

Abstract
A new study reports that genome-wide polygenic risk scores can identify individuals at risk of common complex diseases, such as coronary artery disease or type 2 diabetes, with comparable performance to that of monogenic mutation screens. These findings support the potential clinical utility of genome-wide association study (GWAS)-based risk stratification; however, several issues need to be addressed before this approach can be applied to kidney disease.
The ability to accurately estimate disease risk is central to the success of preventive medicine. Today, risk prediction for most common adult-onset diseases typically relies on a combination of clinical, demographic and lifestyle factors. However, with the exception of well-studied Mendelian mutations involved in monogenic diseases, accurate quantification of genetic risk is not possible for most disorders.
Genome-wide association studies (GWAS) have discovered thousands of genetic variants associated with disease risk, but translation of these findings into clinical benefit has been hindered by the fact that most GWAS loci individually explain a relatively small proportion of overall risk. The concept of genome-wide polygenic risk scores (GPSs) specifically addresses this issue — while correcting for correlation between variants (that is, linkage disequilibrium), the GPS aggregates the individual effects of millions of GWAS variants across the genome. Capturing the cumulative effects of all common variants — including those with weak effects that do not reach statistical significance individually — enables the GPS to maximize the predictive power of GWAS results (Fig. 1).
Fig. 1 |. Construction of a GPS.
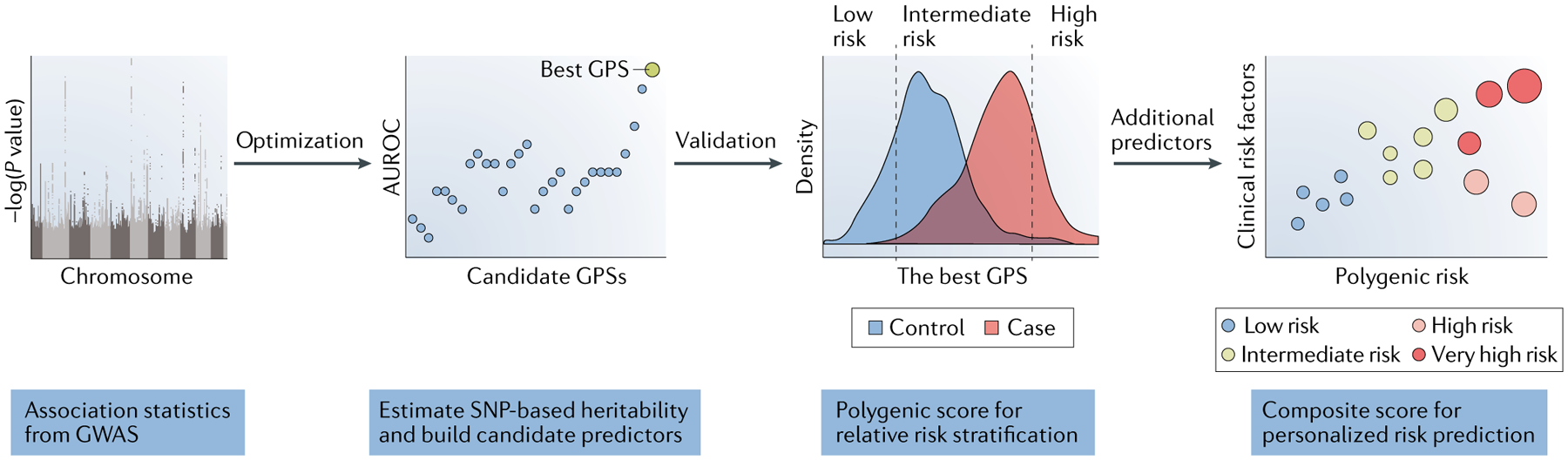
A genome-wide polygenic risk score (GPS) is based on genome-wide association study (GWAS) summary statistics. The optimization step enables selection of the best method according to the genetic architecture of a disease under study. The validation step requires an external cohort and is critical to obtaining reliable metrics of performance. Clinical predictors of absolute risk will require incorporation of additional demographic, clinical or lifestyle factors into composite risk models. AUROC, area under receiver operating characteristic. SNP, single-nucleotide polymorphism.
Khera et al.1 now describe the performance of GPSs for five common complex diseases that have been well studied in large-scale GWAS: coronary artery disease, atrial fibrillation, type 2 diabetes mellitus, inflammatory bowel disease and breast cancer. The researchers derived several candidate predictors based on published GWAS, then optimized predictive methods for each disease using a large validation data set (The UK Biobank phase 1 data set with 120,280 participants), and finally evaluated their performance in an independent testing set (The UK Biobank phase 2 data set with 288,978 participants). This approach identified a considerably larger proportion of population at risk compared to Mendelian mutation screens. For example, the number of individuals with a threefold increased polygenic risk score for coronary artery disease was 20-fold greater than the number that would have been identified by hypercholesterolaemia mutation screens. Individuals in the top 0.5% of polygenic risk score distribution for coronary artery disease have an estimated fivefold increased risk, which would be actionable and would lead to a recommendation of disease screening.
The study by Khera et al. has generated considerable enthusiasm about potential clinical applications of GPSs for a number of complex traits, including in nephrology, where this approach could be potentially used to predict the risk of chronic kidney disease (CKD). However, there are several important factors to consider when applying a GPS approach to any given trait or disease, including the genetic architecture of a disease of interest and the overall magnitude of its single-nucleotide polymorphism (SNP)-based heritability (that is, the genetic risk component that can be captured by SNP arrays); whether available GWAS estimates are of sufficient statistical validity to build a high confidence predictor (that is, whether available GWAS are of sufficient size, SNP coverage and quality for the traits of interest); whether there are adequate external data sets available for the optimization and validation of polygenic predictors; whether other genetic and non-genetic factors should be considered in the interpretation of polygenic risk; and whether the performance of the risk score is generalizable to the target clinical population.
