

Abstract
The vertebrate piggyBac derived transposase 5 (PGBD5) encodes a domesticated transposase, which is active and able to transpose its distantly related piggyBac-like element (pble), Ifp2. This raised the question whether PGBD5 would be more effective at mobilizing a phylogenetically closely related pble element. We aimed to identify the pble most closely related to the pgbd5 gene. We updated the landscape of vertebrate pgbd genes to develop efficient filters and identify the most closely related pble to each of these genes. We found that Tcr-pble is phylogenetically the closest pble to the pgbd5 gene. Furthermore, we evaluated the capacity of two murine and human PGBD5 isoforms, Mm523 and Hs524, to transpose both Tcr-pble and Ifp2 elements. We found that both pbles could be transposed by Mm523 with similar efficiency. However, integrations of both pbles occurred through both proper transposition and improper PGBD5-dependent recombination. This suggested that the ability of PGBD5 to bind both pbles may not be based on the primary sequence of element ends, but may involve recognition of inner DNA motifs, possibly related to palindromic repeats. In agreement with this hypothesis, we identified internal palindromic repeats near the end of 24 pble sequences, which display distinct sequences.
Keywords: domestication, transposition, DNA binding, molecular evolution, transposable element
Graphical Abstract
Introduction
Transposable elements (TEs) are mobile genetic elements present in almost all organisms. Three main types of TEs are found in eukaryote genomes: 1) LTR-retrotransposons and 2) nonLTR-retrotransposons that encode a reverse-transcriptase to replicate an RNA copy into a DNA intermediate which thereafter integrates at a new locus; and 3) transposons with terminal inverted repeats (TIR transposon) at their extremities that code for and use a transposase to move from one locus to another using a DNA molecule as an intermediate [1,2].
Ifp2 is an autonomous TIR transposon, 2,475 bp long, commonly called piggyBac [3]. It is the reference element of the piggyBac family and originated from the cabbage looper (Trichoplusia ni) genome. Ifp2 and the other members of its family, the piggyBac-like elements (pbles), insert into a tetranucleotide TTAA that is duplicated upon insertion [4,5]. They are flanked at their extremities by TIRs approximately 13 bp long. Depending on the pble “species” they can also display sub-terminal inverted repeats (STIR) which are 19 bp in Ifp2 and located internally at 3 and 31 nucleotides distance of 5' and 3' TIR inner ends, respectively [6,7]. An open reading frame (ORF) in the inner region of the element , about 1.8kb long, encodes a pble transposase approximately 600 amino acids long (594 aa in Ifp2 transposase, a.k.a. PB).
TEs can have deleterious effects on the genome of their host through mutagenic consequences of their mobility. TEs can also have a positive effect on their host at the evolutionary scale. They have contributed to the adaptation of gene expression by being a source of novelties to rewire networks of cis-regulating elements. ORFs contained in TEs may also lead to novel genes and functions in the host [8,9], through a process known as molecular domestication or exaptation. While the nucleic acid sequence of the original copies of TEs diverge and degenerate by accumulating mutations over time, the domesticated ORFs derived from TEs are preserved due to selection on their function. Such domesticated ORFs are usually present as single gene copies and conserved at orthologous loci in all related species in which the gene has been domesticated in the common ancestor of the clade.
In vertebrates there are several cases of domesticated genes originating from TIR transposon ORFs [8] and some still encode a transposase that retains its recombinase activity. The RAG1/RAG2 DNA recombinase derived from Transib/protoRAG transposases constitutes a prototypical example [10,11]. RAG1/RAG2 are both required as key actors in vertebrate V(D)J recombination [11-13]. A recent inventory of piggyBac sequences in eukaryotes identified 6 well-characterized pgbd genes in vertebrate genomes [6]: pgbd1-5 genes and the Kobuta gene (hereafter called pgbd6). Pgbd5 appears to be the most ancient case of piggyback ORF domestication and is present in all chordates including cephalochordates and vertebrates, while being absent in tunicates [14,15]. This supports the hypothesis that pgbd5 was the result of a domestication event in the common chordate ancestor of cephalochordates and vertebrates, but was likely lost in the common ancestor of tunicates [16,17]. The activities and roles of pgbd genes in vertebrates remain widely unexplored. For pgbd5, two PGBD5 isoforms of ~523 and 409 amino acids are expressed in vertebrate species (Mm523 and Mm409 in Mus musculus). At least four isoforms are predicted in human cells (Hs554, Hs524, Hs455, Hs435). Hs524 was found to function as a mutator in various human cancers, but its role in healthy tissues remains enigmatic [18]. Interestingly, human Hs524 and Hs455 were found to be transposase isoforms that display a very unusual property: they transpose Ifp2, a pble that is one of the most distantly related element to the vertebrate pgbd5 gene [6,19]. This raises the question of whether human PGBD5 may be more efficient at transposing a pble more closely related to itself than to insect Ifp2.
Here, we investigated this possibility. We first identified the closest pble relative of the pgbd5 gene. We then compared its integration features with those of Ifp2 using PGBD5 as a transposase source. Because no pble close to pgbd5 gene has been deposited so far into public databases [6] and due to the presence of ambiguities among pgbd genes, the closest pble relative of pgbd5 gene was identified in four steps (supplementary data 1). Indeed, identifying the closest pble relatives of PGBD5 was challenging because this gene was the most ancient case of pgbd gene domestication in chordate genomes (~600 million years). In-depth knowledge of features of other pgbd genes and their pble relatives were therefore required to accurately filter sequences. We updated the landscape of pgbd genes in vertebrates using comparative analysis and verification of their domestication signatures. We found 9 pgbd genes, 3 of them being new with reproducible features. Using public databases we identified one to three of the closest pble relatives for each of the 9 pgbd genes. We found that 7 of them originated from distinct pbles, except for pgbd1 and 2 genes. We identified the first pble belonging to the same clade as the pgbd5 gene, Tcr-pble. Finally, we tested whether PGBD5 could transpose or integrate Tcr-pble more efficiently than Ifp2.
Results
Inventory of pgbd genes
Two criteria are used as signatures of domestication: i) the dN/dS ratio of each gene and ii) the interspecific syntenic conservation of their flanking chromosomal regions. The dN/dS ratio indicates whether a gene evolved under neutral selection (ratio close to 1), purifying selection (ratio closer to 0 than 1) or under positive selection (ratio above 1) [20]. For a domesticated gene with a physiological function, some form of selection is expected, most likely purifying selection. Interspecific syntenic conservation of the flanking chromosomal regions of a gene is used to indicate whether its genomic environment was conserved. For a domesticated gene, synteny supported chromosomal conservation between species belonging to the same clade and in which it was originally domesticated.
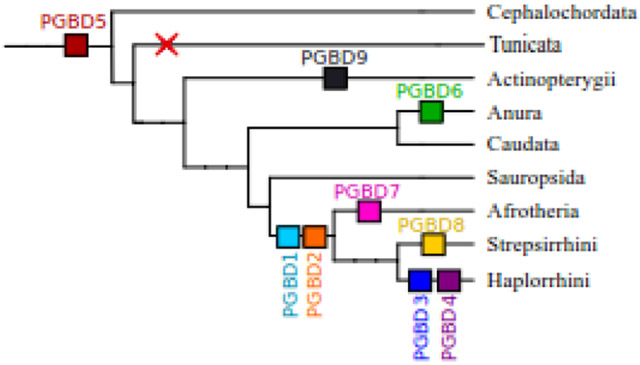
Six pgbd genes were inventoried in chordates. Their gene organization places them in two distinct classes: i) genes involving the apparent fusion of an ORF coding a host protein (Zn-SCAN in pgbd1 gene, an uncharacterized domain in pgbd2 gene, CSB in pgbd3 gene; Fig. 1a) with an ORF coding a transposase-derived protein, and ii) genes derived only from one ORF coding for a transposase derived protein (pgbd4, 5 and 6 genes ; Fig. 1a). These 6 pgbd genes had different numbers of introns. All have been domesticated [6]. The dN/dS ratios for pgbd1 through 6 genes were very low (Fig. 1a), indicating that these genes were under purifying selection. Graphic representations of syntenic environments of pgbd1 to 5 genes were found at http://www.genomicus.biologie.ens.fr. Although three amphibian genomes were sequenced, orthologous regions containing pgbd6 gene in Xenopus tropicalis were not available in the Xenopus laevis and Nanorana parkeri genome models, which prevented us from assessing the syntenic conservation of this gene.
