

Abstract
Recent studies have shown that extra-cellular matrix (ECM) substitutes can have a dramatic impact on cell growth, differentiation and function. However, these ECMs are often applied generically and have yet to be developed for specific cell types. In this study, we developed tissue-specific ECM-based coating substrates for skin, skeletal muscle and liver cell culture. Cellular components were removed from adult skin, skeletal muscle, and liver tissues, and the resulting acellular matrices were homogenized and dissolved. The ECM solutions were used to coat culture dishes. Tissue matched and non-tissue matched cell types were grown on these coatings to assess adhesion, proliferation, maintenance of phenotype and cell function at several time points. Each cell type showed better proliferation and differentiation in cultures containing ECM from their tissue of origin. Although subtle compositional differences in the three ECM types were not investigated in this study, these results suggest that tissue specific ECMs provide a culture microenvironment that is similar to the in vivo environment when used as coating substrates, and this new culture technique has the potential for use in drug development and the development cell-based therapies.
Keywords: ECM (extracellular matrix), cell culture, adhesion, cell proliferation
Introduction
Cell-extracellular matrix (ECM) interactions play a fundamental role in cell growth, organ development, tissue regeneration, and wound healing as well as in malignant growth processes. In vivo, cells attach to proteins and carbohydrate moieties present in the extracellular matrix (ECM). Examples of native forms of ECM include interstitial matrix and basal lamina. These structures provide support and anchorage for cells, segregate tissues from one another and regulate intercellular communication. However, once cells are isolated from tissue and removed from the native matrix, differentiated cells rapidly lose important characteristics when cultured without an adequate supportive microenvironment such as a substrate coating or a feeder layer. Cultured cells are influenced by soluble factors (e.g. growth factors and cytokines), physical factors (e.g. stress and strain) [1, 2] and by the insoluble matrix microenvironment [3]. However, the overwhelming majority of current research on this subject has focused on the use of soluble factors to influence cell growth [4, 5] although numerous groups have shown the importance of providing tissue-specific forms of ECM as a substratum for culturing cells in order to maintain phenotypic and functional characteristics [6–11]. For example, it is particularly difficult to maintain functional hepatocytes in culture, but when these cells are cultured with hormones, growth factors, serum free medium and ECM, the hepato-cellular physiology (including albumin synthesis and urea metabolism) can be maintained [11]. However, ECM cues are multi-factorial and complex, and mimicking them in a culture system is exceedingly difficult. When functional cells, such as liver cells, were cultured in vitro, the influences of hormones, growth factors, serum free medium and ECM, regulated the hepato-cellular physiology [11].
Specific components from the ECM are commonly used as a culture substratum and are commercially available. Individual matrix components (e.g. collagens, firobnectins, laminins) have been used in cell culture for many years and have been shown to have profound effects on cells, both with respect to attachment and survival as well as for the maintenance of various functions [12,13]. Certain tissue extracts enriched in matrix (e.g. Matrigel, extracts from amnions) have well-documented, dramatic effects on cells in culture [14]. However, these extracts are not tissue-specific. Moreover, non-human tissue extracts enriched in matrix, such as Matrigel, cannot be considered for clinical purposes.
Tissue-specific ECM coatings for tissue culture dishes and scaffolds for supporting cell growth have attracted attention in recent years [7]. Subtle differences in ECM composition from one type of tissue to another can affect cellular interactions in a lineage-specific manner. Due to the unique capability of each tissue’s ECM to provide an optimal substrate for specific cell types to attach and grow in vivo, cell culture systems seeking to maintain normal cell function should make use of similar strategies. This type of culture system would provide desirable cell-substrate interactions and would also sustain cell growth while maintaining phenotype and function. It has already been shown that tissue specific ECM can improve the reliability and efficiency of cell culture and stem/ progenitor cell differentiation [7]. Once inexpensive alternatives to conventional coatings are developed, use of this methodology may prove to be more economical for use in production of bio-pharmaceuticals and cell therapy.
The intricate and highly ordered nature of the ECM makes it difficult to reproduce using synthetic or purified components. In an attempt to reconstitute the mature cell niche in vitro, we prepared tissue-specific ECMs from skin, muscle and liver, and used these tissue-specific ECMs to coat culture dishes in which cells from each tissue could be grown. The goals of this study were to demonstrate that specific ECM derived from target tissues can produce an optimal substrate for in vitro culture and to develop optimal ECM-based culture systems for skin, skeletal muscle and liver cells for regenerative therapies and drug development.
2. Materials and Methods
2.1. Skin, skeletal muscle and liver tissue harvest
Institutional Review Board and Institutional Animal Care and Use Committee approvals for this project were obtained for human and animal surgery, including the collection of skeletal muscle, skin and liver tissue samples. Fresh and frozen organs and tissues were used to obtain the ECMs and cells for culture. Skeletal muscle tissue was harvested from the quadriceps and hamstring muscle of adult Fischer 344 × Brown Norway F1 rats. Liver tissues were harvested from the same rats. Skin tissues were harvested from adult swine, provided at no cost by Wayne Farms (Dobson, NC). Human foreskin tissue-derived epidermal and stromal cells were derived from tissues obtained from clinical circumcision procedures. An established human hepatocarcinoma cell line, HepG2, was also used for this study (GeneTex. Inc. San Antonio, TX, http://www.genetex.com).
2.2. Preparation of decellularized ECM
Skeletal muscle
To prepare the decellularized ECM, frozen muscle tissue was thinly sliced using a razor blade. Tissue slices were decellularized through exposure to a series of solutions with continuous stirring at 4°C. First, the tissue was extensively washed in deionized water. The water was changed every 12h for two days. Next, the tissue was incubated in a solution containing 0.05% trypsin and 0.2% ethylene diamine tetra acetic acid (EDTA) for one hour. Then, trypsin activity was quenched by adding DMEM containing 10% fetal bovine serum (FBS) and incubating overnight. The matrix was then incubated in 1% Triton X-100 for the next five days, and the solution was changed daily. Finally, the matrix was rinsed in fresh deionized water for two days followed by one day in PBS.
Skin
The stratum corneum and papillary dermis were removed from thawed swine skin, and the reticular dermis was used to prepare ECM. The decellularization process was similar to the one used for skeletal muscle tissue.
Liver
A tissue perfusion system was used during whole processes of liver tissue decellularization. The rat liver was thawed after being maintained at −80°C for one day. A 22 gauge needle was inserted through the rat liver portal vein and connected to a perfusion pump using silicon tubing. Rat liver tissue was gently flushed with distilled water at a flow rate of 4 ml/min for 48 hours. Subsequently, the liver tissue was treated with 0.05% trypsin soultion for one hour followed by addition of 10%FBS in the culture medium for 24 hours. All liver tissues were treated with 1%Triton X-100 and 0.1% ammonium hydroxide in distilled water for 4–5 days and then washed with distilled water in a perfusion system for 2 days. All the steps were carried out at 4°C unless otherwise mentioned.
2.3. Histological evaluation of decellularized ECM
A small piece of decellularized tissue (3×5×10 mm) from skin, muscle, liver and the respective fresh-frozen tissues, were individually sampled for histology and DNA content to confirm the extent of decellularization. Tissue samples were fixed in 10% phosphate-buffered formalin overnight. The tissues were dehydrated in a graded series of ethanol, soaked in xyelene, and then embedded in paraffin using a Tissue Tek processor. Paraffin -embedded sections were stained with haematoxylin and eosin (H&E), 4,6-diamidino-2-phenylindole (DAPI) (Vector, Burlingame, CA) and Masson’s trichrome to detect cellular structures, nuclei, and the ratio of cell to collagen matrix, respectively. Representative bright field (H&E) and fluorescent (DAPI) micrographs were taken at 100 × magnification.
