

Abstract
While much work has focused on the elucidation of somatic alterations that drive the development of acute leukemias and other hematopoietic diseases, it has become increasingly recognized that germline mutations are common in many of these neoplasms. In this review, we will highlight the different genetic pathways impacted by germline mutations that can ultimately lead to the development of familial and sporadic hematological malignancies, including acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML) and myelodysplastic syndromes (MDSs). Many of the genes disrupted by somatic mutations in these diseases (e.g. TP53, RUNX1, IKZF1, and ETV6) are the same as those which harbor germline mutations in children and adolescents who develop these malignancies. Moreover, the presumption that familial leukemias only present in childhood is no longer true, in large part due to the numerous studies demonstrating germline DDX41 mutations in adults with MDS and AML. Lastly, we will highlight how different cooperating events can influence the ultimate phenotype in these different familial leukemia syndromes.
Table of Contents Summary:
This Review highlights the different genetic pathways impacted by germline mutations that can lead to the development of familial and sporadic hematological malignancies, with a particular focus on recent advances in acute lymphoblastic leukemia, acute myeloid leukemia and myelodysplastic syndromes.
INTRODUCTION
Over the last decade, large scale genomic studies have described the landscape of nearly all major types of cancer in both adults and children1. In addition to providing important diagnostic and prognostic information from the somatic alterations, evaluation of non-tumor or germline material through comprehensive sequencing approaches has transformed our view of how germline variation influences the development of cancer. The incidence of presumably pathogenic germline mutations in children with cancer was estimated to be 8.5% in a large study involving over 1000 children2. These data have been supported by follow-up studies on different pediatric cohorts3,4. Recent studies on over 10,000 adults within The Cancer Genome Atlas (TCGA) data set reported that approximately 8% of adult patients have a pathogenic germline variant5. Many of the patients with these germline mutations lack a family history consistent with a cancer predisposition syndrome2. These findings not only highlight the importance of comprehensive sequencing of normal tissue from all cancer patients, but also that familial cancer syndromes are much more common than previously recognized.
Genes in a number of different pathways are impacted by germline mutations in individuals with a predisposition to hematologic abnormalities and neoplasms. The recent inclusion of the “Myeloid Neoplasms with Germline Predisposition” category in the revised 4th Edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues further emphasizes the increased recognition and importance of germline evaluation in patients with myeloid tumors6. Many of the known germline predisposition mutations affect master transcription factors important in lineage development, such as RUNX1 or GATA2; the genes encoding these transcription factors also harbor recurrent somatic mutations in children and adults with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML)7 (Figure 1). However, it is becoming increasingly clear that these observations now extend to lymphoid neoplasms8–13. Like RUNX1 in myeloid neoplasms, the genes encoding the transcription factors PAX5 and IKZF1 (also known as Ikaros) are examples of genes that may be altered as germline events predisposing to acute lymphoblastic leukemia (ALL), disease-initiating and subtype defining somatic events, or secondary events that cooperate in leukemogenesis.
Figure 1. Association of germline mutations in genes encoding transcription factors with lineage development.
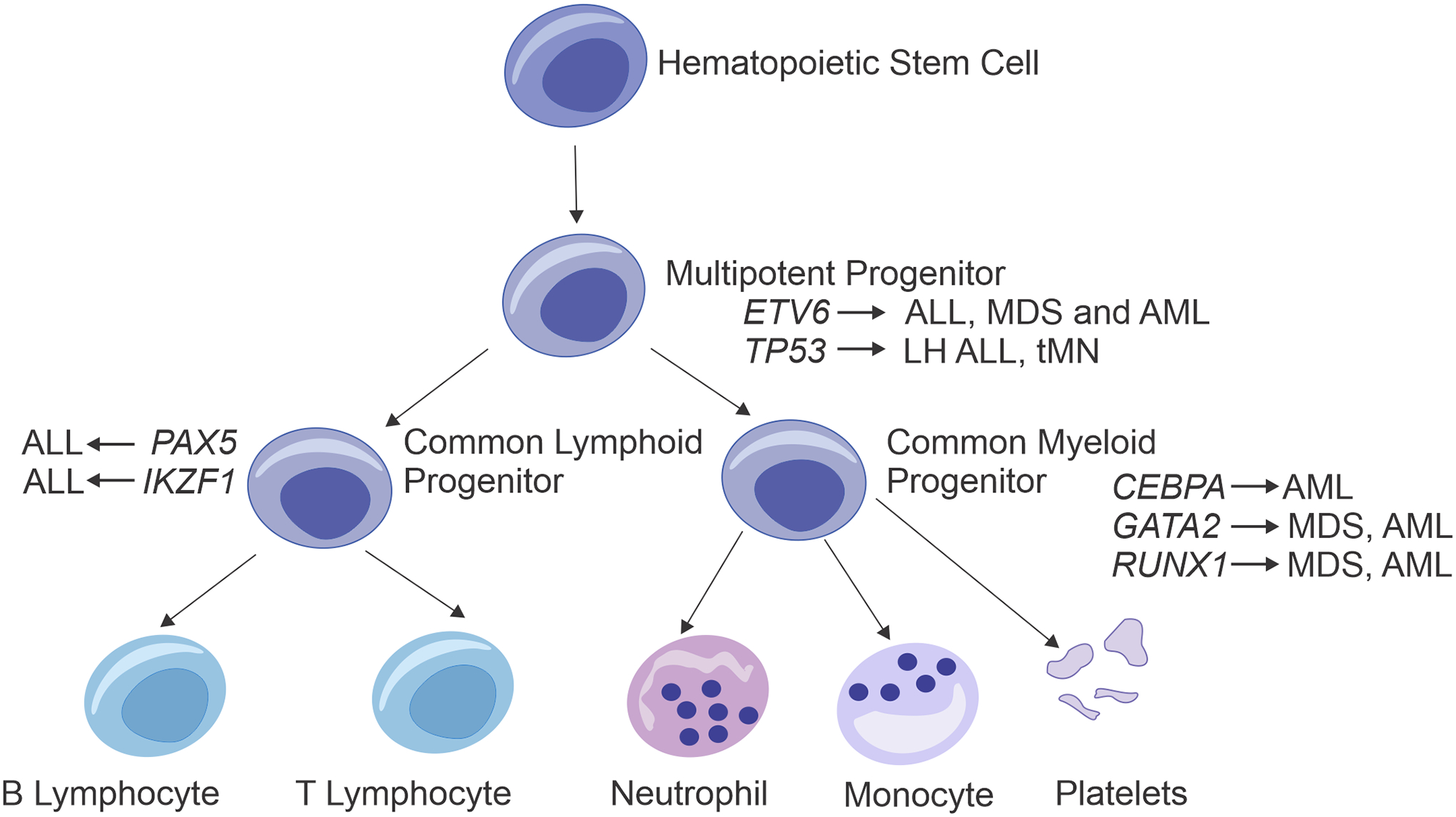
The representative schematic of the hierarchy of hematopoietic development demonstrates the main hematopoietic lineages that are dysregulated in patients with germline mutations in different transcription factors important in hematopoietic growth and differentiation. Germline alterations in ETV6 and TP53 can lead to hematopoietic neoplasms in both myeloid and lymphoid lineages, while disruptions in the genes encoding other hematopoietic transcription factors result in more lineage-restricted malignancies (e.g. B-ALL with PAX5 or AML with CEBPA). tMN: Therapy-related myeloid neoplasm; LH: Low hypodiploid.
Recent comprehensive sequencing studies have expanded the spectrum of predisposition genes in families with leukemia beyond those encoding transcription factors. Not only have these studies opened up new pathways of mechanistic investigation, but they have also shown that clinical suspicion for a familial predisposition syndrome, along with genetic screening, should not be restricted to children or adolescents. For example, germline mutations in the gene encoding the RNA helicase DDX41 define families with an increased risk of MDS and AML that can present in adulthood14. Further, germline mutations in SAMD9L and SAMD9, two interferon-inducible genes implicated in endocytosis, have recently been shown to underlie MDS as well as other multi-system disorders associated with MDS with monosomy 715–19. DDX41, SAMD9 and SAMD9L are involved in innate immunity and antiviral responses, suggesting new pathways that predispose families to hematological malignancies.
This review will address recently described predisposition genes (Table 1) in familial leukemias and hematologic abnormalities, particularly MDS, myeloid proliferations and acute leukemias, as well as the cooperating events that can lead to the fully transformed acute leukemia phenotype. The review is not designed to serve as a comprehensive list of all described predisposition genes in hematological malignancies (such as those important in myeloma or lymphoma) and detailed reviews on specific genes or diseases (i.e. bone marrow failure syndromes) and clinical management are already available20–23. For clarity, we have grouped genes by different differentiation lineages of the resulting hematological malignancies, while recognizing that these categories at times may be somewhat artificial and not truly exclusive.
Table 1.
Common Germline Predispositions in MDS and Acute Leukemias
| Gene or Pathway* | Hematologic Malignancy | Additional Hematologic Abnormality | Other Malignancies | Additional Symptoms | Syndrome|| | Refs. |
|---|---|---|---|---|---|---|
| CEBPA | AML | No common abnormalities noted | No other malignancies are common | None observed | Familial acute myeloid leukemia with mutated CEBPA | 40–42 |
| DDX41 | MDS and AML; less commonly lymphoma | Cytopenia | No other malignancies are common | None observed | Familial AML with mutated DDX41 | 14,63,64 |
| ETV6 | ALL, MDS, AML | Thrombocytopenia and decreased platelet function | No other malignancies are common | None observed | Thrombocytopenia 5 (OMIM 616216) | 144,145,215 |
| GATA2 | MDS and AML | Immunodeficiency, bone marrow failure, monocytopenia and B cell lymphopenia | No other malignancies are common | Lymphedema, pulmonary alveolar proteinosis and congenital deafness | Emberger Syndrome (OMIM 614038) and MonoMac Syndrome (OMIM 614172) | 46,245,246 |
| IKZF1 | B-ALL, less commonly T-ALL | Immunodeficiency | No other malignancies are common | Autoimmunity | Not described | 8,206,207 |
| PAX5 | B-ALL | No common abnormalities noted | No other malignancies are common | None observed | Not described | 12,166,178 |
| RAS–MAPK pathway: CBL, NF1 and PTPN11 | JMML, ALL, AML | JMML-like proliferation with spontaneous regression | Neurofibroma and embryonal rhabdomyosarcoma | Cardiac abnormalities, skin manifestations, growth retardation and facial dysmorphologies | Noonan Syndrome (OMIM 163950); NF1 (OMIM 162200) and other RASopathies | 100;95,104;215 |
| RUNX1 | MDS and AML, less commonly T-ALL | Thrombocytopenia and decreased platelet function | No other malignancies are common | None observed | Familial platelet disorder with propensity for myeloid malignancy (OMIM 601399) | 30,33,36 |
| SAMD9 | MDS and AML with monosomy 7 | Bone marrow failure | No other malignancies are common | Normophosphatemic familiar tumoral calcinosis; congenital adrenal hypoplasia, enteropathy and genital abnormalities | MIRAGE Syndrome (OMIM 617053) and Myelodysplasia and Leukemia Syndrome with Monosomy 7(OMIM 252270) | 16,17,71 |
| SAMD9L | MDS and AML with monosomy 7 | Systemic autoinflammatory disease and bone marrow failure | No other malignancies are common | Ataxia | Ataxia Pancytopenia Syndrome (OMIM 159550) and Myelodysplasia and Leukemia Syndrome with Monosomy 7 (OMIM 252270) | 18,19,70 |
| TP53 | Low hypodiploid ALL, Therapy Related Myeloid Neoplasm | No common abnormalities noted | Osteosarcoma, breast cancer, soft tissue sarcoma, brain tumors and adrenocortical carcinoma | None observed | Li-Fraumeni syndrome (OMIM 151623) | 143,150,181 |
| Trisomy 21 | AML and ALL | Transient abnormal myelopoiesis | No other malignancies are common | Mental retardation, cardiac abnormalities and facial dysmorphologies | Down syndrome (OMIM 190685) | 135 |
Only the major genes and pathways discussed in this Review are included in this table.
Online Mendelian Inheritance in Man (OMIM) numbers are given for each syndrome, when applicable: https://www.omim.org/
Predisposition to MDS and AML
Alterations in transcription factors
RUNX1
Somatic alterations in RUNX1 are among the most common mutations in both adults and children with ALL, AML or MDS and these include recurrent fusions in B lymphoblastic leukemia (B-ALL) (ETV6-RUNX1) and AML (RUNX1-RUNX1T1), as well as loss of function mutations in myeloid and less commonly lymphoid neoplasms24–29. In addition, germline mutations in RUNX1 define familial platelet disorder with predisposition to myeloid malignancy (FDP-MM). FDP-MM is an autosomal dominant disorder characterized by variable penetrance of quantitative and/or qualitative platelet defects with a tendency to develop hematological malignancies. When symptomatic, these patients present with thrombocytopenia and although the platelets are normal in size they have decreased function, typically as a result of decreased dense granules30. The germline mutations in RUNX1, first described in 1999 by Song and colleagues31, result in loss of RUNX1 protein function, by point mutations that produce proteins that act as dominant negatives, as well as nonsense or frameshift mutations and larger deletions30. The overall lifetime risk of progression to a myeloid disease (or rarely T lymphoblastic leukemia (T-ALL)) is 44%7, which most commonly occurs in adulthood32. This risk appears to be higher in patients with missense mutations, presumably due to a higher ability of the resulting proteins to inhibit transactivation of wild-type RUNX1 through a dominant negative activity, as compared to loss of function alleles30,33. However, eventual progression to a frank neoplasm is associated with acquisition of somatic mutations in the remaining wild-type RUNX1 allele, as well as GATA2 mutations34, and less commonly other genes recurrently mutated in AML and MDS, such as CBL (which encodes an E3 ubiquitin ligase), DNMT3A (which encodes a DNA methyltransferase), KRAS and FLT3 (which encodes a receptor tyrosine kinase)30,32,35–37. Notably, patients with FDP-MM also appear to have a higher incidence of clonal hematopoiesis [G] at a younger age35.
