switchers were as follows: Cro/cI: 3BD1, 5W8Y, 2PIJ, 5W8Z; NusG/RfaH: 2OUG and 2LCL, GA/GB: 2LHC, 2LHG, 2KDL, 2JWS, 2JWU, 2KDM, 2LHD, 2LHE; KaiB: 5JYT, 2QKE, 4KSO, 1WWJ, 1R5P. We included all KaiB variants from cyanobacteria presuming that they all switch folds, though this has only been shown explicitly for S. elongatus. We excluded KaiB PDBs 5JWT and 5JWO because they are in complex with KaiC, which is believed to stabilize their fold‐switched form; their apo forms are not available in the PDB. Sequence similarities were calculated using the McLachlan Metric.[ 25 , 37 ] Cartoon diagrams of these sequence‐similar fold switchers (Figure 2) were made using PyMOL.[ 7 ]
FIGURE 2.
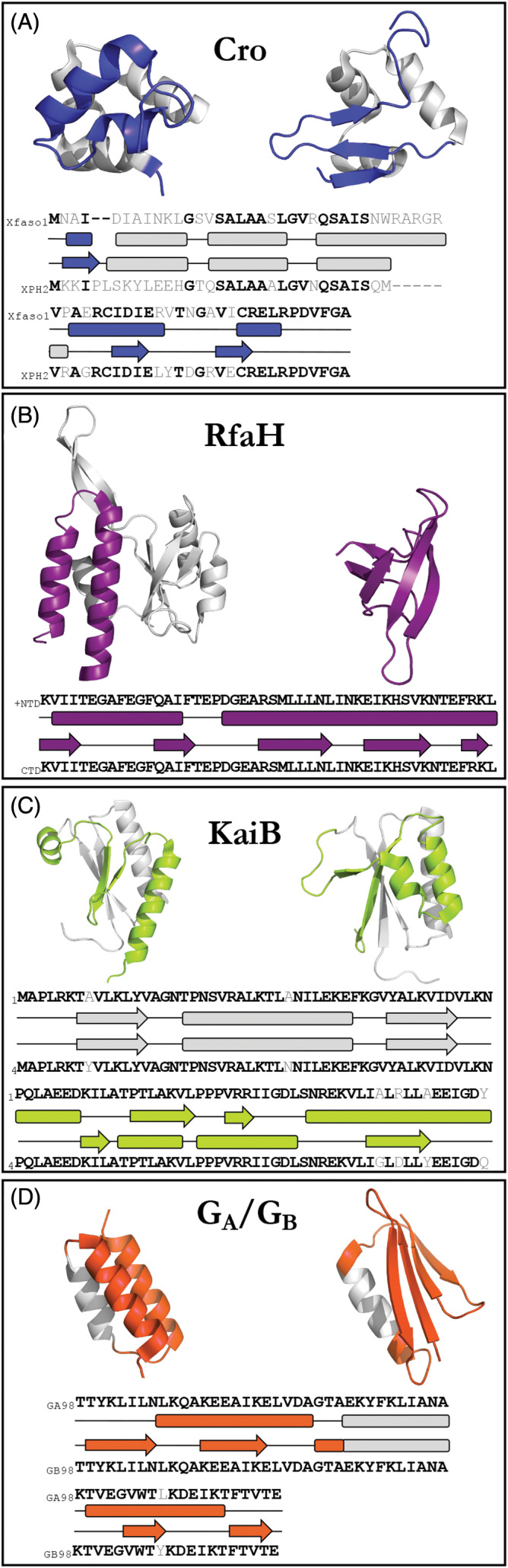
We identified 4 families of sequence‐similar fold switchers: transcription repressors XFaso 1/XPH2 (A) Transcription/translation factor RfaH (B) signaling protein KaiB (C) binding proteins GA98/GB98 (D). Colored regions of each panel are fold‐switched regions. Gray regions are structurally unchanged between the two conformations. Upper/lower sequences and secondary structures correspond to protein structures on the left/right, respectively. Most sequences were too long to fit on a single line. Lower sets of sequences/secondary structures are a continuation of the sequences/secondary structures above them
2.3. Single‐fold protein families
High‐identity single‐fold protein families were acquired by running three rounds of PSI‐BLAST on the whole PDB using three query proteins from different fold families: GB (1GB1, chain A, α/β grasp fold), the Fyn‐SH3 domain (1FYN, chain A, β‐barrel fold), and myoglobin (1MCY, chain A, α‐helical bundle fold) and retrieving all hit sequences with ≥75% aligned identity to their respective queries (allowing for slightly more sequence variation between hits than between hit and query). GB sequences with non‐natural amino acids except selenomethionine were excluded (4KGR, 4KGS, 4KGT, 4OZA, 4OZC, 6 L91, 6L9B, 6L9D, 6LJI). This resulted in 207 unique sequences (37 GB homologs, 36 SH3 homologs, and 134 myoglobin homologs). JPred4 was run on all 207 sequences using its mass submit scheduler (http://www.compbio.dundee.ac.uk/jpred4/api.shtml#massSubmit). PSIPRED and SPIDER3 were run locally on all 207 sequences using both the nr database and JPred4's curated database (more details below). We allowed up to 10 000 alignments for each run and a minimum e‐value of 0.001 for PSIPRED (as suggested by its script runpsipred); no minimum e‐value was imposed for SPIDER3 in accordance with its script run_list.sh. Three iterations of PSI‐BLAST were run for both PSIPRED and SPIDER3. SPIDER3 also requires inputs from HHBlits,[ 38 ] which we ran locally using the Uniref20 library from 2/2016.