Fig. 1. Characteristics of PGBDs in vertebrates.

(a) Domain organization in PGBD proteins. For each domesticated sequence, colored boxes represent domains. Yellow is for Zn-SCAN domain, gray is for an unknown domain, red is for CSB domain, light blue is for SCAN domain and green is for zF-FCS. The orange circle is for CRD or CRD-like domains. Blue is for the catalytic triad with the composition of the residues. The catalytic triad is in pink for PGBD5 because it is not aligned with other domesticated sequences. (b) Distribution of PGBDs in vertebrates. Each coloured square represents a PGBD. PGBD1 and PGBD2 are found in mammals, PGBD3 and PGBD4 are found in haplorrhinis, PGBD5 is present in all vertebrates and cephalochordates but not in tunicates. PGBD6 is specific to anura, PGBD7 is found in Afrotheria, PGBD8 is specific to Strepsirrhini and PGBD9 is found in Actinopterygii.
Five other potential pgbd gene candidates were reported in vertebrates [6]. Some of them were inappropriately identified in databases as genes coding for PGBD2 or PGBD4-like proteins; their domestication signatures have not, so far, been fully verified. Here we have renamed these 5 genes as follows: pgbd7 gene is specific to Afrotherians, pgbd8 gene is specific to Stepsirrhini, and pgbd9, 10 and 11 genes are specific to the Actinopterygii species (called G3 or NeoPGBD, G2 and G1 in [6], respectively; all references are supplied in supplementary Table 1). We studied the domestication signatures of these five candidate genes. One criteria that is sometimes used to identify a domesticated TE is the loss of TIRs. However, presence of TIRs in the region containing the pgbd gene is not sufficient to exclude domestication. Indeed, the pgbd3 andpgbd4 genes were both flanked by sequences corresponding to 5’ and 3’ regions of MER85 and MER75 pble, respectively. This suggests that each shares a common origin with one of these two 2 pbles (supplementary data 2).
dN/dS ratios of PGBDs.
Results showing domain conservation as well as dN/dS ratios are summarized in Fig. 1a. The dN/dS ratio of the pgbd7 gene was calculated using 4 afrotherian protein sequences (for references see supplementary Table 1) yielding a value of 0.209. The dN/dS ratio of the pgbd8 gene was 0.117 and was calculated from 6 protein sequences (for references see supplementary Table 1), while the values of actinopterygian-specific pgbd 9, 10 and 11 genes were 0.079, 0.146, 0.171 respectively. Two genes had a very low dN/dS ratio, 0.052 for the pgbd5 gene and 0.079 for the pgbd9 gene. These genes were the two oldest cases of piggyBac domestication in our analysis. In summary, our results indicated that the 11 pgbd genes had a dN/dS ratio closer to 0 than to 1 indicating purifying selection.
Syntenic conservation of pgbd7 through 11 gene candidates.
Public databases were sufficient to analyze pgbd7, 8 and 9 genes. Despite a lack of proper annotation and limited number and sizes of contigs, we found that pgbd7 gene was located between two genes that code for SCAND3 and TRIM27 in 4 related species (Fig. 2a). For two species (T. m. latirosis and C. asiatica) three more genes (coding for ZSCAN23, GPX6, and GPX5) were found upstream of the pgbd7 gene and two other genes (coding for ZNF311 and HLA) were found downstream. pgbd8 contigs displayed very different sizes as well as many gaps filled with Ns. In two species (M. murinus and P.coquereli), we succeeded in identifying four genes upstream and downstream of the pgbd8 gene (Fig. 2b). In the other species for which shorter contigs were available, we found that the pgbd8 gene was located between the same genes. For the pgbd9 gene four genes were identified in upstream and downstream regions (Fig. 2c). Some of these were duplicated or even triplicated. Pgbd9 seemed to be the only gene among the three potential actinopterygians pgbd genes to display a well-conserved genomic environment. Our analyses of public databases for the pgbd10 and pgbd11 gene candidates were not consistent enough to achieve similar analyses. It should also be noted that the complex situation found in fish genomes, particularly in teleosts, might be due to the presence of giant pbles such as “Teratorn” [21,22] (a fusion of a herpesvirus and a pble). Indeed, these giant pbles contain numerous host and viral genes that are comprised of mobile repeated copies found in the host genome. These results indicated that the pgbd7, 8 and 9 genes all originated from separate domesticated pble ORFs.
Fig. 2. Syntenic conservation around pgbd genes 7, 8 and 9.

Schematic representation of syntenic conservation in the neighbouring genes of pgbd7 (a), pgbd8 (b), and pgbd9 (c) across genomes. The names are those of the proteins encoded by the genes. Orthologous genes are colour-coded similarly. Some genomic regions have been drawn in reversed order for visual purposes – designated “reverse”.
Conclusions.
To date nine vertebrate pgbd genes were shown to display domestication signatures (Fig. 1b). We have provided evidence supporting the existence of three new pgbd genes. Pgbd7 gene is specific to afrotherian species and encodes a protein characterized by the presence of a SCAN domain at its N-terminal and a CRD at its C-terminal (Fig. 1a). The pgbd8 gene is specific to strepsirrhinian species and encodes a PGBD that displays a zF-FCS domain (MYM-type Zinc finger with FCS sequence motif) at its N-terminal and a partial CRD at its C-terminal (noted as CRD-like in Fig. 1a). The pgbd8 gene has been annotated as a MYM2 protein in only one species, Microcebus murinus. While the pgbd8 gene was specific to Strepsirrhini species, our BLAST analyses showed that genes coding for PGBD3 and PGBD4 were not present in all primate species but only in a specific sub-order, the Haplorrhini. No trace of these two genes was found in the current Strepsirrhini data, although the percentage of protein identity between PGBD8 and PGBD4 was about 30% (similarity = 49%). The pgbd9 gene was specific to actinopterygian species.
Identifying the closest pble transposase relative(s) to PGBD5
We focused on pble transposases close to each PGBD. We analysed databases with reciprocal BLAST searches using as a query each PGBD sequence. We then verified the relationship of each pble candidate by plotting phylogenetic trees. Fig. 3 shows a phylogenetic tree summarizing our final results. The protein sequence alignment used to calculate this tree and their references are provided in supplementary data 3 and supplementary Table 1.
Fig. 3. Phylogenetic tree of PBLE and PGBD transposases.

Named in bold refers to domesticated piggyBac transposases (PGBD), names in italics to pbles. The colour of each pble refers to the classification and the grey blocks refer to the PGBDs. Bootstraps above 60 are indicated in red.
We did not find pbles closely related to pgbd1 and pgbd2 genes but we found one element, Ota-pble, located at the root of both genes in the phylogenetic tree (Fig. 3). This suggested that the pgbd1 and pgbd2 genes resulted from a single domestication event or that a single (or two highly related) pble was at the origin of both genes via two closely timed domestication events. For the pgbd3 gene, our analysis confirmed that Smi7-pble [6], Hvu-pble and Lfu-pble were the closest relatives of the pgbd3 genes. For pgbd4 gene, two pbles, Oab1-pble and Pva-pble were identified as the closest relatives. For the pgbd5 gene, we identified a new pble, Tcr-pble, from the genome of the walking stick, Timema cristinae (a phasmid species). Phylogenetic investigations showed that Tcr-pble was the only relative close to the pgbd5 gene found in public databases and its lineage shares the same root as these genes (Fig. 3). Because the pgbd5 gene is the most ancient case of pble domestication, the distance between Tcr-pble and PGBD5 is greater than that observed between other pble/PGBD couples. For PGBD6, two pbles were already identified, Uribo1 and Uribo2 [23]. For PGBD7, two pbles were identified, Smi2-pble and Nvi-pble. For PGBD8, three pbles were identified, Api1-pble, Atr1-pble and Api3-pble. Finally, we identified a new pble from a hemichordate, Ptychodera flava (Pfl-pble), which was the closest relative to PGBD9 together with Cgi-pble and Bgl-pble.
In all, we identified pbles sharing a close common ancestor with each of the 9 pgbd genes, Tcr-pble being currently the pble encoding a transposase that is closest relative of PGBD5 available in databases.