CEBPA
AMLs with biallelic mutations in CEBPA (which encodes CCAAT/enhancer binding protein alpha) are recognized as a unique subtype with good outcome38,39 and it has been shown that nearly 10% of these cases also harbor a germline CEBPA mutation40. These germline CEBPA mutations lead to the development of AML with a near complete penetrance, typically in the second or third decade of life and can occur without a preceding MDS or cytopenic phase [G]7,41. These germline mutations are commonly frameshift or nonsense mutations near the amino terminus of the encoded protein. This results in a truncated 30 kD C-terminal protein through usage of an internal initiation codon that has dominant negative activity38,42. However, C-terminal germline mutations have been reported43, including a recent publication describing a multigenerational family with AML44. Progression to AML is frequently associated with an acquired mutation in the remaining wild-type CEBPA allele40,41. Surprisingly, Tawana and colleagues showed that when these patients have disease recurrence after chemotherapy, they do so with new clones that have a different spectrum of acquired mutations, including new somatic CEBPA mutations, demonstrating that these second leukemias are not true relapses41. This pattern of progression likely underlies the clinical observations that patients with AML and germline CEBPA mutations have a good outcome, yet are prone to second leukemias that continue to be sensitive to chemotherapy, unlike true relapsed disease.
GATA2
Perhaps more so than predisposition syndromes associated with mutations in other transcription factor-encoding genes, patients with germline GATA2 mutations have a marked phenotypic variation with MDS and AML as a main clinical feature. However, these patients can also present with syndromes characterized by mycobacterial infections, monocytopenia [G], B-lymphopenia and pulmonary alveolar proteinosis [G] (MonoMac Syndrome) or primary lymphedema, cutaneous warts and sensorineural deafness [G] (Emberger Syndrome)45. Recent studies have identified germline GATA2 mutations in up to 15% of pediatric MDS cases with isolated bone marrow disease, and these germline variants are more frequent in higher grade MDS46. Most commonly, these germline GATA2 mutations are missense mutations that affect the second zinc finger of the protein or true null mutations that collectively result in loss of protein function and haploinsufficiency47 (Figure 2a). However, other mechanisms of germline GATA2 disruption have been reported, such as disruption of intronic regulatory sequences within intron 4 (ref. 48), synonymous coding alterations that disrupt transcript splicing49,50 and an in-frame insertion that alters the spacing between the GATA2 zinc fingers51. Progression to AML is commonly associated with acquired mutations in the Polycomb group member ASXL1, as well as monosomy 7 (ref. 52) or trisomy 8, although marked clinical variability exists within families53–55, some of which has been linked to patterns of allele-specific expression56.
Figure 2. Genetics of cancer predisposition genes in pediatric myeloid disorders malignancies.
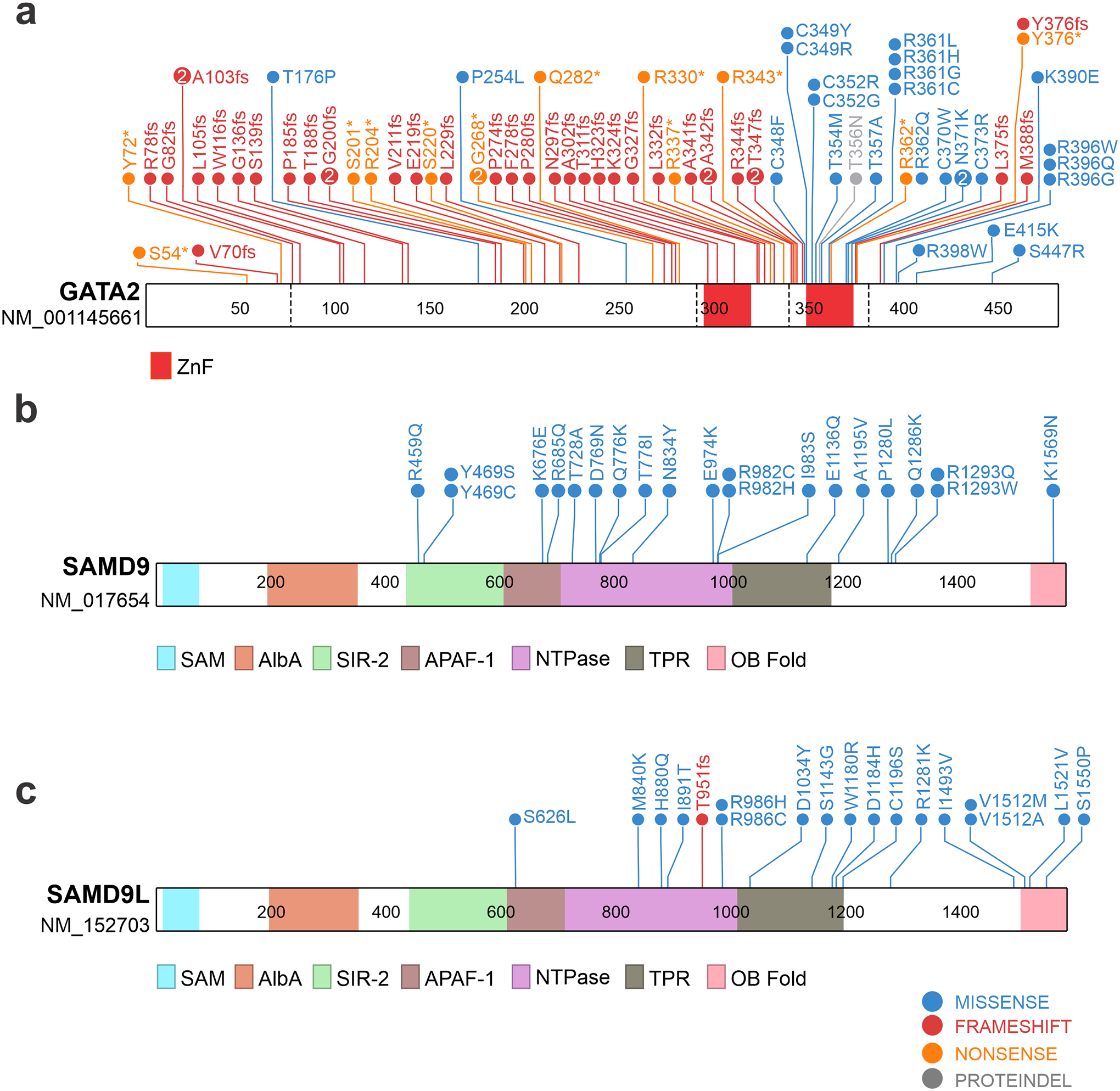
Schematics and domain architectures of proteins encoded by genes that have germline and somatic mutations in pediatric myeloid disorders, including GATA2 (a), SAMD9 (b), and SAMD9L (c) are shown. Refseq identifiers for transcripts used to generate the protein schematics are shown under the gene symbols. Listed germline mutations in GATA2 were collected from ref 46 and from available Pathogenic and Likely Pathogenic variants from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). SAMD9 and SAMD9L variants were taken from refs. 15,17–19,46,70–73. ZnF, zinc finger; NTPase, nucleoside triphosphatase; SAM, sterile alpha motif; AlbA, acetylation lowers binding affinity; SIR-2, silent information regulator 2; APAF-1, apoptotic protease-activating factor, TPR tetratricopeptide repeat; OB Fold, oligonucleotide/oligosaccharide-binding fold. SAMD9 and SAMD9L domain structure obtained from ref. 80.
Alterations in non-transcription factors
DDX41
Members of the DExD/H-box helicase family, including DDX41, have been implicated in numerous aspects of RNA biology57, as well as antiviral immunity via regulation of signaling pathways or serving as sensors for viral nucleic acids58. DDX41 in particular was originally identified as an intracellular DNA sensor in myeloid dendritic cells that binds cytosolic double-stranded DNA and triggers a signaling cascade that ultimately leads to the production of type 1 interferons via the stimulator of interferon genes (STING) pathway59,60. In addition to the well-documented role of DDX41 in innate immunity, somatic and germline mutations in DDX41 have recently been described in patients with myeloid tumors, and less commonly lymphoid malignancies, including follicular lymphoma and Hodgkin lymphoma14,61–64. Unlike the majority of other constitutional disorders, patients with germline DDX41 mutations present in adulthood. In fact, the first described family consisted of a father in his 70s and a brother and sister in their 40s14. Further, there is an estimated 3:1 male predominance in carriers with myeloid tumors65. The reported germline alterations are commonly frameshift mutations, most commonly a D140fs mutation. Emerging data is also suggesting that known germline alleles are associated with different ethnicities14,66, including preliminary data of an A500fs mutation observed in Asian populations67. Somatic missense mutations in DDX41 frequently occur in the second allele in patients with a germline DDX41 loss of function mutation, the most common of which is R525H14. Secondary mutations in TP53 (which encodes p53) have likewise been described in patients with advanced MDS or AML who have germline DDX41 mutations65. Clinically silent carriers have also been noted and due to the rarity of these mutations, the overall disease penetrance is currently not known14,63,64.
While evidence is mounting that DDX41 is important in cell growth, the overall function of this protein in the context of hematopoietic cells and how these mutations lead to hematopoietic neoplasms is still under investigation. Early biochemical studies identified components of the U2 and U5 spliceosome in complex with DDX41 and demonstrated that disruption of DDX41 alters RNA splicing and processing14, which are pathways known to be dysregulated in MDS68. However, non-RNA splicing functions of DDX41, such as its role as a cytosolic censor in the STING pathway to influence interferon signaling, may also be important in the development of MDS in these patients. Functional studies have shown that the common DDX41R525H missense mutation results in a defect in ribosomal biogenesis and cell cycle progression69.
SAMD9 and SAMD9L
Germline mutations in SAMD9 or SAMD9L, two interferon-inducible genes located on chromosome 7, were initially shown to cause a variety of multisystem syndromes characterized by neurological and/or endocrine abnormalities, as well as MDS with monosomy 717,18,70,71. Recent studies have shown that germline mutations in these genes can also be found in isolated and familial pediatric MDS and AML15,16,19,72, as well as inherited bone marrow failure syndromes73 and potentially adult MDS74 with a strong association with whole or partial deletions involving chromosome 7. Although the function of the SAMD9 and SAMD9L proteins in hematopoietic cells is not entirely clear, they appear to be involved in endocytosis and cytokine signaling75, although like DDX41, a role in antiviral response has also been reported76–78. Specifically, SAMD9 and SAMD9L are known host restriction factors for Pox virus infections79. The germline mutations associated with MDS are heterozygous missense (Figure 2b–c) mutations that lead to growth arrest when exogenously expressed in cells. This phenotype is an enhancement of the effects of wild-type SAMD9 or SAMD9L, suggesting that these may be gain of function mutations17. Although little is known about the biochemical activity of the SAMD9 and SAMD9L proteins and their domain structure, these missense mutations do appear to cluster in the second half of the proteins within or near a putative P-loop nucleoside triphosphatase domain19,80 (Figure 2b–c).
Surprisingly, when patients present with monosomy 7, the clone that eventually grows out has consistently lost the copy of chromosome 7 with the mutant allele. This haploinsufficiency of a number of genes on chromosome 7 (e.g. EZH2, SAMD9, SAMD9L, CUX1, and KMT2C (also known as MLL3)) perturbs hematopoiesis and can ultimately lead to MDS and AML81,82. Many other patients that do not develop monosomy 7 acquire additional somatic mutations in SAMD9 or SAMD9L that abrogate the function of the mutant protein (acquired revertant mutations) or duplicate the wild-type allele through a uniparental disomy event15,19,71. These observations suggest that there is strong selective pressure to not express the mutant allele and that the clinical phenotype in these individuals is heavily influenced by the method of mutant inactivation. However, clinically silent carriers of pathogenic germline variants that lack known rescue mutations have been observed, implying that other rescue mechanisms exist or that these germline mutations are not fully penetrant16,19.
To date, there appears to be an increased risk of abnormal blood counts, MDS and AML in children compared to adult mutation carriers, suggesting that developmental stage may influence clinical phenotype. However, adults with hematologic disease have been reported infrequently15. When patients progress to advanced disease they acquire additional somatic mutations in genes commonly mutated in pediatric and adult MDS and AML, including SETBP1 (which encodes SET-binding protein 1), RUNX1, or ETV615,19. In contrast to the presumed gain of function missense mutations in MDS, germline loss of function mutations in SAMD9L have recently been described in children with a systemic autoinflammatory disease, yet no apparent risk of MDS or AML83. Importantly, considering the fairly recent discovery and characterization of these mutations and their associated clinical syndromes, their natural history remains to be determined, especially regarding disease penetrance and phenotypes.
