

Abstract
Proper cardiac Ca2+ homeostasis is essential for normal excitation–contraction coupling. Perturbations in cardiac Ca2+ handling through altered kinase activity has been implicated in altered cardiac contractility and arrhythmogenesis. Thus, a better understanding of cardiac Ca2+ handling regulation is vital for a better understanding of various human disease processes. ‘Striated muscle preferentially expressed protein kinase’ (SPEG) is a member of the myosin light chain kinase family that is key for normal cardiac function. Work within the last 5 years has revealed that SPEG has a crucial role in maintaining normal cardiac Ca2+ handling through maintenance of transverse tubule formation and phosphorylation of junctional membrane complex proteins. Additionally, SPEG has been causally impacted in human genetic diseases such as centronuclear myopathy and dilated cardiomyopathy as well as in common acquired cardiovascular disease such as heart failure and atrial fibrillation. Given the rapidly emerging role of SPEG as a key cardiac Ca2+ regulator, we here present this review in order to summarize recent findings regarding the mechanisms of SPEG regulation of cardiac excitation–contraction coupling in both physiology and human disease. A better understanding of the roles of SPEG will be important for a more complete comprehension of cardiac Ca2+ regulation in physiology and disease.
Keywords: Atrial fibrillation, Cardiomyopathy, Centronuclear myopathy, Excitation–contraction coupling, Heart failure, JPH2, SERCA2a, Striated muscle preferentially expressed protein kinase, Ryanodine receptor
1. Introduction
The regulation of Ca2+ release and re-uptake into the sarcoplasmic reticulum is a key component of normal excitation–contraction coupling in cardiomyocytes. Altered regulation of Ca2+ handling proteins by kinases and phosphatases has been implicated in the pathogenesis of cardiovascular diseases such as heart failure and atrial fibrillation.1–3 Striated muscle preferentially expressed protein kinase (SPEG) is a member of the obscurin (OBSCN) sub-family of the myosin light-chain kinase (MLCK) family, containing two tandem kinase domains both shown to be catalytically active in the heart.4 Studies within the last 5 years have revealed that SPEG is a key regulator of cardiac Ca2+ regulation within the junctional membrane complex and has been causally implicated in both genetic and acquired cardiovascular disease. Thus, knowledge of SPEG may aid in advancing the broader field of cardiac Ca2+ handling and excitation–contraction coupling. We here present an overview of SPEG's key functional domains and regulations as well as mechanisms of SPEG regulation of cardiovascular Ca2+ homeostasis in both physiology and heart disease.
2. SPEG isoforms
In humans, the striated muscle preferentially expressed gene, also referred to as the SPEG complex locus, is localized to chromosome 2q35. The SPEG gene is comprised of 43 exons and produces four distinct protein variants or isoforms that differ both in structure and composition (Figure 1). The first isoform expressed from the SPEG complex locus, aortic preferentially expressed gene-1 (APEG-1), was reported in 1996 as part of a differential mRNA display performed in arterial smooth muscle cells.5 Subsequent studies revealed that the APEG-1 is one of four isoforms [i.e. SPEGB, SPEGA, APEG-1, and brain preferentially expressed gene (BPEG)] generated from the SPEG complex locus.6 The SPEG complex locus contains two transcriptional start sites with alternative splice variations resulting in multiple isoforms that have both temporal and tissue-specific regulation patterns (Figure 1A).6Tissue-specific expression of these isoforms is likely mediated by the desmin (DES) locus control region, located 5′ of SPEG, which, as its name suggests, also concomitantly regulates expression of DES.7 Thus mechanisms that regulate DES expression in striated and smooth muscle may have similar effects on SPEG expression as the gene expression of both are regulated by the same regions of muscle-specific DNase I hypersensitivity predicted to bind a variety of transcription factors.
Figure 1.

Tissue-specific isoforms of SPEG and functional domains. (A) Diagram of the SPEG complex locus (top) along with the gene regions transcribed for each isoform. Black arrows mark alternative transcription start sites. Red-filled boxes are protein coding sequences/exons. Clear boxes code untranslated regions. Tissue-specific transcripts with exons/sequences included are indicated below the gene diagram. Adult tissues expressing high levels of the transcripts are indicated to the left. (B) Diagram of full-length canonical SPEG protein showing key domains including immunoglobulin, fibronectin III and kinase domains. The lines below the diagram indicate which domains are part of the four SPEG isoforms (SPEGβ, SPEGα, APEG-1, and BPEG) marked below. Scale bar of 200 amino acids (a.a.).
SPEGβ is considered the full-length canonical isoform, and mRNA and protein of SPEGβ are found in both heart and skeletal myocytes in adult muscle.6 Each protein is generated from an 11 kb mRNA transcript and weighs a total of 355 kDa. In comparison, SPEGα, another striated muscle-specific isoform, lacks a portion of the N-terminal sequence of SPEGβ with loss of amino acid residues 1-841. SPEGα is translated from a 9 kb mRNA transcript and weighs around 250 kDa.6 There is an approximate 1:1 ratio of these two isoforms in both cardiac and skeletal muscle.6 Recent studies have identified human single-nucleotide polymorphisms located in close proximity to the SPEG gene associated with increased SPEGβ levels in skeletal muscle.8 The authors reporting these findings did not, however, specify whether these isoforms had an effect on expression of SPEGα. Regardless, these findings may provide clues into the specific DNA regions including enhancer elements important for the regulation of skeletal muscle SPEG levels.
