

Abstract
Steroid receptor coactivators (SRCs) possess specific and distinct oncogenic roles in the initiation of cancer and in cancer progression to a more aggressive disease. These coactivators interact with nuclear receptors and other transcription factors to boost transcription of multiple genes which potentiate cancer cell proliferation, migration, invasion, tumor angiogenesis and epithelial mesenchymal transition (EMT). Targeting SRCs using small molecule inhibitors (SMIs) is a promising approach to control cancer progression and metastasis. By high throughput screening analysis, we recently identified SI-2 as a potent SRC SMI. To develop therapeutic agents, SI-10 and SI-12, the SI-2 analogs, are synthesized that incorporate the addition of fluorine atoms to the SI-2 chemical structure. As a result, these analogs exhibit a significantly prolonged plasma half-life, minimal toxicity and improved hERG activity. Biological functional analysis showed that SI-10 and SI-12 treatment (5–50 nM) can significantly inhibit viability, migration and invasion of breast cancer cells in vitro and repress the growth of breast cancer PDX organoids. Treatment of mice with 10 mg/kg/day of either SI-10 or SI-12 was sufficient to repress growth of xenograft tumors derived from MDA-MB-231 and LM2 cells. Furthermore, in spontaneous and experimental metastasis mouse models developed from MDA-MB-231 and LM2 cells respectively, SI-10 and SI-12 effectively inhibited progression of breast cancer lung metastasis. These results demonstrate that SI-10 and SI-12 are promising therapeutic agents and are specifically effective in blocking tumor metastasis, a key point in tumor progression to a more lethal state that results in patient mortality in the majority of cases.
Keywords: Steroid receptor coactivator, SI-10, SI-12, small molecule inhibitor, breast cancer progression and metastasis
Introduction
Steroid receptor coactivators SRC-1, −2 and −3 comprise the SRC (p160) coactivator family. These coactivators interact with nuclear receptors and other transcription factors (TFs), recruiting additional coactivators and general transcription factors to remodel chromatin, assemble general transcription factors and recruit RNA polymerase II to activate gene transcription (1–4). Clinical evidence has shown that SRC-1 and SRC-3 are overexpressed in human breast cancers and their overexpression correlates with Her2 activation, endocrine therapy resistance, disease recurrence and poor prognosis (Fleming et al. 2004; Torres-Arzayus et al. 2004; Redmond et al. 2009). Genetic deletion of SRC-1 or SRC-3 significantly reduces mammary tumor lung metastasis in MMTV-PyMT mouse models (Qin et al. 2008a; Wang et al. 2009), while overexpression of SRC-1 significantly increases mammary tumor lung metastasis in a MMTV-neu mouse model (Qin et al. 2014a). In prostate cancer, SRC-1 amplification in cancer cells is rare. However the expression level of SRC-1 positively correlates with tumor grade (Agoulnik et al. 2005). More commonly, SRC-2 expression is elevated in patients with metastatic prostate cancer, and the frequency is even higher (Taylor et al. 2010). Overexpression of SRC-2 in mouse prostate epithelium resulted in neoplasia (Qin et al. 2014b) and inhibition of SRC-2 expression in murine models severely attenuated the survival, growth and metastasis of prostate cancer (Dasgupta et al. 2015). SRC-3 is overexpressed in prostate cancer as well and its expression positively correlates with elevated PSA expression (Zhou et al. 2005). Mechanistically, SRC-3 interacts with multiple TFs to up-regulate transcription of TNF Receptor Associated factor-4 (TRAF4), pro-inflammatory protein macrophage Migration Inhibitor Factor (MIF), and Matrix Metalloprotease Protein (MMP)-2 and −9. These gene products abrogate p53 function and promote tumorigenesis, angiogenesis and cancer cell invasion (Torres-Arzayus et al. 2004; Qin et al. 2008a; Xu et al. 2009; Yi et al. 2013). Long et al. reported that a splicing isoform of SRC-3, SRC-3Δ4 can mediate Epidermal Growth Factor Receptor (EGFR) and Focal Adhesion Kinase (FAK) interaction, promoting EGF-induced FAK and c-Src phosphorylation and breast cancer cell migration (Long et al. 2010). Conversely, SRC-1 does not potentiate primary tumor growth, but specifically and strongly promotes breast cancer metastasis to the lung (Wang et al. 2009; Qin et al. 2014a). SRC-1 serves as a coactivator for different TFs to up-regulate the expression of Twist1, ITGA5, SDF1 and CSF-1 that drive breast cancer cell epithelial-mesenchymal transition (EMT), migration, invasion, macrophage recruitment and lung metastasis (Kishimoto et al. 2005; Qin et al. 2009, 2011, 2014a). Taken together, these studies strongly support the concept that targeting the expression of the family of SRCs in cancer cells could be a powerful way to control cancer progression and metastasis.
By high-throughput screening analysis, we identified SRC small molecular inhibitors including 2,2’-bis-(Formyl-1,6,7-trihydroxy-5-isopropyl-3-methylnaphthalene (gossypol), the cardiac glycoside bufalin and verrucarin A (Wang et al. 2011, 2014; Yan et al. 2014). These inhibitors were able to reduce cellular protein concentration of SRCs and/or promote SRC-3 degradation in multiple cancer cells, disturb the intrinsic transcription activities of SRCs and inhibit cancer cell growth in vitro and tumor growth in vivo. These studies established the proof-of-principle that SRCs are accessible chemotherapeutic targets. SI-2 was recently identified as a more promising inhibitor candidate which meets all of the criteria of Lipinski’s rule for a drug-like molecule (Song et al. 2016). SI-2 treatment inhibited proliferation of MCF-7 breast cancer cells and strongly downregulated SRC-3 protein levels in cancer cells. In a xenograft mouse model, SI-2 treatment significantly inhibited mammary tumor growth (Song et al. 2016). However, its short half-life in vivo limits the potential of SI-2 as a therapeutic agent (Song et al. 2016).
To develop therapeutic SRC SMIs, we introduced two or three fluorine atoms respectively to the SI-2 core structure to generate two novel SI-2 analogs, SI-10 and SI-12. These new compounds possess many favorable properties, including reduced human ether-a-go-go (hERG) channel activity, better pharmacokinetic properties and improved efficacy, which endow these analogs with the potential to be leading therapeutic agents. In this study, we assessed the pharmacological features of these novel compounds in hERG activity assays, pharmacokinetic (PK) assays and by toxicity testing in vivo. We also systemically evaluated the impact of these compounds on cancer cell malignancy and PDX organoid growth as well as their therapeutic efficacy on tumor growth and metastasis in mouse model systems.