Additional Predisposing Variants
Due to advances in next-generation sequencing approaches, the list of rare variants associated with hematological disorders and an increased risk of MDS and AML is rapidly expanding. This includes recently described mutations in hematopoietic transcription factors like MECOM (also known as EVI1)84,85, as well as variants in genes not directly involved in gene regulation. For example, germline mutations in the 5’ regulatory region of ANKRD26, which encodes a protein that promotes MAPK signaling and megakaryocyte development86, been reported as a cause of thrombocytopenia-2 [G]87–89 and these families have an approximately 30-fold increased risk of developing AML88,90. Further, deficiency in MBD4, which regulates 5-methylcytosine deamination, is associated with early onset AML91. Although beyond the scope of this review, several inherited bone marrow failure syndromes are also associated with variable risks of MDS and AML (Box 1) (ref. 20–23). These mutations target a myriad of cellular pathways, including ribosomal biogenesis, telomere maintenance and DNA repair.
Box 1. Bone Marrow Failure Syndromes.
Inherited bone marrow failure syndromes (IBMFSs) are a heterogeneous group of genetic disorders typically associated with a decreased production of bone marrow cells, specific clinical phenotypes and variable risk of developing MDS or AML. The different types were traditionally distinguished based on the presence of classical physical manifestations, such as the triad of abnormal nails, reticular skin pigmentation, and oral leukoplakia in dyskeratosis congenita247. However, as sequencing approaches are becoming more mainstream, more and more patients with pathogenic variants in genes implicated in IBMFSs are presenting with unusual or non-classic clinical symptoms and the spectrum of genes implicated in IBMFSs are also expanding73,248. Within this group of disorders, Fanconi Anemia, an X-linked or autosomal recessive disorder caused by mutations in a group of genes that repair DNA crosslinks has perhaps the highest risk of progression to MDS or AML (cumulative incidence of AML at age 40 years is ~15%–20% and the cumulative incidence of MDS at age 50 is~ 40%)249. Although less common, the risk of developing a myeloid neoplasm is also increased in other IBMFSs like severe congenital neutropenia, dyskeratosis congenita, Shwachman-Diamond syndrome and Diamond Blackfan anemia.
Predisposition to Myeloid Proliferations
RASopathies
Aberrant activation of the RAS–MAPK pathway is a common finding in hematologic malignancies92. This is especially true for juvenile myelomonocytic leukemia (JMML), a rare but highly aggressive malignancy in childhood characterized by a marked expansion of granulocytic and monocytic cells in the peripheral blood, spleen, bone marrow and other tissues. Mutations in NF1 (which encodes neurofibromin 1), PTPN11 (which encodes the non-receptor protein tyrosine phosphatase SHP2 that regulates signaling through growth factor receptors), NRAS, KRAS or CBL, which are all part of the RAS pathway, have been identified in nearly 90% of JMML cases93–96. While many of these are somatic mutations, a subset of patients present with JMML, or JMML-like proliferations, as part of a constitutional syndrome associated with aberrant activation of the RAS pathway (RASopathy). In particular, children with neurofibromatosis type 1 are 200–500 times more likely to develop JMML, and bone marrow cells from these patients typically demonstrate loss of the wild-type NF1 allele and duplication of the mutant allele (acquired uniparental isodisomy)97,98. Likewise, germline mutations in the gene encoding CBL, which regulates the turnover of components in the RAS pathway, is present in 10–15% of JMML patients99. Like NF1-mutant patients, the mutant CBL allele is duplicated in JMML cells through uniparental isodisomy. Curiously, unlike patients with germline NF1 mutations, those with germline CBL mutations show a high rate of spontaneous resolution of JMML and lack additional genetic alterations associated with disease progression100,101. Despite the decrease in symptoms related to JMML, these patients can ultimately develop vascular complications, including hypertension and optic atrophy96,.
Noonan Syndrome, the most common of the RASopathies, is frequently associated with germline mutations in PTPN11. Somatic PTPN11 mutations are also the most common alteration in JMML, and children with JMML who have somatic PTPN11 mutations have a poor prognosis with higher rates of relapse99. Despite the aggressive nature of neoplasms with somatic PTPN11 mutations, Noonan Syndrome patients with PTPN11 germline mutations commonly develop a JMML-like disease that regresses spontaneously over time102. These clinical differences between the somatic and germline variants, which occur at different positions within PTPN11, are likely due to differences in the phosphatase activity of the resulting SHP2 protein103. Importantly, patients with Noonan Syndrome also have an increased risk of ALL104. In addition to PTPN11, germline mutations in other genes that encode members of the RAS pathway, including RIT1105, RRAS106 and SOS1107 have also been described in patients with Noonan syndrome, some of which also have features of JMML. Likewise, the spectrum of RASopathies continues to expand with germline mutations in BRAF, SHOC2, HRAS and others being associated with distinct clinical syndromes. Despite activation of the same pathway, many of these lack a propensity to develop hematologic malignancies108.
While most studies have focused on the impact of secondary mutations in non-syndromic JMML, it is likely that similar pathways are disrupted in those with constitutional syndromes. This includes deletions of chromosome 7 and acquired mutations in JAK3 (which encodes Janus kinase 3), SETBP1 and components of Polycomb repressive complex 2 (PRC2)109–112 along with distinct patterns of methylation113,114.
Myeloid Abnormalities in Down Syndrome
Children with Down Syndrome (DS) have an increased risk of developing both ALL and AML, however, the risk is much higher for AML with nearly a 150-fold increase in the first 5 years of life115. These AMLs are commonly megakaryoblastic in morphology and both AML and MDS have been described and collectively are grouped as “Myeloid Leukemia associated with DS (ML-DS)” in the current WHO guidelines. The ALLs that develop in children with DS are frequently characterized by alterations in cytokine receptors or kinase signaling pathways (e.g. Philadelphia chromosome-like ALL [G], also known as BCR-ABL1-like or Ph-like ALL), notably with cytokine receptor-like factor 2 (CRLF2) dysregulation116. Unlike non-DS acute megakaryoblastic leukemias (AMKLs), DS patients with AMKL have a favorable outcome117. At birth, approximately 5–10% of children with DS develop Transient Myeloproliferative Disease (TMD), which is characterized by circulating megakaryoblasts, abnormal blood counts and hepatosplenomegaly115. In addition to trisomy 21, patients with both TMD and ML-DS commonly have a somatic GATA1 mutation118,119. These somatic GATA1 mutations result in a truncated GATA1 protein (termed GATA1s) that disrupts GATA1-mediated regulation of other transcription factors important in megakaryocyte development and differentiation119,120. Although TMD commonly resolves spontaneously, nearly 20–30% of children with TMD will develop ML-DS within the first 4 years of life115. The progression from TMD to ML-DS reflects evolution from a TMD clone that has acquired additional somatic mutations, especially in genes that influence the cohesin complex, epigenetic regulation and the RAS pathway121. Despite the clear association of trisomy 21 and ML-DS, the mechanism of how altered dosage of genes on chromosome 21 results in the leukemic phenotype remains under investigation, with particular focus on RUNX1, ERG (which encodes a transcription factor) and DYRK1A (which encodes a dual-specificity kinase) (reviewed in ref. 122).
Other Predisposing Variants for MPNs
Like MDS, some MPNs may be part of a germline predisposition as relatives of individuals with an MPN are more likely to also develop an MPN as compared to the general population123. In fact, certain common polymorphisms associate with an increased risk of MPN. Notably a germline haplotype in the 3’ region of the JAK2 gene (GGCC haplotype, also known as 46/1) is linked with a three to four-fold increase in acquiring the canonical JAK2V617F mutation in cis as well as an overall increase in non-JAK2 mutated MPNs124–127. Despite the association, debate remains regarding the potential mechanistic role for the 46/1 haplotype in the development of MPNs. Potential explanations include the “fertile ground hypothesis” in which cells with the haplotype have a clonal advantage after they also acquire an oncogenic alteration in the JAK–signal transducer and activator of transcription (STAT) pathway and the “hypermutability hypothesis” in which the haplotype leads to genomic instability within the JAK2 locus128. Genome-wide association studies (GWAS) have also shown that polymorphisms in TERT (which encodes telomerase reverse transcriptase), MECOM and in the intergenic region between HBS1L and MYB are associated with an increased risk of MPNs129,130. In addition, germline variants in RBBP6131 (which encodes an E3 ubiquitin ligase), SH2B3 (encoding SH2B adaptor protein 3 (also known as LNK))132,133 and duplications of ATG2B (which is involved in autophagy) and GSKIP (which encodes a glycogen synthase kinase 3β-interacting protein)134 have also been described.
Genetic predisposition to ALL
Historically, outside of the setting of defined cancer predisposition syndromes including DS135, Ataxia-Telangiectasia [G] and Nijmegen breakage syndrome [G]136,137, ALL has been considered to have had a relatively low heritable predisposition, with few familial cases and low twin or sibling concordance. Rarely, constitutional chromosomal rearrangements predispose to ALL, such as the Robertsonian translocation [G] r(15;21) and ring chromosome [G] 21 which are strongly associated with ALL with intrachromosomal amplification of chromosome 21138. However, studies utilizing single nucleotide polymorphism (SNP) array-based case-control GWAS and genome sequencing of families and cohorts of sporadic ALL have shown that non-coding variants subtly influence risk of developing ALL in a manner that may be associated with disease subtype, outcome and genetic ancestry13,139–142. Furthermore, although familial ALL is relatively uncommon, these studies have identified highly deleterious non-silent germline variants, most commonly in genes encoding tumor suppressors and transcription factors, that often predispose to specific subtypes of ALL8,12,143–145, and similarly genes implicated in predisposition to familial ALL commonly also harbor deleterious non-silent germline variants in sporadic ALL that does not have evidence of familial inheritance8,10. Finally, structural variants represent an important class of predisposing genetic alteration146–149, and germline variants may influence anti-leukemic drug response and outcome, as well as disease susceptibility8,150.
Inherited SNPs and ALL susceptibility
GWAS have identified over 10 genetic loci harboring SNPs that are associated with a modest (typically up to 2-fold) increase in ALL risk. The most commonly implicated loci involve genes encoding transcription factors, particularly those known to encode regulators of hematopoietic and early lymphoid development and tumor suppressors: ARID5B, BAK1, CDKN2A, CDKN2B, CEBPE, ELK3, ERG, GATA3, IGF2BP1, IKZF1, IKZF3, LHPP, MYC, PTPRJ, TP63 and the BMI1-PIP4K2A locus9,13,139,140,151–159. Although the risk of ALL associated with a single variant is low, cumulatively the risk genotypes may increase ALL risk up to 9-fold153,154. Several loci are associated with distinct ALL subtypes and ancestral groups, such as GATA3 with Ph-like ALL in those of Hispanic ancestry13,160, TP63 and PTPRJ with ETV6-RUNX1 ALL159 and ERG with TCF3-PBX1 ALL and African-American ancestry9,161. A variant in USP7 (which encodes a deubiquitinase) is associated with risk of T-lineage ALL161; this gene is also somatically altered in T-ALL, particularly in cases with deregulation of the transcription factor TAL1162. The enrichment for associations with loci at or near genes with key roles in early hematopoietic and lymphoid development (IKZF1, IKZF3, CEBPE, ERG)163, many of which are also targeted by deleterious somatic alterations in ALL164, suggests that the variants influence timing or amplitude of expression of transcription factor-encoding genes and thus, lymphoid maturation. This is supported by data correlating SNP genotype with gene expression data in cell lines13,153,156.Alternatively, integration of multimodal data (SNP genotype, gene expression, chromatin accessibility and chromatin conformation) suggests that some SNPs mark promoter or enhancer elements, and may loop to regulate expression of local or more distant loci140. Preliminary data indicates that GATA3 variants, which are associated with Ph-like ALL, and specifically, dysregulating rearrangements CRLF2, may promote rearrangement by creating a strong GATA3 enhancer and perturbing chromatin topology at the CRLF2 locus165.
PAX5
Two reports described germline variants in PAX5 in several kindreds with heritable B-progenitor ALL associated with loss of the second, wild-type copy of PAX5 through deletion of chromosome 9p or the formation of a dicentric chromosome 912,166 (Figure 3a). PAX5 encodes a DNA-binding transcription factor required for B-lymphoid lineage maturation167. PAX5 is a common target of somatic mutation in B-ALL, being altered by chromosomal rearrangement, copy number alterations, or sequence mutations in approximately one third of B-ALL cases, resulting in loss of PAX5 function and arrested B-lymphoid development168–178. PAX5 is involved in somatic chromosomal rearrangements with over 20 partner genes175,178. Focal intragenic, partial and broad deletion of PAX5 is observed across the spectrum of B-ALL cases, with some subtype specificity – for example, focal internal deletions, but not sequence mutations, are present in one third of ETV6-RUNX1 cases168,177. Somatic PAX5 missense, frameshift and nonsense mutations are present across the PAX5 protein, with clustering in the paired domain and C-terminal transactivating domains, several of which define B-ALL subtypes, notably PAX5-P80R178 (Figure 2c).
Figure 3. Genetics of cancer predisposition genes in pediatric acute lymphoblastic leukemia.