2.4. Measurement of DNA content in decellularized ECM
A total of 18 decelluarized ECM along with their respective fresh-frozen tissue samples were used for DNA content evaluation. The tissues were lyophilized after incubation at −80°C for 24 h, weighed and then placed into sterile 1.5 ml micro-centrifuge tubes. Total DNA was isolated from the tissues using a commercially available kit (DNeasyTM, Qiagen, Valencia, CA). The DNA concentration (μg/mg tissue dry weight) was estimated at 280nm using a spectrophotometer (Thermo Spectronic, Biomate 3, Rochester, NY) and normalized to initial dry weight of the sample.
2.5. Preparation of ECM powders
To extract ECM from the decellularized tissues, the samples were sliced into small pieces and rinsed with deionized water for one hour. Citrate buffer (pH 4.3) was used to swell the tissue pieces with constant shaking at 4°C for 48 hours. After the solution was completely drained, the samples were frozen for one day at −80°C prior to lyophylization. Lyophilized samples were powdered using a micro grinder for 15 minutes and dissolved in 2M urea with constant shaking for three days at 4°C. Undissolved material was removed by centrifugation at 6,000 rpm for 20 min and the supernatant from the centrifuged samples further were clarified through a 40 μm filter,prior to dialysis against distilled water for three days. After dialysis, the resulting solution was frozen overnight in 50 ml conical tubes at −80°C and lyophilized.
2.6. ECM coating of tissue culture dishes
For use as a coating material, lyophilized ECM product was dissolved in 0.1% acetic acid. Protein content was determined using the Lowry method [15], and the solution was adjusted to the appropriate protein concentration for coating (0.1–0.8 mg/ml, Table 2). The coating solution was applied to the desired surface for two days in a humidified 37°C, 5% CO2 incubator. After two days, the coating solution was removed and the culture dish rinsed twice with PBS prior to seeding cells.
Table 2.
Effect of ECM coating concentrations on the growth of skeletal muscle cell on day 5
| Muscle-tissue ECM Concentration (Number of samples) |
0% mg/ml (n=3) |
0.05 mg/ml (n=3) |
0.1 mg/ml (n=3) |
0.15 mg/ml (n=3) |
0.2 mg/ml (n=3) |
|---|---|---|---|---|---|
| Cell Number (1×104) Mean ± SD |
4.96±0.814 | 7.70±0.40* | 8.13±0.602* | 8.30±0.70* | 7.86±1.301* |
P< 0.01 compared to control (control, 0% mg/ml)
2.7. Cell adhesion assay
Cell adhesion with skeletal muscle, skin and Hep G2 cells were measured using the Vybrant™ Cell Adhesion Assay Kit (Invitrogen, OR). Briefly, 96-well plates with flat-bottoms were coated with liver, skin, skeletal muscle ECM and collagen at 37°C for 3 h with 30 μl of a 0.05 mg/ml solution of coating material in PBS (pH7.2). The solution was then discarded and nonspecific binding blocked with 100 μl of 5% BSA in PBS for 1 h at 37°C. The wells were rinsed twice with culture media prior to cell seeding. After labeling the cells with 20 μM of calcein AM for 30 min at 37°C to incorporate the fluorescent dye, individual wells were seeded with 200 cells in 100 μl of culture media. Following a 90 min incubation period, fluorescence was measured using a Millipore Cytofluor 2350 plate reader to obtain total fluorescence. Non-adherent cells and media were aspirated, the wells were washed 3 times with culture medium and fluorescence was measured again to obtain adherent fluorescence. The background fluorescence was also measured after removing the cells from the wells as described in the kit. The percentage of adhesion was calculated using the formula: % cell adhesion = [(fluorescence in adherent cells-background fluorescence)/total fluorescence] × 100.
2.8. Effect of ECM concentration on cell growth in vitro
To determine the effect of ECM concentration on coating efficacy for rat skeletal muscle cell growth, ECM solutions ranging in concentration from 0.05 to 0.2 mg/ml were used to coat tissue culture dishes. Cells were seeded in triplicate in a 96 well plate, with 2,500 cells/well cultured in 200 μL of media under standard growth conditions. Media was changed every other day. Cell proliferation was determined using a 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-arboxanilide (MTS) cell proliferation assay kit (Roche Molecular Biochemicals, IN, USA) according to the manufacturer’s instructions.
2.9. Cell growth curve
To evaluate the effect of tissue-specific bioactive components, cell proliferation of each cell type was tested with all of the tissue-derived ECM coatings in a three-by-three factorial matrix. Well-established cell lines of human hepatocyte (Hep G2), rat skeletal muscle progenitor cells and human foreskin-derived epidermal cells were used to determine cell growth kinetics on the different coatings. Collagen-coated and non-coated wells were used as controls. Cells were plated in 24-well culture plates at a concentration of 5,000 cells per well in 2 ml growth medium. Cell number was determined on day 1, 2, 4 and 6 using a Coulter counter (Beckman Coulter, Chaska, Minnesota).
2.10. Immunocytochemistry
To indentify whether tissue specific ECM maintain the cell phenotypes, the cells were assessed for expression of specific cell markers, i.e. albumin and hepatocyte-specific antigen (Dako) for Hep G2; AE1/AE3( Sigma) for skin-derived epidermal cell, vimentin (Sigma) for fibroblasts and desmin and myosin(Sigma) for skeletal muscle cellls. Briefly, cells were fixed with 4% paraformaldehyde for 20 min, washed with PBS three times and incubated for an hour at room temperature with the various antibodies diluted in PBS containing 0.5% Triton-X-100 and 5% goat serum. Cells were washed and incubated with fluorescently labeled anti mouse secondary antibody (IgG2b, Southern Biotech) for an additional hour. Cells were washed, mounted and analyzed using an inverted fluorescent microscope.
2.11. Western blot analysis.
For protein analysis, cultured cell lysate was prepared using a lysis buffer as previously described [16]. Thirty micrograms of proteins were applied to a 10% SDS-Poly acrylamide gel and transferred to PVDF membrane following electrophoresis. Monoclonal antibodies to Hepatocyte-specific antigen (1:100, Dako), albumin (1:2500, Sigma) and β-actin (1:5000, Sigma) were used as the primary antibodies diluted in 5% fat-free milk powder in TBST [10mM Tris (pH7.4), 150mM NaCl, 0.1% Tween-20). After overnight incubations in the primary antibody, the membrane was washed with TBST and incubated with peroxidase-conjugated goat anti-mouse IgG (1:2000, CellSignaling) as the secondary antibody for 1h at room temperature. The membrane was washed well with TBST and protein bands detected using chemiluminescence reagent (Pierce). Images were captured using the Fuji Image Capture system (LAS300).
2. 12. Fluorescence-Activated Cell Sorter Analysis (FACS)
Cells were trypsinized, permeabilized with triton X-100 and stained with antibodies for skin markers [monoclonal AE1/AE3 (1:200, Dako) and goat polyclonal vimentin (1:200, Santa Cruz)], muscle markers [monoclonal desmin (1:50, Santa Cruz) and monoclonal myosin (1:50, Santa Cruz)] and hepatocyte markers [(monoclonal albumin (1:200) and hepatocyte-specific antigen; (1:50)]. Phycoerythrin (PE)- or fluorescein isothiocyanate (FITC)-conjugated isotype were used as controls. The stained cells were analyzed by flow cytometry using a fluorescence-activated cell sorter (FACS Caliber; Becton Dickinson, Mountain View, CA).
2.13. Statistical analysis
For DNA content, adhesion, coating concentration assessment and growth curves, samples were analyzed in replicates, averaged and reported as mean ± standard error of the mean (SEM). Comparisons between treatment groups were made using a single-factor analysis of variance (ANOVA) with a Holm-Sidak post-hoc test for significance at p<0.05 (SigmaStat) for the DNA, cell adhesion, and coating concentration data. A repeated measure ANOVA was used for the growth curve data.
