

Abstract
The highly contagious Coronavirus Disease 2019 (COVID-19) caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), which is a newborn infectious member of the dangerous beta-coronaviruses (β-CoVs) following SARS and MERS‐CoVs, can be regarded as the most significant issue afflicting the whole world shortly after December 2019. Considering CoVs as RNA viruses with a single-stranded RNA genome (+ssRNA), the critical viral enzyme RNA dependent RNA polymerase (RdRp) is a promising therapeutic target for the potentially fatal infection COVID-19. Nicotinamide riboside (NR), which is a naturally occurring analogue of Niacin (vitamin B3), is expected to have therapeutic effects on COVID-19 due to its super close structural similarity to the proven RdRp inhibitors. Thus, at the first phase of the current molecular docking and dynamics simulation studies, we targeted SARS-CoV-2 RdRp. On the next phase, SARS-CoV RdRp, human Angiotensin-converting enzyme 2, Inosine-5’-monophosphate dehydrogenase, and the SARS-CoV-2 Structural Glycoproteins Spike, Nonstructural viral protein 3-Chymotrypsin-like protease, and Papain-like protease were targeted using the docking simulation to find other possible antiviral effects of NR serendipitously. In the current study, the resulted scores from molecular docking and dynamics simulations as the primary determinative factor as well as the observed reliable binding modes have demonstrated that Nicotinamide Riboside and its active metabolite NMN can target human ACE2 and IMPDH, along with the viral Spro, Mpro, PLpro, and on top of all, RdRp as a potential competitive inhibitor.
Keywords: RdRp, Ribavirin, Favipiravir, SARS-CoV-2, Pharmacophore model
Graphical Abstract
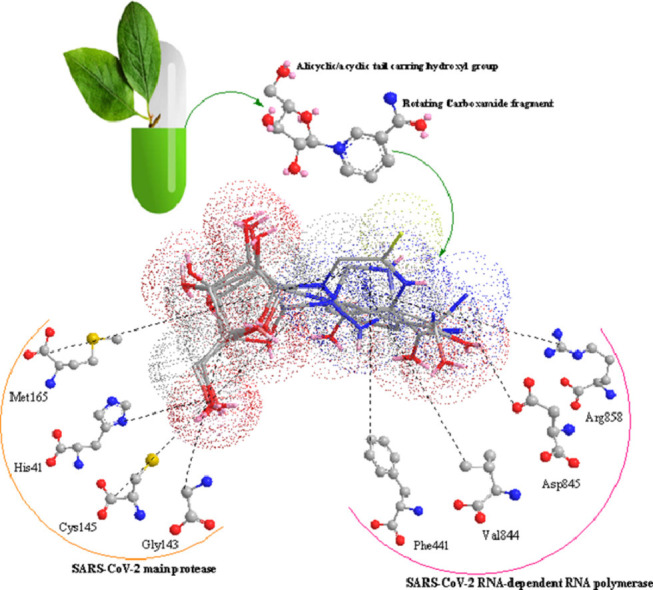
1. Introduction
The potentially fatal infection coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has begun in December 2019 from Wuhan city of China and affected the whole aspects of life all over the world via becoming a pandemic. The capability of SARS-CoV-2 to attack different parts of the body, particularly vital organs i.e., lungs, heart, brain, and kidneys, makes this highly contagious multi-organ infection a serious global crisis [1,2]. Although it seems that with the advent of vaccines, the transmission chains in this viral pandemic have been broken, considering the current concerns about their effectiveness against the mutant variants, the reported adverse effects, the probable long-term side effects [3], [4], [5], [6], and the necessity for saving the infected peoples’ lives, we need to find safe, efficient, and preferably affordable, and on top of all, specific drugs instead of the currently prescribed non-specific therapeutic cocktails [7,8].
In this way, RNA-dependent RNA polymerase (RdRp) is an essential enzyme for viral genome replication and translation; therefore, it can be considered excellent therapeutic target for COVID- 19 treatments. RdRp-targeted antivirals structurally fall into two classes: nucleoside inhibitors (NIs) and non-nucleoside inhibitors (NNIs) [9]. NIs, i.e., Ribavirin, Favipiravir, and the related structural analogues [10], act as purine pseudo bases after phosphorylation and cause irreversible mutations in the viral genome or inhibit the viral RdRp directly, in a concentration-dependent manner [11]. The inhibitory mechanism of NIs is related to bioisosterism and steric similarity with purine nucleoside triphosphates [12]. The NIs binding pocket (enzyme active site) is highly conserved among various RdRp enzymes; therefore, it is not surprising that the above-mentioned broad spectrum NIs, i.e., Favipiravir and Ribavirin, have efficacy against COVID-19 [13].
An essential structural element in nucleotide analogue inhibitors of RdRp is the rotating carboxamide moiety attached to the aromatic core (Scheme 1 ). This structural portion makes the antiviral activity of these compounds possible. The formation of different rotational conformations of the amide bond has been responsible for forming the critical drug-target interactions. Compounds containing a rotatable carboxamide group attached to an aromatic center can mimic the endogenous ligand guanosine triphosphate (GTP) and create base-pair interactions with Cytosine. Such kind functional groups can also create the base-pair interactions with Uracil via mimicking the adenosine triphosphate (ATP) binding properties [14]. Theoretically, the presence of a ribose portion in the chemical structure of NIs, causes incorrect RNA chain elongation, since these compounds are used as alternative substrates for endogenous nucleotides. In this sense, it is worth mentioning that this leads to virus mutagenesis [11]; but it seems not to be crucial for antiviral activity [15].
Scheme 1.

The structural pattern of the broad-spectrum inhibitors of viral RdRp: Favipiravir and Ribavirin (Red: Oxygen, Blue: Nitrogen, Gray: Carbon, Yellow: Fluorine, and Pink: Lone pair of electrons).
Viral DNA polymerase inhibitor Penciclovir with the same structural features as Favipiravir and Ribavirin has revealed clinical efficacy against COVID-19 [13] (Schemes 1,2 ). Based on the molecular docking studies, this efficacy has been associated with the inhibition of SARS-CoV2 RdRp (PDB ID: 6M71) [16]. Besides, the other viral DNA polymerase inhibitors Ganciclovir which was successfully administered for the first reported COVID-19 patient with diabetes [17], and also the clinically approved drug candidate IDX-184 as a direct-acting antiviral drug in numerous theoretical studies [15,18,19], both possess the discussed structural features (Scheme 2), and also the capability to be tightly wrapped to the SARS-CoV-2 RdRp (PDB ID: 6M71), according to the molecular dynamics and docking simulations [20,21]. In this regard, it can be said that all of these antivirals have the structural pattern of Favipiravir and Ribavirin (Schemes 1,2), albeit with an in-flexible and rigid carboxamide portion which allows these compounds to mimic GTP and base pair with Cytosine [14].
Scheme 2.

Structural analogues of Ribavirin and Favipiravir with almost rigid carboxamide moiety.
The naturally occurring vitamin Nicotinamide riboside (NR) with a close structural similarity [22] to NIs (Scheme 3 ) is orally bioavailable and currently sold as a safe supplement [23]. Once NR is entered the cell, NR metabolized into Nicotinamide ribose monophosphate or nicotinamide mononucleotide (NMN) by a phosphorylation step catalyzed by the nicotinamide riboside kinases (NRKs) [24]. NMN structurally corresponds to the monophosphate intermediate of the activated NIs (NIs Tri-Phosphate) [11].
Scheme 3.

The similar structural pattern of NR with the proven broad-spectrum RdRp inhibitors Favipiravir and Ribavirin.