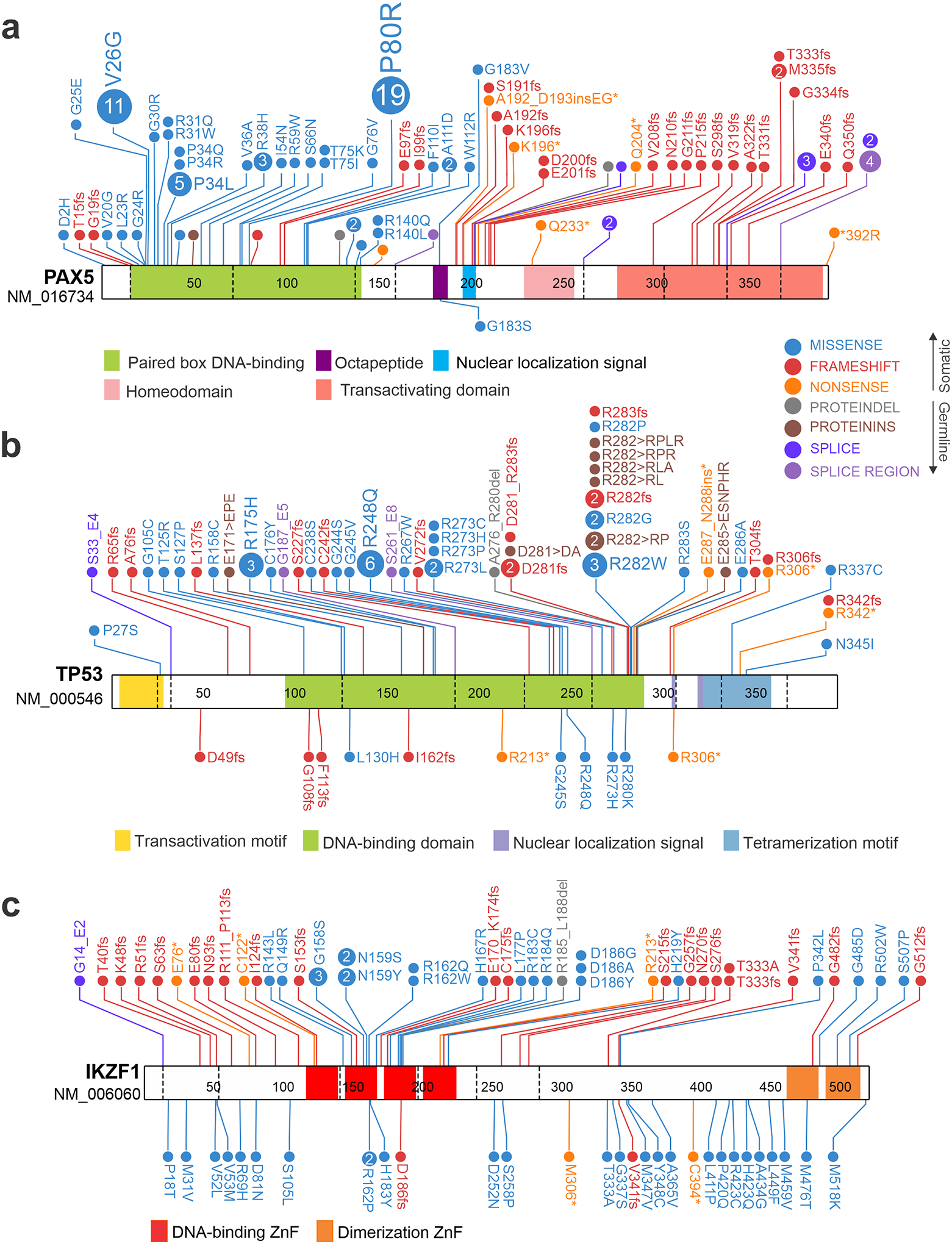
Schematics and domain architectures of proteins encoded by genes harboring germline and somatic mutations in pediatric acute lymphoblastic leukemia (ALL), including PAX5 (a), TP53 (b) and IKZF1 (c). The germline mutations are shown below the protein schematic and the somatic mutations on top. (a) PAX5 somatic variants are predominantly missense variants in the DNA binding domain and truncations that remove the transactivation domain. By contrast, a single residue has been observed mutated in familial ALL, G183S in the octapeptide domain. (b) Germline TP53 variants are shown for hypodiploid ALL, which involve the DNA binding domain and nuclear localization sequences. (c) Differences in distribution in IKZF1 variants are observed between germline and somatic variants. Somatic variants are most commonly N terminal truncating mutations, DNA binding domain missense mutations (most commonly in the second and third DNA-binding zinc fingers (ZnFs), which are required for DNA binding), and distal truncating and C-terminal ZnF missense mutation. In contrast, germline variants are predominantly located outside of ZnFs (possibly as these would not be tolerated as germline events) but in functional assays such as protein localization, adhesion and drug resistance, are commonly as deleterious or more deleterious than somatic variants. Germline data taken from refs 8,12,143; somatic mutation data updated from ref. 180.
A single hotspot at G183 of PAX5 harbors missense alterations, most commonly PAX5-G183S observed in familial ALL12,166 (Figure 3a). Leukemic cells typically exhibit loss of wild type PAX5, either by deletions of 9p (PAX5 is located at 9p13), or by the formation of iso-/dicentric 9q chromosomes, with few other recurrent somatic mutations. G183 is located in the octapeptide domain of PAX5, which may mediate Groucho family -mediated transcriptional repression179. The G183S variant is subtly hypomorphic in assays of transcriptional activation and DNA binding, suggesting that it is tolerable in the germline, heterozygous state, but requires inactivation of the wild type PAX5 allele to promote leukemogenesis178. This model of biallelic PAX5 alteration in leukemogenesis is supported by mouse models, in which chemical or retroviral mutagenesis of Pax5+/− mice as well as B-ALL spontaneously arising in heterozygous Pax5P80R/+ knock in mice is accompanied by deletion or mutation of the wild type Pax5 allele178,180. A minority of cases harbor biallelic alterations of a single gene (PAX5 or IKZF1), and such compound heterozygous mutations result in profound attenuation rather than complete abrogation of activity of a single transcription factor178. It is possible that a residual degree of activity of B-lineage transcription factors is required for maintenance of B cell lineage in B-ALL.
TP53
TP53 is one of the most frequently mutated genes in cancer, particularly in adult-onset somatic cancers, and germline mutations are the defining feature of Li-Fraumeni syndrome [G] (LFS) and predisposition to a diverse range of tumors, particularly sarcomas, central nervous system tumors, adrenocortical carcinoma and breast cancer, and both myeloid and lymphoid leukemia181,182. Within the hematopoietic system, germline TP53 alterations are a hallmark of low hypodiploid ALL143, and to a lesser extent have been associated with AML and therapy-related myeloid neoplasms183,184. Hypodiploid ALL comprises up to 5% of pediatric and 10% of adult B-ALL cases, and is characterized by recurring, stereotyped losses of whole chromosomes occurring in two main patterns: near haploidy with a leukemic cell modal chromosome number of 24–31, and low hypodiploid ALL with 32–39 chromosomes185. Each subtype has distinct additional somatic mutations and deletions, commonly affecting the RAS pathway in near haploid ALL, and IKZF2 and IKZF3 in low hypodiploid and near haploid ALL, respectively185. Low hypodiploid, but not near haploid ALL, has near-universal mutation of TP53150,185,186. Most commonly these are sequence mutations in the DNA-binding and nuclear export domains of the protein (Figure 3b), but structural alterations are also observed2. In pediatric low hypodiploid ALL, approximately half of cases harbor the TP53 variants in remission hematopoietic cells and so these are presumed to be germline variants with loss of wild type TP53 due to chromosome 17 aneuploidy resulting in hemizygosity for mutant TP53143.Germline TP53 structural variants (e.g. deletions) may also be observed2. In contrast, while low hypodiploidy is more frequent in adults, the TP53 alterations are rarely germline187. A broader study of TP53 variants in pediatric ALL has identified germline TP53 variants in approximately 2% of patients, most commonly in low hypodiploid cases, and these variants are associated with poor outcome150. The germline nature of TP53 mutations in low hypodiploid ALL is supported by the identification of TP53-mutation bearing LFS kindreds with diverse tumor types, including low hypodiploid ALL188. Thus, the identification of low hypodiploidy in pediatric ALL should prompt the suspicion of a germline TP53 variant.
IKZF1
IKZF1 shares several similarities with PAX5 in the pathogenesis of ALL: both have diverse germline and somatic alterations; and a role as tumor-initiating as well as cooperating events in leukemogenesis. In addition, germline variants of IKZF1 predispose to immunodeficiency syndromes, and influence therapeutic responsiveness in ALL.
IKZF1 encodes the founding member of the Ikaros family of zinc finger transcription factors, at least three of which (IKZF1, IKZF2 (also known as Helios), and IKZF3 (also known as Aiolos)) have roles in hematopoietic development and malignancy189. IKZF1 is required for the development of all lymphoid cells: T, B, natural killer and dendritic cells190. Somatic alterations of IKZF1 were first identified in BCR-ABL1 positive (Ph+) B-ALL [G] lymphoid leukemia, either de novo B-ALL or lymphoid blast crisis of chronic myeloid leukemia191. Subsequent studies have shown that IKZF1 alterations are enriched as secondary events in kinase-driven B-ALL (Ph+ or Ph-like ALL) and double homeobox 4 (DUX4)-rearranged ALL192–194. In kinase-driven192,195,196, but not DUX4-rearranged B-ALL194,197, IKZF1 alterations are associated with poor outcome198,199. A single missense mutation in the second DNA-binding zinc finger of IKZF1, N159Y, defines a subtype of ALL with distinct gene expression profile, and is considered a leukemia-initiating mutation178,200. Somatic alterations of IKZF1 in ALL are commonly deletions that result in loss of function, or focal internal deletions that act as dominant negative alleles, particularly the IK6 variant that lacks the DNA binding zinc fingers201. Somatic sequence mutations are enriched in the central two DNA-binding zinc fingers and C-terminal zinc fingers, and also commonly result in aberrant nuclear and/or cytoplasmic localization and dominant negative effects (Figure 3c). Collectively, IKZF1 deletions and mutations cooperate with fusion oncoproteins such as BCR-ABL1 in lymphoid leukemogenesis, and drive resistance by activation of self-renewal, stemness and cell-cell and cell-stromal adhesion phenotypes, by derepression of stem cell (e.g. CD90, THY1) and integrin proteins202–204.
Several recent reports have identified germline IKZF1 non-silent mutations in familial and sporadic ALL, and immunodeficiency. We reported a family with multiple individuals with ALL in which affected individuals harbored a germline truncating variant in the second N-terminal zinc finger of IKZF18. Several unaffected carriers were also identified that exhibited features of immunodeficiency. Germline IKZF1 genotyping of almost 5000 children with ALL identified non-silent missense IKZF1 variants not present in control populations in approximately 1% of B-ALL cases. In contrast to somatic variants, the germline missense variants were mostly located outside of the regions of the gene that encode the N- or C-terminal zinc fingers (Figure 3c). Functional assays showed that the majority of germline variants had little direct effect on transcriptional repression, with the exception of truncating mutations, whereas most variants had profound effects on subcellular localization, adhesion, and responsiveness of BCR-ABL1+ leukemic cells to conventional chemotherapy and kinase inhibition; indeed, germline variants were the most deleterious in these assays of all germline and somatic IKZF1 alleles tested. In parallel, multiple studies have identified associations between germline IKZF1 alterations and immunodeficiency and autoimmunity, particularly common variable immunodeficiency, a heterogeneous disorder characterized by humoral immunodeficiency and associated autoimmune phenomena205–208. Germline IKZF1 variants predisposing to immunodeficiency are clustered at N159, a key DNA contact residue of the second zinc finger in the protein, but are commonly serine substitutions, which are rare in ALL; in contrast the ALL subtype-defining somatic IKZF1N159Y variant has not been reported in the germline, suggesting it is either more deleterious and not tolerated as a germline event, or that there are distinct genotype-phenotype associations. The importance of germline variations in the Ikaros family in hematological phenotypes is reinforced by the recent description of germline mutations in the DNA-binding zinc fingers of IKZF5 (also known as Pegasus) in hereditary thrombocytopenia209.
ETV6
ETV6 encodes a transcriptional regulator of hematopoiesis and platelet development210. Somatic rearrangements (most commonly to RUNX1), deletions and sequence mutations are observed in ALL168,192,211. Moreover, second hit alterations of ETV6, particularly deletion, are common in ETV6-RUNX1 rearranged leukemia168,212,213. Germline alterations of ETV6 are recognized in both familial and sporadic ALL. Multiple kindreds with germline ETV6 mutations, ALL, and commonly thrombocytopenia, were described in 2015144,145, with over 20 kindreds now described214. Subsequently, large-scale sequencing of pediatric ALL cohorts identified germline, predominantly missense variants in approximately 1% of ALL cases, which were commonly hyperdiploid10. More complex germline ETV6 alterations have been identified in kindreds with thrombocytopenia or leukemia, including insertion/deletion mutations and structural variants (deletions, rearrangements) that can result in truncation and loss of function of ETV611,146,147,215,216. Such structural variants may not be identified in conventional analysis of coding (e.g. exome) sequencing data, reinforcing the importance of structural genomic analysis of exomes and/or whole genome sequencing data in kindreds with a presumptive heritable predisposition to leukemia147,216. Carriers of ETV6 mutations commonly exhibit mild to moderate thrombocytopenia, which may be associated with an abnormal platelet function and a bleeding diathesis11,144. It is estimated that leukemia arises in approximately 30% of carriers, and is most commonly ALL, but MDS, AML, mixed phenotype acute leukemia, diffuse large B cell lymphoma, myeloproliferative disease and plasma cell myeloma have also been reported. ETV6 variants are clustered in the DNA-binding ETS domain and predicted to be deleterious, and may have dominant negative effects; tested variants usually result in impaired transcriptional repression217.Functional interrogation of ETV6 variants has shown perturbed expression of genes involved in platelet biogenesis and platelet cytoskeletal dynamics, including CDC42 and RHOA218. While this provides a plausible explanation for altered platelet development and function, like many of the other predispositions it is likely that additional germline or somatic genetic alterations drive leukemogenesis219.