Materials and Methods
Cells, animals and PDX tumors
Breast cancer cell lines MDA-MB-231 (HTB-26™), LM2 (HB-204™) and MCF-7 (CRL-3435™) were obtained from the Tissue Culture Core at Baylor College of Medicine. Athymic Nude Foxn1nufemale mice at an age of 5–6 weeks were purchased from ENVICO.com. Fox Chase SCID Beige mice and CD-1 IGS female mice were purchased from Charles River Laboratories. Breast cancer PDX tumors were obtained from the Baylor College of Medicine PDX Core. The 5097 PDX line was collected from a patient who had an estrogen receptor (ER) positive intraductal micropapillary carcinoma with a BRCA2 mutation (Zhang et al. 2013). The tumor was passaged three times in SCID mice before use. The 4013 PDX line was from a triple negative infiltrating ductal carcinoma patient and was transplanted into mice eight times before use (Zhang et al. 2013). Animal protocols were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Binding assessment
The binding of SI-12 to SRC-3 was assayed by NanoBRET Target Engagement Assays, a live-cell based binding assay that uses florescent resonant energy transfer to measure molecular proximity. Briefly, HeLa cells (ATCC, CCL-2™) were transfected with expression vectors for NanoLuc-SRC-3 and NLuc-MDM2 (energy donor) fusion proteins and then treated with different a concentration of a Halo-tag SI-12 Tracer compound (energy acceptor) at 37oC with 5% CO2 for 20h and the binding of the SI-12 tracer compound to the SRC-3 or MDM2 fusion proteins was evaluated by measuring the donor and acceptor emission at an appropriate wavelength using a bioluminescence reader. The reversible binding of SI-12 to the SRC-3 protein was confirmed by adding cold SI-12 (20 μM) to Nluc-SRC-3 expressing cells along with halotag SI-12 tracer compound, followed by an examination of the loss of the Nano-BRET signal at five mins, 90 mins and 24 h after addition of cold SI-12. NanoBRET activity was calculated by the following equation:
Toxicity testing
20 mg/kg/day SI-10 or SI-12 was administered by i.p. injection to CD-1 IGS female mice for two weeks. Vehicle treated mice served as controls. At the end of treatment, 200 μl of blood was drawn from mice by submandibular bleeding and plasma was isolated for a full panel of serum chemistry assays and liver and kidney tissues were collected for histological examination. For each group, five mice were included (see https://www.bigdatarunning.com/5k_percentiles/#M20).
Testing of the therapeutic efficacy of SI-10 and SI-12 in mouse metastasis models
A spontaneous breast cancer lung metastasis model was established with MDA-MB-231-Luc cells as described previously (Dasgupta et al. 2018). In brief, 1.6×106 MDA-MB-231-luc cells were implanted into Nude mice by fat pad injection with mammary tumors palpable two weeks after injection. When tumors reached about 1.0 cm in diameter, mammary tumors were surgically removed, allowing for the remaining metastatic cells to develop into larger tumors in lung. After two weeks of recovery from surgery, mice were treated with 4 mg/kg/day SI-12 by i.p. injection with treatment continuing for five weeks. Lung metastasis was monitored weekly by examining luciferase signal emanating from the MDA-MB-231-Luc cells. At the end of treatment, lung tissues were collected and luciferase activity in lung lysates was measured with vehicle treated mice serving as controls. For each treatment group, 14 mice were included.
Another experimental breast cancer lung metastasis model was established and used for testing by tail vein injection of LM2 cells into nude mice. Briefly, 2×105 of LM2 cells were injected through the tail vein into female nude mice at 8–9 weeks of age. One week after injection, mice were divided into two groups and treated with either vehicle or 10 mg/kg/day of SI-12 and treatment was continued for four weeks. Metastatic growth was monitored bi-weekly by examining luciferase activity emanating from LM2 cells. At the end of treatment, lung tissues were collected and luciferase activity in lung lysates was measured. For each treatment group, at least six mice were included.
Results:
SI-10 and SI-12 exhibit low hERG activity
The FDA criterion for defining a drug as hERG-positive is when its IC50 is less than 1 μM. According to general accepted criteria used to score potential cardiac liabilities, a hERG channel inhibition with IC50 greater than 10 μM is ranked as low and an IC50 within 1–10 μM is considered as moderate. To evaluate if the modification of SI-2 with the addition of fluorine atoms to the core structure could improve hERG channel effects, we synthesized SI-10 and SI-12 using a similar method as described for SI-2 (Song et al. 2016) and measured hERG activity of these SI-2 analogs. We found that the IC50 of SI-2 on hERG activity is 3.78 μM which is within the moderate-inhibition range. The IC50 for SI-10 is more than 10 μM, which was significantly improved compared to SI-2 (Fig. 1A). While SI-12 showed comparable hERG channel inhibition compared with SI-2 with a 1.13 μM IC50 (Fig. 1B), suggesting that SI-12 might also possess moderate hERG inhibition similar to SI-2.
Fig. 1. hERG activity and interaction of compound with SRC-3.

A&B. hERG activity of SI-10 and SI-12 was evaluated using a manual patch-clamp system. Percentage of current inhibition was calculated as described in methods. C. The binding of SI-12 to SRC-3 fusion protein was assayed by NanoBRET Target Engagement Assays. The binding to fusion protein MDM2 served as a negative control. D. Reversible binding of SI-12 to SRC-3 protein was confirmed by adding cold SI-12 to Nluc-SRC-3 expressing cells along with halotag SI-12 tracer compound. The loss of Nano-BRET signal was examined at different time point after addition of halotag SI-12 tracer compound.
SI-10 and SI-12 pharmacokinetic profiles and oral bioavailability
The pharmacokinetic profiles of SI-10 and SI-12 were evaluated by intravenous (i.v.) and PO routes (Table 1). SI-12 was found to have a slower clearance time (22 mL/min/kg) which is beneficial for therapeutic efficacy. The prolonged terminal half-life (T1/2) after administration is 14.3 h by PO and 22 h by i.v. routes which are significantly improved when compared with the SI-2 rate of 1 h (Song et al. 2016). Furthermore, the AUClast for SI-12 by PO reached 1719 h/ng/ml, suggesting that SI-12 has better bioavailability. F for SI-2 by PO is about 24.6%, suggesting an acceptable oral bioavailability for SI-12.
Table 1.
SI-10 and SI-12 pharmacokinetic parameters
| Compound | SI-10 | SI-12 | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Parameters | Mean | SD | CV(%) | Mean | SD | CV(%) | |
|
|
|||||||
| I.V. (1mg/kg) | CL_obs (mL/min/kg) | 176 | 7 | 4.13 | 22.0 | 3.7 | 16.6 |
| T1/2 (h) | NA | NA | NA | 22.5 | 0.1 | 0.54 | |
| C0 (ng/mL) | 552 | 117 | 21.1 | 1066 | 254 | 23.8 | |
| AUClast (h*ng/mL) | 87.6 | 5.2 | 5.98 | 737 | 119 | 16.1 | |
| AUCinf (h*mg/mL) | NA | NA | NA | 772 | 126 | 16.3 | |
| MRTinf_obs (h) | NA | NA | NA | 6.65 | 0.39 | 5.94 | |
| Vss_obs (L/kg) | 19.5 | 4.6 | 23.6 | 8.72 | 1.01 | 11.6 | |
|
|
|||||||
| PO (10mg/kg) | T1/2 (h) | 9.09 | 1.10 | 12.1 | 14.3 | 1.7 | 11.6 |
| Tmax (h) | 0.25 | 0.00 | 0.00 | 0.25 | 0.00 | 0.00 | |
| Cmax (ng/mL) | 89 | 30 | 33.5 | 217 | 75 | 34.4 | |
| AUClast (h*ng/mL) | 477 | 36 | 7.52 | 1719 | 111 | 6.44 | |
| AUClast/D (h*mg/mL) | 47.7 | 3.6 | 7.52 | 172 | 11 | 6.44 | |
| MRTinf_obs (%) | 11.6 | 0.9 | 7.72 | 18.6 | 1.8 | 9.89 | |
| F (%) | 54.4 | 4.1 | 7.52 | 24.6 | 1.2 | 4.80 | |
PK parameters were estimated by non-compartmental model using WinNonlin 6.1.