2.4. Helix <‐> strand discrepancies and distribution
We generated family‐specific multiple sequence alignments (MSAs) using ClustalOmega[ 39 ] and obtained all pairwise alignments from the resulting MSAs. Secondary structure predictions were re‐registered according to the resulting alignments and compared. Helix <‐> strand discrepancies between the predictions were summed and then normalized by the minimum number of secondary structure annotations (helix and sheet) in the two sequences compared. The fold‐switching distribution in Figure 4 (plotted using Matplotlib[ 40 ]) was generated by averaging the prediction discrepancies between a given sequence X with all predictions from sequences in its family that (a) had experimentally determined structures different from X and (b) had at least 70% sequence similarity with X. This calculation was performed for all sequences X (totaling 19). By contrast, the single‐fold distribution was generated by averaging the prediction discrepancies between a given sequence Y with all predictions from sequences in its family (excluding itself). This calculation was performed for all sequences Y (totaling 207).
FIGURE 4.
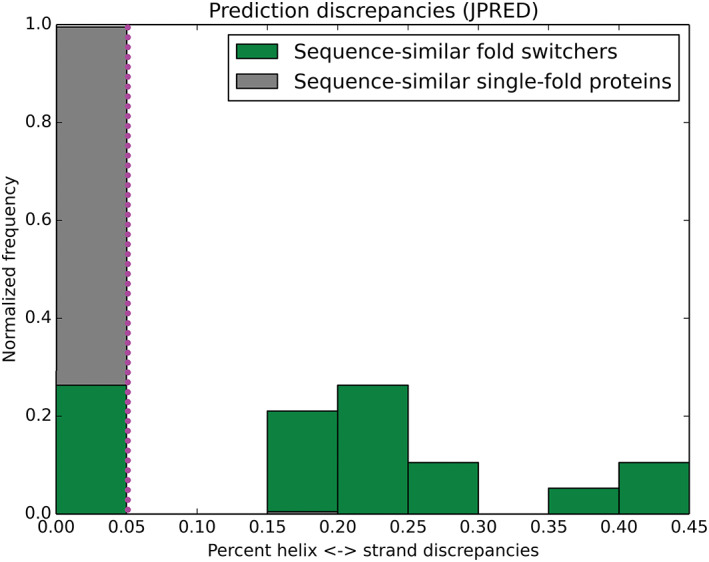
Secondary structure discrepancies from JPred4 are a robust predictor of sequence‐similar fold switchers. The distributions of these discrepancies for sequence‐similar fold switchers (green) and sequence‐similar single‐fold proteins (gray) differ significantly P < 1.8*10−19 (Kolmogorov‐Smirnov test). Most (14/19) of the sequence‐similar fold switchers fall above the 5% discrepancy threshold (dotted magenta line), while <1% (1/207) of sequence‐similar single‐fold proteins fall above it. The Matthews Correlation Coefficient for distinguishing fold switchers from single‐fold proteins at this threshold was 0.82
2.5. Secondary structure predictions and PSI‐BLAST searches using JPred4's sequence database
We downloaded JPred4's sequence database from: http://www.compbio.dundee.ac.uk/jpred/about_RETR_JNetv231_details.shtml
After generating BLAST libraries from JPred4's database, we used it to run three rounds of PSI‐BLAST[ 27 ] locally (blastpgp for PSIPRED and psiblast for SPIDER3) and generate predictions. We allowed up to 10 000 alignments for each run and a minimum e‐value of 0.001 for PSIPRED (as suggested by its script runpsipred); no minimum e‐value was imposed for SPIDER3 in accordance with its script run_list.sh. SPIDER3 also requires inputs from HHBlits,[ 38 ] which we ran locally using the Uniref20 library from 2/2016.
2.6. JPred runs on common and unique sequences
Full JPred4 results (.TAR.GZ archives) for GA98/GB98, full‐length RfaH/RfaH CTD, and XPH1/XPH2 were downloaded from the JPred4 website (http://www.compbio.dundee.ac.uk/jpred/). MSAs from their .align files were compared; sequences that occurred in both .align files were assigned to common profiles, while sequences that occurred in just one .align file were assigned to unique profiles. HMMer profiles of both common and unique profiles were generated by running:
hmmbuild ‐fast ‐gapmax 1.0 ‐wblosum <output_hmm> <input_MSA>
hmmconvert ‐p <input_hmm> <output_prf>
The output prf file was then converted to JPred4's HMM format by transforming elements 1‐24 (x) of its prf matrix using the sigmoid function:
and retaining five significant digits after the decimal point. PSI‐BLAST PSSMs were generated by making libraries from the MSAs of common and unique sequences and running three iterations of PSI‐BLAST (version 2.10) on those libraries using the query sequences for making JPred predictions (i.e., the sequences of GA98/GB98, full‐length RfaH/RfaH CTD, and XPH1/XPH2). These PSSMs were then converted to JPred4 PSSM format using the sigmoid function:
and retaining eight significant digits after the decimal point. The resulting HMMer and PSSM profiles were used as inputs to the jnet 2.3.1 algorithm.
3. RESULTS
3.1. Identifying sequence‐similar fold switchers
First, we sought to identify likely sequence‐similar fold switchers, that is, proteins with >70% sequence similarity but different folds (Methods). To do this, we BLASTed[ 27 ] the amino acid sequences from a culled set of >40 000 non‐redundant PDB entries against one other. Secondary structure annotations of all pairwise alignments with e‐values ≤1e‐04 were compared, and their differences were quantified. Protein pairs were queried for: (a) regions with different annotated secondary structure (Methods) and (b) sequence similarity ≥70%. Global and local RMSDs were then calculated for protein pairs that satisfied these requirements. Local RMSDs were calculated using the regions of the two proteins with different annotated secondary structures (Methods). Protein pairs with local/global RMSDs exceeding 5.0 and 3.0 Å, respectively, were then manually examined. Four protein families fit our criteria (Figure 2 , Methods), all of which have been previously identified.[ 6 , 9 , 41 , 42 ]
Firstly, the Cro protein family has members with two different C‐terminal topologies: one largely composed of α‐helices and the other of β‐strands, respectively[ 22 ] (Figure 2A). Proteins that assume both folds are transcriptional repressors present in bacteriophages, and two forms of sequence analysis‐a PSI‐BLAST search and transitive homology modeling‐suggest that the observed topological differences likely arose from stepwise mutation.[ 22 , 43 ] These structural differences appear to have functional consequences (Table 1): many β‐strand Cro proteins dimerize more strongly in solution, and their cognate operator affinities appear to be stronger than their α‐helical homologs.[ 22 ] This stronger dimerization may have evolved after the evolutionary transition from the α‐helical fold to the β‐strand fold, however, leaving open the question of whether the fold change fostered stronger dimerization or whether the stronger dimerization resulted apart from the change in fold.[ 44 ] The specific Cro variants shown in Figure 2A are Xfaso 1 and XPH2, respectively. We note that another Cro variant, XPH1,[ 42 ] has higher sequence similarity to XPH2, but it also has substantially less helical structure. Thus, we showed Xfaso 1 to highlight structural differences while maintaining a high level of sequence similarity.