Other genes predisposing to ALL
Several case reports have been informative in identifying putative new predisposition genes in ALL. SH2B3 is a negative regulator of JAK–STAT signaling220, and somatic SH2B3 deletions and mutations are observed in B- and T-ALL, particularly in Ph-like B-ALL221–223. Rare kindreds with germline SH2B3 mutations and B-ALL have been identified223. Germline amplification of the neuroblastoma breakpoint region, encompassing MEF2D and BCL9, was reported in an individual with recurrence of MEF2D-BCL9 positive ALL, in which the two tumors had distinct genomic and RNA breakpoints, suggesting a (rare) heritable predisposition to ALL arising from this anomaly149. Germline sequencing of a pediatric patient and both parents (also known as trio sequencing) identified an individual with DS-associated ALL with concurrent JAK2 and STAT3 variants that together, but not alone, induced growth advantage in cell lines224. Analysis of 5 children with recurrent, clonally unrelated leukemia identified two cases with germline TYK2 mutations225. TYK2 is a member of the JAK family and has been implicated in the pathogenesis of T-ALL226. The germline TYK2 variants activated signaling that was abrogated with JAK inhibitors, supporting a tumor-promoting role. Analysis of T-ALL with late relapse (to distinguish recurrence of the primary tumor from a second malignancy) identified germline deletion of LMO2 in a case, suggesting this alteration predisposed to the development of T-ALL148.
Analyses of cohorts of sporadic ALL have identified additional putative leukemia-predisposing variants. Screening of a cohort of high hyperdiploid ALL identified putative pathogenic mutations in the Nijmegen Breakage Syndrome gene NBN, ETV6, FLT3, SH2B3, CREBBP (encoding a histone acetyltransferase somatically mutated in aneuploid ALL, relapsed ALL and lymphoma)227,228, PMS1 (encoding a protein involved in DNA mismatch repair), ABL1 and MYH9 (encoding myosin heavy chain 9). Multiple variants were identified in GRB2-associated-binding protein 2 (GAB2), a SHP2-interacting protein, which led to increased AKT signaling in vitro229. Exome sequencing of a cohort of individuals with DS-associated ALL identified different pathogenic or likely pathogenic variants in 3 of 73 patients, including variants in IKZF1, NBN and the telomere maintenance gene RTEL1230. Heterozygous deleterious mutations in genes in the Fanconi anemia–BRCA pathway have been identified in approximately 10% of T-lineage ALL, several of which were shown to be germline231.
Models of Disease Progression
From the data presented above, it is clear that several themes are emerging for patients with germline predisposition syndromes and the development of hematological malignancies (Figure 3). The development of a hematological malignancy in patients with a germline predisposition (e.g. mutation in DDX41) can occur as late as the 6th or 7th decade of life and thus is no longer restricted to childhood. However, for some predispositions, most notably those involving SAMD9L, SAMD9 or NF1, the risk of hematological malignancies does appear to be restricted to younger ages, suggesting a specific developmental stage may be more susceptible to transformation. This is also true for at least a subset of ALL, where notably, TP53 alterations are nearly universal in low hypodiploid ALL in both children and adults, but are only observed as germline events in children. Another theme is that the different types of germline predispositions are associated with widely variable penetrance for both myeloid and lymphoid leukemias, and the disease phenotypes are likely influenced by both cell intrinsic and cell extrinsic factors, including somatic alterations acquired during disease progression. In addition, although there are rare exceptions, most predisposition syndromes are linked with abnormalities in specific hematopoietic cell lineages and lead to distinct tumor profiles. For example, while germline variants in RUNX1, ANKRD26 and ETV6 all predispose to thrombocytopenia and hematological malignancy, there are striking differences in cancer predisposition: ANKRD26 variants predisposes to myeloid malignancy, ETV6 predominantly to B-lineage ALL, and RUNX1 to myeloid and to a lesser extent T-lineage ALL. Finally, common patterns of disease progression are beginning to emerge in germline predispositions (e.g. RUNX1, PAX5G183S and NF1), which frequently involve inactivation of the remaining wild-type allele and/or somatic acquisition of canonical mutations that define specific hematological malignancy subtypes.
Future Perspectives
The last decade has witnessed remarkable advances in our understanding of the inherited basis of hematological malignancies, particularly in children (Box 2). However, much remains to be learned.
Box 2. Clinical Management and Testing of Children and Families with Predisposition Syndromes.
It is increasingly being recognized that germline alterations are much more common than previously appreciated. Considering that many patients with pathogenic or likely pathogenic mutations lack a suggestive family history of a germline predisposition, unbiased genetic testing on all patients, especially children, is the best approach to accurately catalogue and estimate the frequency of germline alterations. Indeed tumor and germline exome and/or genome sequencing is increasingly used in pediatric cancer diagnosis, as such sequencing commonly identifies “actionable” variants, and patients and families have a desire to understand the potential heritability of cancer.4,250,251 This approach will present a challenge to practicing genetic counselors and oncologists as they integrate these data into patient care and management, especially with the interpretation of variants of undetermined significance that lack experimental documentation of deleteriousness in a given tumor context, which are commonly identified by such broad sequencing studies.
For hematopoietic tumors, genomic DNA isolated from a skin biopsy or other non-hematopoietic source is advisable for germline analysis. In the absence of true germline testing, the germline status of some variants can be inferred from “tumor only” testing from the variant allele frequency, especially if analyzed using serial genetic testing during remission. Importantly, the finding of specific mutations in tumor only testing in children, such as those in TP53 or GATA2, should prompt germline evaluation.
While the majority of mutations identified to date are present within coding exons and suitable panels are available for targeted sequencing of relevant genes, unbiased approaches are still identifying new and novel coding mutations (for example in SAMD9L and DDX41) and non-coding alterations (for example ANKRD26 and intronic GATA2 alterations). Importantly, cascade testing of the immediate family members is critical as mutations detected in the germline of patients may reflect a pattern of familial inheritance, even in those lacking a strong family history of disease, or may result from a de novo germline mutation. For most variants, data regarding the relative frequency of de novo germline variants versus inherited variants are sparse. This clinically important distinction will influence patient and family communication and management, including options for future hematopoietic stem cell transplantation. In particular, recent reports have described donor derived AMLs as a result of the transfer of germline DDX41252,253 or GATA2254 mutations within family members during allogeneic transplantation.
Extent of germline predisposition.
Most studies have examined small numbers of genes, or have limited analysis to the coding exome. In pediatric cancer, it has been described that approximately 8–10% of patients have a heritable predisposition, however that number is biased both by analyses of selected cohorts or patients and panels of genes, despite the interrogation of genome-wide sequencing data2–4,232,233. Until recently, many such germline panels did not include key genes relevant to hematological malignancy (e.g. IKZF1, PAX5 and SAMD9L). The increasing recognition of elevated familial risks for many hematological malignancies234, ascertainment of kindreds lacking a causal mutation from targeted or exome sequencing, and the ability of genome-wide sequence and structural analyses to identify putative predisposition mutations235 strongly suggests that careful analysis of whole genome sequencing data is required to identify new predisposition genes. This poses several challenges, including amassing the optimal cohorts for comparative analyses, utilizing novel genomic analysis approaches to map complex variants, including long read sequencing and optical mapping [G]236, and developing optimal algorithms for detection of putative causal non-coding and structural variants237. Further, larger efforts are needed to accurately define the frequency of these germline mutations across the spectrum of cancer subtypes as many of the new predisposition genes have been reported in small studies focused on rare but informative families. Thus, the absolute risk associated with individual non-silent coding variants is difficult to quantify. This also applies to identification of predisposing silent or non-coding variants, where cohort size directly influences power to detect significant associations in hematological malignancies140. Such large cohorts will also be required to elucidate cooperativity of germline variants in leukemia predisposition, which is suggested by emerging reports of multiple predisposing variants in individuals with ALL, the combinatorial effect of GWAS-identified SNPs in ALL risk, and the common observation of incomplete penetrance of familial leukemia genes.
Mechanistic interpretation.
Our understanding of how germline variants predispose to hematological malignancies, and the basis for genotype-phenotype differences, lags considerably behind variant identification. Improved understanding will require a broad, sustained and intensive effort to build faithful in vitro and in vivo models to study leukemia initiation and progression.
Variant significance and clinical utility.
There are ongoing extensive efforts, including systematic cooperative efforts between the Precision Medicine Taskforce of the American Society of Hematology and the Clinical Genome Resource (ClinGen), to rigorously define criteria for germline variants, and to annotate such variants accordingly238–241. However, these annotation efforts rely on published functional data and in silico prediction of deleteriousness, and the majority of identified germline variants are of unknown significance. One intriguing approach is high-throughput functional testing to systematically characterize a large number of mutations within critical proteins or protein domains. For example, this was rigorously done for BRCA1242 and p53243. In particular the BRCA1 study generated a comprehensive map of functionally important residues in BRCA1 that could be used for the clinical classification of variants of uncertain significance. Perhaps as a model moving forward, ClinGen variant curation expert panels are being established to systemically evaluate the pathogenicity of germline variants. In light of this need the American Society of Hematology convened a ClinGen Myeloid Malignancy Variant Curation Expert Panel that has published their recommendations for germline RUNX1 variants239,244, and will expand to perform similar curation for additional genes. A challenge moving forward will be to match the rate of variant discovery in the clinical sphere with robust functional data to properly assign pathogenicity to novel variants.
Figure 4. Model of disease progression.
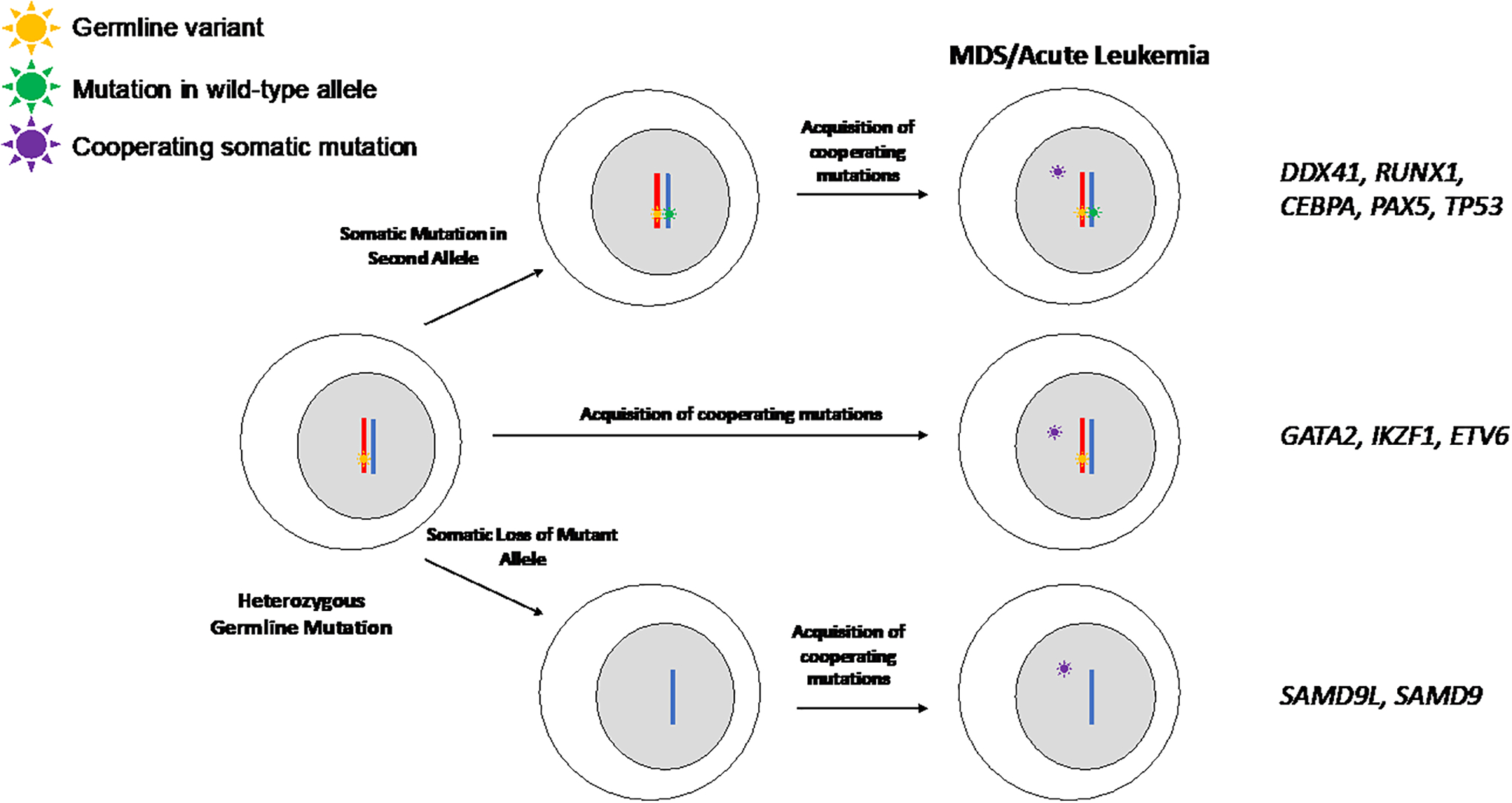
Progression to advanced disease occurs through multiple different and potentially overlapping mechanisms in patients with MDS or acute leukemia predisposition syndromes. The pathways commonly observed for the different germline predispositions (genes highlighted on the right) are shown. Progression to advanced disease in many predisposition syndromes occurs through a step wise process involving loss of the remaining wild-type allele and acquisition of additional cooperating mutations (top), while others appear to maintain the wild-type allele (middle). SAMD9 and SAMD9L are unique in that disease progression is observed when there is outgrowth of monosomy 7 cells that lack the deleterious germline mutation and then cells can acquire additional somatic mutations for the full disease phenotype.