Mobilisation of Ifp2 and Tcr-pble for transposition mediated by PGBD5
Ectopic expression of PGBD5 isoforms can be cytotoxic depending on the cell line and/or their expression levels [24,25]. In order to manage this issue, two integration assays were used to investigate whether PGBD5 would integrate Tcr-pble more efficiently than Ifp2, both in terms of integration rates and the precision of integration events at their insertion sites. The first assay was done in HeLa cells transiently transfected with two plasmids: a source of transposon (pBSK-Ifp2-TIR5'-NeoR-TIR3', pBSK-Tcr-pble-TIR5'-NeoR-TIR3' (supplementary data 4a and b), or pBSK-NeoR as a control), and a source of PGBD5, the murine isoform Mm523 (94% identical to its orthologous human isoform, Hs524). In this assay, the expression of Mm523 was driven by a strong CMV promoter, which can be cytotoxic for transfected HeLa cells [25]. However, NeoR clones could be still obtained at a rate lower than that of the control done with the GFP as a source of protein. The second assay was done by transiently transfecting one plasmid source of transposon in a human rhabdoid tumor G401 cell line which expresses Hs524 endogenously [24]. Two clonal G401 cell lines were used. The first line was lentivirally transduced to constitutively express specific shRNA that suppress the expression of Hs524 PGBD5 [13]. The second line was modified to constitutively express shRNA to target GFP which is not expressed, thereby preserving the endogenous expression of Hs524 and its recombination activities.
Transposon integration rates in both assays.
Integration assays in HeLa cells were monitored separately using transposon-donor plasmids. Two controls were done: the first was done in the presence of GFP instead of Mm523; the second using a source of NeoR cassette void of any transposon sequences (pBSK-NeoR plasmid).
Results confirmed that Mm523 isoform had a negative effect on obtaining NeoR clones since the numbers of clones was one third of the GFP control with pBSK-NeoR (Fig. 4a), and half of GFP controls when Ifp2 and Tcr-pble were used (Fig. 4b,c). However, a significant difference was found between each of both transposons and the NeoR cassette void of any transposon sequences (t-test, p<0.00001). In spite of cytotoxicity induced by ectopic expression of PGBD5, this suggested that a residual integration activity could be detected under these experimental conditions.
Fig. 4. Graphic representations of integration assay results.

(a) rates of chromosomal integration in HeLa cells that were mediated by GFP or Mm523 for a NeoR cassette void of transposon sequences ; (b) rates of chromosomal integration in HeLa cells that were mediated by GFP or Mm523 for a NeoR cassette contained or not in Ifp2 ; (c) rates of chromosomal integration in HeLa cells that were mediated by GFP or Mm523 for a NeoR cassette contained or not in Tcr-pble ; (d) rates of chromosomal integration between both G401 lines (shGFP/shPGBD5) that were mediated by Hs524 for a NeoR cassette contained or not in Ifp2 or Tcr-pble. In each plot, the red lines represented the median and black symbols, the value of each replicate.
Results obtained with the two clonal G401 cell lines and both transposons as donor plasmids were monitored separately. They revealed that the rates of integration events for Ifp2 and Tcr-pble were seven-fold and nine-fold more elevated than that obtained with a source of NeoR cassette void of any transposon sequences, respectively (Fig. 4d). This confirmed that PGBD5 actively integrates DNA fragments containing these elements into chromosomes. This also suggested that the sequence of Tcr-pble ends would be a bit more efficient than those of Ifp2 ends to stimulate integration events (t-test: p<0.0001). We thereafter characterized integration events into chromosomes using populations of NeoR clones from both assays.
Landscapes of insertion junctions among neointegrated transposons.
To prepare chromosomal DNA samples we used ~1800, ~1600, ~1200 and ~1000 NeoR clones from integration assays performed with Ifp2-NeoR and Mm523, Tcr-pble-NeoR and Mm523, Ifp2-NeoR and Hs524, and Tcr-pble-NeoR and Hs524, respectively. Fragment populations of integration junctions were produced by LAM-PCR from NeoR clones chromosomal DNA, sequenced using Illumina Miseq technology. About 3 million paired Miseq reads (250 nucleotides per read) were obtained for each of the 4 samples. Using computer analyses as described in materials and methods we characterized 1,461 and 1,051 transposon/chromosome junctions produced by integration events mediated by Mm523 or Hs524 using Ifp2-NeoR as a transposon source (supplementary Table 3a and 4a) and 1766 and 210 junctions with Tcr-pble-NeoR (supplementary Table 5a and 6a). Junctions at the 5’ and 3’ ends of both transposons were not equally represented in sequence data, perhaps because of different efficiency during DNA fragment amplification in the LAM-PCR.
We further characterized sequence junctions while taking into account the conservation of TSD and TIR sequences, two features required to keep the capacity of neo-inserted elements to be re-remobilized for a new excision and insertion, i.e. to remain “active in mobility” [4,26]. Four kinds of junctions were observed: those displaying i) a full TIR sequence and a putative TTAA TSD (Red bars in Fig. 5), ii) a region containing an intact TIR and a TTAA TSD juxtaposed with a small piece of plasmid backbone (black bars in Fig. 5), iii) no TTAA TSD but a full TIR sequence (blue bars in Fig. 5), and iv) no apparent TTAA TSD and a TIR sequence lacking one or several nucleotides at its outer end (grey bars fin Fig. 5). In each assay, the relative proportions of each of these 4 categories for each transposon end were evaluated. Because of the read length (250 nucleotides), the parameter of minimal alignment used in bwa and lumpy (20 nucleotides) and the window used for both transposons ends (180 and 170 nucleotides at the 5’ end and 175 and 150 nucleotides at the 3’ ends of Ifp2 and Tcr-pble, respectively), these proportions could not be biased by the detection procedure for junctions.
Fig. 5. Features of transposon breakpoints.

Distributions in Ifp2 and Tcr-pble inserted in the presence of Mm523 (a and b) and Hs524 (c and d), and theoretical distribution whether insertions of fragments containing Ifp2-NeoR or Tcr-pble-NeoR occur at random (e and f). Black and grey bars correspond to percentages of breakpoints located within the plasmid backbone flanking the transposon and those located within inner transposon regions (from the position 2 in TIR to the primer used for the LAM-PCR), respectively. Four kinds of junctions were observed: those displaying i) a full TIR sequence and a putative TTAA TSD (Red bars from positions 101 to 104 and 2222 to 2225 in supplementary Table 3a and 4a and 101 to 104 and 731 to 734 in supplementary Table 5a and 6a), ii) a region containing an intact TIR and a TTAA TSD juxtaposed with a small piece of plasmid backbone (black bars from positions 1 to 100 and 2226 to 2301 in supplementary Table 3a and 4a and 1 to 100 and 735 to 833 in supplementary Table 5a and 6a), iii) no TTAA TSD but a full TIR sequence (blue bars from positions 102 to 105 and 2218 to 2221 in in supplementary Table 3a and 4a and 102 to 105 and 727 to 730 in supplementary Table 5a and 6a), and iv) no apparent TTAA TSD and a TIR sequence lacking one or several nucleotides at its outer end (black bars from positions 107 to 178 and 2147 to 2217 in supplementary Table 3a and 4a and 106 to 173 and 669 to 726 in supplementary Table 5a and 6a). In (d), n. a. indicates that data were “not available” because the number of transposon/chromosome junctions was too small to perform statistics.
Using our analysis procedure for detecting transposon/chromosome junctions, we have re-analysed integration assays previously performed in HEK293 cells with transiently transfected plasmid sources of transposon (Ifp2) and PGBD5 (Hs455) [18]. We found 202 breakpoints (supplementary Table7), 66% of them displayed features corresponding to Ifp2 integration into chromosomes by canonical transposition (i.e. with a perfect duplication of the “TTAA” TSD and a full conservation of the TIR sequences). The 33% remaining integration events could be considered as resulting from improper transposition events or alternative mechanisms of chromosomal integration (PGBD5-dependent integration by recombination based on its nuclease activity). When PB was used as a transposase source to transpose Ifp2 into chromosomes, 96-98 % of breakpoints displayed a perfect duplication of the “TTAA” TSD and conservation of the TIR sequences [26,27,28].