TABLE 1.
Functions of sequence‐similar fold switchers
| Function 1 | Function 2 | |
|---|---|---|
| Cro α‐helix/Cro β‐sheet | Often form weaker dimers and have weaker operator‐binding affinities | Often form stronger dimers and have stronger operator‐binding affinities |
| RfaHNTD + CTD/RfaHCTD | Regulates NTD's transcriptional activities by masking its RNAP binding site and binds S10 ribosomal subunit, fostering efficient translation | Binds S10 ribosomal subunit, fostering efficient translation |
| KaiB1/KaiB4 | Binds KaiC CI domain, KaiA, and CikA | Inactive tetramer |
| GA98/GB98 | Binds Human Serum Albumin | Binds Immunoglobulin G |
The second family of sequence‐similar fold switchers are different forms of the bacterial transcriptional regulator RfaH[ 6 ] (Figure 2B). RfaH's N‐terminal domain is a conserved NGN‐like domain that binds RNA polymerase (RNAP), fostering transcriptional readthrough. Its C‐terminal domain (CTD) folds into an α‐helical bundle that occludes the RNAP‐binding site of its NGN‐like domain. RfaH also specifically binds the ops DNA consensus sequence. Remarkably, when RfaH binds both ops and RNAP (known as the Transcription Elongation Complex, or TEC), its CTD refolds into a β‐barrel that binds the S10 ribosomal subunit, fostering efficient protein translation.[ 12 ] Thus, full‐length RfaH is classified as a single‐sequence (extant) fold switcher. Here, however, we focus on two different RfaH sequences: its auto inhibited full‐length form and its C‐terminal domain expressed in isolation. Under physiological conditions and in the absence of the TEC, full‐length RfaH's CTD folds into a stable α‐helical bundle with an undetectable population of its alternative β‐barrel conformation.[ 12 ] In contrast, its CTD expressed in isolation folds into a β‐barrel able to bind the S10 ribosomal subunit[ 6 ] (Table 1), also with no observable evidence of fold switching. Thus, we focus here on RfaH's fold switching as induced by domain insertion and deletion by comparing RfaH's full‐length, uninduced form with its CTD expressed in isolation. To our knowledge, RfaH's CTD is the only known single‐sequence fold switcher that changes from one stable fold to another by including/excluding a neighboring domain. Although domain insertion/deletion is not the biologically relevant trigger for RfaH's fold switch (i.e., binding of TEC and ops), it is relevant to the question of how changes in sequence can drive changes in protein fold and function.
Thirdly, KaiB plays a critical role in circadian clock regulation of the cyanobacterium Synechococcus elongatus.[ 9 ] This circadian rhythm is generated by KaiA, KaiB, and KaiC in the presence of Mg2+ and ATP,[ 45 ] which produce a KaiC phosphorylation/dephosphorylation cycle with a period of approximately 24 hours. Multiple lines of evidence suggest that wild‐type KaiB populates an inactive tetrameric form in high abundance, while also populating a rare active form—a monomer with a different fold. A total of six mutations to wild‐type KaiB (shown as the inactive tetramer “KaiB4” in Figure 2C) stabilized KaiB in the rare monomeric form well enough for its structure to be determined by solution NMR (“KaiB1” in Figure 2C). The engineered protein KaiB1 binds to a KaiC subunit (CI) in two of its phosphorylated forms, inactivating KaiA and allowing KaiC to be dephosphorylated[ 46 ] (Table 1). Like RfaH, wild‐type KaiB switches folds in vivo, making it a single‐sequence fold switcher. Here, however, we compare the sequences of wild‐type (“KaiB4”) with that of the engineered, monomer‐stabilized KaiB1, which differ by 6 amino acids and have different ground states, making them a pair of sequence‐similar fold switchers.
The fourth and final family of sequence‐similar fold switchers, GA and GB, arose from a combination of directed evolution and rational protein design[ 47 ] (Figure 2D). Remarkably, this family ‐ engineered from two domains of Streptococcus (Lancefield group G) protein G ‐ contains variants that are 98% identical in sequence[ 41 , 48 ] (differing at a single position) but maintain their native human serum albumin (HSA) binding (GA98) and IgG binding (GB98) functions (Table 1).
We note that for the specific examples of sequence‐similar fold switchers shown in Figure 2, at least one member of each set did not evolve naturally, but rather was engineered to either increase sequence identity (Cro variants XPH2 and XPH1 [discussed later], GA98, and GB98) or stabilize an alternative conformation (RfaHCTD and KaiB1). We selected these examples to purposely highlight the high degree of sequence identity possible while retaining distinct, structurally characterized, folds. Nevertheless, three of these four protein families have naturally occurring members with different folds and lower (but still quite significant) sequence similarity. Specifically, P22 Cro and λ Cro have different folds but 40% aligned similarity.[ 43 ] Secondly, RfaH has a paralog, NusG, whose CTD assumes a β‐barrel conformation that is not known to switch folds.[ 49 ] In contrast to RfaH, NusG's CTD is involved in Rho‐mediated transcription termination, and it is not known to mask the RNAP binding site of its N‐terminal NGN domain as does RfaH's CTD. Finally, KaiB also has an ortholog in Legionella pneumophila that adopts the same fold as KaiB1; this ortholog forms a dimer. Unlike S. elongatus, L. pneumophila is not known to have a circadian clock, even though it has both KaiB and KaiC orthologs in its genome.[ 50 ] These genes are transcribed under the control of the stress factor RpoC, and they are not expressed in a circadian manner, again unlike their S. elongatus orthologs. Furthermore, L. pneumophila KaiB does not interact with KaiC. Instead, these KaiB and KaiC variants are probably involved in bacterial stress responses to oxidation and changes in environmental sodium concentration.[ 50 ] Sequence alignments of all three sets of homologs can be found in (Table S1).