ACKNOWLEDGEMENTS
The authors thank Ilaria Iacobucci for assistance with figure preparation. The authors are supported by the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, the St. Jude Comprehensive Cancer Center Core Grant CA021765, NHLBI R01 HL144653 (to J.M.K.), the Edward P. Evans Foundation (to J.M.K.), NCI R35 CA197695 (to C.G.M.), the Henry Schueler 19 Foundation (to C.G.M.) and a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (to C.G.M.).
Competing interests:
C.G.M. has received research funding from Loxo Oncology, Pfizer and Abbive; speaking and travel fees from Amgen; holds stock in Amgen; and compensated advisory board participation for Illumina. J.M.K. declares no competing interests.
Glossary
- clonal hematopoiesis
The expansion of a clonal population of hematopoietic cells marked by somatic mutations
- cytopenic phase
A period marked by a decrease in peripheral blood cell counts (anemia: red blood cells, thrombocytopenia: platelets, leukopenia: white blood cells)
- monocytopenia
A decrease in monocytes
- pulmonary alveolar proteinosis
A lung disease characterized by a build-up of proteins and lipids in the functional units of the lung (alveoli) that is part of MonoMac syndrome, which results from GATA2 germline mutations
- sensorineural deafness
A form of hearing loss that results from damage to the inner ear or auditory nerve that is part of Emberger Syndrome, which results from GATA2 germline mutations
- thrombocytopenia-2
An autosomal dominant syndrome resulting from germline mutations in ANKRD26 that is characterized by lifelong mild-to-moderate thrombocytopenia and an increase in the risk for myeloid malignancies
- Philadelphia chromosome-like ALL
A group of B lymphoblastic leukemias that have a global expression profile similar to BCR-ABL1 positive (Ph+) ALL but have alterations in other kinase signaling and cytokine receptor pathways
- Ataxia-Telangiectasia
An autosomal recessive condition caused mutations in the ATM gene. This syndrome is characterized by immunodeficiency, decreased DNA damage repair and neurological abnormalities
- Nijmegen breakage syndrome
An autosomal recessive disorder that leads to chromosomal instability and is clinically characterized by microcephaly, growth retardation, immunodeficiency, and predisposition to cancer
- Robertsonian translocation
A non-reciprocal chromosomal translocation in which the long arms of two distinct acrocentric chromosomes become fused and share a single centromere
- ring chromosome
An abnormal chromosome in which the ends of a single chromosome fuse to form a ring
- Li-Fraumeni syndrome
An autosomal-dominant condition caused by an inherited mutation in TP53 that renders humans highly susceptible to a number of cancers, including breast cancer, leukaemia and sarcoma
- BCR-ABL1 positive (Ph+) B-ALL
A class of hematopoietic disorders driven by the BCR-ABL1 fusion oncoprotein
- optical mapping
An approach to genome assembly in which DNA molecules are fluorescently labeled and imaged to construct maps
References
- 1.The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93, doi: 10.1038/s41586-020-1969-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373, 2336–2346, doi: 10.1056/NEJMoa1508054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; seminal study estimating the prevalence of deleterious germline mutations in childhood cancer, based on interrogation of a panel of genes
- 3.Grobner SN et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327, doi: 10.1038/nature25480 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Parsons DW et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol 2, 616–624, doi: 10.1001/jamaoncol.2015.5699 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang KL et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370 e314, doi: 10.1016/j.cell.2018.03.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405, doi: 10.1182/blood-2016-03-643544 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Godley LA Inherited predisposition to acute myeloid leukemia. Semin Hematol 51, 306–321, doi: 10.1053/j.seminhematol.2014.08.001 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Churchman ML et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 33, 937–948 e938, doi: 10.1016/j.ccell.2018.03.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; identified germline non-silent IKZF1 variants in familial and sporadic ALL, showing that many of the germline variants are more deleterious than previously identified somatic variants.
- 9.Qian M et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood 133, 724–729, doi: 10.1182/blood-2018-07-862946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriyama T et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol 16, 1659–1666, doi: 10.1016/s1470-2045(15)00369-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topka S et al. Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet 11, e1005262, doi: 10.1371/journal.pgen.1005262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; one of several studies that identified germline ETV6 variants associated with ALL and thrombocytopenia.
- 12.Shah S et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 45, 1226–1231, doi: 10.1038/ng.2754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; identification of germline PAX5 variants as causal in familial B-ALL with loss of chromosome 9p leading to hemizygosity of the variant allele
- 13.Perez-Andreu V et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 45, 1494–1498, doi: 10.1038/ng.2803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; example of a GWAS study showing association of germline SNP (in GATA3) associated with specific subtype and ancestry of B-ALL
- 14.Polprasert C et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 27, 658–670, doi: 10.1016/j.ccell.2015.03.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive study evaluating the frequency of germline and somatic DDX41 mutations in myeloid malignancies.
- 15.Pastor VB et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica 103, 427–437, doi: 10.3324/haematol.2017.180778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz JR et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 31, 1827–1830, doi: 10.1038/leu.2017.142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narumi S et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48, 792–797, doi: 10.1038/ng.3569 (2016). [DOI] [PubMed] [Google Scholar]; First description of germline SAMD9 mutations in children with MIRAGE syndrome.
- 18.Tesi B et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 129, 2266–2279, doi: 10.1182/blood-2016-10-743302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong JC et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3, e121086, doi: 10.1172/jci.insight.121086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Used historical samples to demonstrate germline mutations in SAMD9 and SAMD9L in families with Myelodysplasia and Leukemia Syndrome with Monosomy and illuminated the variety of revertant and cooperating mutations that influence clinical phenotypes.
- 20.Mangaonkar AA & Patnaik MM Hereditary Predisposition to Hematopoietic Neoplasms: When Bloodline Matters for Blood Cancers. Mayo Clinic proceedings 95, 1482–1498, doi: 10.1016/j.mayocp.2019.12.013 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Furutani E & Shimamura A Germline Genetic Predisposition to Hematologic Malignancy. J Clin Oncol 35, 1018–1028, doi: 10.1200/jco.2016.70.8644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter CC et al. Recommendations for Surveillance for Children with Leukemia-Predisposing Conditions. Clin Cancer Res 23, e14–e22, doi: 10.1158/1078-0432.CCR-17-0428 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Wegman-Ostrosky T & Savage SA The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol 177, 526–542, doi: 10.1111/bjh.14535 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Faber ZJ et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48, 1551–1556, doi: 10.1038/ng.3709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaidzik VI et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 30, 2160–2168, doi: 10.1038/leu.2016.126 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Grossmann V et al. Prognostic relevance of RUNX1 mutations in T-cell acute lymphoblastic leukemia. Haematologica 96, 1874–1877, doi: 10.3324/haematol.2011.043919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osato M et al. Biallelic and heterozygous point mutations in the runt domain of the AML1/PEBP2alphaB gene associated with myeloblastic leukemias. Blood 93, 1817–1824 (1999). [PubMed] [Google Scholar]
- 28.Zelent A, Greaves M & Enver T Role of the TEL-AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene 23, 4275–4283 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Zhang J et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163, doi: 10.1038/nature10725 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Latger-Cannard V et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J Rare Dis 11, 49, doi: 10.1186/s13023-016-0432-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song WJ et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 23, 166–175, doi: 10.1038/13793 (1999). [DOI] [PubMed] [Google Scholar]; Linked familial platelet disorder with predisposition to AML with germline alterations in RUNX1.
- 32.Antony-Debre I et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia 30, 999–1002, doi: 10.1038/leu.2015.236 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Michaud J et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood 99, 1364–1372 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Brown AL et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv 4, 1131–1144, doi: 10.1182/bloodadvances.2019000901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Churpek JE et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 126, 2484–2490, doi: 10.1182/blood-2015-04-641100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preudhomme C et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 113, 5583–5587, doi: 10.1182/blood-2008-07-168260 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Shiba N et al. CBL mutation in chronic myelomonocytic leukemia secondary to familial platelet disorder with propensity to develop acute myeloid leukemia (FPD/AML). Blood 119, 2612–2614, doi: 10.1182/blood-2011-02-333435 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Pabst T et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 27, 263–270, doi: 10.1038/85820 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Frohling S et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 22, 624–633 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Pabst T, Eyholzer M, Haefliger S, Schardt J & Mueller BU Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol 26, 5088–5093, doi: 10.1200/JCO.2008.16.5563 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Tawana K et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126, 1214–1223, doi: 10.1182/blood-2015-05-647172 (2015). [DOI] [PubMed] [Google Scholar]; Studied diagnosis and relapse leukemia samples from patients with germline CEBPA mutations and demonstrated that disease recurrence is commonly driven by new, independent clones rather than re-emergence of an existing clone present at diagnosis.
- 42.Smith ML, Cavenagh JD, Lister TA & Fitzgibbon J Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med 351, 2403–2407, doi: 10.1056/NEJMoa041331 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Taskesen E et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117, 2469–2475, doi: 10.1182/blood-2010-09-307280 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Pathak A et al. Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica 101, 846–852, doi: 10.3324/haematol.2015.130799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wlodarski MW, Collin M & Horwitz MS GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54, 81–86, doi: 10.1053/j.seminhematol.2017.05.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wlodarski MW et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127, 1387–1397; quiz 1518, doi: 10.1182/blood-2015-09-669937 (2016). [DOI] [PubMed] [Google Scholar]; Definitive and comprehensive study demonstrating the prevalence of germline GATA2 mutations in pediatric MDS.
- 47.Cortes-Lavaud X et al. GATA2 germline mutations impair GATA2 transcription, causing haploinsufficiency: functional analysis of the p.Arg396Gln mutation. J Immunol 194, 2190–2198, doi: 10.4049/jimmunol.1401868 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Hsu AP et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood 121, 3830–3837, S3831–3837, doi: 10.1182/blood-2012-08-452763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wehr C et al. A novel disease-causing synonymous exonic mutation in GATA2 affecting RNA splicing. Blood 132, 1211–1215, doi: 10.1182/blood-2018-03-837336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozyra EJ et al. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia 34, 2673–2687, doi: 10.1038/s41375-020-0899-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cavalcante de Andrade Silva M et al. Breaking the spatial constraint between neighboring zinc fingers: a new germline mutation in GATA2 deficiency syndrome. Leukemia, doi: 10.1038/s41375-020-0820-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshida M et al. Prevalence of germline GATA2 and SAMD9/9L variants in paediatric haematological disorders with monosomy 7. Br J Haematol, doi: 10.1111/bjh.17006 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Micol JB & Abdel-Wahab O Collaborating constitutive and somatic genetic events in myeloid malignancies: ASXL1 mutations in patients with germline GATA2 mutations. Haematologica 99, 201–203, doi: 10.3324/haematol.2013.101303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J & Hickstein DD Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica 99, 276–281, doi: 10.3324/haematol.2013.090217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bodor C et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica 97, 890–894, doi: 10.3324/haematol.2011.054361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al Seraihi AF et al. GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2-mutated MDS/AML. Leukemia 32, 2502–2507, doi: 10.1038/s41375-018-0134-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rocak S & Linder P DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5, 232–241, doi: 10.1038/nrm1335 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Fullam A & Schroder M DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim Biophys Acta 1829, 854–865, doi: 10.1016/j.bbagrm.2013.03.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12, 959–965, doi: 10.1038/ni.2091 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee KG et al. Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell reports 10, 1055–1065, doi: 10.1016/j.celrep.2015.01.039 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Cardoso SR et al. Germline heterozygous DDX41 variants in a subset of familial myelodysplasia and acute myeloid leukemia. Leukemia 30, 2083–2086, doi: 10.1038/leu.2016.124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li R, Sobreira N, Witmer PD, Pratz KW & Braunstein EM Two novel germline DDX41 mutations in a family with inherited myelodysplasia/acute myeloid leukemia. Haematologica 101, e228–231, doi: 10.3324/haematol.2015.139790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewinsohn M et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood 127, 1017–1023, doi: 10.1182/blood-2015-10-676098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sebert M et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood 134, 1441–1444, doi: 10.1182/blood.2019000909 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Quesada AE et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am J Hematol 94, 757–766, doi: 10.1002/ajh.25486 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Polprasert C et al. Novel DDX41 variants in Thai patients with myeloid neoplasms. Int J Hematol 111, 241–246, doi: 10.1007/s12185-019-02770-3 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Takeda J et al. Genetic Predispositions to Myeloid Neoplasms Caused By Germline DDX41 Mutations. Blood 126, 2843–2843, doi: 10.1182/blood.V126.23.2843.2843 (2015). [DOI] [Google Scholar]
- 68.Inoue D, Bradley RK & Abdel-Wahab O Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 30, 989–1001, doi: 10.1101/gad.278424.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadono M et al. Biological implications of somatic DDX41 p.R525H mutation in acute myeloid leukemia. Exp Hematol 44, 745–754 e744, doi: 10.1016/j.exphem.2016.04.017 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Chen DH et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98, 1146–1158, doi: 10.1016/j.ajhg.2016.04.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buonocore F et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest 127, 1700–1713, doi: 10.1172/JCI91913 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwartz JR et al. The genomic landscape of pediatric myelodysplastic syndromes. Nature communications 8, 1557, doi: 10.1038/s41467-017-01590-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; First study to comprehensive profile the genomic landscape of pediatric MDS and described germline mutations in SAMD9 or SAMD9L in nearly 20% of cases.