Xu et al. (2015) have presented a successful Pyridoxine-derived small-molecule inhibitor (DMB220) for Dengue virus RdRp as a novel class of RdRp inhibitors [25]. The enzymatic and molecular modeling results proved that DMB220 competes with the natural nucleoside triphosphate (NTP) substrates for binding to the RdRp active site, which is different from the binding site of classic NNIs. According to the undeniable principle of Structure-Activity Relationship (SAR), we expect common biological effects from similar chemical structures. Therefore, in this study, the potential inhibitory effects of NR, and NMN against SARS-CoV-2 RdRp were studied using molecular docking and molecular dynamics (MD) simulation as a considerable primary step in the identifying of novel therapeutic agents for COVID-19. This research has been based on the structural similarity of NR, with the proven broad-spectrum inhibitors of viral RdRp which are successfully used for the clinical management of COVID-19 patients, i.e., Ribavirin and Favipiravir. Natural substrates Adenosine triphosphate (ATP), Guanosine-triphosphate (GTP), and the active forms of broad-spectrum RdRp inhibitors Favipiravir and Ribavirin were used as the reference substrates. Considering the obtained molecular docking and MD results, NR can be considered as a Nicotinamide-derived small-molecule that can ultimately be wrapped by the SARS-CoV-2 RdRp active site as the competitive inhibitor DMB220.
Along with that, In the next phase of the research, Spro, ACE2 receptor, 3CLpro or the main protease (Mpro), and Papain-like protease (PLpro) which play essential roles in host cell-virus surface interaction for viral entry into the target cells, and processing of translated polyproteins respectively, was used as the molecular docking targets. Furthermore, IMPDH is an essential enzyme in the biosynthesis of guanine nucleotide, that its inhibitors have revealed proven effects against human coronaviruses [26], was the last selected target in this represented molecular docking studies since NR shows the same structural pattern with one major group of IMPDH inhibitors including Ribavirin and Mizoribine (as their monophosphate-activated metabolites) which target the binding site of the natural substrate (inosine monophosphate). The obtained results from the present investigation discuss not only NR as a potential small-molecule drug against COVID-19 but also a pharmacophore model for the rational design or screening of the multi-target anti-SARS-CoV-2 agents.
2. Materials and methods
2.1. Preparation of drug target
The first drug target for the current study is RdRp of SARS-CoV-2 in complex with cofactors (PDB ID: 6M71) [27]. NSP12, which is the RdRp is chain A with 851 amino acids (Supplementary information, Figure S1 (A)). Another drug target is the RdRp structure of SARS-CoV (PDB ID: 6NUR) [28] and identified SARS Coronavirus (SARS-CoV) NSP12 bound to NSP7 and NSP8 cofactors. The NSP12 (chain A) was selected as RdRp, and the cofactors (NSP7 (chain C)) and NSP8 (chains (B) and (D)) were removed from the structure (Figure S1 (B)). COVID-19 main protease in complex with an inhibitor N3 (PDB ID: 6LU7) [29] (chain A) was chosen as another target in this study (Figure S1 (C)). Additionally, we carried out the docking using perfusion SARS-CoV-2 Spike glycoprotein (Spro) with a single receptor-binding domain (chain A) (PDB ID: 6VSB) [30] (Figure S1 (D)). Moreover, the human type II Inosine Monophosphate Dehydrogenase (PDB ID: 1NF7) [31] is another selected target in this investigation (Figure S1 (E)). For the molecular docking, we used SARS-CoV-2 Spike receptor-binding domain bound with ACE2 (PDB ID 6M0J) [32] (Figure S1 (F)). Furthermore, SARS-CoV-2 Papain-like protease (PDB ID: 6WX4) [33], which is essential for virus maturation and its infectivity [34], was used as another protease target in this study (Figure S1 (G)). The three-dimensional structures were selected and obtained from Protein Data Bank (http://www.rscb.org) with proper resolutions.
2.2. Ligand preparation
In this study, a total of fourteen compounds was used as docking ligands to screen and identify the potent antiviral agents for COVID-19 (Table 1 ). PubChem database (www.pubchem.ncbi.nlm.nih.gov/) was used to retrieve the 3D chemical structures of the selected molecules, which were downloaded in .sdf format. Energy minimization was subjected to all conformers. All the minimizations were performed under an RMSD gradient of 0.01 kcal mol−1 and RMS distance of 0.1 Å with MMFF94X force-field. The ligands were converted to (pdbqt) file format as inputs for docking studies.
Table 1.
General information of the selected ligands and targets in this study.
 |
 |
2.3. Molecular docking studies
Molecular docking as a powerful computational modeling tool is a structure-based drug design (SBDD) approach to identify the essential amino acid interactions between the active site of a receptor/enzyme and generated ligands with low energy conformation [35]. In the present study, the computational investigations were performed using AutoDock Vina in all of the docking experiments [36]. MGL Tools 1.5.6 and Autodock Tool (ADT) were used to prepare all the protein and ligands. After removing water molecules or ligands, polar hydrogen atoms and Kollman charges were added to the protein and then saved in PDBQT format. All molecules were docked against the targets. Having a negative value of binding energy determines the stability of the ligand-receptor complex and is surmised to be a better inhibitor. PLIP webserver (Technical University of Dresden) is used to investigate the possible reasons for the examination of the formed complexes [37]. Following the molecular docking studies, we compared the energy gap values between two of the best investigated ligands. The energy gap between HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) values that represent the charge transfer in the chemical reaction [38], can be related to the chemical activity of the molecules and pharmacological activity of the therapeutic compounds [39] and subsequently the tendency to bind to a particular receptor. The energies gap of Nicotinamide riboside and Nicotinamide riboside mono phosphate has been calculated using Density functional theory (DFT) analysis [40] as well as the stabilization energies for NR and NMN. To find reliable results, full geometry optimizations to obtain energy levels of occupied (HOMOs) and unoccupied (LUMOs) orbitals of the most stable conformation of NR and NMN ligands were performed without any symmetry constraints by means of 3–21G basis set, employing the Gaussian package [41].
2.4. Molecular dynamics (MD) simulation studies
In order to validate the docking results, 70 ns Molecular Dynamics (MD) simulation was employed for two main drug-able targets of SARS-Cov-2 RdRp and Mpro in complex with NR. As a computational approach, MD is used to analyze the dynamic behavior of complexes where atoms and molecules interact as a function of time. The top-predicted docking pose of NR with the highest binding score to proteins was used as a starting point for the MD simulation. GROMACS 2019.1 software was used for all simulations applying the AMBER03 force field. All the MD simulation systems were solvated using the SPC water model. The particle mesh Ewald algorithm (PME) [42] was considered for long-range electrostatic interactions during simulation. To regulate the simulation temperature, the Berendsen thermostat process was used for 1ns at 310K. A cubic cell was generated within 0.9 Å on each side of the system and a periodic boundary condition was preferred during the simulation. During the simulation, the Fourier grid spacing, and Coulomb radius were set at 0.16 and 1.2 nm, respectively, and the van der Waals interactions were limited to 1.2 nm. The MD trajectories were saved at every 10 ps for energy stabilization. Overall, 10,000 frames were obtained from each production simulation. Molecular Mechanics/Poisson-Boltzmann Surface Area method was used for binding free energy calculation [43]. Selected snapshots from the last 20 ns MD simulation were used for two protein-ligand complexes. Binding free energy of protein and drug was calculated using the equation below:
| (1) |
where Gcomplex depicts the total free energy of the protein−ligand complex and Gprotein and Gligand are the free energies of the protein and ligand in the solvent, respectively. Moreover, the equation below was used to understand further each energy term, such as Van der Waal forces (Gvdw), electrostatics energy(Gele), polar (Gpol), and non-polar (Gnpol) interactions contributing to the total energy (G), while TS refers to the entropic contribution to the free energy in a vacuum where T and S denote the temperature and entropy, respectively.
