

Abstract
Background
The enlarged vestibular aqueduct (EVA) is the commonest malformation of inner ear accompanied by sensorineural hearing loss in children. Three genes SLC26A4, FOXI1, and KCNJ10 have been associated with EVA, among them SLC26A4 being the most common. Yet, hotspot mutation screening can only diagnose a small number of patients.
Methods
Thus, in this study, we designed a new molecular diagnosis panel for EVA based on multiplex PCR enrichment and next‐generation sequencing of the exon and flanking regions of SLC26A4. A total of 112 hearing loss families with EVA were enrolled and the pathogenicity of the rare variants detected was interpreted according to the American College of Medical Genetics and Genomics (ACMG) guidelines.
Results
Our results showed that 107/112 (95.54%) families carried SLC26A4 biallelic mutations, 4/112 (3.57%) carried monoallelic variants, and 1/112 (0.89%) had none variant, resulting in a diagnostic rate of 95.54%. A total of 49 different variants were detected in those patients and we classified 30 rare variants as pathogenic/likely pathogenic, of which 13 were not included in the Clinvar database.
Conclusion
Our diagnostic panel has an increased diagnostic yield with less cost, and the curated list of pathogenic variants in the SLC26A4 gene can be directly used to aid the genetic counseling to patients.
Keywords: enlarged vestibular aqueduct, multiplex PCR, pathogenic variants, SLC26A4
We designed a new molecular diagnosis panel for EVA based on multiplex PCR enrichment and next‐generation sequencing of the exon and flanking regions of SLC26A4. This strategy can not only greatly save time and reagents, but also provide more accurate diagnostic information for EVA patients.

1. INTRODUCTION
Hearing loss (HL) is one of the most common disabilities that can seriously affect human recognition and communication. HL affects nearly 1–3 in 1000 infants (Morton & Nance, 2006). In China, there are ~0.8 million HL children whose age less than 7 years, and this number increases at a rate of 30,000 cases annually (Zhang et al., 2013).
Sensorineural HL with enlarged vestibular aqueduct (EVA) is the second common in children, and the EVA is the commonest malformation of the inner ear in children (Wemeau & Kopp, 2017). To date, three genes SLC26A4 (OMIM: 605646), FOXI1 (OMIM: 601093; Yang et al., 2007), and KCNJ10 (OMIM: 602208; Yang et al., 2009) have been associated with EVA; yet some studies have raised questions regarding the involvement of the last two genes in EVA. Landa et al. tested mutations in KCNJ10 and FOXI1 for sixty‐eight patients with monoallelic mutations of SLC26A4 and found no evidence for a significant association between mutations of KCNJ10 and FOXI1 with SLC26A4 (Priya Landa et al., 2013). Zhao et al. (2014) also showed that KCNJ10 might not be a contributor to non‐syndromic EVA in the Chinese population.
Mutations in the SLC26A4 gene lead to Pendred syndrome (OMIM: 274600) or deafness, autosomal recessive 4, with EVA (OMIM: 600791). Moreover, SLC26A4 is the second most frequent gene in the Chinese population suffering from HL, which accounts for 5%–14% of the patients with NSHL (Yuan et al., 2009; Zhou et al., 2019). The SLC26A4 gene has 21 exons and encodes Pendrin, a multiple transmembrane protein containing 780 amino acids. Pendrin is expressed in the inner ear, thyroid gland, and kidney and can mediate the transport of Cl−, I−, OH−, and other anions (Royaux et al., 2001). So far, a total of 608 mutations of the SLC26A4 gene have been reported (UniProt, ClinVar, VarSome & PubMed), including missense, nonsense, frameshift, in‐frame indel, start loss, splicing, synonymous and non‐coding, with missense variants being the most common type (339 variants, https://varsome.com/gene/SLC26A4). The mutation spectrum varies in different regions and races; for example, in East Asia c.919‐2A>G and c.2168A>G are the most common variants (Park et al., 2005; Tsukamoto et al., 2003; Wang et al., 2007), whereas in Caucasians the most frequent variant is c.1001+1G >A (Colleen Campbell et al., 2001).
Sanger sequencing is the traditional approach for EVA diagnosis. Nevertheless, this technique is time‐consuming and costly, which limits its clinical application. Population‐specific diagnostic panels have been designed to screen hotspot mutations of SLC26A4 (Yan et al., 2017; Yuan et al., 2012). These methods can only be used to diagnose some of the patients with HL; for many patients only one or no mutation is detected. Recent application of next‐generation sequencing (NGS) has proven to be powerful in identifying pathogenic mutations in EVA patients (Lin et al., 2019; Liu, Wang, et al., 2016; Liu et al., 2020). Multiplex PCR can simultaneously amplify multiple regions in the same reaction tube, thus significantly saving time and reagents, and providing more accurate diagnostic information (Lin et al., 2012). Using multiplex PCR and NGS technology to sequence target regions of associated EVA genes (SLC26A4, FOXI1, and KCNJ10), biallelic variants were detected in 59% (27/46) of cases (Liu, Wang, et al., 2016). Yet, the sample size of the study was small; therefore, the diagnostic rate of sequencing the whole SLC26A4 gene may be inaccurate.
Herein, we designed a new molecular diagnosis panel for EVA on the basis of multiplex PCR enrichment and NGS of the exon and flanking regions of SLC26A4. We recruited 112 families with EVA patients from Henan Province to investigate the detection rate of SLC26A4 whole‐gene sequencing in EVA patients. This genetic diagnostic panel has the power to explore the etiologies of EVA and can be extended to clinical practices (Figure 1).
FIGURE 1.
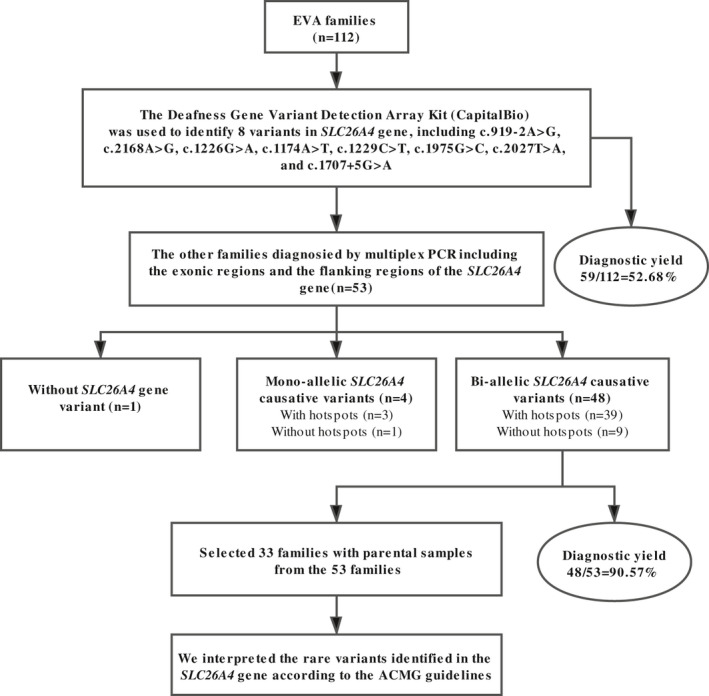
Study design and summary of genetic diagnostic results
2. MATERIALS AND METHODS
2.1. Subject recruitment and clinical evaluation
In this study, 112 hearing loss families with EVA were from Henan Province enrolled at the Department of Otology, the affiliated hospitals of Zhengzhou University between 2018 and 2019. All subjects underwent comprehensive clinical evaluations to ensure the EVA diagnosis and rule out other diseases, which could result in HL, such as otitis media or syndromic HL. Physical examinations included thyroid sonography, functional thyroid tests, a high‐resolution CT scan of the temporal bone, and optional magnetic resonance hydrography (MRH) examination of the inner ear. Detailed audiological examinations were performed in all patients. Otoscopic examination, tympanometry, distortion product otoacoustic emission (DPOAE), and auditory steady‐state response (ASSR), auditory brainstem response (ABR), and pure‐tone audiometry were executed based on the patient's age and degree of coordination. According to the criteria of EVA, the temporal bone CT scan of the patients showed bilateral EVA with the width of the vestibular aqueduct greater than 1.5 mm (Hwang et al., 2015). Patients’ audiological data were assessed based on the HL criteria established by the European Working Group on Genetics of Hearing Impairment and HL degree averaged over 0.5, 1, 2, and 4 kHz was classified into four tiers: mild (20–40 dB HL), moderate (41–70 dB HL), severe (71–95 dB HL), and profound (>95 dB HL).