CKD tends to aggregate in families2; the commonly used creatinine-based measure of kidney function, estimated glomerular filtration rate (eGFR), has a significant SNP-based heritability of 21–31%3. From the perspective of genetic architecture, however, CKD does not represent a single disease, but rather a highly heterogenous group of primary and secondary pathogenic processes. The commonly used eGFR-based definitions of CKD fail to reflect this aetiological heterogeneity. As a result, analyses of the SNP-based heritability of CKD are difficult to interpret, as they probably reflect a mixture of cases with different causes, some more familial than others, at variable stages of the disease trajectory. It is therefore not surprising that even very large population-based genetic studies of renal function typically uncover loci with tiny effects that explain only a minuscule fraction (under 2%) of the overall heritability3–5. For this reason, we feel that GPS-based predictors for CKD are unlikely to be successful, at least based on currently available GWAS data.
One way to circumvent the issue of CKD heterogeneity would be to concentrate GWAS efforts on specific primary kidney diseases that are more aetiologically homogenous. This approach has already proved successful for membranous nephropathy6 and IgA nephropathy7, for which common variants with considerable effect sizes have been found. Nevertheless, because of the requirement for a diagnostic kidney biopsy, such studies are typically several orders of magnitude smaller than those analysed by Khera et al. For example, the GPS for breast cancer was based on a GWAS analysis of 122,977 cases8, whereas the largest GWAS for membranous nephropathy includes only 556 cases6. These staggering differences in power clearly indicate that the available GWAS estimates are simply inadequate to build high-confidence GPSs for primary nephropathies. Accordingly, we feel that investment in more-powerful GWAS for primary kidney disorders is a key prerequisite for the success of this approach in nephrology.
The third problem relates to the lack of external validation data sets that are adequately phenotyped for kidney traits, and that include sufficient numbers of cases with a tissue-based diagnosis. The UK Biobank provides the largest currently available population-based data set with genome-wide genetic information linked to electronic health records9. In clinical practice, however, only a small fraction of CKD patients receive a diagnostic biopsy; thus, even this large cohort of nearly half a million individuals may be of limited utility for studies of primary kidney disease. Moreover, most kidney disorders cannot be reliably defined using International Classification of Disease codes, and require more complex phenotyping algorithms that combine structured laboratory data with information from pathology, radiology and physician reports. Although such algorithms are now in development, the fact remains that sufficiently large real-life data sets for external validation studies are presently not available.
The fourth important issue relates to the fact that the GPS approach can only estimate a relative risk of disease, whereas the absolute risk for an individual will vary with age, lifestyle and other non-genetic factors. Therefore, there remains a great need to further understand the effect of environmental factors on disease risk, and to develop methods to incorporate these additional risk factors into polygenic models. Moreover, none of the Mendelian causes of disease are captured by GPSs, yet rare monogenic causes may explain a substantial fraction of CKD10. Improved understanding of the effects of polygenic background on the penetrance of monogenic disorders is therefore also urgently needed. At the very least, the results by Khera et al. provide a strong rationale for SNP genotyping (and preferably whole-genome sequencing) of all existing prospective cohorts of kidney patients to facilitate the development of more-comprehensive risk models of disease.
The final and perhaps most critical limitation of GPSs relates to the fact that GWAS-based risk models are inherently population-specific, due to differences in SNP allelic frequencies, linkage disequilibrium patterns, and allelic effects between populations. Because most published GWAS for kidney diseases are based on European populations, the accuracy of predictive models based on these studies is expected to be significantly diminished for individuals of non-European ancestry. From a practical standpoint, this limitation means that clinicians must always consider the ancestry of their patients in relationship to the source of the GPS population used to build and validate a predictor. From the public health perspective, this also means that clinical applications of polygenic predictors could potentially contribute to the existing inequalities in health care and further exacerbate disparities in CKD care. The only sensible way to address this critical limitation is to rapidly expand genetic studies of kidney disease to multiethnic settings and understudied populations across the globe.
Acknowledgements
The authors acknowledge support from the following grants from the National Institutes of Health (NIH): Kidney Precision Medicine Project grant number UG3DK114926 from the National Institute of Diabetes and Digestive Kidney Diseases (NIDDK), the Electronic Medical Records and Genomics (eMERGE) Network grant number U01HG8680 from the National Human Genome Research Institute (NHGRI), and the Columbia Clinical and Translational Science Award grant number UL1TR001873 from the National Center for Advancing Translational Sciences (NCATS). Additional sources of funding include the following grants: R01-DK105124 and RC2-DK116690 (NIDDK) and R01-MD009223 (National Institute on Minority Health and Health Disparities (NIMHD)). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox CS et al. Genomewide linkage analysis to serum creatinine, GFR, and creatinine clearance in a community-based population: the Framingham Heart Study. J. Am. Soc. Nephrol 15, 2457–2461 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Gorski M et al. 1000 Genomes-based meta-analysis identifies 10 novel loci for kidney function. Sci. Rep 7, 45040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kottgen A et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet 41, 712–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kottgen A et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet 42, 376–384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanescu HC et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N. Engl. J. Med 364, 616–626 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Kiryluk K et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet 46, 1187–1196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michailidou K et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge T et al. Phenome-wide heritability analysis of the UK Biobank. PLOS Genet. 13, e1006711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lata S et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann. Intern. Med 168, 100–109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]