| (2) |
3. Results and discussion
3.1. The molecular docking studies and binding mode analysis of the selected ligands against SARS-CoV-2 RdRp in complex with cofactors (PDB ID: 6M71)
Natural occurring vitamin NR and its active metabolite (NMN) were structurally similar to Favipiravir and Ribavirin. Thus, according to the Structure-Function Relationship, it was expected to have comparable antiviral activity due to a similar mechanism. Therefore, a molecular docking study was used as the first step for a structure-based investigation of this assumption. According to the binding affinities which are shown in Table 2 , The natural substrate GTP (-7.9 kcal/mol) was found to have the highest affinity among other compounds for the active site of SARS-CoV-2 RdRp (PDB ID: 6M71), followed by ATP (-7.5kcal/mol). Molecular docking studies of Favipiravir riboside, Favipiravir ribosyl monophosphate (Favipiravir-RMP), Favipiravir ribosyl triphosphate (Favipiravir-RTP), and Ribavirin triphosphate against the RdRp of SARS-CoV-2 show their ability to bind to the coronavirus strain RdRp with an approximately equal amount of energy (-7.2 kcal/mol). This study can be considered as the first investigation that a comparison between the binding energy of the activated three-phosphate forms of Ribavirin or Favipiravir and the other structural forms of these drugs such as unchanged prodrug form, mono-phosphate intermediate, etc. are presented. The docking score of NMN (-6.7 kcal/mol) was found to be 0.8 kcal/mol and 2.3 kcal/mol less than that of NR and Nicotinamide, respectively, while the hydrogen bond score of Favipiravir was found to be (-5.2) kcal/mol. Moreover, the binding energy value for Ribavirin (-6.1 kcal/mole) and Ribavirin monophosphate (-6.4) implicate the stability of the complexes (Table 2). 2D interaction diagram of the docking of studied compounds is displayed in (Figure S2). Generally, based on the literature review [15,16,19], and comparison of docking scores of the tested ligands, natural substrates, three-phosphate forms of broad-spectrum RdRp inhibitors, and also the RdRp competitive inhibitor DMB220 (-6.6 kcal/mol), it seems that NMN may create stable ligand-protein interactions with the active site of SARS-Cov-2 RdRp. Besides, Favipiravir riboside, Favipiravir-RMP, Ribavirin, and Ribavirin monophosphate have shown noticeable binding free energy and affinity to RdRp. The possible inhibitory effects of NR on RdRp can proceed via a direct inhibition or competition with purine nucleoside triphosphate GTP. The possible mutagenicity of NR through the incorporation into the viral genome as a false substrate needs further studies since the formation of a triple phosphate active metabolite of NR has not been reported yet. According to this virtual screening, apart from the NR and its active metabolite, DMB220, Favipiravir riboside, Favipiravir-RMP, and Ribavirin monophosphate also deserve more attention and evaluations as potential competitive small molecule inhibitors of (SARS-Cov-2) RdRp.
Table 2.
Molecular docking analysis of studied compounds against SARS-Cov-2 RdRp (6M71).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.4 | PHE442, ALA547, SER549, ARG553 | - | GLN444, ARG555 |
| 2 | NR | -5.9 | ASP845, ARG858 | - | PHE441, VAL844 |
| 3 | NMN | -6.7 | ARG553, ARG555, THR556, THR680, SER682, THR687, ASN691 | ARG624 | ASP623 |
| 4 | ATP | -7.5 | ASP452, THR556, TYR619, CYS622, ASP623, ASP760 | ARG553, ARG555, ARG624 | - |
| 5 | GTP | -7.9 | ARG553, THR556, TYR619, LYS621, CYS622, ASP623 | ARG553, ARG555, ARG55, ASP623, ARG624 | - |
| 6 | Favipiravir | -5.2 | ARG553, THR556, ARG624 | - | - |
| 7 | Favipiravir riboside | -7.1 | ARG553, THR556, TYR619, LYS621, CYS622, ASP623, ARG624 | - | - |
| 8 | Favipiravir-RMP | -7.2 | ARG553, THR556, TYR619, LYS621, CYS622, ASP623, ARG624 | LYS621 | - |
| 9 | Favipiravir-RTP | -7.2 | ARG553, THR556, LYS621, CYS622, ASP623 | ARG553, ARG555, ARG624 | - |
| 10 | Ribavirin | -6.1 | ARG553, THR556, LYS621, CYS622, ASP623, ARG624 | ARG553, ARG624 | - |
| 11 | Ribavirin monophosphate | -6.4 | ARG553, THR556, LYS621, CYS622, ASP623, ARG624 | ARG553, ARG624 | - |
| 12 | Ribavirin triphosphate | -7.2 | ASP452, ARG553, THR556, ASP623, SER682, ASN691, SER759 | ARG553, ARG555, ARG624 | - |
| 13 | DMB220 | -6.6 | ASP452, ARG553, THR556, LYS621, ARG624 | - | - |
3.2. The molecular docking studies and binding mode analysis of the selected ligands against SARS-CoV NSP12 bound to NSP7 and NSP8 cofactors (PDB ID: 6NUR)
Binding interactions of ligands with PDB ID 6NUR are presented in (Figure S3), and the calculated values of the binding free energy and the involved amino acid residues are shown in Table 3 . SARS-CoV RdRp interacts with Nicotinamide, NR, and NMN with dock score values of (-5.2 kcal/mol), (-6.4 kcal/mol), and (-6.8 kcal/mol) respectively. Prior to testing the physiological substrates (ATP and GTP) against SARS-CoV RdRp, these compounds exhibited binding energies between (-8.3 kcal/mol for ATP) and (−9 kcal/mol for GTP). Furthermore, the vast number of interactions between Ribavirin triphosphate and SARS-CoV RdRp gives the complex its stability displays relatively large negative ΔG value (-8.5 kcal/mol). Similarly, Ribavirin (-6.2 kcal/mol) and Ribavirin monophosphate (-7.3 kcal/mol) showed promising results in docking studies, as shown in Table 3. The calculated binding energy for Favipiravir-RMP (-7.8 kcal/mol) represents the efficiency of ligand binding to the designated receptor active site, which is slightly better than the calculated ΔG for Favipiravir-RTP (-7.5 kcal/mol) and Favipiravir riboside (-7.1 kcal/mol). Generally, in this part of our docking study, the high values of the three-phosphate forms of the broad spectrum RdRp inhibitors were expected. However, the high values of the intermediates Favipiravir riboside and Favipiravir-RMP provide new insight into the other possible mechanisms of the antiviral activity of these drugs. The same is valid for some extent about Ribavirin. Moreover, the tested ligands NR and its metabolite NMN have also shown considerable docking scores, which shows this supplement deserves further studies as a potential therapeutic agent in this novel coronavirus infection.
Table 3.