Before being enrolled in this study, all patients were genetically tested using the Deafness Gene Variant Detection Array Kit (CapitalBio). The test kit covers 15 hotspot mutations of four genes, of which eight are hotspot mutations of the SLC26A4 gene, including c.919‐2A>G, c.2168A>G, c.1226G>A, c.1174A>T, c.1229C>T, c.1975G>C, c.2027T>A, and c.1707+5G>A. With the help of this kit, confirmative genetic diagnosis was achieved in 59 (52.68%) of the 112 families. To verify the accuracy of our panel and to diagnose the other 53 families, we performed multiplex PCR enrichment and NGS in all 112 EVA families.
2.2. DNA extraction, multiplex PCR enrichment, and sequencing
A minimum of 2 mL of peripheral blood was obtained from the patients and their parents. The whole blood genomic DNA extraction kit (GenMagBio) was used to extract genomic DNA. NanoDrop One (Thermo Fisher Scientific) was used to measure DNA concentration and purity. The extracted DNA was controlled for quality by 1% agarose gel electrophoresis.
After purity and quality checking, multiplex PCR enrichment was performed to amplify the exonic regions and the flanking region of the SLC26A4 gene following the optimal reaction conditions developed in this study. A total of 22 pairs of primers were designed, synthesized, and assigned into two reaction pools, the primers displayed in Table S1. The first target amplification was carried out with the following cycling program: 99℃ for 2 min, 23 cycles of 99℃ for 15 s and 60℃ for 4 min, then 72℃ for 10 min. In the second round of amplification, the two multiplex PCR products from the first round were mixed and then amplified using a pair of universal primers with index sequences to distinguish different samples. The cycling program for the second round was as follows: 95℃ for 3 min, 4 cycles of 98℃ for 20 s, 60℃ for 15 s, 72℃ for 30 s, then 72℃ for 5 min. The resulting libraries were sequenced on an Illumina MiniSeq sequencer (Illumina Inc.) with the paired‐end of 150 bp.
2.3. Bioinformatics analysis and variant interpretation
Sequencing reads were aligned to GRCh37 using the Burrows‐Wheeler Aligner (BWA; version 0.7.17‐r1188); Single nucleotide variants (SNVs) and short Indels calling were identified using GATK Haplotype Caller software (version 4.1.2; McKenna et al., 2010). The obtained VCF files were annotated using Vcfanno software (version 0.3.1; Pedersen et al., 2016) with external database, including Clinvar (Landrum et al., 2018), 1000 Genomes Project (Genomes Project et al., 2015), gnomAD (Karczewski et al., 2020), ExAC (Lek et al., 2016), and dbNSFP (Liu et al., 2016). According to the information of population frequency and inherited pattern (Paila et al., 2013), the variants were filtered, and the selected variants were interpreted by clinicians and genetic consultants based on the standard and guidelines of genetic variation interpretation of ACMG (Richards et al., 2015) and the ClinGen hearing loss expert group's recommendation on variant interpretation (Oza et al., 2018). The variant nomenclature was based on the SLC26A4 canonical transcript NM_000441.2.
2.4. Sanger sequencing
Sanger sequencing was used to verify the variants in the proband and parents revealed by our panel. NCBI Primer‐last software was used to design the primers, which were synthesized by Sunya Biotechnology Co., Ltd. Sequencing was done by SeqStudio Genetic Analyzer (Applied Biosystems/Life Technologies) after PCR product purification, the results were visualized by Chromas software.
3. RESULTS
3.1. Clinical features of the EVA patients
In this study, 117 patients from 112 families met the diagnostic criteria for EVA. There were 70 males and 47 females; the average age was 7.1 years, ranging from 6 months to 33 years old. The average onset age was 2.4 years, ranging from 0 to 20 years old. In our cohort, the degree of HL was mild to profound in 51 (43.59%) patients, severe in 53 (45.30%) patients, moderate in 11 (9.40%) patients, and mild in 2 (1.71%) patients. Of the 117 patients, 36 (30.77%) patients showed progressive HL, 38 (32.48%) patients were stable, and 43 (36.75%) patients were fluctuating (Table 1).
TABLE 1.
Clinical features of the 112 EVA families
| Demographics | Number of patients (%), n = 117 |
|---|---|
| Gender | |
| Male | 70 (59.83%) |
| Female | 47 (40.17%) |
| Test age (years) | |
| <10 | 91 (77.78%) |
| ≥10 | 26 (22.22%) |
| Onset age (years) | |
| 0–2 | 84 (71.79%) |
| >2 | 33 (28.21%) |
| Degree of HL | |
| Profound | 51 (43.59%) |
| Severe | 53 (45.30%) |
| Moderate | 11 (9.40%) |
| Mild | 2 (1.71%) |
| Evolution of HL | |
| Stable | 38 (32.48%) |
| Progressive | 36 (30.77%) |
| Fluctuating | 43 (36.75%) |
3.2. Genetic examination of SLC26A4 by multiplex PCR combined with NGS
In this study, we designed a diagnostic assay for EVA patients based on multiplex PCR target enrichment and NGS of the SLC26A4 gene (Figure 2). The assays can simultaneously be applied to as many as 96 samples and can be completed in three days (one day for DNA extraction and PCR enrichment, one day for sequencing, and one day for data analysis). For each sample, an average of raw 10 Mbp was generated, with more than 85% of bases having a Phred quality score Q ≥ 30 (Q30). The quality control standards for each sample were as follows: 99% of the clean reads can map to the human genome (GRCh37), and the average sequencing depth of target regions was 1000X, with 100% of target regions having coverage greater than 200X.
FIGURE 2.
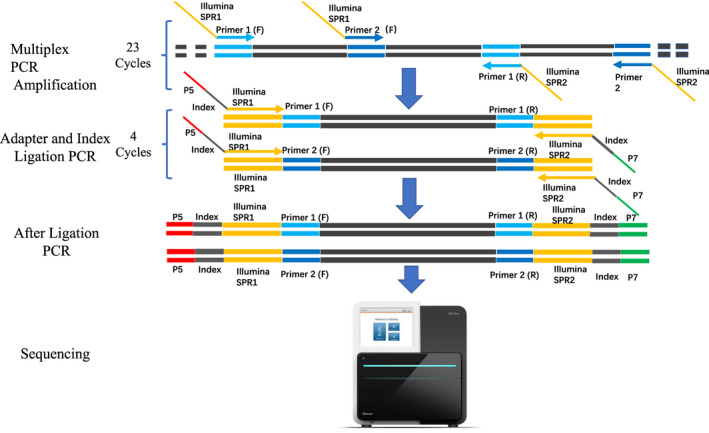
Multiplex PCR target enrichment and sequencing
Among 112 families, 59 families were previously (before enrolling in this study) diagnosed with EVA using CapitalBio Deafness Gene Variant Detection Array Kit. These patients were re‐tested for EVA in this study and our results are consistent with theirs. Among the undiagnosed 53 families, 43 (37.72%) carried monoallelic variants, and 10 (8.77%) had none hotspot variants of the SLC26A4 gene. After screening, genetic diagnosis was achieved for 48 (90.57%) of 53 families. Of the five undiagnosed families, 4 (7.55%) carried monoallelic variants and 1 (1.89%) had no variant of SLC26A4. In total, this NGS panel yielded a diagnostic rate of 95.54% (107/112 families; Table 2). Sanger sequencing was used to verify the 41 mutations (excluding eight hotspot mutations), and the results (Supplemental File 1) were consistent with NGS, indicating that our panel is accurate and reliable. Of the 107 families with a purported genetic diagnosis, mutations in 26 families (24.3%) were confirmed in one or none of the parents, which needs further analysis (Table 2).