- 73.Bluteau O et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 131, 717–732, doi: 10.1182/blood-2017-09-806489 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Nagata Y et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 132, 2309–2313, doi: 10.1182/blood-2017-05-787390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagamachi A et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 24, 305–317, doi: 10.1016/j.ccr.2013.08.011 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Liu J & McFadden G SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol 89, 1925–1931, doi: 10.1128/JVI.02262-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nounamo B et al. An interaction domain in human SAMD9 is essential for myxoma virus host-range determinant M062 antagonism of host anti-viral function. Virology 503, 94–102, doi: 10.1016/j.virol.2017.01.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sivan G, Ormanoglu P, Buehler EC, Martin SE & Moss B Identification of Restriction Factors by Human Genome-Wide RNA Interference Screening of Viral Host Range Mutants Exemplified by Discovery of SAMD9 and WDR6 as Inhibitors of the Vaccinia Virus K1L-C7L- Mutant. MBio 6, e01122, doi: 10.1128/mBio.01122-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meng X et al. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog 14, e1006884, doi: 10.1371/journal.ppat.1006884 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mekhedov SL, Makarova KS & Koonin EV The complex domain architecture of SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol Direct 12, 13, doi: 10.1186/s13062-017-0185-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wong JC et al. Functional evidence implicating chromosome 7q22 haploinsufficiency in myelodysplastic syndrome pathogenesis. eLife 4, doi: 10.7554/eLife.07839 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Honda H, Nagamachi A & Inaba T −7/7q- syndrome in myeloid-lineage hematopoietic malignancies: attempts to understand this complex disease entity. Oncogene 34, 2413–2425, doi: 10.1038/onc.2014.196 (2015). [DOI] [PubMed] [Google Scholar]
- 83.de Jesus AA et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest 130, 1669–1682, doi: 10.1172/JCI129301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ripperger T et al. MDS1 and EVI1 complex locus (MECOM): a novel candidate gene for hereditary hematological malignancies. Haematologica 103, e55–e58, doi: 10.3324/haematol.2017.178723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rio-Machin A et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun 11, 1044, doi: 10.1038/s41467-020-14829-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bluteau D et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest 124, 580–591, doi: 10.1172/JCI71861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Al Daama SA et al. A missense mutation in ANKRD26 segregates with thrombocytopenia. Blood 122, 461–462, doi: 10.1182/blood-2013-03-489344 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Noris P et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood 117, 6673–6680, doi: 10.1182/blood-2011-02-336537 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Pippucci T et al. Mutations in the 5’ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet 88, 115–120, doi: 10.1016/j.ajhg.2010.12.006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perez Botero J et al. ASXL1 mutated chronic myelomonocytic leukemia in a patient with familial thrombocytopenia secondary to germline mutation in ANKRD26. Blood cancer journal 5, e315, doi: 10.1038/bcj.2015.41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanders MA et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood 132, 1526–1534, doi: 10.1182/blood-2018-05-852566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ward AF, Braun BS & Shannon KM Targeting oncogenic Ras signaling in hematologic malignancies. Blood 120, 3397–3406, doi: 10.1182/blood-2012-05-378596 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arico M, Biondi A & Pui CH Juvenile myelomonocytic leukemia. Blood 90, 479–488 (1997). [PubMed] [Google Scholar]
- 94.Niemeyer CM & Kratz CP Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 140, 610–624, doi: 10.1111/j.1365-2141.2007.06958.x (2008). [DOI] [PubMed] [Google Scholar]
- 95.Flotho C et al. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood 111, 966–967; author reply 967–968, doi: 10.1182/blood-2007-09-111831 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Chang TY, Dvorak CC & Loh ML Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood 124, 2487–2497, doi: 10.1182/blood-2014-03-300319 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Stiller CA, Chessells JM & Fitchett M Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70, 969–972, doi: 10.1038/bjc.1994.431 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shannon KM et al. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med 330, 597–601 (1994). [DOI] [PubMed] [Google Scholar]
- 99.Niemeyer CM JMML genomics and decisions. Hematology Am Soc Hematol Educ Program 2018, 307–312, doi: 10.1182/asheducation-2018.1.307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Niemeyer CM et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42, 794–800, doi: 10.1038/ng.641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Genomic and clinical analysis of patients with germline CBL mutations and demonstrated a propensity for spontaneous resolution of hematopoietic abnormalities with prolonged risk for vascular and developmental complications.
- 101.Loh ML et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114, 1859–1863, doi:blood-2009-01-198416 [pii] 10.1182/blood-2009-01-198416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Strullu M et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51, 689–697, doi: 10.1136/jmedgenet-2014-102611 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Kratz CP et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106, 2183–2185, doi: 10.1182/blood-2005-02-0531 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cave H et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet 59, 173–178, doi: 10.1016/j.ejmg.2016.01.003 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Cave H et al. Mutations in RIT1 cause Noonan syndrome with possible juvenile myelomonocytic leukemia but are not involved in acute lymphoblastic leukemia. Eur J Hum Genet 24, 1124–1131, doi: 10.1038/ejhg.2015.273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Flex E et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 23, 4315–4327, doi: 10.1093/hmg/ddu148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roberts AE et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39, 70–74, doi: 10.1038/ng1926 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Kratz CP, Rapisuwon S, Reed H, Hasle H & Rosenberg PS Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157C, 83–89, doi: 10.1002/ajmg.c.30300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stieglitz E et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47, 1326–1333, doi: 10.1038/ng.3400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Genomically characterized serial samples from JMML patients, including diagnosis, relapse and transformation to AML and identified recurrent mutations in genes involved in signal transduction, splicing, PRC2 and transcription.
- 110.Caye A et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47, 1334–1340, doi: 10.1038/ng.3420 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Murakami N et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131, 1576–1586, doi: 10.1182/blood-2017-07-798157 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Stieglitz E et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood 125, 516–524, doi: 10.1182/blood-2014-09-601690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stieglitz E et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nature communications 8, 2127, doi: 10.1038/s41467-017-02178-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lipka DB et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nature communications 8, 2126, doi: 10.1038/s41467-017-02177-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hitzler JK & Zipursky A Origins of leukaemia in children with Down syndrome. Nat Rev Cancer 5, 11–20, doi: 10.1038/nrc1525 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Mullighan CG et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41, 1243–1246, doi:ng.469 [pii] 10.1038/ng.469 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lange BJ et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children’s Cancer Group Studies 2861 and 2891. Blood 91, 608–615 (1998). [PubMed] [Google Scholar]
- 118.Rainis L et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102, 981–986, doi: 10.1182/blood-2002-11-3599 (2003). [DOI] [PubMed] [Google Scholar]
- 119.Wechsler J et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32, 148–152, doi: 10.1038/ng955 (2002). [DOI] [PubMed] [Google Scholar]
- 120.Li Z et al. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet 37, 613–619, doi: 10.1038/ng1566 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Yoshida K et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet 45, 1293–1299, doi: 10.1038/ng.2759 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Mateos MK, Barbaric D, Byatt SA, Sutton R & Marshall GM Down syndrome and leukemia: insights into leukemogenesis and translational targets. Transl Pediatr 4, 76–92, doi: 10.3978/j.issn.2224-4336.2015.03.03 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Landgren O et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 112, 2199–2204, doi: 10.1182/blood-2008-03-143602 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jones AV et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 115, 4517–4523, doi: 10.1182/blood-2009-08-236448 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jones AV et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet 41, 446–449, doi: 10.1038/ng.334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kilpivaara O et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet 41, 455–459, doi: 10.1038/ng.342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Olcaydu D et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet 41, 450–454, doi: 10.1038/ng.341 (2009). [DOI] [PubMed] [Google Scholar]
- 128.Andrikovics H et al. JAK2 46/1 haplotype analysis in myeloproliferative neoplasms and acute myeloid leukemia. Leukemia 24, 1809–1813, doi: 10.1038/leu.2010.172 (2010). [DOI] [PubMed] [Google Scholar]
- 129.Trifa AP et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am J Hematol 93, 100–106, doi: 10.1002/ajh.24946 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Tapper W et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun 6, 6691, doi: 10.1038/ncomms7691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Harutyunyan AS et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood 127, 362–365, doi: 10.1182/blood-2015-09-668673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen Y et al. The Polymorphisms in LNK Gene Correlated to the Clinical Type of Myeloproliferative Neoplasms. PLoS One 11, e0154183, doi: 10.1371/journal.pone.0154183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rumi E et al. LNK mutations in familial myeloproliferative neoplasms. Blood 128, 144–145, doi: 10.1182/blood-2016-04-711150 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Saliba J et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet 47, 1131–1140, doi: 10.1038/ng.3380 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Izraeli S The acute lymphoblastic leukemia of Down Syndrome - Genetics and pathogenesis. Eur J Med Genet 59, 158–161, doi: 10.1016/j.ejmg.2015.11.010 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Pastorczak A, Szczepanski T & Mlynarski W Clinical course and therapeutic implications for lymphoid malignancies in Nijmegen breakage syndrome. European journal of medical genetics 59, 126–132, doi: 10.1016/j.ejmg.2016.01.007 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Schutte P et al. Preexisting conditions in pediatric ALL patients: Spectrum, frequency and clinical impact. European journal of medical genetics 59, 143–151, doi: 10.1016/j.ejmg.2015.12.008 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Kirwan M et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet 90, 888–892, doi: 10.1016/j.ajhg.2012.03.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Trevino LR et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 41, 1001–1005, doi: 10.1038/ng.432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; one of the first studies demonstrating the utility of GWAS to identify susceptibility loci in B-ALL
- 140.Vijayakrishnan J et al. Identification of four novel associations for B-cell acute lymphoblastic leukaemia risk. Nature communications 10, 5348, doi: 10.1038/s41467-019-13069-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Karol SE et al. Genetics of ancestry-specific risk for relapse in acute lymphoblastic leukemia. Leukemia, in press, doi: 10.1038/leu.2017.24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang JJ et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 43, 237–241, doi: 10.1038/ng.763 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Holmfeldt L et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45, 242–252, doi: 10.1038/ng.2532 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; genomic analysis of hypodiploid ALL showing constellations of genomic alterations in low hypodiploid and near haploid ALL, and near-universal TP53 mutation in low hypodiploid ALL, approximately half of which are germline
- 144.Noetzli L et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 47, 535–538, doi: 10.1038/ng.3253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; one of the initial studies documenting the syndrome of germline ETV6 mutation, thrombocytopenia and ALL
- 145.Zhang MY et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 47, 180–185, doi: 10.1038/ng.3177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jarviaho T et al. Predisposition to childhood acute lymphoblastic leukemia caused by a constitutional translocation disrupting ETV6. Blood Adv 3, 2722–2731, doi: 10.1182/bloodadvances.2018028795 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rampersaud E et al. Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv 3, 1039–1046, doi: 10.1182/bloodadvances.2018030635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Szczepanski T et al. Late recurrence of childhood T-cell acute lymphoblastic leukemia frequently represents a second leukemia rather than a relapse: first evidence for genetic predisposition. J Clin Oncol 29, 1643–1649, doi: 10.1200/JCO.2010.30.2877 (2011). [DOI] [PubMed] [Google Scholar]; A study focused on the genomic of relapse in T-ALL identifying a case with germline deletions of LMO2, demonstrating that relapse is due to propagation of a second primary leukemia
- 149.Waanders E et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discovery 1, 96–111, doi: 10.1158/0008-5472.Bcd-19-0041 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Qian M et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36, 591–599, doi: 10.1200/JCO.2017.75.5215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Papaemmanuil E et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41, 1006–1010, doi: 10.1038/ng.430 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Urayama KY et al. Regional evaluation of childhood acute lymphoblastic leukemia genetic susceptibility loci among Japanese. Scientific reports 8, 789, doi: 10.1038/s41598-017-19127-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu H et al. Novel susceptibility variants at 10p12.31–12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105, 733–742, doi: 10.1093/jnci/djt042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Migliorini G et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122, 3298–3307, doi: 10.1182/blood-2013-03-491316 (2013). [DOI] [PubMed] [Google Scholar]
- 155.Sherborne AL et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42, 492–494, doi: 10.1038/ng.585 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hungate EA et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nature communications 7, 10635, doi: 10.1038/ncomms10635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vijayakrishnan J & Houlston RS Candidate gene association studies and risk of childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Haematologica 95, 1405–1414, doi: 10.3324/haematol.2010.022095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wiemels JL et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nature communications 9, 286, doi: 10.1038/s41467-017-02596-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ellinghaus E et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 26, 902–909, doi: 10.1038/leu.2011.302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jain N et al. GATA3 rs3824662A Allele Is Overrepresented in Adult Patients with Ph-like ALL, Especially in Patients with CRLF2 Abnormalities. Blood 130, 1430–1430 (2017).28694326 [Google Scholar]