3. Results
3.1. Histological evaluation of decellularized matrices
The overall scheme of the experimental design in our study is shown in Fig 1. In order to evaluate the efficiency of decellularization, histological staining of tissue sections were carried out. In fresh-frozen control tissues, intense cellular remnants, specifically nuclear material, were obvious in H&E (Figs. 2a,c,e) and DAPI (Figs. 2g,I,k) stained sections. There was minimal inter-fascicular and intra-fascicular space present in the H&E-stained sections of the fresh-frozen muscle and liver tissue ECM prior to processing. After decellularization, few nuclei are evident via H&E staining (Figs. 2b,d,f). DAPI staining revealed the presence of diffuse DNA remaining within the decellularized ECM (Figs. 2 h,j,l). An increase in intra-fascicular and inter-fascicular space after oxidative treatment was observed via H&E staining.
Fig. 1.
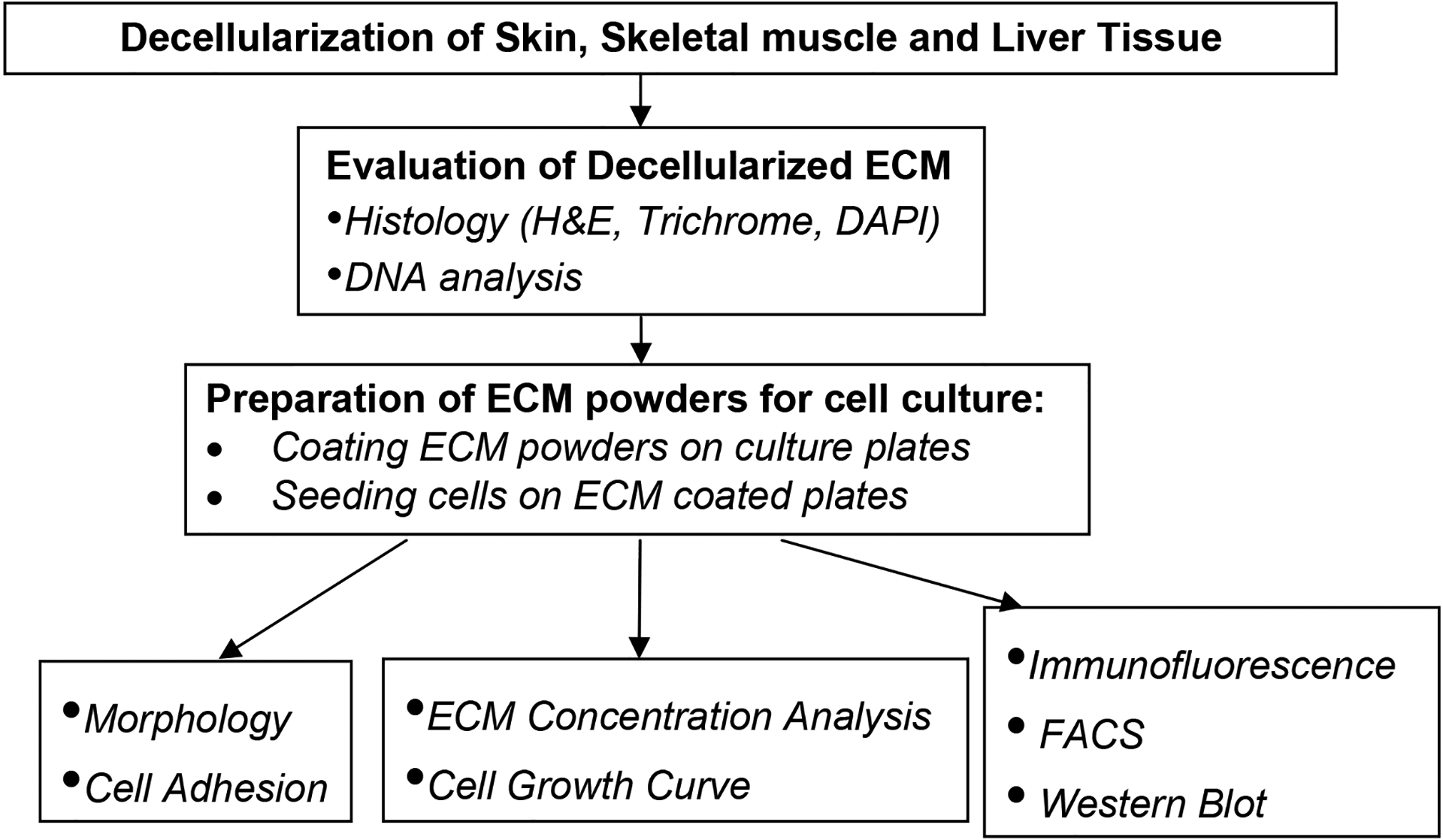
Schematic representation of the experimental design
Fig. 2. Histology of fresh liver, muscle and skin tissue and corresponding decellularized ECM using H&E and DAPI-staining.
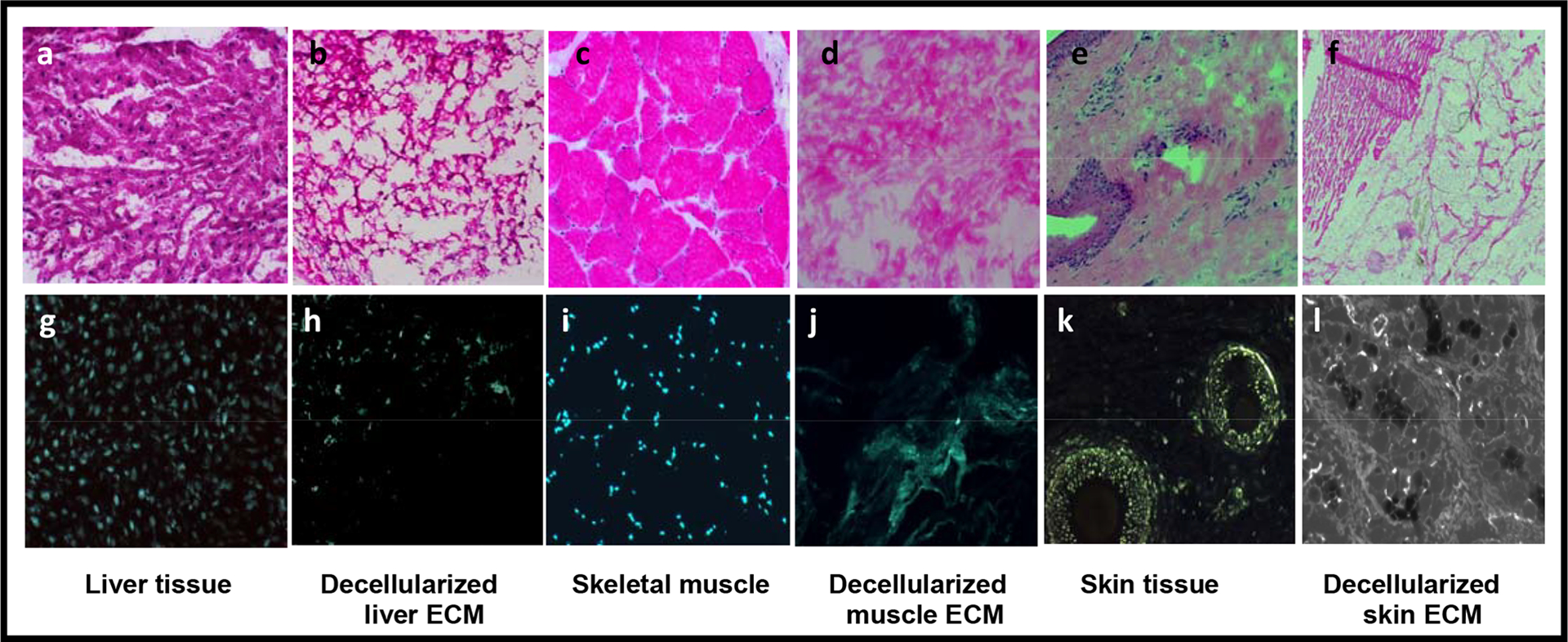
Histology of fresh liver (a,b) and decellularized liver (g,h) fresh muscle (c,d) and decellularized muscle (I,j) and fresh skin (e,f) and decellularized skin (k,l). The top panels show H&E (a-f) and the bottom panels, DAPI staining (g-l). Magnification, 100x. Cellular components are clearly visible in the fresh-frozen tissues (a, c, e) and rare or absent in the decellularized ECM tissues (b, d, f). Well-organized, punctate nuclei were clearly visible in fresh-frozen tissues (g,i,k), whereas efficient cellular removal was noted in the decellularized tissues. Only strands of disrupted DNA and RNA remnants can be seen, indicating that cells were no longer intact (h, j, l).