TABLE 2.
Pathogenic variants of the 117 patients from 112 EVA families
| GT no. | SLC26A4 genotype | No. of patientsa/b/c | GT no. | SLC26A4 genotype | No. of patientsa/b/c | ||
|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | ||||
| 1 | c.919‐2A>G | c.919‐2A>G | 19/2/2 | 32 | c.919‐2A>G | c.754T>C | 1/0/0 |
| 2 | c.919‐2A>G | c.2168A>G | 8/2/1 | 33 | c.919‐2A>G | c.916dup | 0/1/0 |
| 3 | c.919‐2A>G | c.1229C>T | 4/1/0 | 34 | c.919‐2A>G | c.946G>T | 1/0/0 |
| 4 | c.919‐2A>G | c.1226G>A | 3/1/0 | 35 | c.1174A>T | c.87G>C | 1/0/0 |
| 5 | c.919‐2A>G | c.1975G>C | 2/1/1 | 36 | c.1174A>T | c.2027T>A | 0/1/0 |
| 6 | c.919‐2A>G | c.2027T>A | 3/0/1 | 37 | c.1226G>A | c.1336C>T | 1/0/0 |
| 7 | c.919‐2A>G | c.1174A>T | 3/0/0 | 38 | c.1226G>A | wt | 1/0/0 |
| 8 | c.919‐2A>G | c.317C>A | 3/0/0 | 39 | c.1226G>A | c.946G>T | 1/0/0 |
| 9 | c.919‐2A>G | c.589G>A | 3/0/0 | 40 | c.1226G>A | c.2027T>A | 1/0/0 |
| 10 | c.919‐2A>G | c.1299dup | 2/0/0 | 41 | c.1229C>T | c.1707+5G>A | 1/0/0 |
| 11 | c.919‐2A>G | c.1318A>T | 2/0/0 | 42 | c.1229C>T | c.439A>G | 1/0/0 |
| 12 | c.919‐2A>G | c.439A>G | 2/0/0 | 43 | c.1229C>T | c.1975G>C | 1/0/0 |
| 13 | c.916dup | c.1656T>G | 2/0/0 | 44 | c.1264‐12T>A | c.1547dup | 1/0/0 |
| 14 | c.919‐2A>G | wt | 1/1/0 | 45 | c.1264‐6T>G | wt | 0/0/1 |
| 15 | c.2168A>G | c.2168A>G | 1/0/0 | 46 | c.1343C>T | c.1336C>T | 1/0/0 |
| 16 | c.1174A>T | c.1174A>T | 1/0/0 | 47 | c.1586T>G | c.1786C>T | 1/0/0 |
| 17 | c.279T>A | c.279T>A | 0/1/0 | 48 | c.1707+5G>A | c.1336C>T | 1/0/0 |
| 18 | c.919‐2A>G | c.109G>T | 1/0/0 | 49 | c.1975G>C | c.1746del | 1/0/0 |
| 19 | c.919‐2A>G | c.1343C>A | 0/0/1 | 50 | c.2168A>G | c.589G>A | 0/1/0 |
| 20 | c.919‐2A>G | c.1708G>A | 0/1/0 | 51 | c.2168A>G | c.349del | 1/0/0 |
| 21 | c.919‐2A>G | c.1803+1G>A | 0/1/0 | 52 | c.2168A>G | c.1318A>T | 0/1/0 |
| 22 | c.919‐2A>G | c.1985G>A | 0/1/0 | 53 | c.2168A>G | c.846T>A | 0/0/1 |
| 23 | c.919‐2A>G | c.1991C>T | 1/0/0 | 54 | c.2168A>G | c.1174A>T | 1/0/0 |
| 24 | c.919‐2A>G | c.2000T>C | 1/0/0 | 55 | c.2168A>G | c.387del | 1/0/0 |
| 25 | c.919‐2A>G | c.2014G>A | 0/1/0 | 56 | c.2168A>G | c.1975G>C | 1/0/0 |
| 26 | c.919‐2A>G | c.2162C>A | 1/0/0 | 57 | c.249G>A | c.1371C>A | 0/0/1 |
| 27 | c.919‐2A>G | c.2167C>G | 1/0/0 | 58 | c.281C>T | c.1173C>A | 1/0/0 |
| 28 | c.919‐2A>G | c.2174_2177dup | 1/0/0 | 59 | c.697G>C | c.1667A>G | 1/0/0 |
| 29 | c.919‐2A>G | c.227C>T | 0/0/1 | 60 | c.946G>T | c.1229C>T | 0/1/0 |
| 30 | c.919‐2A>G | c.281C>T | 1/0/0 | 61 | wt | wt | 0/1/0 |
| 31 | c.919‐2A>G | c.415+2T>C | 1/0/0 | ||||
Variants are based on SLC26A4 canonical transcript NM_000441.2.
Abbreviations: a,b,c, number of patients with mutations confirmed in both, one, or none of the parental samples; GT no., genotype number; No. of patients, Number of patients with this genotype; wt, wild‐type.
Of the 48 families with achieved genetic diagnosis by our panel, 33 families, which included both parental samples, were selected and enrolled for interpreting the genetic variants identified in the SLC26A4 gene. The 33 families were found to have one allele or none of the common SLC26A4 mutations, including c.919‐2A>G, c.2168A>G, c.1226G>A, c.1174A>T, c.1229C>T, c.1975G>C, c.2027T>A, and c.1707+5G>A covered by CapitalBio Deafness Gene Variant Detection Array Kit (CapitalBio). Our assay further identified a second (27 families) or two rare mutations (6 families) in 33 families (Table S2). A total of 30 rare mutations were found, including 17 missense mutations, 4 nonsense mutations, 4 duplications, 3 deletions, and 2 splicing mutations, one of which is a novel mutation (NM_000441.2:c.2162C>A, p.Thr721Lys).
3.3. Interpretation of genetic variation
We interpreted the genetic variants identified in the SLC26A4 gene according to the standards and guidelines for interpreting genetic variants proposed by the ACMG and the ClinGen hearing loss expert group's recommendation on variant interpretation. The guidelines classified variants relevant to Mendelian diseases into a five‐tier system: “Pathogenic,” “Likely Pathogenic,” “Variants of Uncertain Significance,” “Likely Benign,” and “Benign.” Based on those standards, we classified 26 of 30 variants as “Pathogenic,” three as “Likely Pathogenic” and one as “Variants of Uncertain Significance.” The ACMG classification details and the information on whether the variants were reported in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, last accessed May 2, 2020) and Deafness Variation Database (DVD, http://deafnessvariation‐database.org, last accessed May. 2, 2020) or not, are summarized in Table 3.
TABLE 3.