Molecular docking analysis of studied compounds against SARS-CoV RdRp (6NUR).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -5.2 | TYR456, THR556, ARG624, LYS676, SER682 | - | MET542, ALA558 |
| 2 | NR | -6.4 | TRP617, ASP760, ASP761, ALA762, CYS813, SER814 | - | - |
| 3 | NMN | -6.8 | ASP452, ARG553, ALA554, THR556 | ARG553, ARG624 | TYR455, ARG553 |
| 4 | ATP | -8.3 | TYR456, LYS551, ARG553, THR556, LYS621, ASP623, ARG624, THR680, SER682 | ARG624, LYS676 | - |
| 5 | GTP | -9 | TYR456, ARG553, ALA554, THR556, TYR619, LYS621, CYS622, ASP623, ARG624, SER682 | ASP623, ARG624 | - |
| 6 | Favipiravir | -5.9 | TYR456, THR556, ARG624, LYS676, THR680, SER682 | - | - |
| 7 | Favipiravir riboside | -7.1 | ASP452, TYR456, THR556, ARG624, LYS676, SER682 | - | - |
| 8 | Favipiravir-RMP | -7.8 | TYR456, THR556, ARG624, LYS676, SER682 | ARG553, ARG624 | - |
| 9 | Favipiravir-RTP | -7.5 | ASP452, TYR456, THR556, ASP623, ARG624, THR680, SER682 | ARG553, LYS621, ARG624 | - |
| 10 | Ribavirin | -6.2 | TYR456, LYS545, ARG553, ARG555, THR556, ARG624, SER682 | - | - |
| 11 | Ribavirin monophosphate | -7.3 | TRP617, ASP618, ASP761, ALA762, CYS813, SER814 | - | - |
| 12 | Ribavirin triphosphate | -8.5 | ASP452, TYR456, THR540, THR556, ASP623, GLU665, LYS676, THR680, SER682 | ARG553, ARG624 | - |
3.3. The molecular docking studies and binding mode analysis of the selected ligands against the crystal structure of SARS-CoV-2 Mpro in complex with the inhibitor N3 (PDB ID: 6LU7)
The predicted binding site and 2D interaction of the selected ligands for SARS-CoV-2 main protease (Mpro) or 3CLpro are shown in Figure S4. As presented in Table 4 , Favipiravir-RMP (-7.5 kcal/mol) and NMN (-7.4 kcal/mol) showed a better binding affinity with this target protein, about 0.5 kcal/mol more than Ribavirin monophosphate (-7 kcal/mol). The anticipated binding energies of Nicotinamide and NR for this target are (-4.4 kcal/mol) and (-6.4 kcal/mol), respectively. Similarly, Molecular docking analysis of Favipiravir and Favipiravir riboside against the SARS-CoV-2 Mpro revealed their interactions with binding site residues as shown in Figure S4 which was reflected in the binding energy values (-5 kcal/mole) and (-6.9 kcal/mol), respectively implicates the stability of the generated complexes (Table 4). Furthermore, the negative binding affinity value calculated by AutoDock Vina software for the docking of Ribavirin with SARS-CoV-2 Mpro (-6.3 kcal/mol) shows its ability to bind to the target. In this section of this virtual research, apart from the high-docking-score ligands NMN, Favipiravir-RMP, and Ribavirin monophosphate, the calculated docking scores of NR, Favipiravir riboside, and Ribavirin are comparable with Carmofur (-6.3 kcal/mol) which is the proved Michael acceptor inhibitor of Mpro. In addition, the same as Carmofur, the critical structural features for interaction with essential amino acid residues CYS145 and HIS41 are also present in these three successfully tested ligands [44], [45], [46].
Table 4.
Molecular docking analysis of studied compounds against Mpro (6LU7).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.4 | Met49, TYR54, GLN189, | - | HIS41 |
| 2 | NR | -6.4 | HIS41, LEU141, GLY143, SER144, CYS145 | - | - |
| 3 | NMN | -7.4 | TYR54, GLU166, GLN189 | HIS163 | MET165, GLN189 |
| 4 | Favipiravir | -5.0 | TYR54 | - | - |
| 5 | Favipiravir riboside | -6.9 | PHE140, GLY143, SER144, CYS145, GLU166 | - | - |
| 6 | Favipiravir-RMP | -7.5 | TYR54, LEU141, ASN142, GLU166, GLN189 | - | - |
| 7 | Ribavirin | -6.3 | GLY143, SER144, CYS145, HIS164, GLU166 | - | - |
| 8 | Ribavirin monophosphate | -7.0 | TYR54, ASN142, HIS172, ASP187, GLN189 | HIS163 | - |
| 9 | Carmofur | -6.3 | GLY143, SER144, CYS145 | - | - |
3.4. The molecular dynamics simulation studies of the two main drug-able targets of SARS-Cov-2 RdRp and Mpro in complex with NR
Structural parameters including RMSD, RMSF were used to evaluate the stability of protein-drug complexes. In this study, two docked complexes, including NR against Mpro and RdRp were subjected for MD simulation studies. The Root Mean Square Deviation (RMSD) of the protein backbone was calculated for all frames in the trajectory concerning the reference frame for 70 ns simulation period (Fig. 1 ). The plateau at the terminal part of the curves for the two complexes shows that the systems are in the equilibrated state and shows their stability during the simulation after 50 ns for protein-inhibitor complexes.
Fig. 1.

RMSDs of protein (a) SARS-Cov-2 RdRp (PDB:6M71). (b) SARS-Cov-2 Mpro (PDB:6LU7) in complex with NR.
Additionally, the average fluctuation of each residue was calculated as the root mean square fluctuation (RMSF) to investigate the residual vibration (Fig. 2 ). As can be expected, terminal residues of the main protease, show higher RMSF values, confirming their mobile nature (Fig. 2). RMSF for RdRp_NR showed significant fluctuation, which shows the flexibility of different regions of the protein.
Fig. 2.

The root mean square fluctuation (RMSF) plots of (a) SARS-Cov-2 RdRp (PDB:6M71). (b) SARS-Cov-2 Mpro (PDB:6LU7) in complex with NR.
Moreover, the binding free energy (ΔG) between Mpro_NR and RdRp_NR were calculated using the MM-PBSA method for the last 20 ns stable trajectories, and a total of 200 frames were considered for computation. The estimated value of ΔG is reported in Table 5 . The negative values indicate stable complex formation for inhibitor complexes. The van der Waals energy for compounds RdRp and Mpro was -73.436 and -113.772, respectively, which indicates a more specific molecular shape and stability for Rdrp_inhibitor complexes in comparison with Mpro_inhibitor.
Table 5.
Results of the binding free energy* calculation for in complex with the (a) SARS-Cov-2 RdRp (PDB:6M71). (b) SARS-Cov-2 Mpro (PDB:6LU7) in complex with the NR.
| Compound | ΔG binding energy | ΔG Vdw | ΔG elec | ΔG polar |
|---|---|---|---|---|
| (a) | -68.33838 ± 10.56 | -73.436 ± 17.54 | -88.42± 22.37 | 39.64 ± 18.74 |
| (b) | -45.45 ± 3.03 | -27.17 ± 2.81 | -40.05 ± 4.45 | 24.55 ± 4.8 |
* All energies are in kcal/mol.
3.5. The molecular docking studies and binding mode analysis of the selected ligands against SARS CoV-2 Papain-like protease (PLpro) in complex with peptide inhibitor VIR251 (PDB ID: 6WX4)
Docking simulation methods have been applied to predict the binding mode and the affinity of Nicotinamide, NR, and NMN, within the active/binding site of the receptor of interest (PDB ID: 6WX4). According to the literature, Lopinavir has been chosen as the reference ligand/inhibitor of the SARS-CoV-2 PLpro to interact with the binding pocket residues [33,34]. In Table 6 , we have listed the docking results of these four selected ligands wrapped into the binding/active site of SARS CoV-2 PLpro. Lopinavir has shown the highest binding energy among studied compounds (-7.3kcal/mol). Bothe molecules, NR and NMN presented approximately the same score of binding, (-6.4kcal/mol) and (-6.5kcal/mol) respectively. Nicotinamide has shown a higher score of binding (-4.5 kcal/mol) with the involved binding amino acid residues. Visualization of the molecular interaction of the ligands with the receptor (Figure S5) shows their location and the type of connections in the active site of the protein.