3.2. DNA content.
DNA from fresh-frozen tissue and decellularized ECM scaffolds was isolated using the DNeasy kit. DNA content (μg/mg tissue dry weight) was significantly decreased after the decellularization protocol in all tissue types investigated [2.8±0.3 to 0.2±0.1 in the liver (92±9%), 1.1±0.2 to 0.1±0.03 in muscle (93±7%) and 1.4±0.1 to 0.2±0.02 in skin (89±6%)] (Table 1; p<0.01, mg DNA/mg tissue dry weight ± SEM), indicating that most nuclear material, and therefore cellular material, had been removed from each type of tissue.
Table 1.
DNA content (Mean ± SD) in fresh-frozen tissues and their decellularized ECM
| Fresh-frozen tissue μg/mg tissue dry weight | Decellularized μg/mg tissue dry weight | DNA Clearance ratio (%) μg/mg tissue dry weight | |
|---|---|---|---|
| Liver tissue | 2.78 ±0.27 | 0.21 ±0.03 | 92.22± 9% |
| Muscle tissue | 1.07 ±0.21 | 0.07 ±0.03 | 93.01± 7% |
| Skin tissue | 1.43 ±0.12 | 0.16 ±0.02 | 89.06 ± 6% |
3.2. Effect of ECM concentration on cell growth
All three types of cells were plated at a density of 2500 cells/well in a 96 cell plate coated with the various ECMs. On day 5 of cell culture, the number of rat skeletal muscle cells significantly increased on all four concentrations (0.05, 0.1, 0.15, and 0.2 mg/ml) of muscle ECM coating compared to non-coating (Table 2, p<0.05). However, cell numbers were not significantly different among the four different muscle ECM coating concentrations, indicating that the concentration of the ECM used for coating did not affect subsequent cell growth in vitro. Henceforth, 0.05 mg/ml was used for all subsequent coating experiments, to conserve material.
3.3. Cell adhesion
Analyzed with adhesion assay kit, liver cell population significantly increased on all four types of coatings when compared to the non-coating (Table 3, p<0.05), whereas the cell populations were not significantly different among the four types of coating. Similarly, number of skin and muscle cells cultured on four different ECM coatings significantly increased compared to non-coating (Table 3, p<0.05), while all four ECM coatings promoted liver and skin cell adhesion. These data indicate that tissue ECM markedly enhanced cell attachment but there was no obvious tissue specificity for any of these cells.
Table 3.
Effect of various tissue-specific ECM coatings (0.05mg/ml) on cell adhesion
| Skin ECM | Muscle ECM | Liver ECM | Collagen | Non-coating | |
|---|---|---|---|---|---|
| HepG2 cells | 26.2±1.3** | 26.2±1.1** | 26.6±1.5 ** | 25.6±1.3** | 21.8±1.3 |
| Human foreskin cells | 23.3±3.2 ** | 21.1±2.2** | 22.6±2.3** | 22.6±2.7** | 16.8±3.1 |
| Rat Muscle cells | 19.3±2.3 | 20.0±1.8 * | 19.8±2.7 | 18.2±1.5 | 17.7±2.3 |
p< 0.05;
p< 0.01 compared to the control group (non-coating)
Number in bold indicate cells cultured on their original ECM coating.
3.4. Cell proliferation
Tissue specific ECM coatings enhanced cell proliferation in ECM cross-activity assays as shown in Fig 3. Each of the three cell types was cultured on the four different coatings as well as non-coated plates. Each culture had a similar cell proliferation pattern up to day 4 of culture. Thereafter, each cell type appeared to proliferate best on the ECM derived from its tissue of origin.
Fig. 3. Growth curve of skeletal muscle, skin and liver cells on tissue-specific ECM coatings.
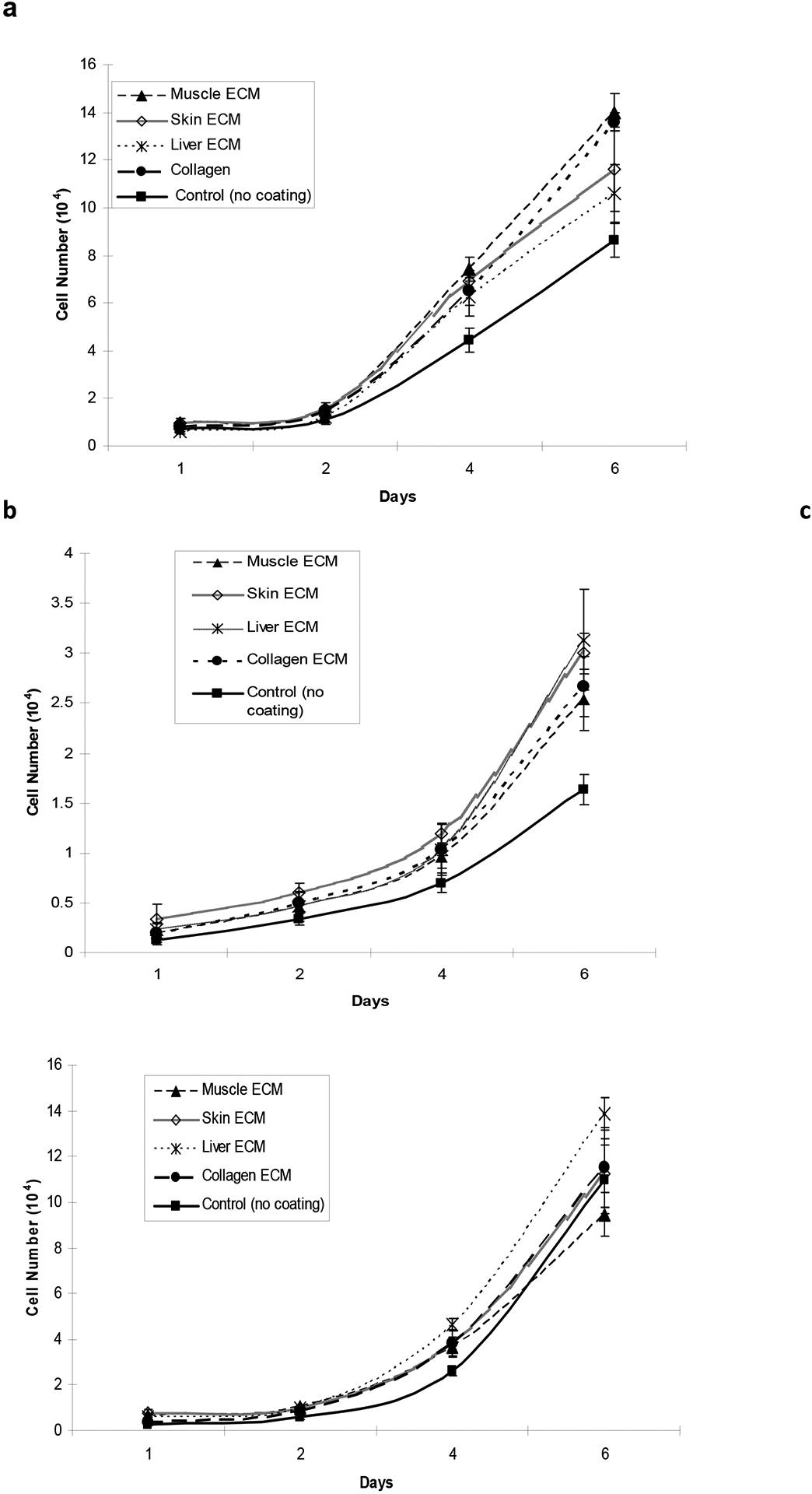
(a) Skeletal muscle cells, (b) skin cells and (c) liver cells were cultured on different tissue-specific ECM coatings for 6 days in 24 well plates. Media was replaced every other day and cells were counted using a Coulter counter on day 1, 2, 4 and 6. Growth and proliferation of each cell type was maximum on ECM derived from its tissue of origin.