ACMG classification of rare variants in the SLC26A4 gene
| No. | Variants | Patients | Reference | Classification | |||
|---|---|---|---|---|---|---|---|
| ClinVar | DVDa | ACMG | ACMG criteria | ||||
| 1 |
c.87G>C p.Glu29Asp |
1 | NI | P | LP |
PM2: Not found in gnomAD PM3: Pathogenic mutation confirmed in trans in one patient PM5: Another pathogenic missense variant (c.85G>C, p.Glu29Gln) at the same codon PP4: Patient phenotype highly specific for gene |
|
| 2 |
c.349del Frameshift |
2 | Pang, Chai, Chen, et al. (2015), Pang, Chai, He, et al. (2015), Wang et al. (2007), Zhao et al. (2014) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2_Supporting: gnomAD exome East Asian allele frequency = 0.000461 <0.0007 PM3_Strong: Pathogenic mutation confirmed in trans in one patient and phase unknown in four patients PP4: Patient's phenotype highly specific for gene |
| 3 |
c.589G>A p.Gly197Arg |
3, 4, 5 | Liu, Wang, et al. (2016), Zhao et al. (2014) | P | P | P |
PM2: gnomAD exome East Asian allele frequency = 0.00005437 <0.00007 PM3_VreyStrong: Pathogenic mutation confirmed in trans in one patient and phase unknown in seventeen patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 4 |
c.1586T>G p.Ile529Ser |
6 | Gao et al. (2016), Huang et al. (2011), Qi Li and Yuan (2012), Zhao et al. (2014) | P/LP | P | P |
PM2: Not found in gnomAD PM3_VreyStrong: Pathogenic mutation phase unknown in eight patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 5 |
c.1786C>T Stop‐gain |
6 | Liu, Wang, et al. (2016) | NI | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3: Pathogenic mutation confirmed in trans in one patient PP4: Patient's phenotype highly specific for gene |
| 6 |
c.317C>A p.Ala106Asp |
7, 8‐1, 8‐2 | Campbell (2001) | NI | P | P |
PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in 3 patients and phase unknown in two patients PP1: Segregation in one affected relative PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 7 |
c.2167C>G p.His723Asp |
9 | Yao et al. (2015), Yuan et al. (2009), Zhao et al. (2014) | NI | P | P |
PM2: gnomAD genomes East Asian allele frequency = 0.00005437 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in four patients PM5: Another missense pathogenic variant (c.2168A>G, p.His723Arg) at the same codon PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 8 |
c.946G>T Stop‐gain |
10, 11 | Gao et al. (2016), Huang (2011), Zhao et al. (2014) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in six patients PP4: Patient's phenotype highly specific for gene |
| 9 |
c.2174_2177 dup Frameshift |
12 | Chai et al. (2013), Yao, Li, et al. (2013) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: gnomAD genomes East Asian allele frequency = 0.000007959 <0.00007 PM3_Strong: Pathogenic mutation confirmed in trans in one patient and phase unknown in two patients PP4: Patient's phenotype highly specific for gene |
| 10 |
c.281C>T p.Thr94Ile |
13 | Zhao et al. (2014) | P | P | P |
PM2: gnomAD genomes East Asian allele frequency = 0.00005437 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in six patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 11 |
c.1318A>T Stop‐gain |
14, 15 | (Zhao et al., 2014) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in four patients PP4: Patient's phenotype highly specific for gene |
| 12 |
c.916dup Frameshift |
16‐1, 16‐2 |
Gao et al. (2016), Han et al. (2017), Jiang et al. (2015, 2017) |
P/LP | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2_Supporting: gnomAD East Asian allele frequency = 0.0001631 <0.00007 PM3_Strong: Pathogenic mutation in six patients phase unknown PP1: Segregation in one affected relative PP4: Patient's phenotype highly specific for gene |
| 13 |
c.1656T>G p.Ser552Arg |
16‐1, 16‐2 | Chen et al. (2016) | NI | NI | LP |
PM2: Not found in gnomAD PM3_Strong: Pathogenic mutation confirmed in trans in two patients and phase unknown in one patient PP1: Segregation in one affected relative PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 14 |
c.2162C>A p.Thr721Lys |
17 | NI | NI | LP |
PM2: Not found in gnomAD PM3: Pathogenic mutation phase known in one patient PM5: Another pathogenic missense variant (c.2162C>T, p. Thr721Met) at the same codon PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
|
| 15 |
c.109G>T Stop‐gain |
18 | Yuan et al. (2009), Zhao et al. (2014) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_Strong: Pathogenic mutation confirmed in trans in one patient and phase unknown in two patients PP4: Patient's phenotype highly specific for gene |
| 16 |
c.439A>G p.Met147Val |
19, 30‐1, 30‐2 | Hosoya et al. (2019), Huang (2011), Lee et al. (2015), Tsukamoto et al. (2003), Zhao et al. (2014) | A | P | P |
PP1: Segregation in one affected relative PM2_Supporting: gnomAD East Asian allele frequency = 0.0001087 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in three patients and phase unknown in six patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 17 |
c.387del Frameshift |
20 | Wang et al. (2007), Zhao et al. (2014) | NI | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_Strong: Pathogenic mutation confirmed in trans in one patient and phase unknown in two patients PP4: Patient's phenotype highly specific for gene |
| 18 |
c.1336C>T Stop‐gain |
21, 22, 23 | Zhao et al. (2014) | P/LP | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: gnomAD exome allele frequency = 0.000007089 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in three patients and phase unknown in six patients PP4: Patient's phenotype highly specific for gene |
| 19 |
c.1343C>T p.Ser448Leu |
22 | Gao et al. (2016), Chen and Liu (2014), Lai et al. (2007), Liu, Wang, et al. (2016), Liu, Wu, et al. (2016), Wu et al. (2008), | LP | P | P |
PM2_Supporting: gnomAD genomes East Asian allele frequency = 0.0001088 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in 10 patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 20 |
c.2000T>C p.Phe667Ser |
24 | Chen et al. (2012), Chen and Liu (2014), Leilei Zhao et al. (2018), Liu, Wang, et al. (2016), Zhao et al. (2014) | NI | P | P |
PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in three patients and phase unknown in five patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 21 |
c.1547dup Frameshift |
25 | Chen and Liu (2014), Gao et al. (2016), Liu, Wang, et al. (2016), Zhang et al. (2016) | LP | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2_Supporting: gnomAD East Asian allele frequency = 0.000272 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in two patients and phase unknown in five patients PP4: Patient's phenotype highly specific for gene |
| 22 |
c.1264‐12T>A aberrant splicing |
25 | Wu et al. (2016) | NI | P | P |
PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in one patient and phase unknown in seven patients PP4: Patient's phenotype highly specific for gene |
| 23 |
c.754T>C p.Ser252Pro |
26 | Duan et al. (2017), Liu, Wang, et al. (2016) | NI | P | P |
PM2: gnomAD genomes East Asian allele frequency = 0.00005437 <0.00007 PM3_VeryStrong: Pathogenic mutation confirmed in trans in five patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 24 |
c.415+2T>C aberrant splicing |
27 | Zhao et al. (2014) | NI | LP | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in one patient and phase unknown in six patients PP4: Patient's phenotype highly specific for gene |
| 25 |
c.1746del Frameshift |
28 | Wang et al. (2007), Yao, Chen, et al. (2013), Yao, Li, et al. (2013), Zhao et al. (2014) | P | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in four patients and phase unknown in one patient PP4: Patient's phenotype highly specific for gene |
| 26 |
c.1667A>G p.Tyr556Cys |
29 | Lopez‐Bigas et al. (2002), Wang et al. (2017) | P/LP | P | P |
PM2: gnomAD genomes allele frequency = 0.00001594 <0.00007 PM3_Strong: Homozygous confirmed in trans in four patients and pathogenic mutation phase unknown in one patient PP1_Strong: Segregation in three affected relatives and one unaffected relative PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 27 |
c.697G>C p.Val233Leu |
29 | Hu et al. (2007), Huang et al. (2018) | VUS | P | LP |
GnomAD genomes East Asian allele frequency = 0.001353 >0.0007, not apply to PM2 PM3_Strong: Pathogenic mutation confirmed in trans in one patient and phase unknown in 4 patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 28 |
c.1173C>A p.Ser391Arg |
31 | Gao et al. (2016), Huang et al. (2011), Liu, Wang, et al. (2016), Zhao et al. (2014) | LP | P | P |
PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in one patient and phase unknown in six patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 29 |
c.1991C>T p.Ala664Val |
32 | Han et al. (2017), Huang et al. (2011), Zhao et al. (2014) | NI | P | P |
PM2: Not found in gnomAD PM3_VeryStrong: Pathogenic mutation confirmed in trans in one patient and phase unknown in six patients PP3: REVEL score >0.7 PP4: Patient's phenotype highly specific for gene |
| 30 |
c.1299dup Frameshift |
33‐1, 33‐2 | NI | P | P |
PVS1: Null variant in the gene with established LOF as a disease mechanism PP1: Segregation in one affected relative PM2: Not found in gnomAD PM3_Strong: Pathogenic mutation phase known in two patients PP4: Patient's phenotype highly specific for gene |
|
Variants are based on SLC26A4 canonical transcript NM_000441.2.