Table 6.
Molecular docking analysis of studied compounds against PLpro (6WX4).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.5 | ARG166, TYR273 | - | THR301 |
| 2 | NR | -6.4 | GLY163, ASP164, GLN269, GLY271, TYR273 | - | TYR268 |
| 3 | NMN | -6.5 | GLY163, ASP164, TYR264, TYR268, CYS270, GLY271, TYR273, THR301 | - | TYR264, TYR268 |
| 4 | Lopinavir | -7.3 | TYR268, GLN269 | - | LEU162, PRO247, PRO248, ASN267, TYR268, THR301 |
3.6. The molecular docking studies and binding mode analysis of the selected ligands against human type II Inosine Monophosphate Dehydrogenase (IMPDH) (PDB ID: 1NF7)
We set out docking simulations to examine the binding of NMN, Favipiravir-RMP, and Ribavirin monophosphate on 1NF7. In addition to the latter, Mizorbine monophosphate (the active metabolite of immunosuppressant agent Mizorbine) was added to this part of the research as the reference substrate [47]. 2D visualization of the interaction between the protein with studied molecules can be seen in Figure S6. Docking analysis yields high binding affinities, ranging from (-8.6 kcal/mol) to (-8.9 kcal/mol) for all studied ligands (Table 7 ). The vast number of hydrogen interactions with IMPDH in the binding pocket, as shown in Figure S6, causes the stability of the complexes. The inhibition of IMPDH prevents interleukin-6 production [48]. This effect also can suppress virus replication [49]. Therefore, IMPDH inhibitors deserve more attention as COVID-19 therapeutic agent [26]. In this regard, this docking study hypothesizes that the compound NR as monophosphate metabolite may be effective in COVID-19 patients’ management via inhibition of IMPDH. Based on the experimental studies, this docking research proposes that the mechanism of anti-COVID-19 efficiency of Ribavirin [19] may occur through the inhibitory effects on IMPDH. Furthermore, Favipiravir showed weaker ligand-protein interactions, and about 0.3 and 0.1 kcal/mol higher free energy of binding than Mizoribine and Ribavirin, respectively, which these docking results are consistent with the results of experimental researches [50].
Table 7.
Molecular docking analysis of studied compounds against IMPDH (1NF7).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | Hydrogen bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | NMN | -8.7 | SER68, PRO69, ASN303, ARG322, GLY326, SER327, GLY328, ASP364, GLY365, SER388, TYR411 | - | - |
| 2 | Favipiravir-RMP | -8.6 | SER68, ASP274, SER275, ASN303, GLY328, MET414, GLY415 | ARG322 | - |
| 3 | Mizoribine monophosphate | -8.9 | SER68, ASN303, GLY326, SER327, GLY328, SER329, ILE330, ASP364, GLY365, GLY366, ILE367, GLY387, SER388, TYR411 | - | - |
| 4 | Ribavirin monophosphate | -8.7 | SER68, ASN303, ARG322, GLY324, GLY326, SER327, GLY328, SER329, ASP364, GLY366, SER388, TYR411 | - | - |
3.7. The molecular docking studies and binding mode analysis of the selected ligands against crystal structure of SARS-CoV-2 Spike receptor-binding domain bound with ACE2 (PDB ID: 6M0J)
In the current study, Nicotinamide, NR, Favipiravir, and Ribavirin were evaluated against the interface region of SARS-CoV-2 Spro-RBD (receptor binding domain) in complex with the host ACE2 receptor (Fig. 3 ) to find the probable molecular interactions [51]. According to the literature, and considering the target-ligand complex structures of the utilized PDB IDs in our research, the grid coordinate points were predefined. Regarding the PDB ID 6M0J, the interface region between the spike RBD and human ACE2 has been taken and targeted. The 2D interactions of the studied molecules with different protein residues of spike and ACE2 can be found in Figures S7, S8. The docking results (inserted into Tables 8 and 9 ) show the calculated binding data of the four above-mentioned molecules yielded binding affinities ranging from (-4.8 kcal/mol) to (-6 kcal/mol). Among these studied molecules, NR and Ribavirin yielded higher binding affinities towards these targets. As shown in Table 8, NR and Ribavirin have free energy values of -6 kcal/mol and -5.8 kcal/mol, respectively, against ACE2. According to the reflected data in Table 9, the mentioned compounds possess free energy values of -5.8 kcal/mol and -5.5 kcal/mol against Spike receptor respectively. It seems the lower binding affinities of Favipiravir (-4.8 kcal/mol against both Spike and ACE2 receptors) and Nicotinamide (with free binding energy values of -4.4 kcal/mol against Spike and -4.9 kcal/mol against ACE2) to be a natural consequence of their small sizes. As it can be seen in Fig. 4 , the selected ligands bind at the interface region of Spike-RBD and ACE2 and having the potential to inhibit the virus entry inside the host cells. Briefly, in this section of our docking study, we used PDB ID 6M0J (Spike-ACE2 complex) and the junction portion between the Spro-RBD and human ACE2 receptor. In the first phase, we eliminated the Spro as a ligand, then targeted the ACE2 and vice versa at the next phase (Figures S7 and S8). According to the resulted scores, NR can be considered as a primitive ligand for the human ACE2 receptor; therefore, a protective agent against SARS-Cov-2 induced infection via inhibition of the viral entrance.
Fig. 3.

2D interaction of studied ligands with SARS-CoV-2 Spro and human ACE2 receptors. A: Spike + Ligands. B: ACE2 + Ligands. C: Ligands at ACE2 and Spike interaction region.
Table 8.
Molecular docking analysis of studied compounds against human ACE2 (6M0J).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.9 | ASP350, ARG393 | - | PHE390 |
| 2 | NR | -6 | ASP350, TYR385, ARG393, ASN394 | - | PHE40, PHE390 |
| 3 | Favipiravir | -4.8 | GLY352, ARG393 | - | - |
| 4 | Ribavirin | -5.8 | TYR385, ARG393 | - | - |
Table 9.
Molecular docking analysis of studied compounds against SARS-CoV-2 Spro (6M0J).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.4 | TYR453 | - | ARG403, TYR495 |
| 2 | NR | -5.8 | TYR453, SER494, GLY496, ASN501 | - | ARG403, PHE497, TYR505 |
| 3 | Favipiravir | -4.8 | GLY496, ASN501 | - | - |
| 4 | Ribavirin | -5.5 | ARG403, TYR449, TYR453, GLN498 | - | - |
Fig. 4.

The interactions are demonstrating binding positions of NR after docking studies. (A): NR against SARS-CoV-2 RdRp (6M71). (B): NR against SARS-Coronavirus NSP12 bound to NSP7 and NSP8 cofactors (6NUR). (C): NR against the SARS-CoV-2 main protease (6LU7). (D): NR against SARS CoV-2 Papain-like protease (6WX4). (E): NR against SARS-CoV-2RBD-ACE2 (6M0J). (F): NR against SARS-CoV-2 spike receptor (6M0J). (G): NR against Perfusion SARS-CoV-2 spike receptor (6VSB).