The bioavailability (F%) was calculated as following:
AUClast-PO/AUCINF-PO > 80%: F=(AUCINF-PO*DoseIV)/(mean AUCINF-IV*DosePO)
AUClast-PO/AUCINF-PO ≤ 80% or AUCINF was not available: F=(AUClast-PO*DoseIV)/(mean AUClast-IV*DosePO)
The pharmacokinetic profile for SI-10 showed less stability compared with SI-12. The clearance of SI-10 is very fast and CL obs reached 176 ml/min/kg. The T1/2 is 9.09±1.10h by PO dosing and the maximum concentration peak in plasma is 89±30 ng/ml, indicating its stability in plasma is significantly improved compared with SI-2 but the improvement remains less than SI-12. Also, AUClast by PO administration is less than 500 h*ng/ml (477h*ng/ml), even though the F reached 54.4% which may be due to the low basal AUClast level by i.v. administration (Table 1). These results indicated that both SI-12 and SI-10 have an improved T1/2 but distinct AUClast values. SI-12 exhibited a substantially better pharmacokinetic profile than SI-10 and its oral availability is acceptable.
SI-12 specifically binds to SRC-3 in a reversible manner
Next, we performed NanoBRET Target Engagement Assays to determine if a HaloTag-SI-12 compound exhibits direct interaction with a NanoLuc-SRC-3 fusion protein in HeLa cells. From this assay, we found that the mBU interaction value (see Methods) reached about 25 when treated with 20 μM HaloTag-SI-12 (Fig. 1C). On the contrary, the binding of HaloTag-SI-12 to the control NanoLuc-MDM2 fusion protein was extremely low with an mBU of less than two when treated with the same amount of HaloTag-SI-12 (Fig. 1C). To confirm if the binding of SI-12 to SRC-3 is reversible, 20 μM of untagged SI-12 was added to pre-formed NanoLuc-SRC-3 with 20 μM HaloTag-SI-12 binding complexes in HeLa cells. The reduced signal found from this competition assay between HaloTag-SI-12 and NanoLuc-SRC-3 from the addition of SI-12 was reduced from 220 to 30 mBUs after 24 hours (Fig. 1D). These results indicate that SI-12 can specifically bind to SRC-3 protein in living cells.
SI-10 and SI-12 possess minor toxicity in mice
To evaluate drug toxicity, high doses of 20 mg/kg/day SI-10 or SI-12 were administrated to female CD-1 IGS mice by i.p. injection. This dose is two-fold higher than that used in our mouse xenograft models described below. The liver is the major organ responsible for drug metabolism (Ware & Khetani 2017). Standard H&E staining showed that in SI-10 and SI-12 treated liver tissues, liver structure and liver lobes surrounding the portal area and central venule look normal and similar to the livers from vehicle treated mice (Fig. 2A). No obvious structural differences were observed in the kidneys of compound treated mice. Consistent with this, a full panel serum chemistry analysis revealed that compared with vehicle treated mice, liver AST and ALT enzyme levels in mice are comparable in four out of five mice treated with SI-10 and SI-12. Only one mouse in each group exhibited higher levels of these two enzymes than control group (Fig. 2B&C). No obvious difference was observed in ALP levels in drug treated mice compared with control mice (Fig. 2D). The kidney excretes a variety of metabolic waste products into the urine and also plays a critical role in drug metabolism. H&E staining showed that kidney tissues from SI-10 and SI-12 treated mice exhibited normal histological structures (data not shown). Accordingly, BUN levels in compound treated mice are comparable to that of vehicle treated mice (Fig. 2F).
Fig. 2. Toxicity testing with SI-10 and SI-12.
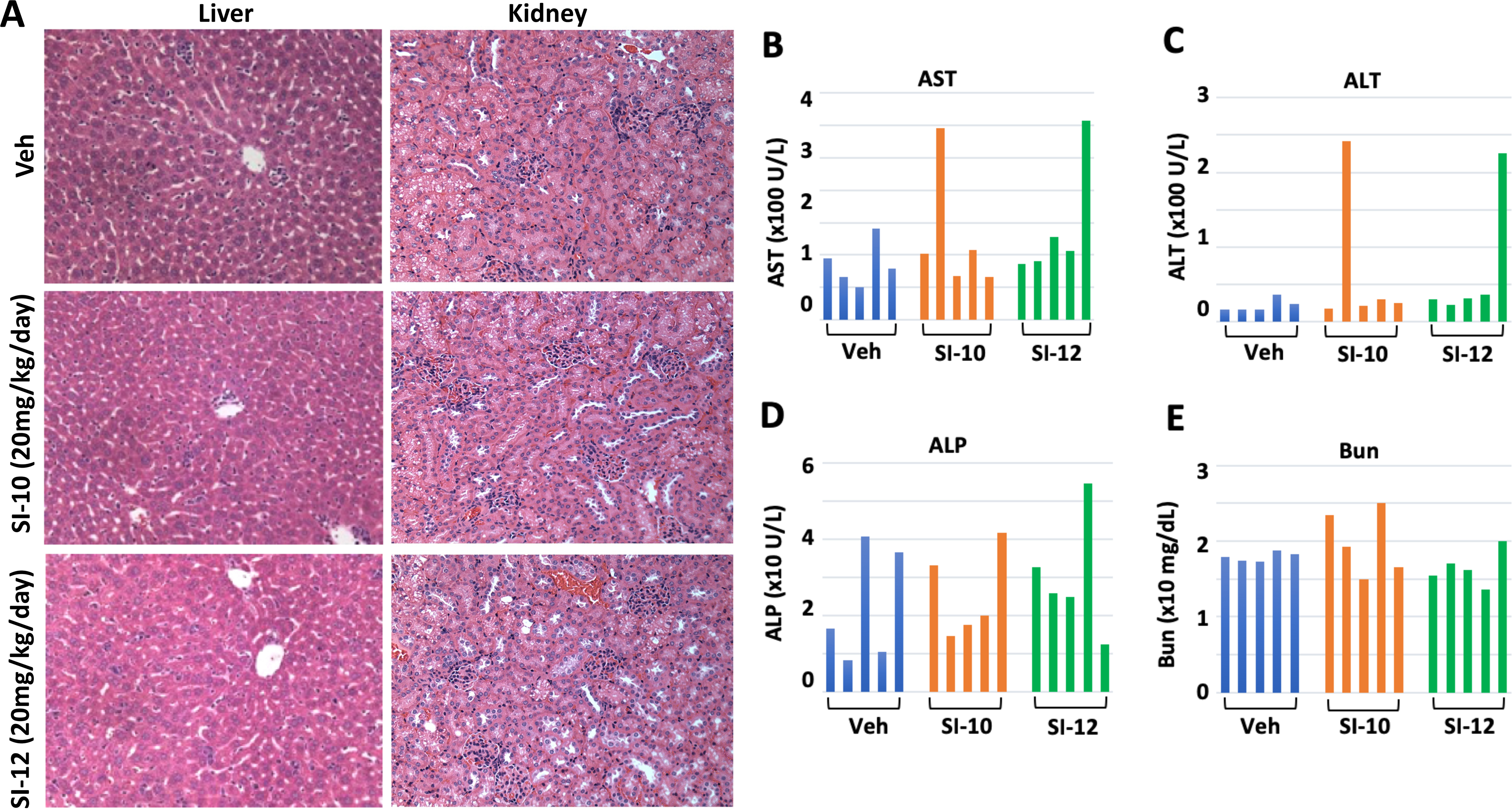
A. H&E staining with liver and kidney tissues from SI-10/SI-12 (20mg/kg/day) treated mice. B.C &D showed liver enzyme AST, ALT and ALP levels in mouse plasma after treatment for 2 weeks. E. Bun levels in mouse plasma were examined after 2-week treatment. For each group, 5 mice were included.