- 161.Qian M et al. Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J Natl Cancer Inst 111, 1350–1357, doi: 10.1093/jnci/djz043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu Y et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49, 1211–1218, doi: 10.1038/ng.3909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Somasundaram R, Prasad MA, Ungerback J & Sigvardsson M Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126, 144–152, doi: 10.1182/blood-2014-12-575688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roberts KG & Mullighan CG The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Cold Spring Harbor perspectives in medicine, doi: 10.1101/cshperspect.a034835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang H et al. Non-coding germline GATA3 variants alter chromatin topology and contribute to pathogenesis of acute lymphoblastic leukemia. bioRxiv, 2020.2002.2023.961672, doi: 10.1101/2020.02.23.961672 (2020). [DOI] [Google Scholar]
- 166.Auer F et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28, 1136–1138, doi: 10.1038/leu.2013.363 (2014). [DOI] [PubMed] [Google Scholar]
- 167.Nutt SL, Heavey B, Rolink AG & Busslinger M Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401, 556–562 (1999). [DOI] [PubMed] [Google Scholar]
- 168.Mullighan CG et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446, 758–764 (2007). [DOI] [PubMed] [Google Scholar]
- 169.Kuiper RP et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 21, 1258–1266, doi:2404691 [pii] 10.1038/sj.leu.2404691 (2007). [DOI] [PubMed] [Google Scholar]
- 170.An Q et al. Variable breakpoints target PAX5 in patients with dicentric chromosomes: a model for the basis of unbalanced translocations in cancer. Proc Natl Acad Sci U S A 105, 17050–17054, doi: 10.1073/pnas.0803494105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bousquet M et al. A novel PAX5-ELN fusion protein identified in B-cell acute lymphoblastic leukemia acts as a dominant negative on wild-type PAX5. Blood 109, 3417–3423, doi:blood-2006-05-025221 [pii] 10.1182/blood-2006-05-025221 (2007). [DOI] [PubMed] [Google Scholar]
- 172.Cazzaniga G et al. The paired box domain gene PAX5 is fused to ETV6/TEL in an acute lymphoblastic leukemia case. Cancer Res 61, 4666–4670 (2001). [PubMed] [Google Scholar]
- 173.Coyaud E et al. Wide diversity of PAX5 alterations in B-ALL: a Groupe Francophone de Cytogenetique Hematologique study. Blood 115, 3089–3097, doi: 10.1182/blood-2009-07-234229 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Kawamata N et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood 111, 776–784, doi: 10.1182/blood-2007-05-088310 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nebral K et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 23, 134–143, doi:leu2008306 [pii] 10.1038/leu.2008.306 (2009). [DOI] [PubMed] [Google Scholar]
- 176.Strehl S, Konig M, Dworzak MN, Kalwak K & Haas OA PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17, 1121–1123, doi: 10.1038/sj.leu.2402923 (2003). [DOI] [PubMed] [Google Scholar]
- 177.Schwab C et al. Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1, 1473–1477, doi: 10.1182/bloodadvances.2017006734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gu Z et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51, 296–307, doi: 10.1038/s41588-018-0315-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; large scale genomic analysis of B-ALL identifying new subtypes driven by diverse PAX5 alterations, including a single mutation defining one subgroup (PAX5P80R)
- 179.Eberhard D, Jimenez G, Heavey B & Busslinger M Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. Embo J 19, 2292–2303 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dang J et al. Pax5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood 125, 3609–3617, doi: 10.1182/blood-2015-02-626127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Malkin D et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238 (1990). [DOI] [PubMed] [Google Scholar]
- 182.Guha T & Malkin D Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harbor perspectives in medicine 7, a026187, doi: 10.1101/cshperspect.a026187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zebisch A et al. Acute myeloid leukemia with TP53 germ line mutations. Blood 128, 2270–2272, doi: 10.1182/blood-2016-08-732610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Link DC et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. Jama 305, 1568–1576, doi: 10.1001/jama.2011.473 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Harrison CJ et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125, 552–559, doi: 10.1111/j.1365-2141.2004.04948.x (2004). [DOI] [PubMed] [Google Scholar]
- 186.Groeneveld-Krentz S et al. Aneuploidy in children with relapsed B-cell precursor acute lymphoblastic leukaemia: clinical importance of detecting a hypodiploid origin of relapse. Br J Haematol 185, 266–283, doi: 10.1111/bjh.15770 (2019). [DOI] [PubMed] [Google Scholar]
- 187.Muhlbacher V et al. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93%. Genes Chromosomes Cancer 53, 524–536, doi: 10.1002/gcc.22163 (2014). [DOI] [PubMed] [Google Scholar]
- 188.Swaminathan M et al. Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb Mol Case Stud 5, doi: 10.1101/mcs.a003210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen Q et al. Multiple functions of Ikaros in hematological malignancies, solid tumor and autoimmune diseases. Gene 684, 47–52, doi: 10.1016/j.gene.2018.10.045 (2019). [DOI] [PubMed] [Google Scholar]
- 190.Georgopoulos K et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell 79, 143–156, doi: 10.1016/0092-8674(94)90407-3 (1994). [DOI] [PubMed] [Google Scholar]
- 191.Mullighan CG et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110–114, doi:nature06866 [pii] 10.1038/nature06866 (2008). [DOI] [PubMed] [Google Scholar]
- 192.Mullighan CG et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360, 470–480, doi: 10.1056/NEJMoa0808253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Den Boer ML et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10, 125–134 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang J et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48, 1481–1489, doi: 10.1038/ng.3691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Martinelli G et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol 27, 5202–5207, doi:JCO.2008.21.6408 [pii] 10.1200/JCO.2008.21.6408 (2009). [DOI] [PubMed] [Google Scholar]
- 196.van der Veer A et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123, 1691–1698, doi: 10.1182/blood-2013-06-509794 (2014). [DOI] [PubMed] [Google Scholar]
- 197.Zaliova M et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28, 182–185, doi: 10.1038/leu.2013.282 (2014). [DOI] [PubMed] [Google Scholar]
- 198.Dorge P et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98, 428–432, doi: 10.3324/haematol.2011.056135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Slayton WB et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J Clin Oncol, Jco2017767228, doi: 10.1200/jco.2017.76.7228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Li JF et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A 115, E11711–e11720, doi: 10.1073/pnas.1814397115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Iacobucci I et al. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: implications for a new mechanism of resistance. Blood 112, 3847–3855, doi: 10.1182/blood-2007-09-112631 (2008). [DOI] [PubMed] [Google Scholar]
- 202.Churchman ML et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 28, 343–356, doi: 10.1016/j.ccell.2015.07.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Joshi I et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol 15, 294–304, doi: 10.1038/ni.2821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Virely C et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia 24, 1200–1204, doi:leu201063 [pii] 10.1038/leu.2010.63 (2010). [DOI] [PubMed] [Google Scholar]
- 205.Boutboul D et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest, doi: 10.1172/jci98164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kuehn HS et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med 374, 1032–1043, doi: 10.1056/NEJMoa1512234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; identification of IKZF1 germline mutations in the region encoding the N-terminal zinc finger as a cause of B-lineage immunodeficiency
- 207.Yoshida N et al. Germline IKAROS mutation associated with primary immunodeficiency that progressed to T-cell acute lymphoblastic leukemia. Leukemia 31, 1221–1223, doi: 10.1038/leu.2017.25 (2017). [DOI] [PubMed] [Google Scholar]
- 208.Hoshino A et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. The Journal of allergy and clinical immunology 140, 223–231, doi: 10.1016/j.jaci.2016.09.029 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Lentaigne C et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood 134, 2070–2081, doi: 10.1182/blood.2019000782 (2019). [DOI] [PubMed] [Google Scholar]
- 210.Hock H & Shimamura A ETV6 in hematopoiesis and leukemia predisposition. Semin Hematol 54, 98–104, doi: 10.1053/j.seminhematol.2017.04.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shurtleff SA et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 9, 1985–1989 (1995). [PubMed] [Google Scholar]
- 212.Raynaud S et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 87, 2891–2899 (1996). [PubMed] [Google Scholar]
- 213.Papaemmanuil E et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 46, 116–125, doi: 10.1038/ng.2874 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Di Paola J & Porter CC ETV6-related thrombocytopenia and leukemia predisposition. Blood 134, 663–667, doi: 10.1182/blood.2019852418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Melazzini F et al. Clinical and pathogenic features of ETV6-related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica 101, 1333–1342, doi: 10.3324/haematol.2016.147496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Paulsson K et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 107, 21719–21724, doi: 10.1073/pnas.1006981107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nishii R et al. Molecular Basis of ETV6-Mediated Predisposition to Childhood Acute Lymphoblastic Leukemia. Blood, doi: 10.1182/blood.2020006164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Poggi M et al. Germline variants in ETV6 underlie reduced platelet formation, platelet dysfunction and increased levels of circulating CD34+ progenitors. Haematologica 102, 282–294, doi: 10.3324/haematol.2016.147694 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Karastaneva A et al. Novel phenotypes observed in patients with ETV6-linked leukaemia/familial thrombocytopenia syndrome and a biallelic ARID5B risk allele as leukaemogenic cofactor. Journal of medical genetics 57, 427–433, doi: 10.1136/jmedgenet-2019-106339 (2020). [DOI] [PubMed] [Google Scholar]
- 220.Bersenev A, Wu C, Balcerek J & Tong W Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest 118, 2832–2844, doi: 10.1172/jci35808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Roberts KG et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371, 1005–1015, doi: 10.1056/NEJMoa1403088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Roberts KG et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22, 153–166, doi: 10.1016/j.ccr.2012.06.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Perez-Garcia A et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood 122, 2425–2432, doi: 10.1182/blood-2013-05-500850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lin M et al. JAK2 p.G571S in B-cell precursor acute lymphoblastic leukemia: a synergizing germline susceptibility. Leukemia 33, 2331–2335, doi: 10.1038/s41375-019-0459-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Waanders E et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia 31, 821–828, doi: 10.1038/leu.2016.277 (2017). [DOI] [PubMed] [Google Scholar]
- 226.Sanda T et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer discovery 3, 564–577, doi: 10.1158/2159-8290.Cd-12-0504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mullighan CG et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–239, doi:nature09727 [pii] 10.1038/nature09727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pasqualucci L et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195, doi: 10.1038/nature09730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.de Smith AJ et al. Predisposing germline mutations in high hyperdiploid acute lymphoblastic leukemia in children. Genes Chromosomes Cancer 58, 723–730, doi: 10.1002/gcc.22765 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Winer P et al. Germline variants in predisposition genes in children with Down syndrome and acute lymphoblastic leukemia. Blood Adv 4, 672–675, doi: 10.1182/bloodadvances.2019001216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pouliot GP et al. Fanconi-BRCA pathway mutations in childhood T-cell acute lymphoblastic leukemia. PLoS One 14, e0221288, doi: 10.1371/journal.pone.0221288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wilson CL et al. Estimated number of adult survivors of childhood cancer in United States with cancer-predisposing germline variants. Pediatr Blood Cancer 67, e28047, doi: 10.1002/pbc.28047 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Oberg JA et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 8, 133, doi: 10.1186/s13073-016-0389-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sud A et al. Analysis of 153 115 patients with hematological malignancies refines the spectrum of familial risk. Blood 134, 960–969, doi: 10.1182/blood.2019001362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Diets IJ et al. High Yield of Pathogenic Germline Mutations Causative or Likely Causative of the Cancer Phenotype in Selected Children with Cancer. Clin Cancer Res 24, 1594–1603, doi: 10.1158/1078-0432.Ccr-17-1725 (2018). [DOI] [PubMed] [Google Scholar]
- 236.Pastor S et al. Optical mapping of the 22q11.2DS region reveals complex repeat structures and preferred locations for non-allelic homologous recombination (NAHR). Scientific reports 10, 12235, doi: 10.1038/s41598-020-69134-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jenko Bizjan B et al. Challenges in identifying large germline structural variants for clinical use by long read sequencing. Comput Struct Biotechnol J 18, 83–92, doi: 10.1016/j.csbj.2019.11.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ritter DI et al. A case for expert curation: an overview of cancer curation in the Clinical Genome Resource (ClinGen). Cold Spring Harb Mol Case Stud 5, a004739, doi: 10.1101/mcs.a004739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Luo X et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv 3, 2962–2979, doi: 10.1182/bloodadvances.2019000644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Mullighan CG The ASH Agenda for Hematology Research: a roadmap for advancing scientific discovery and cures for hematologic diseases. Blood Adv 2, 2430–2432, doi: 10.1182/bloodadvances.2018025403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rehm HL et al. ClinGen--the Clinical Genome Resource. N Engl J Med 372, 2235–2242, doi: 10.1056/NEJMsr1406261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Findlay GM et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222, doi: 10.1038/s41586-018-0461-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Boettcher S et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604, doi: 10.1126/science.aax3649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wu D et al. How I curate: applying American Society of Hematology-Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel rules for RUNX1 variant curation for germline predisposition to myeloid malignancies. Haematologica 105, 870–887, doi: 10.3324/haematol.2018.214221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hahn CN et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 43, 1012–1017, doi: 10.1038/ng.913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hirabayashi S, Wlodarski MW, Kozyra E & Niemeyer CM Heterogeneity of GATA2-related myeloid neoplasms. Int J Hematol 106, 175–182, doi: 10.1007/s12185-017-2285-2 (2017). [DOI] [PubMed] [Google Scholar]
- 247.Dokal I Dyskeratosis congenita in all its forms. Br J Haematol 110, 768–779 (2000). [DOI] [PubMed] [Google Scholar]
- 248.Savage SA & Dufour C Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol 54, 105–114, doi: 10.1053/j.seminhematol.2017.04.004 (2017). [DOI] [PubMed] [Google Scholar]
- 249.Alter BP et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150, 179–188, doi: 10.1111/j.1365-2141.2010.08212.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rusch M et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nature communications 9, 3962, doi: 10.1038/s41467-018-06485-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Howard Sharp KM et al. Factors Associated with Declining to Participate in a Pediatric Oncology Next Generation Sequencing Study. JCO Precis Oncol 4, 202–211, doi: 10.1200/po.19.00213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Berger G et al. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia 31, 520–522, doi: 10.1038/leu.2016.310 (2017). [DOI] [PubMed] [Google Scholar]
- 253.Kobayashi S et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia 31, 1020–1022, doi: 10.1038/leu.2017.44 (2017). [DOI] [PubMed] [Google Scholar]
- 254.Galera P et al. Donor-derived MDS/AML in families with germline GATA2 mutation. Blood 132, 1994–1998, doi: 10.1182/blood-2018-07-861070 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]