Deafness variation database.
In this study, we also identified a novel mutation of SLC26A4 (NM_000441.2: c.2162C>A, p.Thr721Lys), which has not been reported in the Deafness Variation Database, 1000 Genomes Project database, the Human Gene Mutation Database, and the gnomAD database. It is a missense mutation, located in a highly conserved region among mammals. Interestingly, at the same codon, a pathogenic mutation (c.2162C>T, p.Thr721Met) has been reported, which impairs the protein function of SLC26A4 (Ishihara et al., 2010). We interpreted this novel mutation as “Likely Pathogenic” according to the ACMG guidelines (Table 3).
4. DISCUSSION
In this study, SLC26A4 mutation analysis was performed in 112 hearing loss families with EVA. The first‐pass screening was performed by CapitalBio Deafness Gene Variant Detection Array Kit (CapitalBio), including eight hotspots of the SLC26A4 gene, c.919‐2A>G, c.2168A>G, c.1226G>A, c.1174A>T, c.1229C>T, c.1975G>C, c.2027T>A, and c.1707+5G>A. Confirmed genetic diagnoses were achieved in 59 (52.68%) of the 112 families. We performed multiplex PCR enrichment and NGS in all 112 families to verify the accuracy of our panel and to further analyze SLC26A4 mutations. Our results showed that 48 (90.57%) out of 53 families carried biallelic mutations of the SLC26A4 gene, 4 (7.55%) carried monoallelic mutations and one (1.89%) had no variant of the SLC26A4 gene. Of the 112 EVA families, 59/112 (52.68%) were diagnosed by hotspots screening, and 107/112 (95.54%) by multiplex PCR enrichment and NGS. Our results suggested that compared with hotspot mutations screening, multiplex PCR enrichment and NGS significantly improved the diagnostic rate of patients with EVA.
Besides increasing the diagnostic rate of EVA patients, our test strategy (trio sequencing) and the following variant interpretation improved the accuracy of variant pathogenicity interpretation. To date, 608 mutations of the SLC26A4 gene have been reported worldwide. There are many mutations detected in EVA patients; yet, the mutations have not been tested or verified in the parents. Given that the phase of these mutations remained unknown, it is difficult to determine whether the mutations caused HL in patients. Moreover, the reported pathogenicity of the SLC26A4 gene mutations in EVA patients has not been interpreted according to the ACMG guidelines, and the pathogenicity needs to be curated. In our study, we collected and sequenced samples of probands and parents, and analyzed the sequencing results at the same time, which helped detect the pathogenic variants of patients.
Several NGS panels targeting the exon and flanking regions of EVA‐related genes have been designed for diagnosing EVA patients. Liu, Wang, et al. (2016) designed a panel based on multiplex PCR and NGS for sequencing of exon regions of EVA pathogenic genes, including SLC26A4, FOXI1, and KCNJ10, and found biallelic potential pathogenic variants in 27/46 (59%) patients (Liu, Wang, et al., 2016). More recently, Lin et al. (2019) developed an NGS panel targeting the entire length of three genes (SLC26A4, FOXI1, and KCNJ10), as well as the exons of 10 other EVA‐related genes. They performed tests in 50 EVA families, which were negative for two hotspot mutations (c.919‐2A>G and c.2168A>G), and the diagnostic yield for this panel is 84.7% (Lin et al., 2019). The diagnostic yield of our panel was higher compared to those of two panels but is close to another that reported a 90% diagnostic rate, which sequenced the 21 exons of the SLC26A4 gene by PCR and Sanger sequencing in 107 Chinese EVA patients (Wang et al., 2007). Two reasons may explain the higher diagnostic yields of our panel: (a) a trio‐sequencing strategy was exploited for most of the patients, which is helpful for the detection and pathogenicity interpretation of mutations; (b) the cohort in our study (mainly Chinese in Henan Province) may have a higher detection rate of SLC26A4 mutations, as discovered by a molecular epidemiology study (Yuan et al., 2012).
The multiplex PCR has a few limitations. First, multiplex PCR enrichment and NGS can only detect single nucleotide variants (SNVs) and small indels, but not copy number variants (CNVs). Indeed, several CNVs in the SLC26A4 gene have been reported (Anwar et al., 2009; Hu et al., 2007; Pang, Chai, He, et al., 2015; Pera et al., 2008; Sloan‐Heggen et al., 2016) and CNVs are a common cause of non‐syndromic HL (Shearer et al., 2014). Allele dropout represents the other risk of misdiagnosis for multiplex PCR based methods (Blais et al., 2015). Cautious primer design avoiding to have polymorphism sites at the ends of primers could decrease the risk of misdiagnosis. One solution would be to long‐distance PCR and NGS of the whole SLC26A4 gene, which can detect all types of variants.
In conclusion, we designed an assay based on multiplex PCR enrichment and NGS to identify variants of the SLC26A4 gene in EVA patients, showing that the diagnostic rate was 95.54% for 112 families. The assay is fast and economical; it requires only a small amount of sequencing data (10 Mbp) and can be completed in three days. This new assay can be used as a first‐round test for EVA patients with satisfactory diagnosis yields in clinical applications.
CONFLICTS OF INTERESTS
The study is funded by the Collaborative Innovation Project of Zhengzhou jointly sponsored by Zhengzhou University and Sanglin Biotechnogy Ltd. Zengguang Yang and Yingtao Tian are employees of Sanglin Biotechnogy Ltd.
AUTHOR CONTRIBUTIONS
Study design: Yongan Tian, Hongen Xu, and Wenxue Tang. Patient phenotypic analysis and genetic counseling: Xiaohua Li, Wei Lu, Ying Shi, Yan Zhang, Hui Zhang, Chang Jiang, Ying Xu, Bei Chen, Jun Liu. Next‐generation sequencing and Sanger sequencing: Yongan Tian, Sen Zhang, Huanfei Liu, Ruijun Li, Yingtao Tian, Beiping Zeng, Tong Li, Qianyu Lin, and Wenxue Tang. Data analysis and variant interpretation: Hongen Xu, Zengguang Yang, Danhua Liu. Writing and review of the original draft of the manuscript: Yongan Tian, Hongen Xu, Danhua Liu, Bei Chen, Jun Liu, and Wenxue Tang. All authors have read and approved the final manuscript.