3.8. The molecular docking studies and binding mode analysis of the selected ligands against perfusion SARS-CoV-2 Spike glycoprotein (PDB ID: 6VSB)
Here the molecular docking studies were carried out for the Nicotinamide, NR, Favipiravir, and Ribavirin against coronavirus Spro to identify the probable molecular interactions serendipitously (Figure S9). According to the resulted scores, all molecules showed binding interactions with SARS-CoV-2 Spro, as can be seen in Table 10 . The highest binding affinity was seen for NR (-5.7 kcal/mol), about 1kcal/mol better than Ribavirin (-5.6 kcal/mol), while Nicotinamide shows the lowest binding affinity (-4.5kcal/mol) towards SARS-CoV-2 Spro. Similarly, the negative free energy calculation indicates the interaction of Favipiravir with SARS-CoV-2 Spro (-5.2 kcal/mole). The number of hydrogen bonds and amino acid residues of the target interacting with each molecule are given in Table 10.
Table 10.
Molecular docking analysis of studied compounds against SARS-CoV-2 Spro (6VSB).
| Ligand No | Ligands | Free Energy of Binding (kcal/mol) | H-bond | Salt Bridge | Hydrophobic |
|---|---|---|---|---|---|
| 1 | Nicotinamide | -4.5 | SER373 | - | PHE338, PHE342, LEU368 |
| 2 | NR | -5.7 | GLY339, ASP364, SER371, SER373 | - | PHE338, PHE342, LEU368 |
| 3 | Favipiravir | -5.2 | VAL341, ASN354, ALA397, SER399 | - | - |
| 4 | Ribavirin | -5.6 | ALA344, TRP436, ASN440 | - | - |
Generally, Considering the docking results of NR and the active metabolite NMN (Table 11 ) and the obtained binding modes (Fig. 4), which are confirmed by the obtained results from MD evaluations at the main target of our research SARS-CoV-2 RdRp, NR can represent a structural pattern for an efficient anti- SARS-CoV-2 scaffold and as a potential therapeutic agent for the management of COVID-19 patients deserves further studies. In this research, NMN revealed the comparable energy values versus the best endogenous ligand GTP against the main chosen targets SARS-CoV, and SARS-CoV-2 RdRp enzymes. NMN and GTP also share several hydrogen bonds involving amino acids at the related active sites (Table 11, Rows 1a-2c). Regarding the protease enzymes, NMN displayed hydrophobic and hydrogen bonding interactions with the critical amino acid residues (Cys145 and His41 of Mpro, and Asp164, Tyr264, Tyr268, Tyr273, and Thr301 of PLpro). However, the exact nature of these observed interactions cannot be identified and estimated only through this virtual evaluation (Table 11, Rows 3a-4c).
Table 11.
The molecular docking analysis of the best/reference ligand of each selected target versus NR and its active metabolite at a glance.
| No | Targets | Ligands | Confirmed/Proposed active/binding sites involved amino acids |
Free Energy of Binding (kcal/mol) | ||
|---|---|---|---|---|---|---|
| H-bond | Salt Bridge | Hydrophobic | ||||
| 1a | 6M71 | GTP* | ARG553, THR556, TYR619, LYS621, CYS622, ASP623 | ARG553, ARG555, ARG55, ASP623, ARG624 | - | -7.9 |
| 1b | NR | ASP845, ARG858 | - | PHE441, VAL844 | -5.9 | |
| 1c | NMN | ARG553, ARG555, THR556, THR680, SER682, THR687, ASN691 | ARG624 | ASP623 | -6.7 | |
| 2a | 6NUR | GTP* | TYR456, ARG553, ALA554, THR556, TYR619, LYS621, CYS622, ASP623, ARG624, SER682 | ASP623, ARG624 | - | -9 |
| 2b | NR | TRP617, ASP760, ASP761, ALA762, CYS813, SER814 | - | - | -6.4 | |
| 2c | NMN | ASP452, ARG553, ALA554, THR556 | ARG553, ARG624 | TYR455, ARG553 | -6.8 | |
| 3a | 6LU7 | Carmofur* | GLY143, SER144, CYS145 | - | - | -6.3 |
| 3b | NR | HIS41, LEU141, GLY143, SER144, CYS145 | - | - | -6.4 | |
| 3c | NMN | TYR54, GLU166, GLN189 | HIS163 | MET165, GLN189 | -7.4 | |
| 4a | 6WX4 | Lopinavir* | TYR268, GLN269 | - | LEU162, PRO247, PRO248, ASN267, TYR268, THR301 | -7.3 |
| 4b | NR | GLY163, ASP164, GLN269, GLY271, TYR273 | - | TYR268 | -6.4 | |
| 4c | NMN | GLY163, ASP164, TYR264, TYR268, CYS270, GLY271, TYR273, THR301 | - | TYR264, TYR268 | -6.5 | |
| 5a | 1NF7 | Mizoribine monophosphate* | SER68, ASN303, GLY326, SER327, GLY328, SER329, ILE330, ASP364, GLY365, GLY366, ILE367, GLY387, SER388, TYR411 | - | - | -8.9 |
| 5b | NMN | SER68, PRO69, ASN303, ARG322, GLY326, SER327, GLY328, ASP364, GLY365, SER388, TYR411 | - | - | -8.7 | |
| 6 | 6M0J-spro | NR | TYR453, SER494, GLY496, ASN501 | - | ARG403, PHE497, TYR505 | -5.8 |
| 7 | 6M0J-ACE2 | NR | ASP350, TYR385, ARG393, ASN394 | - | PHE40, PHE390 | -6 |
| 8 | 6VSB | NR | GLY339, ASP364, SER371,SER373 | - | PHE338, PHE342, LEU368 | -5.7 |
* Reference ligands and the identical involved amino acids between the reference ligands and the studied ligands can be found in bold.
The optimized geometry and the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of NR and NMN were calculated, and the related diagrams are presented in Figures S10 and S11. The most relevant HOMO and LUMO orbitals required in electronic transitions in α and β spin states might be designated for comparative reactivity assignment. Although the calculated energy gaps for NR and NMN ligands were 232 and 184 kcal, respectively, NMN had shown a slightly higher activity than NR, which is following the slightly narrower band energy gap of NMN obtaining from the docking study (Table 2, Table 3, Table 4 and 6).
Considering the approximately identical interactions of NMN and Mizoribine monophosphate against the IMPDH, it also raises the need for further investigation into this possible mechanism (Table 11, Rows 5a and 5b). Besides, the resulted data from the molecular docking studies of NR against Spro and ACE2 receptors by targeting their interface region has demonstrated the potential effectiveness of the discussed structural pattern in this research (Table 11, Rows 6-8).
The experimental studies support molecular docking studies on a particular target at molecular levels, such as mass spectroscopy of ligand-target complexes to characterize the interactions, and the binding modes [52], further x-ray crystallography identifications following successful cellular assessments. This strategy can determine the best compounds and their binding modes, which can be used for future in silico investigations and virtual screening approaches.
In the current research, among the selected targets of SARS-CoV-2, the main protease has revealed confirmed engagement with specific ligands through covalent and, or other kinds of chemical interactions with the catalytic pair amino acid residues Cys145 as the nucleophilic center and His41 as the general acid/base or a π–π interaction creator group (Table 11 and Fig. 4). These established binding modes can be considered as the best critical and determinative structural features of the existing peptide-like and small molecule Mpro inhibitors [52], [53], [54], [55], [56]. The defined binding modes have been determined for the investigated ligands consistent with their affinity energy values (Table 4). On the other hand, although Favipiravir and Ribavirin both are distinguished via mutagenic and non-mutagenic antiviral activity through viral RdRp inhibition by their three-phosphate metabolites, based on the resulted data from the current research, different assumed mechanisms of action are probable for these drugs [57,58]. Here, the binding affinity and the docking energy of different non-three phosphate forms of Favipiravir and Ribavirin were also investigated and discussed. These theoretical results raise the possibility that the only active form of these drugs may not be their triphosphate metabolites, and, it creates a new insight about the other possible mechanisms of antiviral activity of Favipiravir and Ribavirin, that seems necessary to be subject for further investigations.