3.2. JPred4 predicts fold switching in three out of four families of sequence‐similar fold switchers
We then turned to developing a computational approach that predicts whether or not two proteins are sequence‐similar fold switchers from their sequences. To do this, we first tested whether homology‐based secondary structure predictors could suggest different secondary structures for known pairs of sequence‐similar fold switchers. Previous work suggests that discrepancies between homology‐based secondary structure predictions and experimentally predicted protein structures can indicate protein fold switching.[ 5 , 21 ] Here, we sought to expand that work by comparing secondary structure predictions between two similar sequences with different folds, potentially obviating the need for a solved protein structure.
To do this, we ran three high‐performing, homology‐based secondary structure prediction algorithms—JPred4,[ 29 ] PSIPRED,[ 51 ] and SPIDER3[ 30 ]—on all solved structures of known pairs of sequence‐similar fold switchers. We then quantified α<‐>β prediction discrepancies between these pairs, because previous work suggested that these differences occur significantly more frequently for single‐sequence (extant) fold‐switching proteins than for single‐fold proteins.[ 21 ] Significant prediction differences are evident for the GA/GB family, Cro, and full‐length RfaH/RfaH CTD, but not for KaiB (Figure 3).
FIGURE 3.
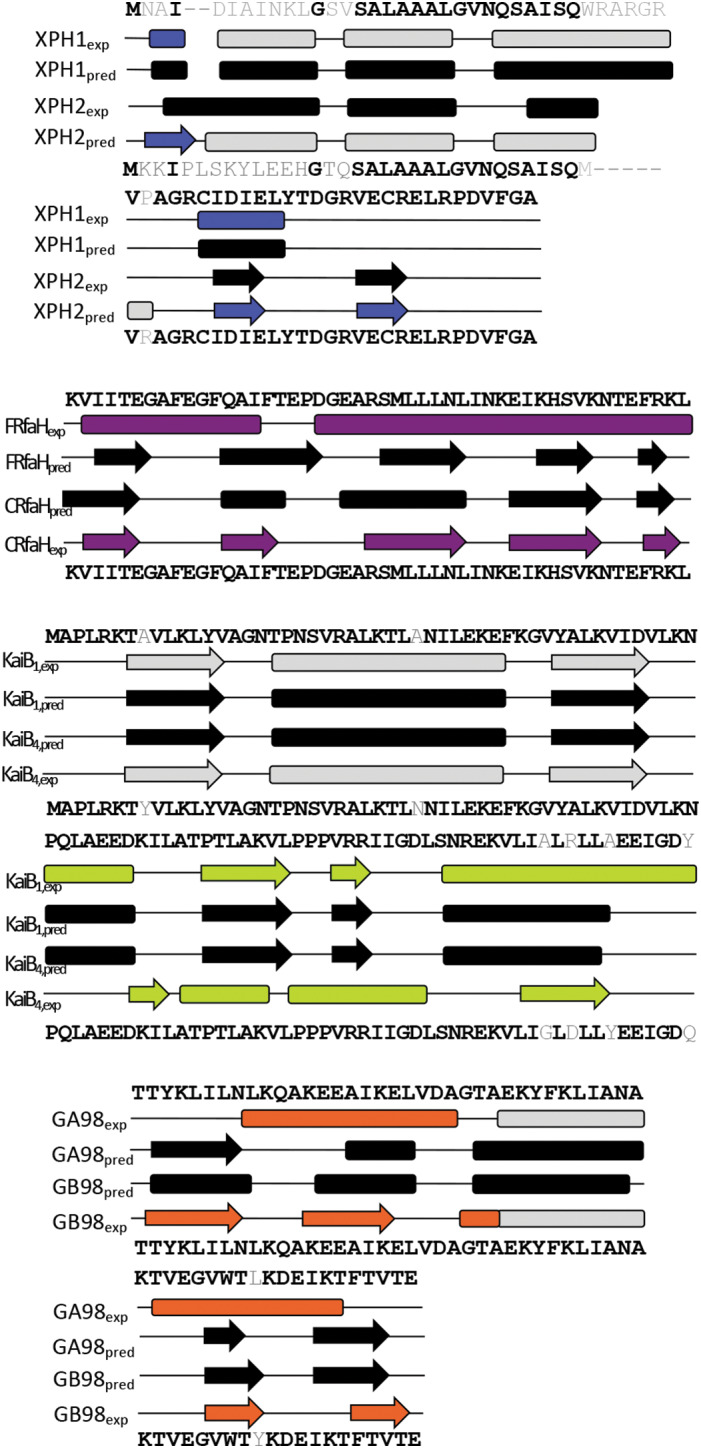
Discrepancies between JPred4 secondary structure predictions are apparent for sequence‐similar fold switchers, except KaiB. Predictions are shown in black; experimentally determined secondary structures are shown according to the color scheme of Figure 2. Identical residues are shown in black; non identical ones are shown in gray
3.3. α<‐>β prediction discrepancies from JPred4 are significantly higher for sequence‐similar fold switchers than for single‐fold protein families
Table 2 shows that JPred4 yields significantly higher α<‐>β discrepancies for sequence‐similar fold switchers than the previously published value of 6% for single‐fold proteins.[ 21 ] The other two predictors gave much lower frequencies of α<‐>β discrepancies for sequence‐similar fold switchers, mostly within error of 0%. To test whether JPred4 systematically yields higher levels of α<‐>β discrepancies for sequence‐similar fold switchers than would be expected by chance, we ran it on three sets of sequence‐similar single‐fold protein families (β‐barrel, α/β grasp, and α‐helical bundle topologies) with solved structures and ≥70% pairwise identity. Only one sequence (3FIL, GB family) yielded significant α<‐>β prediction discrepancies among the three families, totaling 207 nonredundant sequences (Table 2).