ETHICAL COMPLIANCE
This study was conducted following the Declaration of Helsinki, developed by the World Medical Association, and the experimental protocol was approved by the Medical Ethics Committee of The Second Affiliated Hospital of Zhengzhou University (Approval No. 2018008). Written informed consent was obtained from all the participants (for minors, guardians signed the consent).
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank all the family members for their participation in this study. We also thank the Supercomputing Center of Zhengzhou University for providing computational and storage resources. The English in this manuscript has been proofread by Ryan Tang, who is a native speaker of English.
Tian, Y., Xu, H., Liu, D., Zhang, J., Yang, Z., Zhang, S., Liu, H., Li, R., Tian, Y., Zeng, B., Li, T., Lin, Q., Wang, H., Li, X., Lu, W., Shi, Y., Zhang, Y., Zhang, H., Jiang, C., … Tang, W. (2021). Increased diagnosis of enlarged vestibular aqueduct by multiplex PCR enrichment and next‐generation sequencing of the SLC26A4 gene. Molecular Genetics & Genomic Medicine, 9, e1734. 10.1002/mgg3.1734
Yongan Tian and Hongen Xu are co‐first authors.
Funding information
The study is funded by the Collaborative Innovation Project of Zhengzhou (Zhengzhou University) (grant no. 18XTZX12004), the Medical Science and Technology Projects in Henan (grant no. SBGJ2018043) to WT, and the Joint Project of Medical Science and Technology Research in Henan Province (grant no. LHGJ20190317) to HX.
Contributor Information
Bei Chen, Email: fccchenb@zzu.edu.cn.
Jun Liu, Email: ljent0371@126.com.
Wenxue Tang, Email: twx@zzu.edu.cn.
DATA AVAILABILITY STATEMENT
The data of this study are available from the corresponding authors upon reasonable request. The novel or reclassified variants have been submitted to the ClinVar database (SCV001572597‐SCV001572611).
REFERENCES
- Anwar, S., Riazuddin, S., Ahmed, Z. M., Tasneem, S., Khan, S. Y., Griffith, A. J., Friedman, T. B., & Riazuddin, S. (2009). SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred's syndrome in Pakistanis. Journal of Human Genetics, 54(5), 266–270. 10.1038/jhg.2009.21 [DOI] [PubMed] [Google Scholar]
- Blais, J., Lavoie, S. B., Giroux, S., Bussières, J., Lindsay, C., Dionne, J., Laroche, M., Giguère, Y., & Rousseau, F. (2015). Risk of misdiagnosis due to allele dropout and false‐positive PCR artifacts in molecular diagnostics: Analysis of 30,769 genotypes. The Journal of Molecular Diagnostics, 17(5), 505–514. 10.1016/j.jmoldx.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Campbell, C., Cucci, R. A., Prasad, S., Green, G. E., Edeal, J. B., Galer, C. E., Karniski, L. P., Sheffield, V. C., & Smith, R. J. H. (2001). Pendred syndrome DFNB4 and PDS SLC26A4 identification of eight novel mutations and possible genotype phenotype correlations. Human Mutation, 17(5), 403–411. 10.1002/humu.1116 [DOI] [PubMed] [Google Scholar]
- Chai, Y., Huang, Z., Tao, Z., Li, X., Li, L., Li, Y., Wu, H., & Yang, T. (2013). Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. American Journal of Medical Genetics. Part A, 161A(9), 2226–2233. 10.1002/ajmg.a.36068 [DOI] [PubMed] [Google Scholar]
- Chen, K., Wang, X., Sun, L., & Jiang, H. (2012). Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngology Head and Neck Surgery, 146(6), 972–978. 10.1177/0194599812439670 [DOI] [PubMed] [Google Scholar]
- Chen, K., Zong, L., Liu, M., Wang, X., Zhou, W., Zhan, Y., Cao, H., Dong, C., Tang, H., & Jiang, H. (2014). Developing regional genetic counseling for southern Chinese with nonsyndromic hearing impairment a unique mutational spectrum. Journal of Translational Medicine, 12, 64. 10.1186/1479-5876-12-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., Dong, C., Wang, Q., Zhong, Z., Qi, Y., Ke, X., & Liu, Y. (2016). Targeted next‐generation sequencing successfully detects causative genes in Chinese patients with hereditary hearing loss. Genetic Testing and Molecular Biomarkers, 20(11), 660–665. 10.1089/gtmb.2016.0051 [DOI] [PubMed] [Google Scholar]
- Duan, S. H., Ma, J. L., Yang, X. L., & Guo, Y. F. (2017). Simultaneous multigene mutation screening using SNPscan in patients from ethnic minorities with nonsyndromic hearingimpairment in Northwest China. Molecular Medicine Reports, 16(5), 6722–6728. 10.3892/mmr.2017.7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Z., Lu, Y. U., Ke, J., Li, T., Hu, P., Song, Y. U., Xu, C., Wang, J., Cheng, J., Zhang, L., Duan, H., Yuan, H., & Ma, F. (2016). Application of SNPscan in genetic screening for common hearing loss genes. PLoS One, 11(10), e0165650. 10.1371/journal.pone.0165650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project, C , Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., Marchini, J. L., McCarthy, S., McVean, G. A., & Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, R., Li, L., Duan, L., Xia, Y., Kuyaxi, P., Zhao, J., Zhao, Q. I., Zhang, H., & Chen, Y. U. (2017). Efficiency of microarray and SNPscan for the detection of hearing loss gene in 71 cases with nonsyndromic hearing loss. Medicine, 96(25), e7149. 10.1097/MD.0000000000007149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya, M., Saeki, T., Saegusa, C., Matsunaga, T., Okano, H., Fujioka, M., & Ogawa, K. (2019). Estimating the concentration of therapeutic range using disease‐specific iPS cells: Low‐dose rapamycin therapy for Pendred syndrome. Regenerative Therapy, 10, 54–63. 10.1016/j.reth.2018.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H., Wu, L., Feng, Y., Pan, Q., Long, Z., Li, J., Dai, H., Xia, K., Liang, D., Niikawa, N., & Xia, J. (2007). Molecular analysis of hearing loss associated with enlarged vestibular aqueduct in the mainland Chinese: A unique SLC26A4 mutation spectrum. Journal of Human Genetics, 52(6), 492–497. 10.1007/s10038-007-0139-0 [DOI] [PubMed] [Google Scholar]
- Huang, B., Han, M., Wang, G., Huang, S., Zeng, J., Yuan, Y., & Dai, P. (2018). Genetic mutations in non‐syndromic deafness patients in Hainan Province have a different mutational spectrum compared to patients from Mainland China. International Journal of Pediatric Otorhinolaryngology, 108, 49–54. 10.1016/j.ijporl.2018.02.015 [DOI] [PubMed] [Google Scholar]
- Huang, S., Han, D., Yuan, Y., Wang, G., Kang, D., Zhang, X., Yan, X., Meng, X., Dong, M., & Dai, P. U. (2011). Extremely discrepant mutation spectrum of SLC26A4 between Chinese patients with isolated Mondini deformity and enlarged vestibular aqueduct. Journal of Translational Medicine, 9, 167. 10.1186/1479-5876-9-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, M., Marovich, R., Shin, S. S., Chi, D., & Branstetter, B. F. (2015). Optimizing CT for the evaluation of vestibular aqueduct enlargement: Inter‐rater reproducibility and predictive value of reformatted CT measurements. Journal of Otology, 10(1), 13–17. 10.1016/j.joto.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara, K., Okuyama, S., Kumano, S., Iida, K., Hamana, H., Murakoshi, M., Kobayashi, T., Usami, S., Ikeda, K., Haga, Y., Tsumoto, K., Nakamura, H., Hirasawa, N., & Wada, H. (2010). Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hearing Research, 270(1–2), 110–118. 10.1016/j.heares.2010.08.015 [DOI] [PubMed] [Google Scholar]