The number of perfect transposon/chromosome junctions in Mm523 assays done with Ifp2 and Tcr-pble was 6- to 40-fold lower than when Ifp2 was transposed by PB (Fig. 5a and b). A similar observation was obtained in G401 cells expressing endogenous Hs524 (Fig. 5c and d), but with a lower rate of perfect transposon/chromosome junctions. This indicated that numerous integration events did not result from canonical transposition events. In order to verify the possibility that these profiles mainly resulted from integration events due to non-canonical activity, the breakpoint distribution at each end was compared to that of a theoretical breakpoint distribution (Fig. 5e and f) using Chi-squared tests. Whatever the profile of transposon/chromosome junctions, p-values were all <<0.01, indicating that the profiles did not result from random integration events. Interestingly, the numbers of breakpoint junctions were found to be dramatically more elevated within transposon sequences than in the flanking plasmid regions, except for the 3’ end of Tcr-pble. This suggested that numerous events might result from non-canonical transposition events during which wounds at transposon ends would occur (Fig. 6). To confirm the occurrence of such improper transposition events, we identified 5' and 3' junctions of integration events that occurred exactly into the same chromosomal insertion site in each dataset. We found 24, 5, 12 and 1 chromosomal insertion sites in which pairs of transposon/chromosome junctions occurred at both ends and in inverse orientations for IfP2-NeoR and Mm523, Tcr-pble-NeoR and Mm523, IfP2-NeoR and Hs524, and Tcr-pble-NeoR and Hs524, respectively (supplementary Table 3b to 6b). Each of these junction pairs putatively described both junctions of a single insertion event. Detailed examination of read alignment in the bam file using IGV [29] revealed four cases of putative single integration events in which the pble-NeoR transposon was inserted into a duplicated TSD corresponding to duplicated GATC (2 events), CATG (1 event) or C (1 event) motifs.
Fig. 6.

Signatures of proper and improper DNA integration in a pble transposition system (graphic derived from Figure 1 in https://doi.org/10.1101/015289). piggyBac transposition is initiated by nicks at the transposon ends. The exposed 3’OHs then attack the complementary strand 4 nt inside the flanking donor DNA to form the hairpins on the transposon end. The hairpins on the transposon ends are nicked at the 3’ transposon ends. In a proper integration event (pathway 1), transposon ends thereafter attack the TTAA target sequence at staggered positions, forming covalent links between the 3’ end of the transposon and the 5’ ends of the target site. The single strand gap between the 3’ ends of the target DNA and the 5’ ends of the transposon are repaired to generate the four bp TTAA target sequence duplication. In an improper integration event, transposon ends thereafter attack a random target sequence at staggered positions, forming covalent links between the 3’ end of the transposon and the 5’ ends of the target site. The single strand gap between the 3’ ends of the target DNA and the 5’ ends of the transposon are repaired to generate a non-canonical target sequence duplication (pathway 2). This process can also lead to wounds at transposon ends (pathway 3).
Finally, we observed that the rate of junctions resulting from canonical transposition events (Fig. 5, red bars) was lower with Tcr-pble than Ifp2 in both transposition assays. This suggested that Tcr-pble was not a better substrate for canonical transposition with Mm523 and Hs524 than Ifp2. Nevertheless, results of transposition assays (Fig. 4) supported that Tcr-pble was a better substrate than Ifp2 to actively stimulate the integration of the NeoR cassette by non-canonical transposition and/or alternative mechanisms of DNA integration.
Discussion
The murine and human PGBD5 isoforms Mm523 and Hs524 are able to stimulate the integration of Ifp2 and Tcr-pble in HeLa and G401 cells with no apparent differences, at least under our experimental conditions. They can do it by canonical transposition, but in most cases by other mechanisms, such as PGBD5-dependent recombination based on its nuclease activity. Our findings provide new insights into two distinct questions. The first concerns the identity of the closest pble relatives at the origin of each pgbd gene in vertebrates, with specific attention to those related to pgbd5 gene. The second concerns the features of Ifp2 and Tcr-pble ends that stimulate the recombination activities of Mm523 and Hs524.
Domestication of pble transposase genes during evolution of vertebrates
Our study of the origins of pgbd genes showed that different elements of the piggybac family were domesticated at least 8 or 9 different times over a period spanning over 600 million years of evolution in vertebrates. All pgbd genes are under purifying selection pointing at physiological relevance for species in which they were domesticated. New cases of pble domestication should be characterized as further vertebrate genomes are sequenced. We do not anticipate any discovery of domestication cases as ancient as pgbd1, 2 and 5 genes in vertebrate genomes since sufficient data are available to characterize such ancient genes. However, more recent domestication cases might well be discovered in specific lineages. This was exemplified here with pgbd3 and pgbd4 genes that were previously considered specific to the primate lineage [6], while they were in fact specific to a sub-clade of the Haplorhini. Interestingly, we identified the Strepsirrhini as another primate sub-clade in which pgbd3 and pgbd4 genes were absent. We provided evidence for three new pgbd genes, pgbd7, pgbd8, and pgbd9 and identified pbles close to the common ancestor of all pbles. We identified Tcr-pble as the closest relative of pgbd5 gene. To summarize, Figure 3 provides a comprehensive analysis of phylogenetic relationships between all pbles available to date.
The paradigm of specific interactions of PGBD5 with Ifp2 and Tcr-pble ends.
In spite of very limited sequence identity between the terminal motifs (Fig. 7) and regions at ends of Ifp2 and Tcr-pble, both Ifp2 and Tcr-pble ends can stimulate PGBD5 for recombination, including transposition. Since specific recognition by PGBD5 of both pble ends may not rely on their primary sequence, it might be guided by another feature located within the terminal regions. For more than a decade, it has been known that there is another conserved motif in each pble terminal region, an inner inverted repeat (IIR) [30,31]. Here, we observed in the 24 pbles used in Fig. 3 that they display little identity between their terminal motifs (supplementary data 5a), but 100% of them have IR of at least 6 nucleotides and separated by at least 4 nucleotides at both ends (supplementary data 5b). The distance between these IRs and TIR varied at both ends from 0 to 360 nucleotides, with one of both IRs per pble always being close to TIR. Each pble could display the same IR sequence at both ends, thus corresponding to what was previously called subterminal inverted repeats (STIR; Fig. 8a), or different IR sequence corresponding to internal IR (IIR; Fig. 8b). Previous studies have demonstrated that the regions spanning from the outer end to the complete STIR at both ends of Ifp2 were required to obtain a minimal Ifp2 element that can be actively transposed by PB [32,33,34]. These STIR and IIR might therefore be an important component for the binding of pble and PGBD transposases to their cognate DNA targets. An interesting property of these IRs is that they could extrude a non-B cruciform structure when a DNA torsion, such as a negative superhelicity, was applied to DNA [35-37]. This could be correlated with the fact that Ifp2 has substantially decreased transposition efficiency when integration assays were monitored using a linear Ifp2 donor plasmid rather than in a circular and negatively superhelical configuration [38].
Fig. 7. Gapped local alignment of the 5’ and 3’ extremities of Tcr-pble and Ifp2.

The alignment was performed by GLAM2, and the logo was generated by Skyline. The black rectangles indicate similar nucleotides between the two ends. The stars indicate the similarities between the two pble.
Fig. 8. IR organization at pble ends.

(a) IR organization in pbles displaying TIRs (black triangles) and STIR (double triangles in green). (b) IR organization in pbles displaying TIRs (black triangles) and IIR (double triangles in orange or purple).
Further investigation will be required to determine whether other pbles are able to stimulate transposition and recombination events mediated by PGBD5, to define the critical role of inner IRs within pble ends, and whether STIR and IIR can switch between a B and non-B DNA configuration. If non-pbles sequences displaying such features are interspersed in chromosomes, a part of them might be bound by PGBD5 and trigger various recombination events such as deletions, inversions or fusion of chromosomal arms. Elucidating how IRs and their secondary structure affect the binding of PGBD5 to chromosomal DNA will require detailed structural and biochemical studies.
Materials and Methods
Data mining
The identification of pbles was done with a suite of tools. To annotate pbles, we used reciprocal blast searches to find homologous sequences, taking into account both the length of the alignment, its identity and e-value to filter results. This was used to launch BLASTP and TBLASTN. The query set consisted of 7 sequences: human PGBD1-5, PGBD5 from the cephalochordate Branchiostoma floridae (Bfl-PGBD5), PGBD5 from the lamprey Petromyzon marinus (Pma-PGBD5), and KOBUTA from the Xenopus sp. We performed BLASTP against the NCBI nr and transcriptome shotgun assembly (TSA) nr, RepBase [39] and protein data supplied in Bouallègue et al., 2017. TBLASTN was used on NCBI WGS, NT/NR and TSA databases. Due to the large number of sequenced genomes, an important number of orthologues and paralogues of each PGBD was present in databases. In order to manage this issue, two queries per search were used for TBLASTN analyses: all species except mammals for pgbd1-2 and all species except primates for pgbd3-4. To identify the closest pble to pgbd5, we targeted as subject all species except vertebrate genomes. For TBLASTN only sequences with an e-value between 0 and 1e-80 and with a complete transposase were retained. To verify if sequences belong to the piggyBac family, we performed HMM searches (hmmsearch) with HMMER 3.2.1 and translation following by Pfam domain searches. After verification, nucleotide hits were extended to 3 kb on each side and extracted with EFetch. TIRs were identified using custom scripts. These sequences were translated using the ExPASy Translate tool (https://web.expasy.org/translate/).