TABLE 2.
α<‐>β prediction discrepancies (mean ± SD) for fold switchers (top 3 rows) and single‐fold proteins (bottom 3 rows) a
| JPred4 (%) | PSIPRED (%) | SPIDER3 (%) | |
|---|---|---|---|
| RfaH b | 44 | 4 | 0 |
| Cro | 19 ± 2 | 0 ± 0 | 11 ± 8 |
| KaiB | 0 ± 0 | 0 ± 1 | 0 ± 0 |
| GA/GB | 17 ± 2 | 8 ± 8 | 3 ± 2 |
| SH3‐like c | 0 ± 0 | 4 ± 2 | 0 ± 0 |
| Myoglobin‐like c | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| α/β‐grasp c | 1 ± 2 | 4 ± 2 | 9 ± 6 |
Shaded entries exceed the reported error of 6% for homology‐based secondary structure predictors.
Error for RfaH could not be determined because only one measurement of α<‐>β prediction discrepancies could be taken.
Families of sequence‐similar fold switchers.
3.4. JPred4 robustly classifies sequence‐similar fold switchers
Because JPred4 appeared to discriminate between sequence‐similar fold switchers and sequence‐similar single‐fold proteins, we tested its robustness as a classifier. To do this, we compared the distributions of α/β prediction discrepancies for sequence‐similar fold switchers with the three sets of sequence‐similar single‐fold proteins described previously (Figure 4). We found that the distributions were very different, with P < 1.8*10−19 (Kolmogorov‐Smirnov test). The percentage of α<‐>β prediction discrepancies in 206/207 single‐fold proteins fell below a 5% threshold. In contrast, the proportion of α<‐>β prediction discrepancies exceeded the 5% threshold for all sequence‐similar fold switchers, except for those within the KaiB fold family. In light of this observation, we calculated the Matthews Correlation Coefficient[ 52 ] for fold switchers and single‐fold proteins at a 5% threshold and found it to be 0.82 (good true positive detection and excellent true negative detection). This result suggests that α<‐>β prediction discrepancies calculated from JPred4 can potentially predict novel sequence‐similar fold switchers from their sequences alone.
3.5. JPred4's robustness results from its sequence database and HMMer profiles
Our previous results demonstrate that JPred4—but neither PSIPRED nor SPIDER3—can robustly identify fold switchers from their sequences. Thus, we sought to determine the factors that contribute to JPred4's discriminatory power. We noted that, while all three methods base their predictions upon position‐specific scoring matrices (PSSMs) generated from PSI‐BLAST searches, JPred4 differs from both PSIPRED and SPIDER3 in two ways: (a) it runs PSI‐BLAST on a curated version of Uniprot90 that diminishes sequence redundancy,[ 53 ] and (b) it supplements the PSI‐BLAST‐generated PSSM with HMMer profiles[ 31 ] from specially generated multiple sequence alignments.[ 53 ] Therefore, we hypothesized that these distinguishing factors might contribute to JPred4's ability to discriminate between sequence‐similar fold switchers and single‐fold proteins ‐ in sharp contrast to the two other algorithms.
First, we sought to determine whether JPred4's curated database improved its ability to identify sequence‐similar fold switchers. To do this, we ran PSIPRED and SPIDER3 using the PSSMs generated by running three rounds of PSI‐BLAST on the JPred4 database (Methods). Upon using this database, PSIPRED produced significant α<‐>β discrepancies in all fold families except for GA/GB, and SPIDER3 produced significant α<‐>β discrepancies for the Cro fold family (Table 3). Thus, changing the sequence database had a significant effect on PSIPRED's ability to predict fold switching and a more modest effect on SPIDER3.
TABLE 3.
Percent α<‐>β discrepancies of PSIPRED and SPIDER3 using the JPred4 sequence database
| PSIPRED with JPred4 DB (%) | SPIDER3 with JPred4 DB (%) | |
|---|---|---|
| Cro | 10 ± 4 | 17 ± 1 |
| RfaH a | 21 | 0 |
| KaiB | 1 ± 1 | 0 ± 0 |
| GA/GB | 1 ± 1 | 7 ± 7 |
| SH3‐like b | 0 ± 0 | 0 ± 0 |
| Myoglobin‐like b | 0 ± 0 | 0 ± 0 |
| α/β‐grasp b | 4 ± 2 | 6 ± 5 |
Error for RfaH could not be determined because only one measurement of α<‐>β prediction discrepancies could be taken.
Shaded entries exceed the reported error of 6% for homology‐based secondary structure predictors.
Since neither PSIPRED nor SPIDER3 could predict fold switching of the GA/GB family‐even with JPred4's curated sequence database‐we hypothesized that HMMer profiles might have contributed to JPred4's ability to discriminate between the highly identical sequences in this family. JPred4's overall prediction (jnetpred) is derived from two separate components: a prediction based on a HMMer profile (JNETHMM), and a separate prediction based on a PSI‐BLAST‐generated PSSM (JNETPSSM). Upon examining JPred4's jnetpred predictions for all members of the GA/GB family, we found that the region of the protein with differing secondary structure predictions (helix/strand 1) was positively correlated with the JNETHMM prediction (Pearson Correlation Coefficient: 0.63) and negatively correlated with the JNETPSSM prediction (Pearson Correlation Coefficient: −0.68). Thus, we conclude that HMMer profiles contribute to JPred4's ability to discriminate between the GA/GB folds. Taken together, our results demonstrate that both JPred4's sequence database and its use of HMMer profiles contribute to its effectiveness at detecting secondary structure differences between sequence‐similar fold switchers.