SI-10 and SI-12 inhibit breast cancer cell viability, migration and invasion
It has been shown that SRCs up-regulate expression of cell cycle related genes and drive proliferation in both normal and cancer cells (Xu et al. 2009, 2010; Dasgupta et al. 2018). To examine the effect of SI-10 and SI-12 inhibition of SRCs, Western blotting was performed with MCF-7 cells treated by different dosage of SI-10 or SI-12. We found that SRC-3 levels decreased in compound treated cells (Fig. 3A). To further evaluate the effect of these compounds on cancer cell growth, MTT assays were performed on multiple breast cancer cell lines treated with compound. We found that the growth of all tested cell lines was inhibited after 72 h treatment with a range of SI-12 concentrations. The IC50 value for SI-12 is 7.5 nM for MCF-7 cells and 17.5 nM for MDA-MB-453 cells. For LM2 and MDA-MB-231 cells, their IC50 values were 40 nM and 75 nM respectively (Fig. 3B). SI-10 showed similar inhibition toward viability of breast cancer cells and IC50s were 25nM and 35nM for MCF-7 and MDA-MB-453 and 100nM and 125nM for LM2 and MDA-MB-231, respectively (Fig. 3B). Previous reports have shown that SRCs potentiate cancer cell migration and invasion capability, therefore promoting cancer progression and metastasis (Yuan et al. 2007; Qin et al. 2008a; Xu et al. 2009; Fu et al. 2011; Walsh et al. 2014). To assess the effect of SI-12 on breast cancer cell motility, MDA-MB-231 and LM2 cells were pre-treated with 50nM SI-12 for two days and then tested in a motility assay on 96-well plates pre-coated with fibronectin and fluorescent beads (Fig. 3C). Images show that the migration tracts created by SI-12 treated MDA-MB-231 or LM2 cells is shortened and narrow compared with vehicle treated control wells. The migration area made by SI-12 treated cells was reduced about 50% which is substantially smaller than the control cells, indicating that SI-12 can repress cancer cell migration. Similar results were obtained with less motile MCF-7 cells. Migration track area left by SI-12 treated MCF-7 cells was reduced about 50% compared with control cells.
Fig. 3. SI-10 and SI-12 inhibits growth, motility and invasiveness of breast cancer cells.

A. Western blotting showed that SI-10 and SI-12 treatment down regulated SRC-3 protein levels in MCF-7 cells. B. Viability assay was performed with different dosage of SI-10 and SI-12 in 4 breast cancer cell lines. C. Images showed reduced motilities of breast cancer cells on fibronectin (FN) under SI-12 treatment. D. Invasion assay was performed with metastatic MDA-MB-231 and LM2 cells pre-treated by 50 nM of SI-10 or SI-12 and images showed tumor cells invading through Matrigel coated membrane. E. The invading cell number significantly reduced in SI-10 and SI-12 treated group. F. Combined SI-10 or SI-12 with BEZ235 treatment exhibited stronger inhibition on LM2 invasion capability compared to single drug treatment. *p<0.05, **p<0.01
The effects of SI-10 and SI-12 on cancer cell invasiveness was assessed by placing cells in invasion chambers that were previously treated with SI-10 or SI-12 (Fig. 3D). After compound treatment for 48h, fewer MDA-MB-231 or LM2 cells invaded through the Matrigel-coated PET membranes (see Methods). Compared to the vehicle treated group, the invading cell percentages decreased about 50% and 40% for SI-10 and SI-12 treated MDA-MB-231 cells, respectively. Similarly, the percentage of invading cells was reduced about 30–40% in compound treated LM2 cells (Fig. 3D). To evaluate if combination treatment could improve inhibitory effect, we pre-treated LM2 cells with SI-10 or SI-12 alone or in combination with mTOR inhibitor BEZ235. We found that the invading cell number dropped to 20% with combined SI-12/SI-10 and BEZ235 treatment (Fig. 3E), indicating that combined treatment could achieve a stronger inhibition on invasion of LM2 cells.
SI-10 and SI-12 represses growth of breast cancer PDX organoids
Organoid culture systems used to culture normal and tumor epithelial cells in an in-gel suspension can be used to recapture the diversity of disease and tumor-stromal cell interactions that have been developed for drug screening and testing (Sachs et al. 2018; Ooft et al. 2019). To test the responsiveness of breast cancer PDX organoids to SI-12 treatment, we used three different dosages of SI-12 to treat ER+ (5097) and triple negative (4013) PDX organoids for two weeks. We found that 62.5 nM of SI-12 is sufficient to repress organoid formation and growth in both PDX lines. Both organoid number and size decreased in SI-12 treated organoids compared to the vehicle treated group. With an increase in SI-12 dosage, organoid number and size decreased further. Only a small number of organoids of reduced size were observed in both lines after 250 nM of SI-12 treatment for two weeks (Fig. 4A).
Fig. 4. SI-12 significantly inhibited growth of organoids developed from breast cancer PDX tumors.