- Jiang, Y. I., Huang, S., Deng, T., Wu, L., Chen, J., Kang, D., Xu, X., Li, R., Han, D., & Dai, P. U. (2015). Mutation spectrum of common deafness‐causing genes in patients with non‐syndromic deafness in the xiamen area, China. PLoS One, 10(8), e0135088. 10.1371/journal.pone.0135088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, J., Lee, J. S., Cho, K. J., Yu, S., Yoon, J. H., Yung Gee, H., & Choi, J. Y. (2017). Genetic predisposition to sporadic congenital hearing loss in a pediatric population. Scientific Reports, 7, 45973. 10.1038/srep45973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P., Gauthier, L. D., Brand, H., Solomonson, M., Watts, N. A., Rhodes, D., Singer‐Berk, M., England, E. M., Seaby, E. G., Kosmicki, J. A., … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, C. C., Chiu, C. Y., Shiao, A. S., Tso, Y. C., Wu, Y. C., Tu, T. Y., & Jap, T. S. (2007). Analysis of the SLC26A4 gene in patients with Pendred syndrome in Taiwan. Metabolism, 56(9), 1279–1284. 10.1016/j.metabol.2007.05.01 [DOI] [PubMed] [Google Scholar]
- Landa, P., Differ, A.‐M., Rajput, K., Jenkins, L., & Bitner‐Glindzicz, M. (2013). Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in pendred syndrome/enlarged vestibular aqueducts. BMC Medical Genetics, 14, 85. 10.1186/1471-2350-14-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Jang, W., Karapetyan, K., Katz, K., Liu, C., Maddipatla, Z., Malheiro, A., McDaniel, K., Ovetsky, M., Riley, G., Zhou, G., … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. J., Yoo, J. E., Namkung, W., Cho, H.‐J., Kim, K., Kang, J. W., Yoon, J.‐H., & Choi, J. Y. (2015). Thick airway surface liquid volume and weak mucin expression in pendrin‐deficient human airway epithelia. Physiol Rep, 3(8), e12480. 10.14814/phy2.12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leilei Zhao, L. H., Wang, X., Yating, D. U., Wang, X., Cheng, X., Zhao, L., & Li, Y. (2018). Analysis of genotypes and audiological characteristics of children with SLC26A4 gene deaf mutation. Journal of Clinical Otorhinolaryngology Head and Neck Surgery, 32(11), 836–840. 10.13201/j.issn.1001-1781.2018.11.009 [DOI] [PubMed] [Google Scholar]
- Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., O’Donnell‐Luria, A. H., Ware, J. S., Hill, A. J., Cummings, B. B., Tukiainen, T., Birnbaum, D. P., Kosmicki, J. A., Duncan, L. E., Estrada, K., Zhao, F., Zou, J., Pierce‐Hoffman, E., Berghout, J., … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X., Tang, W., Ahmad, S., Lu, J., Colby, C. C., Zhu, J., & Yu, Q. (2012). Applications of targeted gene capture and next‐generation sequencing technologies in studies of human deafness and other genetic disabilities. Hearing Research, 288(1–2), 67–76. 10.1016/j.heares.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.‐H., Wu, C.‐C., Lin, Y.‐H., Lu, Y.‐C., Chen, C.‐S., Liu, T.‐C., Chen, P.‐L., & Hsu, C.‐J. (2019). Targeted next‐generation sequencing facilitates genetic diagnosis and provides novel pathogenetic insights into deafness with enlarged vestibular aqueduct. The Journal of Molecular Diagnostics, 21(1), 138–148. 10.1016/j.jmoldx.2018.08.007 [DOI] [PubMed] [Google Scholar]
- Liu, X., Wu, C., Li, C., & Boerwinkle, E. (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Wang, L., Feng, Y., He, C., Liu, D., Cai, X., Jiang, L. U., Chen, H., Liu, C., Wu, H., & Mei, L. (2016). A new genetic diagnostic for enlarged vestibular aqueduct based on next‐generation sequencing. PLoS One, 11(12), e0168508. 10.1371/journal.pone.0168508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Wen, J., Sang, S., Mei, L., He, C., Jiang, L. U., Huang, S., & Feng, Y. (2020). Next‐generation sequencing‐based mutation analysis of genes associated with enlarged vestibular aqueduct in Chinese families. European Archives of Oto‐Rhino‐Laryngology, 277(12), 3331–3339. 10.1007/s00405-020-06050-3 [DOI] [PubMed] [Google Scholar]
- Lopez‐Bigas, N., Melchionda, S., de Cid, R. , Grifa, A., Zelante, L., Govea, N., & Estivill, X. (2002). Erratum: Identification of five new mutations of PDS/SLC26A4 in Mediterranean families with hearing impairment. Human Mutation, 20(1), 77–78. 10.1002/humu.9043 [DOI] [PubMed] [Google Scholar]
- McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., & DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, C. C., & Nance, W. E. (2006). Newborn hearing screening a silent revolution. New England Journal of Medicine, 354(20), 2151–2164. 10.1056/NEJMra050700 [DOI] [PubMed] [Google Scholar]
- Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., Siegert, R. K., Shen, J., Chapin, A., Boczek, N. J., Schimmenti, L. A., Murry, J. B., Hasadsri, L., Nara, K., Kenna, M., Booth, K. T., Azaiez, H., Griffith, A., Avraham, K. B., Kremer, H., … Abou Tayoun, A. N. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Human Mutation, 39(11), 1593–1613. 10.1002/humu.23630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila, U., Chapman, B. A., Kirchner, R., & Quinlan, A. R. (2013). GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Computational Biology, 9(7), e1003153. 10.1371/journal.pcbi.1003153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, X., Chai, Y., Chen, P., He, L., Wang, X., Wu, H., & Yang, T. (2015). Mono‐allelic mutations of SLC26A4 is over‐presented in deaf patients with non‐syndromic enlarged vestibular aqueduct. International Journal of Pediatric Otorhinolaryngology, 79(8), 1351–1353. 10.1016/j.ijporl.2015.06.009 [DOI] [PubMed] [Google Scholar]
- Pang, X., Chai, Y., He, L., Chen, P., Wang, X., Li, L., Jia, H., Wu, H., & Yang, T. (2015). A 7666‐bp genomic deletion is frequent in Chinese Han deaf patients with non‐syndromic enlarged vestibular aqueduct but without bi‐allelic SLC26A4 mutations. International Journal of Pediatric Otorhinolaryngology, 79(12), 2248–2252. 10.1016/j.ijporl.2015.10.015 [DOI] [PubMed] [Google Scholar]
- Park, H.‐J., Lee, S.‐J., Jin, H.‐S., Lee, J. O., Go, S.‐H., Jang, H. S., Moon, S.‐K., Lee, S.‐C., Chun, Y.‐M., Lee, H.‐K., Choi, J.‐Y., Jung, S.‐C., Griffith, A. J., & Koo, S. K. (2005). Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clinical Genetics, 67(2), 160–165. 10.1111/j.1399-0004.2004.00386.x [DOI] [PubMed] [Google Scholar]
- Pedersen, B. S., Layer, R. M., & Quinlan, A. R. (2016). Vcfanno: Fast, flexible annotation of genetic variants. Genome Biology, 17(1), 118. 10.1186/s13059-016-0973-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera, A., Villamar, M., Vinuela, A., Gandia, M., Meda, C., Moreno, F., & Hernandez‐Chico, C. (2008). A mutational analysis of the SLC26A4 gene in Spanish hearing‐impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. European Journal of Human Genetics, 16(8), 888–896. 10.1038/ejhg.2008.30 [DOI] [PubMed] [Google Scholar]