3.6. Sensitive sequence crowdsourcing contributes to JPred4's ability to predict sequence‐similar fold switchers
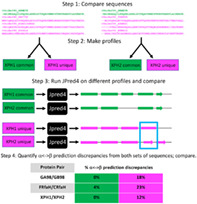
Although our previous results pinpoint features of JPred4 that allow it to predict different secondary structures for sequence‐similar fold switchers, they do not explain the precise ways in which the features contribute. We hypothesized that JPred4's PSI‐BLAST searches yield unique hits, even for highly identical query sequences—with these unique hits causing it to generate different secondary structure predictions. To test this hypothesis, we developed the following strategy (Figure 5):
Compare the multiple sequence alignments (MSAs) that JPred4 generates for a given pair of sequence‐similar fold switchers.
Separate sequences that are common among both alignments and sequences that are unique to a specific alignment; make profiles from these different sets of sequences.
Run JPred4 on profiles generated from the common sequences and unique sequences.
Quantify α<‐>β prediction discrepancies from both sets of sequences and compare.
FIGURE 5.
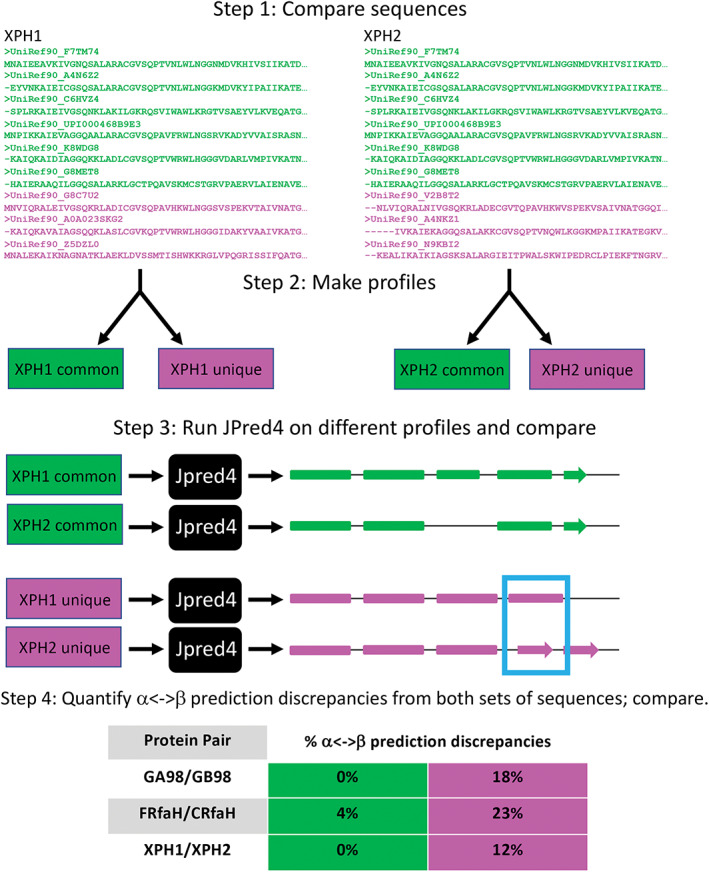
A strategy for determining whence different JPred4 secondary structure predictions arise. Step 1 shows a subset of sequences from the multiple sequence alignments (MSAs) that JPred4 generates from PSI‐BLAST searches. Green sequences were in MSAs from both XPH1 and XPH2; magenta sequences were unique to either XPH1 or XPH2. Step 2 depicts the common and unique sequences being converted to profiles. We generated these profiles from HMMer[ 32 ] and PSI‐BLAST[ 27 ] using the same methodology as JPred4 (Methods). In step 3, we input profiles into JPred4 and compare their secondary structure predictions; α‐helices are rounded rectangles, and β‐strands are arrows. A region of α‐helix <‐> β‐strand discrepancy is highlighted in a blue box. Percent α‐helix <‐> β‐strand discrepancies are quantified and compared in Step 4. JPred4 predicts substantially higher levels of α‐helix <‐> β‐strand discrepancies from unique sequence profiles than from common ones
To assess the robustness of this strategy, we tested it on the most similar pairs of sequence‐similar fold switchers from all 3‐fold families for which JPred4 predicted significant α<‐>β discrepancies. In all three cases, JPred4 predicted substantially larger α<‐>β discrepancies from the unique sequence profiles than from the common profiles (Figure 5). Given that all protein pairs with α<‐>β prediction discrepancies from JPred4 had at least one unique sequence in at least one alignment (Table S2), these results strongly suggest that JPred4 associates highly similar query sequences with unique hits using highly sensitive PSI‐BLAST searches. The profiles generated from these searches cause it to output different secondary structure predictions. We note that different sequence hits do not guarantee a different secondary structure prediction, however. For instance, all members of the KaiB family (Table S2) yield exactly the same secondary structure predictions in spite of hitting different sequences in PSI‐BLAST. Further study will be needed to assess why JPred4 does not yield different predictions for sequence‐similar KaiB variants.
4. DISCUSSION
Although most proteins with high levels of sequence identity tend to adopt the same folds and perform the same functions, a number of exceptions have emerged,[ 3 , 5 , 16 ] including sequence‐similar fold switchers, defined here to have sequences with ≥70% aligned similarity but regions with different secondary structure (α‐helices in one conformation replaced by β‐strands in the other). Accurately predicting sequence‐similar fold switchers from their genomic sequences would be useful because there are biologically relevant examples of proteins with highly similar sequences that assume different secondary structures associated with disease[ 54 ] or different cellular behaviors.[ 55 ]
This paper reports only a handful of sequence‐similar fold switchers: the only ones that we could identify with large shifts from α‐helix<‐>β‐sheet. Nevertheless, we developed a promising sequence‐based predictor that predicts fold switching in three of the four families of sequence‐similar fold switchers analyzed here. To our knowledge, this is the first sequence‐based predictor of sequence‐similar fold switchers to be reported in the literature. Furthermore, although several previous studies have made successful predictions of the conformations of the GA/GB family of sequence‐similar fold switchers,[ 20 , 56 ] to our knowledge, this is the first study that successfully predicts fold changes in three diverse fold families using the same sequence‐based approach.