Sequence alignment and phylogeny
In order to determine the relationships between pbles and PGBDs, and because the DD[D/E] domain of the Tnp_1_7 transposase displayed an elevated divergence between the most distantly related piggyBac transposases, the alignment was done taking into account the secondary structure, residue type, position conservation, position reliability and residue hydrophobicity of the protein sequences using the PRALINE pipeline [40]. We choose PRALINE because structure is more conserved than sequence [41,42]. Junctions between conserved blocks in the alignment were verified and corrected in some case by hand. For the visualization (supplementary data 3), the alignment was shaded using BoxShade server (http://www.ch.embnet.org/software/BOX_form.html). The phylogenetic tree is based on an alignment covering approximately 400 residues. The tree was generated by a Pthreads version called raxmlHPC-PTHREADS-SSE3 Version 8.2.12 of RaXML [43]. One thousand bootstraps were used to infer branch support and we used the model of amino acid substitution PROTGAMMABLOSUM62.
Substitution rate analyses
Amino acid alignments were performed using MAFFT [44] Version 7.407 (--reorder -- adjustdirection settings). Nucleotide sequences of protein-coding genes were extracted and were aligned by using PAL2NAL Version 14 [45] according to the corresponding amino acid alignment. A phylogenetic tree was constructed using the protein sequence alignments with RaxML. To estimate the selection pressure on pgbd genes, we estimated the ratio (ω; so-called dN/dS) of the rate of nonsynonymous substitutions to the rate of synonymous substitutions using PAML version 4.9 [46]. We used different parameters in the CODEML control file: CodonFreq = 2 for estimating the codon frequencies using F3X4 model, runmode = 0 for evaluation of the tree topologies and model = 0 for a single ω value across all branches, model = 0 is for one omega ratio for all sites.
Synteny analyses
The annotations available at NCBI were used to recover annotated genes around pgbd genes in the selected species. From these genes, proteins were recovered to run TBLASTN against selected contig/genomes. Only hits displaying more than 80% of the protein length with an identity percentage greater than 60 were used. For pgbd7 gene, we used the genome annotations of Trichechus manatus latirosis (GCF_000243295.1, TriManLat1.0), Orycteropus afer afer (GCF_000298275.1, OryAfe1.0), Loxodonta africana (GCF_000001905.1, Loxafr3.0) and Chrysochloris asiatica (GCF_000296735.1, ChrAsi1.0). For pgbd8 gene, only two genomes were annotated : Microcebus murinus (GCF_000165445.2, Mmur_3.0) and Propithecus coquereli (GCF_000956105.1, Pcoq_1.0). For pgbd9 gene we used genome annotations of Austrofundulus limnaeus (GCF_001266775.1, AstBur1.0), Clupea harengus (GCF_000966335.1), Haplochromis burtoni (GCF_000239415.1), Oreochromis niloticus (GCF_001858045.2), Pundamilia nyererei (GCF_000239375.1, PunNye1.0), and Takifugu rubripes (GCF_901000725.2, fTakRub1.2).
Searches of introns and loss of TIR
The presence of introns was analysed by BLAST analyses between the proteins available in public databases and their coding genes. The analysis of the presence of TIR was done with an in-house script (https://github.com/Leelouh/G2TIR).
Sequence analysis of the pble
For each PGBD, pble TIR were analysed. For PGBD3 we used TIRs from Smi7-pble, Hvu-pble, Lfu-pble, and MER85, for PGBD4, those of Oab-pble, Pva-pble, Pxu1-pble, and MER75. For PGBD5 we compared Tcr-pble and Ifp2 (supplementary data 6). For PGBD7 we used Smi2-pble and Nvi-pble. For PGBD8 we used Api1-pble, Atr1-pble and Api3-pble. For PGBD9 we used Pfl-pble, Cgi-pble, and Bgl-pble. For each element, 70 bp were extracted from both pble extremities. These were aligned using GLAM2 [47] a tool that can discover conserved sequence motifs containing indels. The ends of each pble were also analysed with non-B DB [48] that predicts the presence of DNA structure from a primary sequence. The identity between all pairs of pble sequences (supplementary Table 2) was calculated by global pairwise alignment "end to end" using stretcher [49].
Cloning of cDNA coding for the Mm523 PGBD5 isoform
A single mouse brain (strain C57Bl6) was used for total RNA extraction using Tri-reagent (Sigma-Aldrich, St-Louis, MO, USA). cDNA synthesis was carried out using Omniscript RT kit and oligo dT primers (Qiagen, Valencia, CA, USA). PCR primers with appropriate flanking restriction sites were synthesized by Eurofins Genomics, Ebersberg, Germany. PCR was performed with Phusion High-Fidelity PCR Master Mix (ThermoScientific). Following agarose gel electrophoresis, PCR fragments were extracted (QIAquick gel extraction kit, Qiagen), submitted to enzyme restriction (EcoRI/XbaI for the long N-term isoform and EcoRI/XhoI for the short N-term isoform), purified (QIAquick PCR purification kit, Qiagen) and kept for cloning. Their sequence identity was verified by Sanger sequencing (Eurofins Genomics, Ebersberg, Germany). The primers used to amplify Mm523 (Accession N°: XM_006530804.1) were supplied in supplementary data 4d.
Integration assays
Plasmid expression for transposases.
The plasmid pCS2-Mm523 encodes two myc tagged PGBD5 isoforms different in size (523 and 409 amino acid residues, respectively). The cDNA was inserted into the multi-cloning site of a modified pCS2 vector with an in-frame N-term 5XMyc tag [50].
Plasmids donors of transposons.
The plasmid pBSK-Ifp2-TIR5'-NeoR-TIR3' (supplementary data 4b) was built by introducing the Ifp2 5’ and 3’ terminal regions (262 and 400 bp, respectively) into the pBluescript SK plasmid (pBSK). A cassette (NeoR) containing a simian virus 40 (SV40) promoter, the neomycin phosphotransferase ORF and a terminator was cloned between both transposon ends as described [51]. The transposon Tcr-pble (2467 bp) was synthesized by ATG:biosynthetic, Merzhausen, Germany (supplementary data 4c). NeoR was cloned using a BamHI site that was added into its sequence during the DNA synthesis. The plasmid pBSK-NeoR was built by cloning the NeoR cassette into the multi-cloning site of a pBSK plasmid was as described [52].
Integration assay in HeLa cells.
Assays were monitored as described [51]. Briefly, each sample of 100000 cells in a well of a 24-well plates of plaque assays was co-transfected with JetPEI (Polyplus- transfection, Illkirch-Graffenstaden) and 400 ng DNA plasmid and with equal amounts of donor of NeoR cassette included or not within a transposon and transposase sources (1:1 ratio). Two days post-transfection, each cell sample was transferred to a cell culture dish (100 mm diameter) and selected with a culture medium containing 800 μg/mL G418 sulfate (Eurobio Scientific, Les Ulis) for 15 days. After two washing with 1X saline phosphate buffer, cell clones were fixed and stained overnight with 70% EtOH-0.5% methylene blue and colonies > 0.5 mm in diameter were counted. Experiments were performed at least twice in triplicate.
Integration assay in G401 cells.
Assays were monitored as described [13]. Two clonal cell G401 lines were used. The first line was lentivirally transduced to constitutively express specific shRNA suppressing the expression of Hs524 PGBD5 [13]. The second line was modified as a control to constitutively express shRNA to target GFP which is not expressed, thereby preserving the endogenous expression of Hs524 PGBD5. Briefly, each sample of 100000 cells in a well of a 24-well plates of plaque assays was transfected with jetOptimus and 500 ng DNA plasmid (pBSK-IFP2-TIR5'-NeoR-TIR3', pBSK-Tcr-pble-TIR5'-NeoR-TIR3' or pBSK-NeoR) as recommended by the supplier (Polyplus- transfection, Illkirch-Graffenstaden). Two days post-transfection, each cell sample was transferred to a cell culture dish (100 mm diameter) and selected with a culture medium containing 2 mg/mL G418 sulfate (Eurobio Scientific, Les Ulis) for 15 days. After two washing with 1X saline phosphate buffer, cell clones were fixed and stained overnight with 70% EtOH-0.5% methylene blue and colonies > 0.5 mm in diameter were counted. Experiments were performed at least twice in triplicate.