Skeletal muscle cells showed a significant increase in cell number when cultured on muscle ECM (1.4×105 cells) and collagen type I coatings (1.3×105 cells) compared to skin (1.1 ×105 cells) and liver ECM (1.0×105 cells) coatings. Muscle cell growth is the poorest on uncoated plates (0.88 ×105 cells) on day 6 (Fig. 3a, p<0.05). Foreskin cells grew best on skin (3.2 ×104 cells) or liver ECM (3×104 cells) coatings rather than muscle ECM (2.6×104 cells) and collagen coatings (2.7×104 cells). The growth of these cells was also poorest on un-coated plates (1.7 ×104 cells) on day 6 (Fig. 3b, P<0.05). HepG2 grew best on liver ECM coatings (1.38 ×105cells) and poorest on collagen coated (1.15 ×105 cells) and non-coated plates (1.10 ×105 cells) (Fig 3c, P<0.05).
3.5. Cell differentiation
Morphology and immunocytochemistry
Each type of cell grew on 4 different coatings (skin, muscle, liver ECM and collagen) and non-coating plates, respectively. At day 5 of incubation the majority of rat skeletal muscle cells were highly differentiated, as evident by their multinucleated morphology. Human foreskin derived cell cultures consisted of epidermal and stromal cells. The HepG2 cell population consisted of clusters in archetypal morphology. Although each cell type had a similar appearance when grown on the four coatings and on non-coated plates, the tissue-specific ECM coatings maintained a high proliferative capacity of cultured cells (Data not shown). Immunofluorescence analysis showed that each cell type grown on its ECM of origin maintained its phenotype and strongly expressed its specific cell markers (Fig. 4).
Fig. 4. Morphology and immunofluorescence analysis of cells cultured on tissue-specifc ECM coatings.
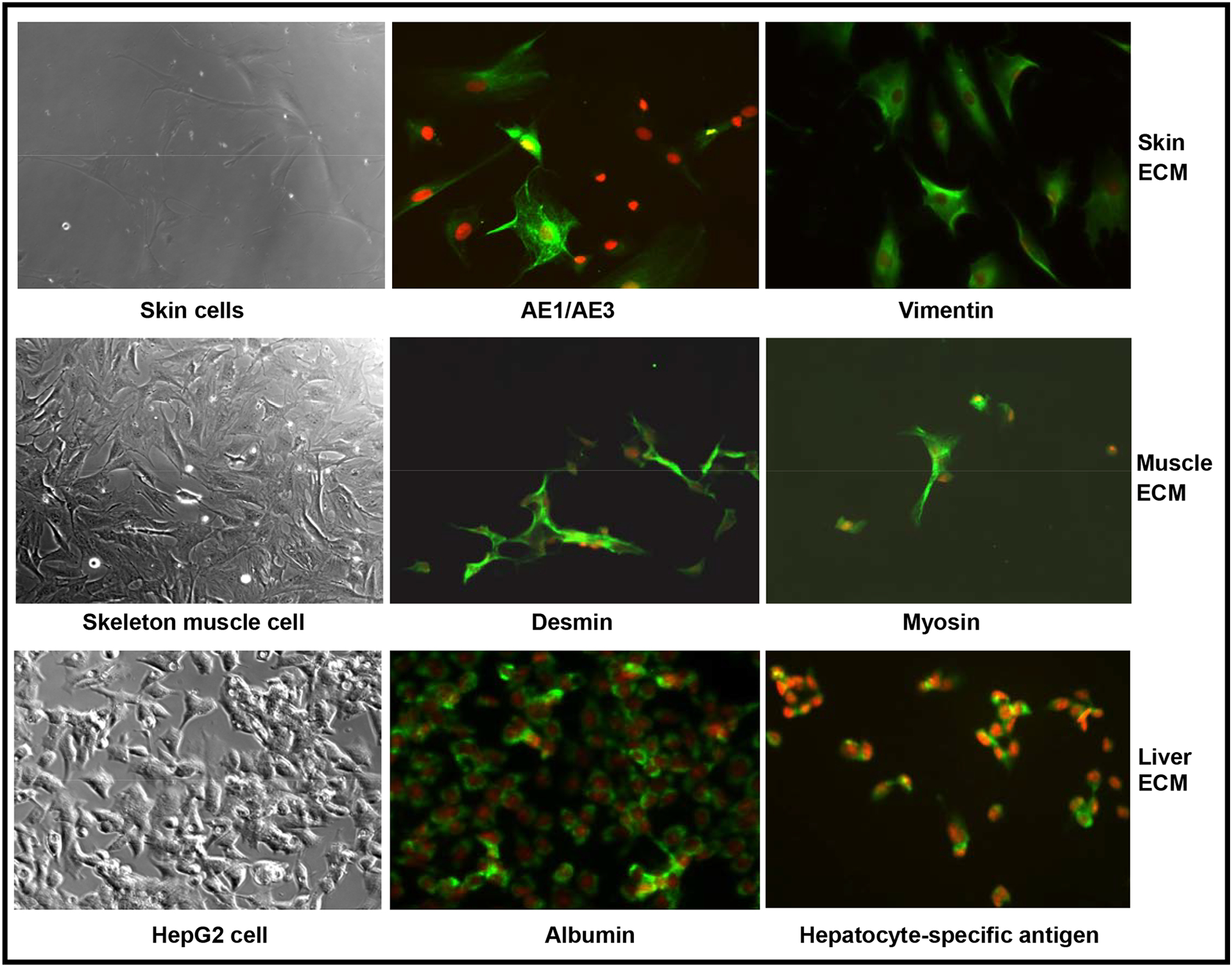
Cells were cultured on their respective tissue-specific ECM for 5 days. The cells were stained for specific protein markers by immunofluorescence. The top row ( AE1/AE3 and vimentin) for skin cells, middle row (desmin and myosin) for skelton muscle cells, and low row (albumin and hepatocyte-specific antigen) for Hep G2 cells depict fluorescent images. Magnification, X100
Flow Cytometry
Fluorescence-assisted cell scanning (FACS) analysis showed that muscle-specific proteins were strongly expressed in higher percentages of muscle derived cells when these cells were cultured on muscle ECM coating when compared to cells grown on other coatings and on non-coated plates (Table 4). On days 3 of culture, about 65% were positive for desmin, 45% positive for desmin, while on 14 of culture, about 45% of the cellswere positive for desmin and 8% positive for myosin. These findings suggest that the number of cells on coated muscle ECM expressing desmin and myosin decreased with time. However, in the presence of the muscle ECM microenvironment, the cells expressing muscle cell markers are at highest percentages and the decrease in expression level was not as rapid when compared to other environments.
Table 4.