Further work remains to increase the accuracy of our method since it did not identify fold switching in any members of the KaiB family. These false negatives demonstrate that our method will not identify all existing sequence‐similar fold switchers. Nevertheless, the robustness of our statistics suggests that similar sequences are likely to assume different folds if their JPred4‐derived secondary structure predictions have significant α‐helix <‐> β‐sheet discrepancies (especially >20%, since there were no false positives at or above that threshold, Figure 4). Thus, we believe that this method is sufficient to foster the discovery of more sequence‐similar fold switchers.
We anticipate that the additional information gained by identifying more fold switchers—through this method and others—will inform more robust and generalizable predictions in the future. Currently our method, though fairly robust, has three clear limitations. Firstly, it requires a collection of diverse homologs for a given protein to yield good predictions. Thus, it would likely not yield robust predictions for orphan proteins, for instance. Secondly, it can only identify fold switchers whose conformational differences involve substantial changes in secondary structure from α‐helix <‐> β‐sheet. Other types of fold switching—such as shifts in β‐sheet register observed in Lymphotactin[ 10 ] or the extension of secondary structures observed in β‐pores[ 57 ]—will not be detected. Thirdly, this method uses a homology‐based algorithm as the basis of its predictions. While JPred4 is a state‐of‐the‐art secondary structure predictor, it can only be as good as current knowledge of protein structure. Indeed, our method uses the inconsistencies in Jpred4 predictions to identify fold switchers. Furthermore, JPred4, like all other homology‐based secondary structure predictors, runs on the assumption that proteins can adopt only one stable secondary structure configuration, though this is not the case for single‐sequence fold switchers. The way forward is to identify and experimentally characterize more sequence‐similar and single‐sequence fold switchers. Then more informed, physically based methods can be developed.
5. CONCLUSIONS
Our results show that the conformational variability of sequence‐similar fold switchers can be predicted from their sequences with considerable accuracy using the rapid homology‐based secondary structure predictor JPred4. This method can potentially identify new sequence‐similar fold switchers from hundreds of millions of genomic sequences. Furthermore, we are optimistic that this method can serve as a steppingstone to develop an accurate, high‐throughput algorithm for predicting more single‐sequence fold‐switching proteins from their sequences.
CONFLICT OF INTEREST
The authors declare no competing interests
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGEMENTS
We thank George Rose, Marius Clore, and Brian Matthews for helpful discussion. This work utilized the computational resources of the NIH HPS Biowulf cluster (http://hpc.nih.gov). This work was supported in part by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Kim AK, Looger LL, Porter LL. A high‐throughput predictive method for sequence‐similar fold switchers. Biopolymers. 2021;112:e23416. 10.1002/bip.23416
Funding information Howard Hughes Medical Institute; National Institutes of Health
DATA AVAILABILITY STATEMENT
The data and code that support the findings of this study are openly available on GitHub at https://github.com/ncbi/Sequence-similar-fold-switchers.
REFERENCES
- 1. Jeffery C. J., Trends Biochem. Sci. 1999, 24, 8. [DOI] [PubMed] [Google Scholar]
- 2. Murzin A. G., Science 2008, 320, 1725. [DOI] [PubMed] [Google Scholar]
- 3. Wright P. E., Dyson H. J., Nat. Rev. Mol. Cell Biol. 2015, 16, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim and Porter , Functional and Regulatory Roles of Fold‐Switching Proteins, Structure 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Porter L. L., Looger L. L., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berman H. M., Battistuz T., Bhat T. N., Bluhm W. F., Bourne P. E., Burkhardt K., Feng Z., Gilliland G. L., Iype L., Jain S., Fagan P., Marvin J., Padilla D., Ravichandran V., Schneider B., Thanki N., Weissig H., Westbrook J. D., Zardecki C., Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899. [DOI] [PubMed] [Google Scholar]
- 7. The PyMOL Molecular Graphics System , Version 2.0 Schrödinger, LLC.
- 8. Burmann B. M., Knauer S. H., Sevostyanova A., Schweimer K., Mooney R. A., Landick R., Artsimovitch I., Rosch P., Cell 2012, 150, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang Y. G., Cohen S. E., Phong C., Myers W. K., Kim Y. I., Tseng R., Lin J., Zhang L., Boyd J. S., Lee Y., Kang S., Lee D., Li S., Britt R. D., Rust M. J., Golden S. S., LiWang A., Science 2015, 349, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuinstra R. L., Peterson F. C., Kutlesa S., Elgin E. S., Kron M. A., Volkman B. F., Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai M., Huang Y., Shen Y., Li M., Mizuuchi M., Ghirlando R., Mizuuchi K., Clore G. M., Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 25446–25455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zuber P. K., Schweimer K., Rosch P., Artsimovitch I., Knauer S. H., Nat. Commun. 2019, 10, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milton R. H., Abeti R., Averaimo S., DeBiasi S., Vitellaro L., Jiang L., Curmi P. M., Breit S. N., Duchen M. R., Mazzanti M., J. Neurosci. 2008, 28, 11488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lei Y., Takahama Y., Microbes. Infect. 2012, 14, 262. [DOI] [PubMed] [Google Scholar]
- 15. Xue B., Dunker A. K., Uversky V. N., J. Biomol. Struct. Dyn. 2012, 30, 137. [DOI] [PubMed] [Google Scholar]
- 16. Jaffe E. K., Trends Biochem. Sci. 2005, 30, 490. [DOI] [PubMed] [Google Scholar]