A. Organoids developed from 5097 line (ER+ human breast cancer PDX line) or 4013 line (Triple negative human breast cancer PDX line) were treated with different dosage of SI-12 for two weeks. Images were taken at the end of treatment and organoid number was counted and analyzed accordingly. B. Combination treatment with SI-12 and CDK4/6 inhibitor abemaciclib (Abe) was tested with organoids from 5097 ER+ PDX tumors. C. Combined SI-12 and BEZ235 (mTOR inhibitor) treatment was performed with organoids from 4013 triple negative PDX tumors and organoid number was counted at the end of treatment. D. Bar graph showed inhibition of single SI-10 or combined SI-10 and Abe treatment on growth of ER+ 5079 PDX organoids. E. Bar graph showed inhibition of single SI-10 or combined SI-10 and BEZ235 treatment on growth of ER- 4013 PDX organoids. *p<0.05, **p<0.01, *** P<0.001
The CDK4/6 inhibitor Abemaciclib has been approved by FDA for combination treatment with anti-estrogen therapy in patients with advanced breast cancer (O’Leary et al. 2016). Here, we sought to test if combined SI-12 and Abemaciclib treatment could achieve a better inhibition on organoid growth and treated organoids from the 5097 line with SI-12 at a 10nM or 100nM concentration for two weeks in combination with 10 nM or 100 nM Abemaciclib. The 5097 PDX line was collected from an ER positive breast cancer patient with lung metastasis (Zhang et al. 2013). We found that 10 nM SI-12 or Abemaciclib treatment reduced organoid numbers compared with the vehicle control. Based on organoid numbers, the combined treatment with SI-12 and Abemaciclib showed stronger inhibition on organoid formation than single treatment with either one at the same concentration. Under high dose treatment with 100 nM with both compounds, very few organoids of a smaller size were observed after two weeks of treatment (Fig. 4B).
SRC-3 has been reported to promote cancer proliferation through its potentiation of the mTOR pathway (Zhou et al. 2003). Because of this, we sought to determine if SI-12 would have a synergistic inhibitory effect on organoid growth in combination with an mTOR inhibitor. We treated organoids developed from the triple negative breast cancer PDX 4013 line (Zhang et al. 2013) with SI-12 in combination with the mTOR inhibitor BEZ235 for two weeks. As expected, single compound treatment with either SI-12 (10 nM) or BEZ235 (2 nM) could repress organoid formation (Fig. 4C). However, combined treatment with SI-12 and BEZ235 exhibited better inhibition of organoid growth than either compound alone. 10 nM or 100 nM SI-12 treatment in combination with 10 nM BEZ235 completely blocked organoid growth. No organoids formed after two weeks of treatment with this combination of compounds (Fig. 4C). SI-10 showed a similar inhibitory effect on growth of PDX 5079 and 4013 organoids with a single SI-10 treatment (100nM) significantly repressing organoid growth. However, combined treatment with SI-10 with either Abe in ER+ 5079 organoids or BEZ235 in ER- 4013 organoids was able to achieve stronger inhibition (Fig. 4D).
SI-12 inhibits growth of xenograft breast tumors in vivo
To assess the therapeutic potential of SI-10 and SI-12 in an in vivo setting, mammary xenograft tumors were established with MDA-MB-231 cells by fat pad injection into the fourth pair of mammary glands of SCID mice. Mammary tumors were palpable two weeks after injection and on the third week after tumor cell injection, tumors reached 0.3–0.5 cm in diameter at which time treatment was started. Tumor length and width were measured weekly during treatment. After one week of treatment with 10 mg/kg/day SI-10 or SI-12 by i.p. injection, tumor volume in the drug treated group was significantly smaller than in the control group and the difference in tumor volume between treated and untreated groups was continuously evident. At the end of the experiment, tumor volume in the drug treated group was reduced more than 60% compared to the vehicle control group. SI-10 and SI-12 exhibited similar tumor growth inhibition efficacy (Fig. 5A). Using a 3.0 cm diameter criteria as an ethical end point for scoring mouse tumor mortality, mouse survival was calculated (Fig. 5B). Compared with the vehicle control group, SI-12 treatment significantly prolonged mouse survival. Similar improvements in mouse survival were observed in SI-12 treated LM2 xenograft tumor models (Fig. 5C&D). After four weeks of SI-12 treatment, tumor volume was reduced more than 50% in the LM2 xenograft model. These results indicate that 10 mg/kg/day SI-10 and SI-12 treatment can significantly repress the growth of xenograft breast tumors.
Fig. 5. SI-10/SI-12 inhibited xenograft tumor growth and prolonged survival in mice.

A. Tumor growth curve from SCID mice harbor MDAMB-231 xenograft tumors treated with 10mg/kg/day of SI-10 or SI-12. B. Survival curve of SI-12 treated SCID mice harbor MDA-MB-231 xenograft tumors. C. Tumor growth curve from a LM2 xenograft mouse model with SI-12 (10mg/kg/day) treatment. D. IHC for Ki67, cleaved caspase 3 and CD31 was performed with MDA-MB-231 xenograft tumors collected from SI-10/SI-12 treated mice. Positive cell number and microvascular structures were counted and shown in bar graph. For each marker examined, 8 tumor samples from 8 mice were included. At least 5 different area was counted under microscopy for each tumor sample. *p<0.05, **p<0.01, *** P<0.001
To examine if SI-10 and SI-12 treatment has an effect on tumor cell proliferation and angiogenesis, immunostaining was performed for Ki67, cleaved caspase 3 and CD31 on tumor tissue sections collected at the end of treatment. We found that more than half of the tumor cells in vehicle treated tumors exhibited Ki67 positive staining. However, Ki67 positive cell numbers were much lower in the tumors of both SI-10 or SI-12 treated animals. We counted Ki67 positive cell numbers and total cell numbers for eight different tumor samples for each group. We found that in the MDA-MB-231 xenograft model, the percentage of Ki67 positive cells is about 20% in SI-10 treated tumors and 23% in SI-12 treated tumors, which is substantially lower than the 55% Ki67 positive cells found in vehicle treated tumors. These results indicate that drug treatment significantly inhibits the proportion of proliferating cells in tumors.
To determine if SI-10 and SI-12 induce apoptosis in tumor cells, we did immunostaining for cleaved caspase-3 in tumor sections from vehicle and compound-treated mice. We observed more cleaved caspase-3 positive cells in SI-10 and SI-12 treated tumors compared to the vehicle controls. The positive cell percentage is 4–5% in MDA-MB-231 tumors, which is markedly higher than the 2% found in MDA-MB-231 control tumors (Fig. 5D). These results indicate that 10 mg/kg/day SI-10 or SI-12 treatment significantly induced apoptosis in tumor cells.
To assess the impact of SI-12 on tumor angiogenesis, we examined the expression of CD31, an endothelial cell marker, by IHC in SI-12 and SI-10 treated tumors. We found that CD31 positive cell numbers are low in SI-10 or SI-12 treated MDA-MB-231 tumors compared to vehicle treated tumors. Moreover, quantitative analysis of CD31 staining revealed that in MDA-MB-231 xenograft tumors, microvascular density (MVD) was reduced by 50% or 40% in SI-10 or SI-12 treated tumors respectively versus vehicle controls (Fig. 5D). These results reveal that 10 mg/kg/day SI-10 or SI-12 treatment can inhibit tumor angiogenesis.