FACS analysis of cells cultured on different ECM coatings for tissue-specific protein expression.
| Muscle-ECM | Skin-ECM | Liver-ECM | Collagen | Non-coating | ||
|---|---|---|---|---|---|---|
| Rat skeleton muscle cells | ||||||
| Day 3 | Desmin | 65.32% | 23.98% | 16.20% | 23.11% | 18.33% |
| Day 14 | Desmin | 45.17% | 15.44% | 42.17% | 40.85% | 26.83% |
| Foreskin cells | ||||||
| Day 3 | AE1/AE3 | 37.53% | 41.82% | 39.17% | 39.99% | 40.30% |
| Day 14 | AE1/AE3 | 30.60% | 40.43% | 31.38% | 36.30% | 31.27% |
| Liver Cells | ||||||
| Day 3 | Albumin | 59.90% | 82.36% | 95.62% | 83.68% | 59.10% |
| Day 14 | Albumin | 83.25% | 96.83% | 93.52% | 97.86% | 61.70% |
Numbers in bold indicate cells cultured on their original ECM coating
FACS analysis confirmed the expression of AE1/AE3 and vimentin in the human skin-derived epidermal and stromal cell cultures [17]. On days 3 and 14 of culture, AE1/AE3 expressing cells represented about 42% and 40%, respectively, of the total skin-derived cells when cultured on the skin ECM coating (Table 4). However, the percentage of cells expressing AE1/AE3 was significantly reduced at day 14 when skin cells were cultured on other coatings or non-coated plates. Unsurprisingly, skin-derived fibroblasts expressing vimentin grew well on any coating. The results from FACS analysis indicated that skin ECM coatings sustained skin-derived cells, particularly the epidermal cell phenotypes, better than other coatings when cultured for longer periods of time.
FACS analysis verified that liver-specific proteins (albumin and hepatocyte-specific antigen) were present in the human hepatoma cell line, HepG2 (Table 4). The percentage of albumin positive cells (96%) was significantly higher on the liver ECM coating group than on any other ECM day 3 of culture. Importantly, 94% of the total liver cells stained positive for albumin when cultured on the liver ECM coating on day 14 of culture. Additionally, cells staining positive for hepatocyte-specific antigen represented 23% and 31% of total liver cells grown on the liver ECM coating on days 3 and 14 of culture, respectively. The percentage of hepatocyte-specific antigen positive cells was significantly higher on the liver ECM coating compared to the other coatings on days 3 and 14 of culture (Table 4). In general, the liver ECM coating stably maintained liver cell phenotype.
Western blot analysis
Immunoblotting data further confirmed expression of albumin and hepatocyte-specific antigen in cultured HepG2 cells. The increased amounts of albumin protein and, to a lesser extent, hepatocyte-specific antigen, were observed when HepG2 cells were cultured either on liver, muscle or skin ECM coatings (Fig. 5). This was significantly higher than the levels observed when the cells were cultured on collagen-coated or non-coated plates.
Fig. 5. Immunoblot cell extracts from HepG2 cells cultured on tissue-specific ECM coatings.
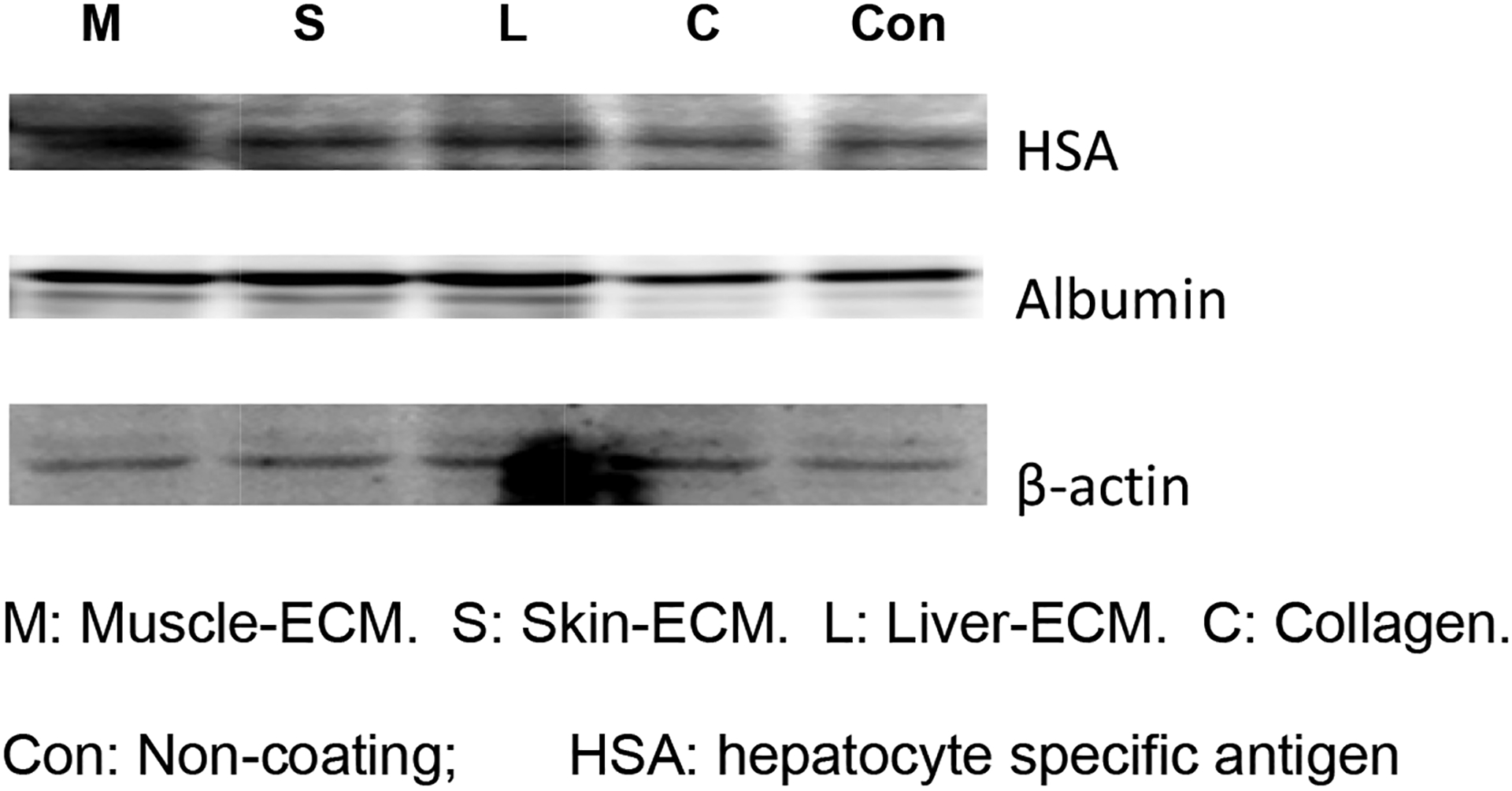
HepG2 cells were cultured on different tissue-specific ECM coatings for 14 days before protein lysates were prepared. Proteins were electrophoresed and transferred to a membrane, which was probed with antibodies to HAS, albumin and β-actin (loading control). Higher expression of liver-specific proteins are visible when cells are cultured on ECM versus control (no ECM coating) plates.
4. Discussion
Although remarkable advances have been made in cell culture techniques that have allowed for the proliferation and maintenance of specific cell phenotypes and functions, the current conservative culture system does not offer the ability to enhance cell expansion while retaining cellular functionality for multiple culture passages or in long-term cultures [18, 19]. The development of new culture conditions, including modification of culture surface coatings, would be beneficial in culturing primary cells [20]. In this study, we have demonstrated that the specificity of tissue-derived ECM as a culture surface coating promoted cell proliferation of three cell types and enhanced specific differentiation of skeletal muscle, and allow skin and liver cell types to maintaine their phenotypes. These findings indicate that tissue specific ECM coatings provide desirable cell-substrate interactions that support cell expansion and sustain cell function in primary cell cultures, which would be important in the fields of cell-based tissue engineering, drug development and cancer research.