4. Conclusion
Regardless of the extensive researches, in the real battlefield against SARS-CoV-2, a broad spectrum of medicinal agents such as antibiotics, antimalarial drugs, non-specific antivirals, anticoagulants, etc., have been prescribed as therapeutic cocktails to save people's lives. At the primitive phase of a higher level, computational drug design and discovery approaches rationalize our attempts to find COVID-19 treatment via virtually targeting the SARS CoV-2 structural and functional constituents in screening processes. In the current study, the resulted scores from molecular docking and dynamics simulations as the primary determinative factor as well as the observed reliable binding modes have demonstrated that Nicotinamide Riboside and its active metabolite NMN can target human ACE2 and IMPDH, along with the viral Spro, Mpro, PLpro, and on top of all, RdRp as a potential competitive inhibitor. Besides, Nicotinamide Riboside is one of the significant nicotinamide adenine dinucleotide (NAD+) precursors, which possess significant health benefits such as anti-inflammatory effects by reducing the expression of inflammatory markers such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 that are responsible for the severity of COVID-19. Thus, the well-tolerated commercially available diary supplement Nicotinamide Riboside can be considered, as a structural analogue of Ribavirin and Favipiravir riboside and its active metabolite NMN, may be beneficial against COVID-19 as potential multi-functional therapeutic agents to be prescribed and, or naturally occurred multi-target scaffolds for further studies.
CRediT authorship contribution statement
Zohreh Esam: Conceptualization, Visualization, Writing – original draft. Malihe Akhavan: Methodology, Data curation. Maryam lotfi: Software, Validation, Investigation. Ahmadreza Bekhradnia: Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.131394.
Appendix. Supplementary materials
References
- 1.Kadali R.A., Janagama R., Peruru S., Malayala S.V. Side effects of BNT162b2 mRNA COVID-19 vaccine: A randomized, cross-sectional study with detailed self-reported symptoms from healthcare workers. Int. J. Infect. Dis. 2021 May 1;106:376–381. doi: 10.1016/j.ijid.2021.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poland G.A., Ovsyannikova I.G., Crooke S.N. SARS-CoV-2 vaccine development: current status. InMayo Clinic Proceedings. Elsevier; 2020 Jul 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noh J.Y., Jeong H.W., Shin E.C. SARS-CoV- 2 mutations, vaccines, and immunity: implication of variants of concern. Signal Transduct. Target. Ther. 2021 May 22;6(1):1–2. doi: 10.1038/s41392-021-00623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostoff R.N., Briggs M.B., Porter A.L., Spandidos D.A., Tsatsakis A. COVID‑19 vaccine safety. Int. J. Mol. Med. 2020 Nov 1;46(5):1599–1602. doi: 10.3892/ijmm.2020.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernández A.F., Calina D., Poulas K., Docea A.O., Tsatsakis A.M. Safety of COVID-19 vaccines administered in the EU: Should we be concerned? Toxicol. Rep. 2021 Jan 1;8:871–879. doi: 10.1016/j.toxrep.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anand P., Stahel V.P. Review the safety of Covid-19 mRNA vaccines: a review. Patient safety in surgery. 2021 Dec;15(1):1–9. doi: 10.1186/s13037-021-00291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asakura H., Ogawa H. Potential of heparin and nafamostat combination therapy for COVID-19. J. Thromb. Haemost. 2020 Jun;18(6):1521–1522. doi: 10.1111/jth.14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.S. El Kantar, B. Nehmeh, P. Saad, G. Mitri, C. Estephan, M. Mroueh, E. Akoury, R.I Taleb, Derivatization and combination therapy of current COVID-19 therapeutic agents: a review of mechanistic pathways, adverse effects, and binding sites. Drug Discovery Today. 2020 Aug 12. doi: 10.1016/j.drudis.2020.08.002. [DOI] [PMC free article] [PubMed]
- 9.K.B.Pandeya, A. Ganeshpurkar, M.K., Mishra Natural RNA Dependent RNA Polymerase Inhibitors: Molecular Docking Studies of Some Biologically Active Alkaloids of Argemone mexicana. Medical Hypotheses. 2020 Jun 1:109905. doi: 10.1016/j.mehy.2020.109905. [DOI] [PMC free article] [PubMed]
- 10.Wang G., Wan J., Hu Y., Wu X., Prhavc M., Dyatkina N., Rajwanshi V.K., Smith D.B., Jekle A., Kinkade A., Symons J.A. Synthesis and anti-influenza activity of pyridine, pyridazine, and pyrimidine C-nucleosides as favipiravir (T-705) analogueues. J. Med. Chem. 2016 May 26;59(10):4611–4624. doi: 10.1021/acs.jmedchem.5b01933. [DOI] [PubMed] [Google Scholar]
- 11.Vanderlinden E., Vrancken B., Van Houdt J., Rajwanshi V.K., Gillemot S., Andrei G., Lemey P., Naesens L. Distinct effects of T-705 (favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob. Agents Chemother. 2016 Nov 1;60(1):6679–6691. doi: 10.1128/AAC.01156-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arshadi S., Bekhradnia A.R., Ebrahimnejad A. Feasibility study of hydrogen-bonded nucleic acid base pairs in gas and water phases — A theoretical study. Can. J. Chem. 2011;89:1403–1409. doi: 10.1139/v11-124. [DOI] [Google Scholar]
- 13.W. Zhang, P. Stephen, J.F. Thériault, R. Wang, S.X. Lin, Novel Coronavirus Polymerase and Nucleotidyl-Transferase Structures: Potential to Target New Outbreaks. The journal of physical chemistry letters. 2020 May 11. doi: 10.1021/acs.jpclett.0c00571. [DOI] [PubMed]
- 14.Qiu L., Patterson S.E., Bonnac L.F., Geraghty R.J. Nucleobases and corresponding nucleosides display potent antiviral activities against dengue virus possibly through viral lethal mutagenesis. PLoS Negl.Trop. Dis. 2018 Apr 19;12(4) doi: 10.1371/journal.pntd.0006421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.A.A. Elfik, Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life sciences. 2020 Feb 28:117477. doi: 10.1080/07391102.2020.1761882. [DOI] [PMC free article] [PubMed]
- 16.W. Zhang, P. Stephen, J.F. Thériault, R. Wang, S.X. Lin, Novel Coronavirus Polymerase and Nucleotidyl-Transferase Structures: Potential to Target New Outbreaks. The journal of physical chemistry letters. 2020 May 11. doi: 10.1021/acs.jpclett.0c00571. [DOI] [PubMed]
- 17.Han X., Fan Y., Wan Y.L., Shi H. A diabetic patient with 2019-nCoV (COVID-19) infection who recovered and was discharged from hospital. J. Thorac. Imaging. 2020 May 1:35. doi: 10.1097/RTI.0000000000000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life sciences. 2020 Feb 28:117477. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed]
- 19.Ribavirin E.A.A., Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life sciences. 2020 Mar 25:117592. doi: 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed]
- 20.Choudhury S., Moulick D., Saikia P., Mazumder M.K. Evaluating the potential of different inhibitors on RNA-dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2: A molecular modeling approach. Med. J. Armed Forces India. 2020 May 30 doi: 10.1016/j.mjafi.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day F.R., Forouhi N.G., Ong K.K., Perry J.R. Season of birth is associated with birth weight, pubertal timing, adult body size and educational attainment: a UK Biobank study. Heliyon. 2015 Oct 1;1(2):e00031. doi: 10.1016/j.heliyon.2015.e00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchbauer G., Klinsky A., Weiß-Greiler P., Wolschann P. Ab Initio molecular electrostatic potential grid maps for quantitative similarity calculations of organic compounds. Molecular model. annual. 2000 Apr;6(4):425–432. [Google Scholar]