SI-10 and SI-12 repressed mammary tumor lung metastasis in Nude mice
Previous reports show that elevated SRC-1 and SRC-3 expression in breast cancer cells strongly promotes tumor metastasis (Qin et al. 2008a, 2009; Wang et al. 2009; Xu et al. 2009; Long et al. 2010; Qin et al. 2014a). To test if SI-12 treatment has an impact on breast cancer metastasis, we developed a spontaneous metastasis mouse model using MDA-MB-231 cells as described in the Methods section, treating animals with 4 mg/kg/day SI-12 for five weeks. Bioluminescent imaging of metastatic tumor cells revealed that in the vehicle treated group, luciferase signal emanating from LM2 cells were observed in the lung area of eight out of 14 mice five weeks after treatment, indicating the eight mice developed breast cancer lung metastasis. However, only two out of the 14 mice in the SI-12 treated group had luciferase signals in lung area (Fig. 6A). In line with bioluminescent image data, luciferase activities in lung tissue lysates from SI-12 treated mice were much lower than in the vehicle control group (Fig. 6B), indicating that 4 mg/kg/day SI-12 treatment can inhibit spontaneous breast cancer lung metastasis.
Fig. 6. SI-12 significantly inhibits breast cancer metastasis in mouse models.

A. Bioluminescent images showed progression of breast cancer lung metastasis w/o SI-12 (4mg/kg/day) treatment in spontaneous metastasis model developed from fat pad injection of MDA-MB-231 cells. B. Luciferase activities in lung lysis were examined at the end of treatment. C. Bioluminescent images showed progression of metastasis growth established from tail vein injection of LM2 cells w/o SI-12 (10mg/kg/day) treatment. D. Luciferase activity in lung was monitored during SI-12 treatment in LM2 lung metastasis mice. E. Luciferase activities in lung lysis from LM2 model was examined at the end of treatment. *p<0.05, **p<0.01
We also tested SI-12 using a LM2 tail vein tumor cell injection metastatic model. One week after tail vein injection of LM2 cells, 10 mg/kg/day SI-12 was administered to mice by i.p. injection. After treatment for two weeks, metastatic tumors in the lungs were detected by bioluminescent imaging that continued to grow larger in almost all vehicle treated mice. Conversely, three out of seven mice in SI-12 treated animals showed a decline in lung metastasis growth, while the other four mice did not show a response to SI-12 treatment. At the end of four weeks of treatment, all vehicle treated mice exhibited extensive lung metastasis, while six out of seven mice treated with SI-12 showed slow metastatic growth or no detectable metastases in their lungs (Fig. 6C). Metastatic growth was repressed by more than 70% with SI-12 treatment (Fig. 6D). Consistent with this, luciferase activity in lung tissue lysates from SI-12 treated mice was significantly lower than in the vehicle control group (Fig. 6E). These results clearly show that SI-12 treatment can inhibit breast cancer lung metastasis in the LM2 tail vein metastasis model.
Discussion:
A number of SRC small molecular inhibitors have been identified by high throughput screening (Wang et al. 2011, 2014; Yan et al. 2014; Song et al. 2016). Among them, SI-2 was identified as a promising candidate for further development due to its potent action at low nanomolar concentrations. However, its shorter half-life and moderate hERG activity limited its potential as a leading drug candidate (Song et al. 2016). To develop improved SMIs based on the SI-2 scaffold, we introduced two or three fluorine atoms to the SI-2 chemical structure to generate SI-10 and SI-12, respectively. The half-life of SI-10 and SI-12 by PO is increased to 9.09 ±1.10h and 14.3±1.7h to that of SI-2. This improvement enhances the efficacy of SI-10 and SI-12 and reduces the need for frequent drug administration. Indeed, the IC50 for SI-10 and SI-12 was reduced to 8.5 nM and 7 nM respectively in MCF-7 cells, which is significantly lower than that of SI-2 at 22 nM in the same cells (Song et al. 2016). Moreover, once daily treatment with SI-10 or SI-12 for four weeks was able to effectively repress the growth of xenograft tumors, while twice daily treatment was required for SI-2 (Song et al. 2016).
hERG activity is an important safety parameter for drug development (Fermini & Fossa 2003). Many potential pharmaceutical drugs in the market were not moved forward for further development due to potential cardiac toxicity. In a previous study from our laboratory, bufalin was identified as a potent SRC SMI with an IC50 of 3.2 nM for suppressing MCF-7 cell growth (Wang et al. 2014). However, its strong cardiac glycoside activity prevented its further development as a therapeutic drug (Wang et al. 2014; Song et al. 2015). In contrast, SI-2 has a much reduced hERG activity and its closely related derivatives have an even better hERG profile. SI-10 showed much improved hERG activity with a >10μM hERG IC50 and is considered as a weak hERG inhibitor.
SRCs play critical roles in a number of cancer pathways related to proliferation, cancer progression and metastasis. Targeting SRC expression or function using SMIs could be a powerful approach to inhibit these key cancer cell attributes responsible for the ultimate manifestation of cancer in its lethal metastatic form (Xu et al. 2009; D’Ambrogio et al. 2013; Otto & Sicinski 2017; Dasgupta et al. 2018). Firstly, SI-10 and SI-12 can potently decrease tumor cell proliferation and primary tumor as indicated by reduced Ki67 staining during SRC inhibition; these SMIs also beneficially interfere with angiogenesis in the tumor itself. For instance, SRC-1 has been shown to promote tumor angiogenesis through up-regulation of HIF-1α-mediated VEGF-A expression (Qin et al. 2015). We observed that 10 mg/kg/day SI-10 and SI-12 treatment dramatically reduced microvascular density in xenograft tumors, suggesting that these SRC SMIs have anti-angiogenesis activity; more studies will be required to understand the mechanism by which this occurs.
SRCs have been implicated in driving breast cancer metastasis (Qin et al. 2008b, 2014a; Wang et al. 2009; Han & Crowe 2010; Walsh et al. 2014). The impact of SRC SMIs on metastasis had not been reported previously. Here, we tested the efficacy of SI-12 on metastasis using two distinct mouse models. In a spontaneous lung metastasis model using mice with surgically resected MDA-MB-231 tumors, we observed a decrease in metastases in SI-12 treated mice. Furthermore, in another metastatic mouse model using tail vein injected LM2 cells, four weeks of SI-12 treatment significantly repressed metastatic growth in the lungs of these animals.
Organoid in-gel culture systems using PDX tumor epithelial cells has become an important platform for more efficient compound screening and tumor-stromal cell interactions (Weeber et al. 2017). These organoids are capable of recapturing the histological makeup of the original human primary tumor, while also retaining DNA copy number and sequence variations contained within the original tumor (Duarte et al. 2018; Sachs et al. 2018). In this study, we evaluated SI-10 and SI-12 in two PDX organoid models developed from an ER positive breast cancer patient (5097 line) and a triple negative breast cancer patient (4013 line) respectively. We found that SI-10 and SI-12 treatment significantly inhibited organoid growth at low nanomolar concentrations in both organoid models, indicating that these compounds shows promise as an anti-cancer agent in tumor cells growing in the context of adjacent stromal cells and more closely mimics tumors in an in vivo state.