Currently, commercially available bio-synthetic and natural bio-materials are commonly used for coating culture surfaces. Biodegradable synthetic polymers such as poly-L-Lysine and poly-L-Ornithine have been used to provide coatings that promote the attachment of various anchorage dependent cell types. One problem with the use of these types of polymeric materials is that they are degraded over a period of time by proteases from the serum in culture medium [21]. Natural polypeptide-based cell attachment factors such as collagen [22, 23], fibronectin [23, 24], laminin [25]and Matrigel [26] have been effectively employed for culture in certain cell lines and primary cell culture. Although many differentiated cell types grow well on these generic coatings, stem and progenitor cell characteristics as well as some terminally differentiated cell characteristics can easily be lost after isolation and culture from the host tissues even in the presence of these materials. Furthermore, synthetic ECM coatings, mentioned above, are non-specific and their expense precludes their large scale usage for clinical translation. There is no evidence to suggest that these are optimal biomaterial systems for maintaining primary cell phenotypes, or for mediating the differentiation of tissue-specific progenitor cells to their target fate, especially for cells involved in complex physiological processes. Additionally, generic culture substrates can induce unwanted differentiation pathways, resulting in heterogeneous cell populations.
In this study, we focused on tissue specific ECM as an optimal substrate for cell growth and maintenance of cell phenotype, especially for the study and eventual generation of differentiated cells. The goal of this study was to investigate the effect of tissue matched and non-tissue matched interactions between the cells obtained from skin, skeletal muscle and liver and ECM extracts developed from these same tissues. Our data showed that specificity of tissue-derived ECM as a culture surface coating enhanced cell viability and proliferation, and maintained stable lineage specific differentiation of skeletal muscle. Although these ECM did not promote the skin and liver cells differentiation, respectively, skin ECM maintained skin cell’s phenotype, same as liver ECM for Hep G2. Each cell type grew best on ECM from its tissue of origin. For example, the number of skeletal muscle cells significantly increased when the muscle cells were cultured on muscle ECM and collagen coating compared to other ECMs. A greater number of muscle cells expressed the muscle-specific proteins myosin and desmin when cultured on muscle ECM for up to two weeks. Similarly, skin cell numbers remarkably increased; more cells expressed the epidermal cell protein, AE1/AE3, in skin cells when skin cells were cultured on skin ECM coatings; Hep G2 cell population statistically increased, more cells expressed the albumin and hepatocyte specific antigens in liver cells when Hep G2 cells were grown on liver ECM coatings. In contrast, cell growth rates and number of cells expressing their specific proteins was not high enough when the cells were cultured on mismatched ECMs of either liver, skin or muscle tissues. Therefore, our experiments suggest that tissue specific decellularized ECM coatings provide a bio-mimetic environment that allows for the growth of more bio-comparable in vivo-like cell phenotypes, and that they may be especially useful for culture of completely differentiated cells and stem/progenitor cells. Each tissue-specific natural bio-matrix coating provided a culture surface, similar to the environment existing in vivo, for the growth of cells from that specific tissue. Interestingly, the matrix substratum can also significantly improve cell attachment and growth in vitro, but we did not find any obvious tissue specificity for these cells. In addition, the matrix effect was not concentration dependent. Although decellularized, each type of ECM still remains optimal substrate for cell growth in vitro. Through the decullularization processes, more than 90% of cellular material was removed from skeletal muscle, skin and liver tissues. Tissue specific ECM used as coating materials may have several other benefits, including 1) decellularized ECM decreases the potential risk of human cell exposure to animal cellular compounds in culture; 2) tissue-specific ECM can promote proliferation of cells while maintaining cell phenotype; 3) the bio-materials, isolated from fresh animal organs, are cost-effective and 4) the process of decellularization and coating are relatively simple.
ECM consists of multiple components such as collagens, elastin, adhesion proteins (i.e. fibronectin, laminin, nidogen) and proteoglyans (PGs) such as glycosaminoglycans (i.e. chondroitin sulfate-PGs, dermatan sulfate-PGs, heparin sulfate-PGs, heparin-PGs) [27]. The subset of these insoluble factors enables precise control of self-replication, proliferation and differentiation modes [7]. The cell microenvironment dictates the type of ECM receptor, or integrins, the cell expresses. Cells expressing these receptors bind to their respective ECM, thereby promoting proliferation or differentiation of the cell. For example, epithelial, muscle and nerve cells bind to Type IV collagen whereas liver cells binding to Type 1 collagen also leads to its differentiation [7]. Proteoglycan, on the other hand, bind cations and water molecules as well as are involved in regulating the movement of signaling molecules through the matrix. Although those ECM compounds enhance cell attachment, proliferation and differentiation, the precise mechanism of the cell-ECM interactions to modulate cell signaling pathways is unclear yet. Thus, cell culture using acellular matrix is more relevant physiologically than culture systems that do not use these matrices. More importantly, tissue-specific matrix is used, rather than a universal matrix that is prepared either from synthetic compounds or from individual matrix proteins purified from natural sources. Because the ECM contained in each tissue type in the body has subtle differences that may provide important cues for the cells that reside there. Mature cells are known to lose their phenotypes at an early stage in the culture process, and ECM derived from the tissue of their origin may prevent that loss. Even though these subtle compositional differences were not examined in the present study, the data presented demonstrate that tissue-specific matrix components significantly promote growth rates while maintaining phenotypes of skin, muscle and liver cells. One potential mechanism for the retention of mature cell characteristics may be the ability of the tissue-specific ECM to sequester most of the endogenously produced growth factors that modulate cell replication and differentiation state. Several studies have demonstrated that cell-derived ECM (for example, Matrigel) is unique in its ability to preserve differentiated cell properties, including functional ones, and prompt cell proliferation [7, 18, 20]. This type of ECM has been used as a substrate for mimicking in vivo micro-environments as well as for culture surface coating to prompt cell adhesion [8, 28], proliferation [9], migration [12, 29] and differentiation [30–32]. It appears that the tissue-specific ECMs used in this study have similar functions to cell-derived ECM, including preservation of cell function and phenotype. This suggests that establishment of a unique cell culture dish coated with tissue-specific ECM proteins and the appropriate combination of growth factors would facilitate the culture and maintenance of skin, muscle and liver cell cultures for development of therapeutic applications.
In this study, all three cell types proliferated and maintained cell phenotype and functionality when seeded on a tissue-specific ECM substrate instead of on non-matrix, cross-matched matrix, or a generic matrix system. Our data suggests that the maintenance of a specific cell phenotype is controlled by elements of the tissue-specific microenvironment, which mainly consists of ECM proteins associated with growth factors. The exact nature of the specific ECM components required to coat culture surfaces is not completely understood. Certain specific molecules may be present in these ECM derivatives, and tissues that presumably contain more complex ECM structures (e.g. liver), appear to demonstrate greater responses to tissue matched ECM, whereas those with less complex matrices (e.g. muscle) appear to have similar characteristics whether they are grown on tissue specific ECM or on generic collagen. This reinforces the hypothesis that the more complex the ECM, the more specific the microenvironment for cell growth and function. The precise nature of these differences is being explored in an ongoing study.
Conclusions
Synthetic and natural biomaterials have commonly been used as substrates for culturing primary mammalian cells. While isolated differentiated cell types grow well in culture, their phenotypic and functional characteristics may change during long-term culture without an inadequate supportive microenvironment or feeder layer. This study demonstrated that tissue-specific matrix components cause significant differences in adhesion efficiencies, growth rates, and morphology and phenotypes of skin, muscle and liver cells. Tissue specific ECM allows mature or differentiated cells to maintain their phenotype. As a culture surface substrate, tissue-specific biomaterials could result in improvements in current cell culture techniques. Moreover, this could result in improved results from cell-based assays and allow faster translation of novel therapeutic strategies to the clinic.