- 17. Ambroggio X. I., Kuhlman B., J. Am. Chem. Soc. 2006, 128, 1154. [DOI] [PubMed] [Google Scholar]
- 18. Wei K. Y., Moschidi D., Bick M. J., Nerli S., McShan A. C., Carter L. P., Huang P. S., Fletcher D. A., Sgourakis N. G., Boyken S. E., Baker D., Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sfriso P., Duran‐Frigola M., Mosca R., Emperador A., Aloy P., Orozco M., Structure 2016, 24, 116. [DOI] [PubMed] [Google Scholar]
- 20. Tian P., Best R. B., PLoS Comput. Biol. 2020, 16, e1008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mishra S., Looger L. L., Porter L. L., Protein Sci. 2019, 28, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newlove T., Konieczka J. H., Cordes M. H., Structure 2004, 12, 569. [DOI] [PubMed] [Google Scholar]
- 23. Baase W. A., Liu L., Tronrud D. E., Matthews B. W., Protein Sci. 2010, 19, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Safadi S. S., Shaw G. S., Biochemistry 2007, 46, 14162–9. [DOI] [PubMed] [Google Scholar]
- 25. Rost B., Protein Eng. 1999, 12, 85. [DOI] [PubMed] [Google Scholar]
- 26. O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith‐White B., Ako‐Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., Chetvernin V., Choi J., Cox E., Ermolaeva O., Farrell C. M., Goldfarb T., Gupta T., Haft D., Hatcher E., Hlavina W., Joardar V. S., Kodali V. K., Li W., Maglott D., Masterson P., McGarvey K. M., Murphy M. R., O'Neill K., Pujar S., Rangwala S. H., Rausch D., Riddick L. D., Schoch C., Shkeda A., Storz S. S., Sun H., Thibaud‐Nissen F., Tolstoy I., Tully R. E., Vatsan A. R., Wallin C., Webb D., Wu W., Landrum M. J., Kimchi A., Tatusova T., DiCuccio M., Kitts P., Murphy T. D., Pruitt K. D., Nucleic Acids Res. 2016, 44, D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped B. L. A. S. T., Nucleic Acids Res. 1997, 25, 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buchan D. W. A., Jones D. T., Nucleic Acids Res. 2019, 47, W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Drozdetskiy A., Cole C., Procter J., Barton G. J., Nucleic Acids Res. 2015, 43, W389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heffernan R., Paliwal K., Lyons J., Singh J., Yang Y., Zhou Y., J. Comput. Chem. 2018, 39, 2210. [DOI] [PubMed] [Google Scholar]
- 31. Eddy S. R., Bioinformatics 1998, 14, 755. [DOI] [PubMed] [Google Scholar]
- 32. Potter S. C., Luciani A., Eddy S. R., Park Y., Lopez R., Finn R. D., Nucleic Acids Res. 2018, 46, W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoon B. J., Curr. Genomics 2009, 10, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang G., Dunbrack R. L. Jr., Bioinformatics 2003, 19, 1589. [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W., Sander C., Biopolymers 1983, 22, 2577. [DOI] [PubMed] [Google Scholar]
- 36. Pearson W. R., Methods Enzymol. 1990, 183, 63. [DOI] [PubMed] [Google Scholar]
- 37. Gribskov M., McLachlan A. D., Eisenberg D., Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Remmert M., Biegert A., Hauser A., Soding J., Nat. Methods 2011, 9, 173. [DOI] [PubMed] [Google Scholar]
- 39. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J., Thompson J. D., Higgins D. G., Mol. Syst. Biol. 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hunter J. D., Comput. Sci. Eng. 2007, 9, 90. [Google Scholar]
- 41. Alexander P. A., He Y., Chen Y., Orban J., Bryan P. N., Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumirov V. K., Dykstra E. M., Hall B. M., Anderson W. J., Szyszka T. N., Cordes M. H. J., Protein Sci. 2018, 27, 1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roessler C. G., Hall B. M., Anderson W. J., Ingram W. M., Roberts S. A., Montfort W. R., Cordes M. H., Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. LeFevre K. R., Cordes M. H., Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Partch C. L., J. Mol. Biol. 2020, 432, 3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tseng R., Goularte N. F., Chavan A., Luu J., Cohen S. E., Chang Y. G., Heisler J., Li S., Michael A. K., Tripathi S., Golden S. S., LiWang A., Partch C. L., Science 2017, 355, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alexander P. A., He Y., Chen Y., Orban J., Bryan P. N., Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 11963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He Y., Chen Y., Alexander P. A., Bryan P. N., Orban J., Structure 2012, 20, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang J. Y., Mooney R. A., Nedialkov Y., Saba J., Mishanina T. V., Artsimovitch I., Landick R., Darst S. A., Cell 2018, 173, 1650–1662.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loza‐Correa M., Sahr T., Rolando M., Daniels C., Petit P., Skarina T., Gomez Valero L., Dervins‐Ravault D., Honore N., Savchenko A., Buchrieser C., Environ. Microbiol. 2014, 16, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones D. T., J. Mol. Biol. 1999, 292, 195. [DOI] [PubMed] [Google Scholar]
- 52. Matthews B. W., Biochim. Biophys. Acta 1975, 405, 442. [DOI] [PubMed] [Google Scholar]
- 53. Cuff J. A., Barton G. J., Proteins 1999, 34, 508. [DOI] [PubMed] [Google Scholar]
- 54. Lei X., Kou Y., Fu Y., Rajashekar N., Shi H., Wu F., Xu J., Luo Y., Chen L., J. Mol. Biol. 2018, 430, 1157. [DOI] [PubMed] [Google Scholar]
- 55. Muller S. I., Friedl A., Aschenbrenner I., Esser‐von Bieren J., Zacharias M., Devergne O., Feige M. J., Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sikosek T., Krobath H., Chan H. S., PLoS Comput. Biol. 2016, 12, e1004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Podobnik M., Savory P., Rojko N., Kisovec M., Wood N., Hambley R., Pugh J., Wallace E. J., McNeill L., Bruce M., Liko I., Allison T. M., Mehmood S., Yilmaz N., Kobayashi T., Gilbert R. J., Robinson C. V., Jayasinghe L., Anderluh G., Nat. Commun. 2016, 7, 11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The data and code that support the findings of this study are openly available on GitHub at https://github.com/ncbi/Sequence-similar-fold-switchers.