- 23.Trammell S.A., Schmidt M.S., Weidemann B.J., Redpath P., Jaksch F., Dellinger R.W., Li Z., Abel E.D., Migaud M.E., Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016 Oct 10;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martens C.R., Denman B.A., Mazzo M.R., Armstrong M.L., Reisdorph N., McQueen M.B., Chonchol M., Seals D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD (+) in healthy middle-aged and older adults. Nat. Commun. 2018;9(1):1286. doi: 10.1038/s41467-018-03421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu H.T., Colby-Germinario S.P., Hassounah S., Quashie P.K., Han Y., Oliveira M., Stranix B.R., Wainberg M.A. Identification of a pyridoxine-derived small-molecule inhibitor targeting dengue virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2016 Jan 1;60(1):600–608. doi: 10.1128/AAC.02203-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed]
- 27.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020 May 15;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirchdoerfer R.N., Ward A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 cofactors. Nat. Commun. 2019 May 28;10(1):1–9. doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X., Zhang B., Jin Z., Yang H., Rao Z. The crytal structure of 2019-nCoV main protease in complex with an inhibitor N3. RCSB Protein Data Bank. 2020 doi: 10.2210/pdb6LU7/pdb. [DOI] [Google Scholar]
- 30.Berry J.D., Jones S., Drebot M.A., Andonov A., Sabara M., Yuan X.Y., Weingartl H., Fernando L., Marszal P., Gren J., Nicolas B. Development and characterisation of neutralising monoclonal antibody to the SARS-coronavirus. J. Virol. Methods. 2004 Sep 1;120(1):87–96. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu T.Y., Peng Y., Pelleymounter L.L., Moon I., Eckloff B.W., Wieben E.D., Yee V.C., Weinshilboum R.M. Pharmacogenetics of the mycophenolic acid targets inosine monophosphate dehydrogenases IMPDH1 and IMPDH2: gene sequence variation and functional genomics. Br. J. Pharmacol. 2010 Dec;161(7):1584–1598. doi: 10.1111/j.1476-5381.2010. 00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020 May;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 33.W. Rut, Z, Lv, M Zmudzinski, S. Patchett, D. Nayak, S.J. Snipas, F. El Oualid, M. Bekes, T.T. Huang, M. Drag, S.K. Olsen, Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv. 2020 Jan. doi: 10.1101/2020.04.29.068890. [DOI] [PMC free article] [PubMed]
- 34.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020 Apr;92(4):418–423. doi: 10.1016/j.bmcl.2020.127377. [27]Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg & Med Chem Lett. 2020 Jul 2:127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg & Med Chem Lett. 2020 Jul 2 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonio C., Chetan C., Gorman A.M., Afshin S., Eriksson L.A. Merits and pitfalls of conventional and covalent docking in identifying new hydroxyl aryl aldehyde like compounds as human IRE1 inhibitors. Scientific Reports (Nature Publisher Group) 2019 Dec 1;9(1) doi: 10.1038/s41598-019-39939-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vina A. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading Trott, Oleg; Olson. Arthur J. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.George J., Prasana J.C., Muthu S., Kuruvilla T.K., Saji R.S. Evaluation of vibrational, electronic, reactivity and bioactivity of propafenone—A spectroscopic, DFT and molecular docking approach. Chemical Data Collections. 2020 Apr 1;26 doi: 10.1016/j.cdc.2020.100360. [DOI] [Google Scholar]
- 39.Majeed A., Hussain W., Yasmin F., Akhtar A., Rasool N. Virtual Screening of Phytochemicals by Targeting HR1 Domain of SARS-CoV-2 S Protein: Molecular Docking, Molecular Dynamics Simulations, and DFT Studies. Biomed. Res. Int. 2021;2021 doi: 10.1155/2021/6661191. Article ID 6661191, 19 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tikhonov S.A., Sidorin A.E., Samoilov I.S., Borisenko A.V. Vovna VI. Photoelectron spectra and electronic structure of boron diacetate formazanates. Spectrochim. Acta Part A. 2020 Sep 5;238 doi: 10.1016/j.saa.2020.118441. [DOI] [PubMed] [Google Scholar]
- 41.Bekhradnia A., Norrby P.-O. New insights into the mechanism of iron-catalyzed cross-coupling reactions. Dalton Transactions. 2015;44:3959–3962. doi: 10.1039/c4dt03491k. [DOI] [PubMed] [Google Scholar]
- 42.Hou T., Wang J., Li Y., Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011 Jan 24;51(1):69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993 Jun 15;98(12):10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 44.Sebastian S., Sven S., Joachim HV. Adasme Melissa F, and Schroeder Michael. Plip: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X., Duan Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020 Jun;27(6):529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 46.Li X.F., Zhang L., Duan Y., Yu J., Wang L., Yang K., Liu F., You T., Liu X., Yang X., Bai F. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 47.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020 Jul 2 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saijo M., Morikawa S., Fukushi S., Mizutani T., Hasegawa H., Nagata N., Iwata N., Kurane I. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)-associated coronavirus. Antiviral Res. 2005 Jun 1;66(2-3):159–163. doi: 10.1016/j.antiviral.2005.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugiyama E., Ikemoto M., Taki H., Maruyama M., Yamashita N., Kobayashi M. Mizoribine, an inhibitor of inosine monophosphate dehydrogenase, inhibits interleukin-6 production by freshly prepared rheumatoid synovial cells. Modern rheumatol. 2001 Mar 1;11(1):28–33. doi: 10.1007/s101650170040. [DOI] [PubMed] [Google Scholar]
- 50.Hu J., Ma L., Wang H., Yan H., Zhang D., Li Z., Jiang J., Li Y. A novel benzo-heterocyclic amine derivative N30 inhibits influenza virus replication by depression of Inosine-5’-Monophospate Dehydrogenase activity. Virol. J. 2017 Dec;14(1):1–9. doi: 10.1186/s12985-017-0724-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gurung A.B., Ali M.A., Lee J., Farah M.A., Al-Anazi K.M. Identification of potential SARS-CoV-2 entry inhibitors by targeting the interface region between the spike RBD and human ACE2. J. Infection and Public Health. 2020 Dec 21 doi: 10.1016/j.jiph.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X., Qin S., Chen S., Li J., Li L., Wang Z., Wang Q., Lin J., Yang C., Shui W. A ligand-observed mass spectrometry approach integrated into the fragment based lead discovery pipeline. Sci. Rep. 2015 Feb 10;5:8361. doi: 10.1038/srep08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanhed A.M., Patel D.V., Teli D.M., Patel N.R., Chhabria M.T., Yadav M.R. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol. Divers. 2020 Jul 31:1–9. doi: 10.1007/s11030-020-10130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X.F., Zhang L., Duan Y., Yu J., Wang L., Yang K., Liu F., You T., Liu X., Yang X., Bai F. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 55.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X., Duan Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020 Jun;27(6):529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 56.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorganic & medicinal chemistry letters. 2020 Jul 2:127377. doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed]
- 57.Furuta Y., Takahashi K., Shiraki K., Sakamoto K., Smee D.F., Barnard D.L., Gowen B.B., Julander J.G., Morrey J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009 Jun 1;82(3):95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomar S., Kaur R., Singh V.A. InViral Polymerases. Academic Press; 2019 Jan 1. Structure–Function Relationship of Negative-Stranded Viral RNA Polymerases: Prospectives for Antiviral Therapy; pp. 43–67. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.