- Qi Li, Q.‐W.‐Z., Yuan, Y.‐Y. et al (2012). Identification of SLC26A4 c.919‐2A>G compound heterozygosity in hearing‐impaired patients to improve genetic counseling. Journal of Translational Medicine, 10, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier‐Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royaux, I. E., Wall, S. M., Karniski, L. P., Everett, L. A., Suzuki, K., Knepper, M. A., & Green, E. D. (2001). Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proceedings of the National Academy of Sciences of the United States of America, 98(7), 4221–4226. 10.1073/pnas.071516798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer, A. E., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E., Clark, E. T., Nishimura, C. J., Black‐Ziegelbein, E. A., & Smith, R. J. (2014). Copy number variants are a common cause of non‐syndromic hearing loss. Genome Medicine, 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan‐Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., Ephraim, S. S., Shibata, S. B., Booth, K. T., Campbell, C. A., Ranum, P. T., Weaver, A. E., Black‐Ziegelbein, E. A., Wang, D., Azaiez, H., & Smith, R. J. H. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genetics, 135(4), 441–450. 10.1007/s00439-016-1648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, K., Suzuki, H., Harada, D., Namba, A., Abe, S., & Usami, S. (2003). Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese. European Journal of Human Genetics, 11(12), 916–922. 10.1038/sj.ejhg.5201073 [DOI] [PubMed] [Google Scholar]
- Wang, Q.‐J., Zhao, Y.‐L., Rao, S.‐Q., Guo, Y.‐F., Yuan, H., Zong, L., Guan, J., Xu, B.‐C., Wang, D.‐Y., Han, M.‐K., Lan, L., Zhai, S.‐Q., & Shen, Y. (2007). A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clinical Genetics, 72(3), 245–254. 10.1111/j.1399-0004.2007.00862.x [DOI] [PubMed] [Google Scholar]
- Wang, R., Han, S., Khan, A., & Zhang, X. (2017). Molecular analysis of twelve Pakistani families with nonsyndromic or syndromic hearing loss. Genetic Testing and Molecular Biomarkers, 21(5), 316–321. 10.1089/gtmb.2016.0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemeau, J. L., & Kopp, P. (2017). Pendred syndrome. Best Practice & Research Clinical Endocrinology & Metabolism, 31(2), 213–224. 10.1016/j.beem.2017.04.011 [DOI] [PubMed] [Google Scholar]
- Wu, C.‐C., Lee, Y.‐C., Chen, P.‐J., & Hsu, C.‐J. (2008). Predominance of genetic diagnosis and imaging results as predictors in determining the speech perception performance outcome after cochlear implantation in children. Archives of Pediatrics and Adolescent Medicine, 162(3), 269–276. 10.1001/archpediatrics.2007.59 [DOI] [PubMed] [Google Scholar]
- Wu, H., Feng, Y., Jiang, L. U., Pan, Q., Liu, Y., Liu, C., He, C., Chen, H., Liu, X., Hu, C., Hu, Y., & Mei, L. (2016). Application of a new genetic deafness microarray for detecting mutations in the deaf in China. PLoS One, 11(3), e0151909. 10.1371/journal.pone.0151909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, D., Xiang, G., Chai, X., Qing, J., Shang, H., Zou, B., Mittal, R., Shen, J., Smith, R. J. H., Fan, Y.‐S., Blanton, S. H., Tekin, M., Morton, C., Xing, W., Cheng, J., & Liu, X. Z. (2017). Screening of deafness‐causing DNA variants that are common in patients of European ancestry using a microarray‐based approach. PLoS One, 12(3), e0169219. 10.1371/journal.pone.0169219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T., Gurrola, J. G.2nd, Wu, H., Chiu, S. M., Wangemann, P., Snyder, P. M., & Smith, R. J. (2009). Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. American Journal of Human Genetics, 84(5), 651–657. 10.1016/j.ajhg.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T., Vidarsson, H., Rodrigo‐Blomqvist, S., Rosengren, S. S., Enerback, S., & Smith, R. J. (2007). Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). American Journal of Human Genetics, 80(6), 1055–1063. 10.1086/518314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, G., Chen, D., Wang, H., Li, S., Zhang, J., Feng, Z., & Ma, D. (2013). Novel mutations of SLC26A4 in Chinese patients with nonsyndromic hearing loss. Acta Oto‐Laryngologica, 133(8), 833–841. 10.3109/00016489.2013.777160 [DOI] [PubMed] [Google Scholar]
- Yao, G., Li, S., Chen, D., Wang, H., Zhang, J., Feng, Z., Guo, L., Yang, Z., Yang, S., Sun, C., Zhang, X., & Ma, D. (2013). Compound heterozygous mutations of SLC26A4 in 4 Chinese families with enlarged vestibular aqueduct. International Journal of Pediatric Otorhinolaryngology, 77(4), 544–549. 10.1016/j.ijporl.2013.01.002 [DOI] [PubMed] [Google Scholar]
- Yao, J., Qian, X., Bao, J., Wei, Q., Lu, Y., Zheng, H., Cao, X., & Xing, G. (2015). Probing the effect of two heterozygous mutations in codon 723 of SLC26A4 on deafness phenotype based on molecular dynamics simulations. Scientific Reports, 5, 10831. 10.1038/srep10831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Y., Guo, W., Tang, J., Zhang, G., Wang, G., Han, M., Zhang, X., Yang, S., He, D. Z. Z., & Dai, P. U. (2012). Molecular epidemiology and functional assessment of novel allelic variants of SLC26A4 in non‐syndromic hearing loss patients with enlarged vestibular aqueduct in China. PLoS One, 7(11), e49984. 10.1371/journal.pone.0049984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Y., You, Y., Huang, D., Cui, J., Wang, Y., Wang, Q., Yu, F., Kang, D., Yuan, H., Han, D., & Dai, P. U. (2009). Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. Journal of Translational Medicine, 7, 79. 10.1186/1479-5876-7-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F., Xiao, Y., Xu, L., Zhang, X., Zhang, G., Li, J., Lv, H., Bai, X., & Wang, H. (2016). Mutation analysis of the common deafness genes in patients with nonsyndromic hearing loss in Linyi by SNPscan assay. BioMed Research International, 2016, 1302914. 10.1155/2016/1302914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., Wang, P., Han, B., Ding, Y., Pan, L., Zou, J., Liu, H., Pang, X., Liu, E., Wang, H., Liu, H., Zhang, X., Cheng, X., Feng, D., Li, Q., Wang, D., Zong, L., Yi, Y., Tian, N., … Wang, Q. (2013). Newborn hearing concurrent genetic screening for hearing impairment‐a clinical practice in 58,397 neonates in Tianjin, China. International Journal of Pediatric Otorhinolaryngology, 77(12), 1929–1935. 10.1016/j.ijporl.2013.08.038 [DOI] [PubMed] [Google Scholar]
- Zhao, J., Yuan, Y., Huang, S., Huang, B., Cheng, J., Kang, D., Wang, G., Han, D., & Dai, P. U. (2014). KCNJ10 may not be a contributor to nonsyndromic enlargement of vestibular aqueduct (NSEVA) in Chinese subjects. PLoS One, 9(11), e108134. 10.1371/journal.pone.0108134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Li, C., Li, M., Zhao, Z., Tian, S., Xia, H., Liu, P., Han, Y., Ren, R., Chen, J., Jia, C., & Guo, W. (2019). Mutation analysis of common deafness genes among 1,201 patients with non‐syndromic hearing loss in Shanxi Province. Molecular Genetics & Genomic Medicine, 7(3), e537. 10.1002/mgg3.537 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data of this study are available from the corresponding authors upon reasonable request. The novel or reclassified variants have been submitted to the ClinVar database (SCV001572597‐SCV001572611).