Recovery of integration sites.
LAM-PCR and Illumina libraries.
Integration assays were done to produce cell populations containing integrated copies of the donor transposon. Fifteen days post-transfection, cell clones were harvested for genomic DNA preparation using the DNeasy kit (Qiagen, Hilden, Germany). Linear amplification-mediated PCR (LAM-PCR) was performed to amplify the vector-genomic DNA junctions of Ifp2 and Tcr-pble vectors as described [53]. All PCRs were done using the high fidelity Q5 DNA Polymerase (New England Biolabs, Ipswich, MA). For both approaches, 1 μg DNA was used for two rounds of 50 linear amplification using a biotinylated primer anchored near one end of the NeoR cassette to enrich DNA species containing transposon-chromosomal DNA junctions (for sequences of (B)-NeoR 5’ and 3’ primers, supplementary data 4e). One reaction was done per ends. The single-stranded products were immobilized on streptavidin-coated magnetic beads (Dynabeads M-280 Streptavidin, Invitrogen, Carlsbad, CA). All subsequent steps were performed on the magnetic bead-bound DNA. Two washes with water followed each step. Second strand synthesis was performed with random hexamer primers (Roche, Basel, Switzerland) using Klenow DNA polymerase (New England Biolabs, Ipswich, MA). The double-stranded DNA was split into two batches and subjected to restriction digests with DpnI for the first one and PciI, NcoI and BspHI for the second one using restriction enzymes. The DNA fragments with a CG-3’ or a CATG-3' overhang ends were ligated to linkers displaying appropriate overhang ends and made from annealed oligonucleotides (supplementary data 4e).
To increase the specificity of the full process, a first PCR was done using one biotinylated primer anchored within the 5’ or 3’ region of the transposon donor and one primer anchored within the linker (for sequences of (B)-TIR-UTR 5’ and 3’, and LC1 primers, see supplementary data 4e). PCR products were immobilized on streptavidin-coated magnetic beads and purified as described above. Next, the bead-bound DNA was subjected to a nested PCR using nested primers anchored within transposon ends and within linkers (supplementary data 4e). Final PCR products were purified, quantified and gathered in equimolar DNA amounts for each transposon vector (4 populations of LAM-PCR products) before to be used to make Illumina libraries using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® and NEBNext Multiplex Oligos for Illumina (New England Biolabs, Ipswich, MA). Fragment size selection, library-quality control and Illumina sequencing (MiSeq 250 nucleotides, TruSeq SBS Kit v3) were achieved at the Plateforme de Séquençage Haut Debit I2BC (Gif-sur-Yvette, France). DNA quantities were monitored at various steps in the procedure with the Qubit® dsDNA (Molecular Probes, Eugene, USA).
Computer analysis.
Trimmomatic [54] was used to filter Miseq reads using default parameters, excepted for SLIDINGWINDOW:5:20 and MINLEN:100. The purpose of the following steps was to recover chromosome-inserted DNA fragment junctions taking into account the plasmid backbone regions located 100 bp upstream and downstream the pble-NeoR transposon. Filtered reads were first mapped against the sequence of plasmid backbone less the 100 bp regions flanking on both sides the pble-NeoR transposon with bwa-mem using default parameters [55]. The unmapped reads were then extracted using SAMtools view with parameters -b -f 4 [56] and bamToFastq from the BEDTools suite using default parameters [57]. Recovered unmapped reads were aligned using bwa-mem against a bwa bank gathering the sequences of hg38 chromosomes plus those of the Ifp2-NeoR or Tcr-pble transposons flanked by the 100 bp plasmid backbone regions on both sides (supplementary data 4e). Default parameters were used except for -w 1 and -r 1. The bam file resulting from each dataset alignment was analysed with Lumpy in order to identify split reads [58] . The used parameters were -e -mw 2 -tt 0.0 and back_distance:20,weight:1,id:lumpy_v1,min_mapping_threshold:20. Structural variants (SV) characterized by “BND” for the broken end notations and displaying for each of them an SV with two positions, one genomic and one on the transposon, were extracted using a house python program (https://github.com/Leelouh/lumpy2site). Results were filtered taking into account a difference below 3 between the transposon breakpoint calculated by Lumpy and the maximal spread of read alignments in the transposon donor sequence for each integration event. Each TSD nucleotide motif at the insertion site was obtained after extracting 10 bp sequences before and after the breakpoint in the chromosome sequences.
Statistical analysis
Values in graphics were medians, quartiles 1 and 3 and spread of experiments done in triplicate. Shapiro-Wilk test was used to confirm the normality of each set of samples, t-test to analyse distribution differences between experimental samples, Chi2 tests to analyse differences between an experimental distribution and a theoretical one, and logarithmic distribution test to analyse enrichment using free tools and tutorials available at http://www.anastats.fr/outils.php.
Data repository
All raw and processed data are available through the European Nucleotide Archive under accession number PRJEB36229, PRJEB36230, PRJEB41045 and PRJEB41052. Files describing the annotation of insertion sites copies in the hg38 release are supplied as Supplementary Tables 3 to 7.
Supplementary Material
Supplementary data 1. Graphic representation of the data mining process to inventory pgbd genes and their closest pble relatives.
Supplementary data 2. Localization of PGBD3 and PGBD4 ORFs and respectively MER85 and MER75. Fasta sequences of localization of PGBD3 and PGBD4 genes. PGBD3 sequence is reversed (complementary strand). ORF of genes is underlined in green and TIR of the element are in bold. For PGBD3, the first bold sequence corresponds to position 1 to 43 of MER85 and the second to the positions 39 to 140. For PGBD4, the first sequence is for positions 68 to 341 of MER75 and the second one is the positions 379 to 514.
Supplementary data 3. Sequence alignment of pble transposases and PGBD proteins used to calculate the phylogenetic tree shown inFigure 4. The black shade is for residues which are present in more than 60% at a position. Yellow is for the position of the catalytic triad. The blue line is for β-sheet and red line for α-helix.
Supplementary data 4. Transposon donor sequences and primers used in the study.
Supplementary Table 1. References of pble or PGBD sequences used in this study. For each sequence: name in this manuscript, accession number or location, names in the databases, species, lineage, and nucleotide and protein sequences for the new entries.
Supplementary Table 2. Identity rates between each pair of pble sequences used in Figure 4. MER75 and MER85 were not included in this analysis because they were miniature pbles. Rates typed in red were above 0.5.
Supplementary Table 3. (a) Inventory of Ifp2-chromosome junctions resulting from integration events mediated by Mm523 in HeLa cells. Transposon breakpoints are located at positions 102 and 2220, 103 and 2219, 104 and 2218, and 105 and 2217 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 2221, 100 and 2222, 99 and 2223, and 98 and 2224 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non-canonic duplicated TSD was highlighted in green.
Supplementary Table 4. (a) Inventory of Ifp2-chromosome junctions resulting from integration events mediated by Hs524 in G401 cells. Transposon breakpoints are located at positions 102 and 2220, 103 and 2219, 104 and 2218, and 105 and 2217 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 2221, 100 and 2222, 99 and 2223, and 98 and 2224 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical in the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 5. (a) Inventory of Tcr-pble-chromosome junctions resulting from integration events mediated by Mm523 in HeLa cells. Transposon breakpoints are located at positions 102 and 730, 103 and 729, 104 and 728, and 105 and 727 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 731, 100 and 732, 99 and 733, and 98 and 734 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 6. (a) Inventory of Tcr-pble-chromosome junctions resulting from integration events mediated by Hs524 in G401 cells. Transposon breakpoints are located at positions 102 and 730, 103 and 729, 104 and 728, and 105 and 727 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 731, 100 and 732, 99 and 733, and 98 and 734 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 7. Inventory of 3’Ip2-chromosome junctions resulting from integration events mediated by Hs455 in HEK293 cells. Transposon breakpoints located at positions 3611, 3612, 3613 and 3614 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotidic motif (IUPAC nucleotide code; blue bars in Figure 10a). Those at positions 3616, 3617, 3618, and 3619 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events.