Abemaciclib is a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and cyclin-dependent kinase 6, able to induce cell cycle arrest of breast cancer cells from G1 to the S phase (Brower 2014). It was approved as a breakthrough therapy for breast cancer in 2015 for hormone receptor positive metastatic breast cancer (O’Leary et al. 2016). In this study, we found that combined SI-10 or SI-12 with abemaciclib treatments could achieve a stronger inhibitory effect on growth of organoids developed from 5079 PDX tumors collected from a heavily pre-treated patient with advanced breast cancer who had received neoadjuvant docetaxel as well as exemestane therapies, suggesting that drug combinations with SRC-SMIs might be promising regimens for treatment in vivo.
The PI3K/AKT/mTOR signaling pathway plays a critical role in regulating the cell cycle and is frequently upregulated in breast cancers. PIK3CA or AKT1 mutations and PTEN loss are common, leading to activation of the pathway (Saal et al. 2007). Studies have shown that SRCs promote breast and prostate cancer cell proliferation and malignancy in part through the PI3K/AKT/mTOR pathway (Xu et al. 2009; Dasgupta et al. 2015). This evidence points to the likelihood that targeting of SRCs and PI3K/AKT/mTOR in combination would attain better therapeutic outcomes. Here, we tested SI-10 and SI-12 in combination with BEZ235, a dual PI3K/mTOR inhibitor, in triple negative breast cancer PDX organoids. As expected, combination treatment showed much stronger inhibition of organoid growth than single agent treatment.
A key challenge in moving candidate cancer therapeutic agents to the clinic is that compounds with potent cytotoxic effects toward cancer cells grown under traditional 2D culture conditions in vitro frequently show less efficacy in xenograft tumor models. This can be due to a variety of factors related to drug bioavailability, metabolism and clearance in vivo. In addition to these factors, cancer cells grown as xenograft tumors interact with stromal cells and proliferate as complex 3D structures that alter the properties of the tumor cells themselves often rendering them more resistant to anti-cancer agents. Added to this is the fact that while most cancer drug enabling studies utilize tumor cells grown as primary tumors in host mice, but especially for breast and prostate cancers, metastatic disease is responsible for the majority of patient mortality as primary tumors are usually surgically resected. In light of these factors, here we investigated SI-10 and SI-12 in organoid and mouse metastatic disease models more pertinent to human breast cancer disease. Ultimately, by finding that these SRC inhibitors can elicit anti-cancer activities in 2D cell culture, in in vitro organoid models and in both primary and metastatic mouse cancer models, we have been able to position these agents among our lab’s lead SRC targeting anti-cancer agents with strong therapeutic potential.
Supplementary Material
Acknowledgments
We acknowledge Breast Center PDX Core to provide PDX 5079 and 4013 tumor lines. We also thank Dr. Hong Yong, Dr. Weizhang Tong, Dr. Xiaoli Qi and Dr. Yunfeng Ding for sharing experiment reagents and technique assistance during performing this work.
Funding
This work was supported by funding from the Department of Defense Breast Cancer Research Program (BC120894), the Cancer Prevention and Research Institute of Texas (RP100348 and RP150700), and from the National Institutes of Health (HD08818 and HD07857) to BWO; the Adrienne Helis Malvin Foundation to DML and The Dan L Duncan Comprehensive Cancer Center Pilot Award at Baylor (DLDCCC) to LQ. LQ, JC, DL, PJ, YY, JX, JW, BWO and DML all hold stock in a virtual company created within Baylor College of Medicine that has no commercial funding.
Footnotes
Conflicts of interest statement
LQ, JC, DL, PJ, YY, JX, JW, BWO and DML all hold stock in a virtual company created within Baylor College of Medicine that has no commercial funding that was formally associated with Coactigon, Inc. JW, BWO and DML hold ownership interest in patents for compounds described in this study.
References
- Agoulnik IU, Vaid A, Bingman WE, Erdeme H, Frolov A, Smith CL, Ayala G, Ittmann MM & Weigel NL 2005. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Research. (doi: 10.1158/0008-5472.CAN-04-3541) [DOI] [PubMed] [Google Scholar]
- Brower V 2014Cell cycle inhibitors make progress. Journal of the National Cancer Institute. (doi: 10.1093/jnci/dju221) [DOI] [PubMed] [Google Scholar]
- D’Ambrogio A, Nagaoka K & Richter JD 2013. Translational control of cell growth and malignancy by the CPEBs. Nature Reviews Cancer. (doi: 10.1038/nrc3485) [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Putluri N, Long W, Zhang B, Wang J, Kaushik AK, Arnold JM, Bhowmik SK, Stashi E, Brennan CA et al. 2015Coactivator SRC-2’dependent metabolic reprogramming mediates prostate cancer survival and metastasis. Journal of Clinical Investigation. (doi: 10.1172/JCI76029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, Choi JM, Jung SY, Coarfa C, Westbrook TF et al. 2018Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature. (doi: 10.1038/s41586-018-0018-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AA, Gogola E, Sachs N, Barazas M, Annunziato S, R De Ruiter J, Velds A, Blatter S, Houthuijzen JM, Van De Ven M et al. 2018BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nature Methods. (doi: 10.1038/nmeth.4535) [DOI] [PubMed] [Google Scholar]
- Fermini B & Fossa AA 2003. The impact of drug-induced QT interval prolongation on drug discovery and development. Nature Reviews Drug Discovery. (doi: 10.1038/nrd1108) [DOI] [PubMed] [Google Scholar]
- Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill ADK & Young LS 2004. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; A predictive role for SRC-1. Journal of Clinical Pathology. (doi: 10.1136/jcp.2004.016733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L & Xu J 2011. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Research 21. (doi: 10.1038/cr.2010.118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS & Crowe DL 2010. Steroid receptor coactivator 1 deficiency increases MMTV-neu mediated tumor latency and differentiation specific gene expression, decreases metastasis, and inhibits response to PPAR ligands. BMC Cancer. (doi: 10.1186/1471-2407-10-629) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto H, Wang Z, Bhat-Nakshatri P, Chang D, Clarke R & Nakshatri H 2005. The p160 family coactivators regulate breast cancer cell proliferation and invasion through autocrine/paracrine activity of SDF-1α/CXCL12. Carcinogenesis. (doi: 10.1093/carcin/bgi137) [DOI] [PubMed] [Google Scholar]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ & O’Malley BW 1999. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. (doi: 10.1016/S0092-8674(00)80711-4) [DOI] [PubMed] [Google Scholar]
- Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, Mancini MA, Tsai SY, Tsai MJ & O’Malley BW 2010. SRC-3Δ4 Mediates the Interaction of EGFR with FAK to Promote Cell Migration. Molecular Cell. (doi: 10.1016/j.molcel.2010.01.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna NJ & O’Malley BW 2002. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. (doi: 10.1016/S0092-8674(02)00641-4) [DOI] [PubMed] [Google Scholar]