Acknowledgements,
The study has been supported by NIH STTR 1R41EB005900-01A1. The authors would like to thank Dr. Lola M Reid for her valuable comments and Dr. Jennifer Olson for editorial assistance with this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol 1997;59:551–571. [DOI] [PubMed] [Google Scholar]
- 2.Syedain ZH, Weinberg JS, Tranquillo RT. Cyclic distension of fibrin-based tissue constructs: evidence of adaptation during growth of engineered connective tissue. Proc Natl Acad Sci USA 2008; 105:6537–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funderburgh ML, Mann MM, Funderburgh JL. Keratocyte phenotype is enhanced in the absence of attachment to the substratum. Mol Vis 2008;14:308–317. [PMC free article] [PubMed] [Google Scholar]
- 4.Duffy DM, Garrett SM, Ellis SE, Scott TR. Influence of supramammary lymph node extract on in vitro cell proliferation. Cell Prolif 2008;41:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida A, Kanno H, Watabe D, Akasaka T, Sawai T. The role of heparin-binding EGF-like growth factor and amphiregulin in the epidermal proliferation of psoriasis in cooperation with TNFalpha. Arch Dermatol Res 2008;300:37–45. [DOI] [PubMed] [Google Scholar]
- 6.Reid LM, Fiorino AS, Sigal SH, Brill S, Holst PA. Extracellular matrix gradients in the space of Disse: relevance to liver biology. Hepatology 1992;15:1198–1203. [DOI] [PubMed] [Google Scholar]
- 7.McClelland R, Wauthier E, Uronis J, Reid L. Gradients in the liver’s extracellular matrix chemistry from periportal to pericentral zones: influence on human hepatic progenitors. Tissue Eng Part A 2008;14:59–70. [DOI] [PubMed] [Google Scholar]
- 8.Lallier TE, Yukna R, Moses RL. Extracellular matrix molecules improve periodontal ligament cell adhesion to anorganic bone matrix. J Dent Res 2001;80:1748–1752. [DOI] [PubMed] [Google Scholar]
- 9.Huet C, Pisselet C, Mandon-Pepin B, Monget P, Monniaux D. Extracellular matrix regulates ovine granulosa cell survival, proliferation and steroidogenesis: relationships between cell shape and function. J Endocrinol 2001;169:347–360. [DOI] [PubMed] [Google Scholar]
- 10.Fujita M, Spray DC, Choi H, Saez J, Jefferson DM, Hertzberg E, et al. Extracellular matrix regulation of cell-cell communication and tissue-specific gene expression in primary liver cultures. Prog Clin Biol Res 1986;226:333–360. [PubMed] [Google Scholar]
- 11.Enat R, Jefferson DM, Ruiz-Opazo N, Gatmaitan Z, Leinwand LA, Reid LM. Hepatocyte proliferation in vitro: its dependence on the use of serum-free hormonally defined medium and substrata of extracellular matrix. Proc Natl Acad Sci U S A 1984;81:1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everitt EA, Malik AB, Hendey B. Fibronectin enhances the migration rate of human neutrophils in vitro. J Leukoc Biol 1996;60:199–206. [DOI] [PubMed] [Google Scholar]
- 13.Shirahashi H, Wu J, Yamamoto N, Catana A, Wege H, Wager B, et al. Differentiation of human and mouse embryonic stem cells along a hepatocyte lineage. Cell Transplant 2004;13:197–211. [DOI] [PubMed] [Google Scholar]
- 14.Yamasaki C, Tateno C, Aratani A, Ohnishi C, Katayama S, Kohashi T, et al. Growth and differentiation of colony-forming human hepatocytes in vitro. J Hepatol 2006;44:749–757. [DOI] [PubMed] [Google Scholar]
- 15.Lopez JM, Imperial S, Valderrama R, Navarro S. An improved Bradford protein assay for collagen proteins. Clin Chim Acta 1993;220:91–100. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, McNeill E, Tian H, Soker S, Andersson KE, Yoo JJ, et al. Urine derived cells are a potential source for urological tissue reconstruction. J Urol 2008;180:2226–2233. [DOI] [PubMed] [Google Scholar]
- 17.Smith KJ, Graham JS, Skelton HG, Hamilton T, O’Leary T, Okerberg CV, et al. Sensitivity of cross-reacting antihuman antibodies in formalin-fixed porcine skin: including antibodies to proliferation antigens and cytokeratins with specificity in the skin. J Dermatol Sci 1998;18:19–29. [DOI] [PubMed] [Google Scholar]
- 18.Spray DC, Ginzberg RD, Morales EA, Gatmaitan Z, Arias IM. Electrophysiological properties of gap junctions between dissociated pairs of rat hepatocytes. J Cell Biol 1986;103:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kropp BP, Zhang Y, Tomasek JJ, Cowan R, Furness PD 3rd, Vaughan MB, et al. Characterization of cultured bladder smooth muscle cells: assessment of in vitro contractility. J Urol 1999;162:1779–1784. [PubMed] [Google Scholar]
- 20.Spray DC, Fujita M, Saez JC, Choi H, Watanabe T, Hertzberg E, et al. Proteoglycans and glycosaminoglycans induce gap junction synthesis and function in primary liver cultures. J Cell Biol 1987;105:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodhi M, Naqvi T, Opperman G, inventors. Polymeric coatings and methods for cell attachment. US Patent No. 028253, 2006. [Google Scholar]
- 22.Chen Y, Li Y, Zhang S, Song Y, Ye N, Hua P, et al. Mass culture of anchorage-dependent animal cells with newly born calf skin collagen membrane and microcarrier. Chin J Biotechnol 1992;8:123–130. [PubMed] [Google Scholar]
- 23.Bengali Z, Rea JC, Shea LD. Gene expression and internalization following vector adsorption to immobilized proteins: dependence on protein identity and density. J Gene Med 2007;9:668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol 1989;109:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran T, McNeill KD, Gerthoffer WT, Unruh H, Halayko AJ. Endogenous laminin is required for human airway smooth muscle cell maturation. Respir Res 2006;7:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelain F, Bottai D, Vescovi A, Zhang S. Designer self-assembling Peptide nanofiber scaffolds for adult mouse neural stem cell 3-dimensional cultures. PLoS ONE 2006;1:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Hernandez A, Amenta PS. The extracellular matrix in hepatic regeneration. Faseb J 1995;9:1401–1410. [DOI] [PubMed] [Google Scholar]
- 28.Salber J, Grater S, Harwardt M, Hofmann M, Klee D, Dujic J, et al. Influence of different ECM mimetic peptide sequences embedded in a nonfouling environment on the specific adhesion of human-skin keratinocytes and fibroblasts on deformable substrates. Small 2007;3:1023–1031. [DOI] [PubMed] [Google Scholar]
- 29.Guerrero-Esteo M, Lastres P, Letamendia A, Perez-Alvarez MJ, Langa C, Lopez LA, et al. Endoglin overexpression modulates cellular morphology, migration, and adhesion of mouse fibroblasts. Eur J Cell Biol 1999;78:614–623. [DOI] [PubMed] [Google Scholar]
- 30.Mergenthaler HG, Dormer P. Hemopoiesis in human micro long-term bone marrow culture with preformed extracellular matrix. Haematologica 1990;75:12–16. [PubMed] [Google Scholar]
- 31.de Jong-Hesse Y, Kampmeier J, Lang GK, Lang GE. Effect of extracellular matrix on proliferation and differentiation of porcine lens epithelial cells. Graefes Arch Clin Exp Ophthalmol 2005;243:695–700. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang A, Ng R, Yang ST. Long-term culturing of undifferentiated embryonic stem cells in conditioned media and three-dimensional fibrous matrices without extracellular matrix coating. Stem Cells 2007;25:447–454. [DOI] [PubMed] [Google Scholar]