Highlights.
Nine domesticatedpiggyBac transposases of different ages are present in vertebrates
The closestpble relatives of these 9 domesticated transposases were identified
The closestpiggyBac-like transposon relative was Tcr-pble
We confirm that the domesticated piggyBac transposase PGBD5 triggers Ifp2 mobility
PGBD5 mediates proper and improper transposition events with Ifp2 and Tcr-pble
Acknowledgments
This work was supported by the C.N.R.S., the I.N.R.A., and the GDR CNRS 2157. It also received funds from a research program grants from the Ligue Nationale Contre le Cancer, the Merck foundation, and the French National Society of Gastroenterology. Laura Helou holds a PhD fellowship from the Region Centre Val de Loire. We acknowledge the high-throughput sequencing facilities of I2BC for its sequencing and bioinformatics expertize. Alex Kentsis is a consultant for Novartis and is supported by the National Cancer Institute grants R01 CA214812 and P30 CA008748. Yves Bigot, who was in charge of the achievement of this project does not have to thank the French National Research Agency for its financial support but he kindly thanks it for the excellent reviews embellished with arguments based on scientific and cultural novelties in the expertise of his yearly application file during the last decade. Our thanks to Peter Arensburger at California Polytechnic University in Pomona CA (Cal Poly Pomona) for reviewing the manuscript for legibility.
List of abbreviations:
- CMV
Human cytomegalovirus
- CRD
Cystein-rich domain
- GFP
green fluorescent protein
- LTR
long terminal repeats
- NeoR
Neomycin resistance
- ORF
Open reading frame
- pble
piggyBac-like element
- PGBD or pgbd
“piggyBac derived transposase” protein or gene
- STIR
sub-terminal inverted repeats
- SV40
simian virus 40
- TE
transposable element
- TIR
terminal inverted repeats
- TSD
target site duplication
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none'
References
- 1.Piégu B, Bire S, Arensburger P, Bigot Y (2015) A survey of transposable element classification systems - A call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. Evol 86, 90–109. [DOI] [PubMed] [Google Scholar]
- 2.Goerner-Potvin P, Bourque G (2018) Computational tools to unmask transposable elements. Nat. Rev. Genet 19, 688–704. [DOI] [PubMed] [Google Scholar]
- 3.Fraser MJ, Smith GE, Summers MD (1983) Acquisition of host cell DNA sequences by baculoviruses: Relationship between host DNA insertions and FP mutants of Autographa californica and Galleria mellonella nu- clear polyhedrosis viruses. J. Virol 47, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitra R, Fain-Thornton J, Craig NL (2008) piggyBac can bypass DNA syn- thesis during cut and paste transposition. EMBO J. 27, 1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra R, Li X, Kapusta A, Mayhew D, Mitra RD, Feschotte C, Craig NL (2013) Functional characterization of piggyBat from the bat Myotis lucifugus unveils an active mammalian DNA transposon. Proc. Nat. Acad. Sci. USA 110, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouallègue M, Rouault JD, Hua-Van A, Makni M, Capy P (2017) Molecular Evolution of piggyBac Superfamily: From Selfishness to Domestication. Gen. Biol. Evol 9, 323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morellet N, Li X, Wieninger SA, Taylor JL, Bischerour J, Moriau S, Lescop E, Bardiaux B, Mathy N, Assrir N, Bétermier M, Nilges M, Hickman AB, Dyda F, Craig NL, Guittet E (2018) Sequence-specific DNA binding activity of the cross-brace zinc finger motif of the piggyBac transposase. Nucl. Acids. Res 46, 2660–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinzelle L, Izsvák Z, Ivics Z (2009) Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell. Mol. Life. Sci 66, 1073–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joly-Lopez Z, Bureau TE (2018) Exaptation of transposable element coding sequences. Curr. Opin. Genet. Dev 49, 34–42. [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Tao X, Yuan S, Zhang Y, Li P, Beilinson HA, Zhang Y, Yu W, Pontarotti P, Escriva H, Le Petillon Y, Liu X, Chen S, Schatz DG, Xu A (2016) Discovery of an Active RAG Transposon Illuminates the Origins of V(D)J Recombination. Cell. 166, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Cheng TC, Huang G, Lu Q, Surleac MD, Mandell JD, Pontarotti P, Petrescu AJ, Xu A, Xiong Y, Schatz DG (2019) Transposon molecular domestication and the evolution of the RAG recombinase. Nature. 569, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapitonov VV, Jurka J (2005) RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS. Biol 3, e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hencken CG, Li X, and Craig NL (2012). Functional characterization of anactive Rag-like transposase. Nat. Struct. Mol. Biol 19, 834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar A, Sim C, Hong YS, Hogan JR, Fraser MJ, Robertson HM, Collins FH (2003) Molecular evolutionary analysis of the widespread piggyBac transposon family and related “domesticated” sequences. Mol. Genet. Genom 270, 173–180. [DOI] [PubMed] [Google Scholar]
- 15.Pavelitz T, Gray LT, Padilla SL, Bailey AD, Weiner AM (2013) PGBD5: a neural-specific intron containing piggyBac transposase domesticated over 500 million years ago and conserved from cephalochordates to humans. Mob. DNA 4, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delsuc F, Brinkmann H, Chourrout D, Philippe H (2006) Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 439, 965–968. [DOI] [PubMed] [Google Scholar]
- 17.Putnam NH, Butts T, Ferrier DEK, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, Benito-Gutierrez EL, Dubchak I, Garcia-Fernandez J, Gibson-Brown JJ, Grigoriev IV, Horton AC, de Jong PJ, Jurka J, Kapitonov VV, Kohara Y, Kuroki Y, Lindquist E, Lucas S, Osoegawa K, Pennacchio LA, Salamov AA, Satou Y, Sauka-Spengler T, Schmutz J, Shin-I T, Toyoda A, Bronner-Fraser M, Fujiyama A, Holland LZ, Holland PW, Satoh N, Rokhsar DS(2008) The amphioxus genome and the evolution of the chordate karyotype. Nature. 453, 1064–1071. [DOI] [PubMed] [Google Scholar]
- 18.Henssen AG, Henaff E, Jiang E, Eisenberg AR, Carson JR, Villasante CM, Ray M, Still E, Burns M, Gandara J, Feschotte C, Mason CE, Kentsis A (2015) Genomic DNA transposition induced by human PGBD5. Elife. 4, e10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henssen AG, Koche R, Zhuang J, Jiang E, Reed C, Eisenberg A, Still E, MacArthur IC, Rodríguez-Fos E, Gonzalez S, Puiggròs M, Blackford AN, Mason CE, de Stanchina E, Gönen M, Emde AK, Shah M, Arora K, Reeves C, Socci ND, Perlman E, Antonescu CR, Roberts CWM, Steen H, Mullen E, Jackson SP, Torrents D, Weng Z, Armstrong SA, Kentsis A (2017) PGBD5 promotes site-specific oncogenic mutations in human tumors. Nat. Genet 49, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kryazhimskiy S, Plotkin JB (2008) The Population Genetics of dN/dS. PLoS. Genet 4, e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue Y, Saga T, Aikawa T, Kumagai M, Shimada A, Kawaguchi Y, Naruse K, Morishita S, Koga A, Takeda H (2017) Complete fusion of a transposon and herpesvirus created the Teratorn mobile element in medaka fish. Nat. Commun 8, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arkhipova IR, Yushenova IA (2019) Giant Transposons in Eukaryotes: Is Bigger Better? Gen. Biol. Evol 11, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hikosaka A, Kobayashi T, Saito Y, Kawahara A (2007) Evolution of the Xenopus piggyBac transposon family TxpB: domesticated and untamed strategies of transposon subfamilies Mol. Biol. Evol 24, 2648–2656. [DOI] [PubMed] [Google Scholar]
- 24.Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A (2017) Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci. Transl. Med 9, eaam9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helou L, Beauclair L, Dardente H, Arensburger P, Buisine N, Jaszczyszyn Y, Guillou F, Lecomte T, Kentsis A, Bigot Y (2020) The C-terminal domain of piggyBac transposase is not required for DNA transposition. J. Mol. Biol in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elick TA, Lobo N, Fraser MJ Jr. (1997) Analysis of the cis-acting DNA elements required for piggyBac transposable element excision. Mol. Gen. Genet 255, 605–610. [DOI] [PubMed] [Google Scholar]
- 27.Li MA, Pettitt SJ, Eckert S, Ning Z, Rice S, Cadiñanos J, Yusa K, Conte N, Bradley A (2013) The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol. Cell. Biol 33, 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Mayhew D, Chen X, Johnston M, Mitra RD (2012) "Calling cards" for DNA-binding proteins in mammalian cells. Genetics. 190, 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu M, Sun ZC, Hu CL, Zhang GF, Han ZJ (2008) An active piggyBac-like element in Macdunnoughia crassisigna. Insect. Sci 15:521–528. [Google Scholar]