- O’Leary B, Finn RS & Turner NC 2016. Treating cancer with selective CDK4/6 inhibitors. Nature Reviews Clinical Oncology. (doi: 10.1038/nrclinonc.2016.26) [DOI] [PubMed] [Google Scholar]
- Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, Schipper L, Hoes L, Vis DJ, van de Haar J et al. 2019Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Science Translational Medicine. (doi: 10.1126/scitranslmed.aay2574) [DOI] [PubMed] [Google Scholar]
- Otto T & Sicinski P 2017. Cell cycle proteins as promising targets in cancer therapy. Nature Reviews Cancer. (doi: 10.1038/nrc.2016.138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O’Malley BW & Xu J 2008a. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Molecular and Cellular Biology 28. (doi: 10.1128/MCB.00579-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O’Malley BW & Xu J 2008b. The AIB1 Oncogene Promotes Breast Cancer Metastasis by Activation of PEA3-Mediated Matrix Metalloproteinase 2 (MMP2) and MMP9 Expression. Molecular and Cellular Biology. (doi: 10.1128/mcb.00579-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Z, Chen H & Xu J 2009. The steroid receptor coactivator-1 regulates Twist expression and promotes breast cancer metastasis. Cancer Research. (doi: 10.1158/0008-5472.CAN-08-4389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Chen X, Wu Y, Feng Z, He T, Wang L, Liao L & Xu J 2011. Steroid receptor coactivator-1 upregulates integrin α 5 expression to promote breast cancer cell adhesion and migration. Cancer Research 71. (doi: 10.1158/0008-5472.CAN-10-3453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu YL, Toneff MJ, Li D, Liao L, Gao X, Bane FT, Tien JCY, Xu Y, Feng Z et al. 2014aNCOA1 directly targets M-CSF1 expression to promote breast cancer metastasis. Cancer Research. (doi: 10.1158/0008-5472.CAN-13-2639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lee HJ, Wu SP, Lin SC, Lanz RB, Creighton CJ, DeMayo FJ, Tsai SY & Tsai MJ 2014b. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. Journal of Clinical Investigation. (doi: 10.1172/JCI76412) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Xu Y, Xu Y, Ma G, Liao L, Wu Y, Li Y, Wang X, Wang X, Jiang J et al. 2015NCOA1 promotes angiogenesis in breast tumors by simultaneously enhancing both HIF1a- and AP-1-mediated VEGFa transcription. Oncotarget 6. (doi: 10.18632/oncotarget.4341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond AM, Bane FT, Stafford AT, Mcllroy M, Dillon MF, Crotty TB, Hill AD & S.Young L 2009. Coassociation of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clinical Cancer Research. (doi: 10.1158/1078-0432.CCR-08-1649) [DOI] [PubMed] [Google Scholar]
- Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, Koujak S, Ferrando AA, Malmström P, Memeo L et al. 2007Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proceedings of the National Academy of Sciences of the United States of America. (doi: 10.1073/pnas.0702507104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H et al. 2018A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell. (doi: 10.1016/j.cell.2017.11.010) [DOI] [PubMed] [Google Scholar]
- Song X, Zhang C, Zhao M, Chen H, Liu X, Chen J, Lonard DM, Qin L, Xu J, Wang X et al. 2015Steroid Receptor Coactivator-3 (SRC-3/AIB1) as a Novel Therapeutic Target in Triple Negative Breast Cancer and Its Inhibition with a Phospho-Bufalin Prodrug. PLoS ONE 10. (doi: 10.1371/journal.pone.0140011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Chen J, Zhao M, Zhang C, Yu Y, Lonard DM, Chow DC, Palzkill T, Xu J, O’Malley BW et al. 2016Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proceedings of the National Academy of Sciences of the United States of America. (doi: 10.1073/pnas.1604274113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ et al. 1997Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. (doi: 10.1038/38304) [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B et al. 2010Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell. (doi: 10.1016/j.ccr.2010.05.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Arzayus MI, De Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR & Brown M 2004. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. (doi: 10.1016/j.ccr.2004.06.027) [DOI] [PubMed] [Google Scholar]
- Walsh CA, Bolger JC, Byrne C, Cocchiglia S, Hao Y, Fagan A, Qin L, Cahalin A, McCartan D, McIlroy M et al. 2014Global gene repression by the steroid receptor coactivator SRC-1 promotes oncogenesis. Cancer Research 74. (doi: 10.1158/0008-5472.CAN-13-2133) [DOI] [PubMed] [Google Scholar]
- Wang S, Yuan Y, Liao L, Kuang SQ, Tien JCY, O’Malley BW & Xu J 2009. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proceedings of the National Academy of Sciences of the United States of America. (doi: 10.1073/pnas.0808703105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG & O’Malley BW 2011. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Molecular Endocrinology. (doi: 10.1210/me.2011-1222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F et al. 2014Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Research. (doi: 10.1158/0008-5472.CAN-13-2939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware BR & Khetani SR 2017. Engineered Liver Platforms for Different Phases of Drug Development. Trends in Biotechnology. (doi: 10.1016/j.tibtech.2016.08.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber F, Ooft SN, Dijkstra KK & Voest EE 2017. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chemical Biology. (doi: 10.1016/j.chembiol.2017.06.012) [DOI] [PubMed] [Google Scholar]
- Xu J, Wu RC & O’Malley BW 2009. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nature Reviews Cancer. (doi: 10.1038/nrc2695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JM, Chen XA & Liu ZL 2010. The Cooperative Function of Nuclear Receptor Coactivator 1 (NCOA1) and NCOA3 in Placental Development and Embryo Survival. Molecular Endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Yu Y, Chow DC, Palzkill T, Madoux F, Hodder P, Chase P, Griffin PR, O’Malley BW & Lonard DM 2014. Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS ONE. (doi: 10.1371/journal.pone.0095243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P, Xia W, Wu RC, Lonard DM, Hung MC & O’Malley BW 2013. SRC-3 coactivator regulates cell resistance to cytotoxic stress via TRAF4-mediated p53 destabilization. Genes and Development. (doi: 10.1101/gad.203760.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Qin L, Liu D, Wu RC, Mussi P, Zhou S, Songyang Z & Xu J 2007. Genetic screening reveals an essential role of p27kip1 in restriction of breast cancer progression. Cancer Research. (doi: 10.1158/0008-5472.CAN-07-0083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M et al. 2013A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Research. (doi: 10.1158/0008-5472.CAN-12-4081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Hashimoto Y, Kwak I, Tsai SY & Tsai M-J 2003. Role of the Steroid Receptor Coactivator SRC-3 in Cell Growth. Molecular and Cellular Biology. (doi: 10.1128/mcb.23.21.7742-7755.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, Ittmann M, Tsai SY & Tsai MJ 2005. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Research. (doi: 10.1158/0008-5472.CAN-04-4076) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.