- 31.Daimon T, Mitsuhira M, Katsuma S, Abe H, Mita K, Shimada T (2010) Recent transposition of yabusame, a novel piggybac-like transposable element in the genome of the silkworm, Bombyx mori. Genome. 53:585–593. [DOI] [PubMed] [Google Scholar]
- 32.Solodushko V, Bitko V, Fouty B (2014) Minimal piggyBac vectors for chromatin integration. Gene. Ther 21:1–9. [DOI] [PubMed] [Google Scholar]
- 33.Troyanovsky B, Bitko V, Pastukh V, Fouty B, Solodushko V (2016) The Functionality of Minimal PiggyBac Transposons in Mammalian Cells. Mol. Ther. Nucleic. Acids 5, e369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Luo W, Veach RA, Hickman AB, Wilson MH, Dyda F (2020) Structural basis of seamless excision and specific targeting by piggyBac transposase. Nat Commun 11, 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brázda V, Laister RC, Jagelská EB, Arrowsmith C (2011) Cruciform structures are a common DNA feature important for regulating biological processes. BMC. Mol. Biol 12, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaushik M, Kaushik S, Roy K, Singh A, Mahendru S, Kumar M, Chaudhary S, Ahmed S, Kukreti S (2016) A bouquet of DNA structures: Emerging diversity. Biochem. Biophys. Rep 5, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guiblet WM, Cremona MA, Cechova M, Harris RS, Kejnovská I, Kejnovsky E, Eckert K, Chiaromonte F, Makova KD (2018) Long-read sequencing technology indicates genome-wide effects of non-B DNA on polymerization speed and error rate. Gen. Res 28, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakanishi H, Higuchi Y, Kawakami S, Yamashita F, Hashida M (2011) Comparison of piggyBac transposition efficiency between linear and circular donor vectors in mammalian cells. J. Biotechnol 154, 205–208. [DOI] [PubMed] [Google Scholar]
- 39.Bao W, Kojima KK, Kohany O (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simossis VA, Heringa J, (2003) The PRALINE online server: Optimising progressive multiple alignment on the web. Comput. Biol. Chem 27, 511–519. [DOI] [PubMed] [Google Scholar]
- 41.Sander C, Schneider R (1991) Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 9, 56–68. [DOI] [PubMed] [Google Scholar]
- 42.Rost B (1999) Twilight zone of protein sequence alignments. Prot. Engin 12, 85–94. [DOI] [PubMed] [Google Scholar]
- 43.Stamatakis A, 2014. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 20, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucl. Acids. Res 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- 47.Frith MC, Saunders NF, Kobe B, Bailey TL (2008) Discovering sequence motifs with arbitrary insertions and deletions. PLoS. Comput. Biol 4, e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, Halusa GN, Volfovsky N, Yi M, Luke BT, Bacolla A, Collins JR, Stephens RM (2013) Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucl. Acids. Res 41, D94–D100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Myers EW, Miller W (1988) Optimal alignments in linear space. Comput. Appl. Biosci 4, 11–17. [DOI] [PubMed] [Google Scholar]
- 50.Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P (2002) Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. USA 99, 7728–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bire S, Ley D, Casteret S, Mermod N, Bigot Y, Rouleux-Bonnin F (2013) Optimization of the piggyBac transposon using mRNA and insulators: toward a more reliable gene delivery system. PLoS. One 8, e82559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bire S, Casteret S, Piégu B, Beauclair L, Moiré N, Arensbuger P, Bigot Y (2016) Mariner Transposons Contain a Silencer: Possible Role of the Polycomb Repressive Complex 2. PLoS. Genet 12, e1005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartholomae CC, Glimm H, von Kalle C, Schmidt M (2012) Insertion site pattern: global approach by linear amplification-mediated PCR and mass sequencing. Meth. Mol. Biol 859:255–265. [DOI] [PubMed] [Google Scholar]
- 54.Bolger AM, Lohse M, Usadel B (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Layer RM, Chiang C, Quinlan AR, Hall IM (2014) LUMPY: a probabilistic framework for structural variant discovery. Gen. Biol 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1. Graphic representation of the data mining process to inventory pgbd genes and their closest pble relatives.
Supplementary data 2. Localization of PGBD3 and PGBD4 ORFs and respectively MER85 and MER75. Fasta sequences of localization of PGBD3 and PGBD4 genes. PGBD3 sequence is reversed (complementary strand). ORF of genes is underlined in green and TIR of the element are in bold. For PGBD3, the first bold sequence corresponds to position 1 to 43 of MER85 and the second to the positions 39 to 140. For PGBD4, the first sequence is for positions 68 to 341 of MER75 and the second one is the positions 379 to 514.
Supplementary data 3. Sequence alignment of pble transposases and PGBD proteins used to calculate the phylogenetic tree shown inFigure 4. The black shade is for residues which are present in more than 60% at a position. Yellow is for the position of the catalytic triad. The blue line is for β-sheet and red line for α-helix.
Supplementary data 4. Transposon donor sequences and primers used in the study.
Supplementary Table 1. References of pble or PGBD sequences used in this study. For each sequence: name in this manuscript, accession number or location, names in the databases, species, lineage, and nucleotide and protein sequences for the new entries.
Supplementary Table 2. Identity rates between each pair of pble sequences used in Figure 4. MER75 and MER85 were not included in this analysis because they were miniature pbles. Rates typed in red were above 0.5.
Supplementary Table 3. (a) Inventory of Ifp2-chromosome junctions resulting from integration events mediated by Mm523 in HeLa cells. Transposon breakpoints are located at positions 102 and 2220, 103 and 2219, 104 and 2218, and 105 and 2217 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 2221, 100 and 2222, 99 and 2223, and 98 and 2224 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non-canonic duplicated TSD was highlighted in green.
Supplementary Table 4. (a) Inventory of Ifp2-chromosome junctions resulting from integration events mediated by Hs524 in G401 cells. Transposon breakpoints are located at positions 102 and 2220, 103 and 2219, 104 and 2218, and 105 and 2217 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 2221, 100 and 2222, 99 and 2223, and 98 and 2224 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical in the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 5. (a) Inventory of Tcr-pble-chromosome junctions resulting from integration events mediated by Mm523 in HeLa cells. Transposon breakpoints are located at positions 102 and 730, 103 and 729, 104 and 728, and 105 and 727 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 731, 100 and 732, 99 and 733, and 98 and 734 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 6. (a) Inventory of Tcr-pble-chromosome junctions resulting from integration events mediated by Hs524 in G401 cells. Transposon breakpoints are located at positions 102 and 730, 103 and 729, 104 and 728, and 105 and 727 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotide motif (IUPAC nucleotide code; blue bars in Figure 8a). Those at positions 101 and 731, 100 and 732, 99 and 733, and 98 and 734 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events. (b) Inventory of chromosomal site in which Ifp2 insertions were found with both 5’ and 3’ extremities. Integration events corresponding to proper transposon ends were typed in black while those with improper ends were typed in red. Their non canonic duplicated TSD was highlighted in green.
Supplementary Table 7. Inventory of 3’Ip2-chromosome junctions resulting from integration events mediated by Hs455 in HEK293 cells. Transposon breakpoints located at positions 3611, 3612, 3613 and 3614 had a TSD displaying a DTAA, DDAA, DDBA or DDBB nucleotidic motif (IUPAC nucleotide code; blue bars in Figure 10a). Those at positions 3616, 3617, 3618, and 3619 (red bars in Figure 8a) displayed a TTAA TSD with upstream or downstream 0 nucleotide, one nucleotide, a dinucleotide or a trinucleotide that were identical to the plasmid backbone and the chromosome. As the origin of this specific trait could not be differentiated with a probability threshold below 1%, all these junctions were considered in the analysis as originating from proper integration events.