The smaller APEG-1 and BPEG isoforms are preferentially expressed in arterial smooth muscle and brain respectively.6APEG-1 expression has been seen to be inversely correlated with smooth muscle cell dedifferentiation.5 However, in contrast to striated muscle-specific SPEG isoforms, little is yet known of the physiological significance of either of these two isoforms with no brain or vascular phenotype yet detected in Speg germline knockout mouse lines (Table 1).
Table 1.
SPEG knockin and knockout mouse models
| Animal model | Phenotype | Cellular and molecular findings | Ref |
|---|---|---|---|
|
Speg germline knockout Deletion of Exons 8-10 |
Normal baseline in heterozygous knockout but with reduced LV ejection fraction after pressure overload Dilated cardiomyopathy in homozygous knockout 98% death by postnatal day 2 in knockout |
Speg expressed in embryonic atria and ventricle Dysregulation of myofibril and sarcomere structure in knockout Reduced cardiac tropomyosin phosphorylation in knockout |
9 , 10 |
|
Cardiac-specific SPEG conditional knockout SPEGfl/fl (Floxed Exon 9) × αMHC-MerCreMer |
Enhanced atrial fibrillation inducibility 2 weeks post-tamoxifen injection Dilated cardiomyopathy and heart failure with reduced fractional shortening and ejection fraction by 8 weeks post-tamoxifen injection Premature death as early as 4 weeks post-tamoxifen injection with 100% death by 24 weeks |
T-tubule structure disruption Reduced SR Ca2+ load and steady state Ca2+ transient amplitude Reduced SERCA2a activity and increased RyR2 SR Ca2+ Leak Reduced SPEG mediated JPH2, SERCA2a-T484, and RyR2-S2367 phosphorylation |
4 , 11–13 |
|
SPEG3A KI mice SPEG-S2461A-S2462A-T2463A knockin |
Decreased cardiac ejection fraction Left ventricular dilation |
Decreased cardiac SERCA2a activity and oligomerization Reduced SERCA2a-T484 phosphorylation |
11 |
|
Atrial-specific SPEG conditional knockout SPEGfl/fl (Floxed Exon 9) + AAV9-ANF-Cre |
Increased inducibility of atrial fibrillation with rapid atrial pacing |
Increased Ca2+ spark frequency in atrial cardiomyocytes Reduced RyR2-S2367 phosphorylation |
12 |
3. SPEG functional domains
Members of the MLCK family contain a variety of structural protein-binding domains in addition to serine/threonine kinase domains. These domains can work together in order to perform their downstream physiological functions.14 SPEG is similar to other members of the MLCK family in this regard with a variety of immunoglobulin and fibronectin III protein-binding domain. In addition, SPEG contains two serine/threonine kinase domains similar to other members of the OBSCN sub-family of the MLCK branch.
3.1 SPEG serine/threonine kinase domains
SPEG is composed of tandem myosin light chain kinase regions, each bearing a conserved serine-threonine kinase domain.11 Similar to other catalytically active kinases, both domains contain a Asp-Phe-Gly (DFG) motif at the N-terminus of the activation loop as well as several other invariant amino acid residues.14 Mutation of these conserved residues has been successfully performed in order to abolish kinase activity in vitro.11 The first kinase domain located in the central region of the protein—also referred to as SPEG-1—has been shown to phosphorylate junctophilin-2 (JPH2). although the specific serine or threonine residue on JPH2 remains unknown.4,11 The C-terminal kinase domain—referred to also as SPEG-2—phosphorylates cardiac sarco-endoplasmic reticulum ATPase-2a (SERCA2a) at the T484 residue.11 Finally, SPEG has also been shown to phosphorylate the ryanodine receptor type-2 (RyR2) at S2367, but the specific kinase domain involved is not known.12 The physiological role of these phosphorylation events in striated muscle will be discussed later.
The catalytic regions of many kinase domains in the MLCK family, including SPEG-1, are followed by an auto-regulatory domain (ARD) that contains a CaM-binding motif.15 True CaM autoregulated kinase domains help translate increased Ca2+ levels into phosphorylation events resulting in a range of downstream consequences including changes in sarcomeric contractility, ion channel function, and gene transcription.16 However, some of these ARDs may be regulated by separate signalling pathways necessary for cardiovascular function. For instance, the ARD of titin is actually physically removed from the catalytic site through passive stretching of cardiac muscle during diastole, thus allowing increased end-diastolic volume to translated into enhanced contractility as described in the well-known Frank–Starling Law.17 Future research will be needed to determine whether the SPEG-1 catalytic kinase domain is activated through Ca2+/CaM or through an alternative mechanism. Given that in vitro assays reveal that SPEG-1 is capable of autophosphorylation as well as phosphorylation of JPH2 in the absence of Ca2+ or CaM, it appears that Ca2+/CaM binding to the ARD is not necessary for catalytic activity.4,6
In contrast to many other kinase domains of the MLCK family, SPEG-2 does not appear to contain an autoregulatory domain.15 At least in ventricle, SPEG-2 catalytic activity is enhanced by protein kinase B (PKB)-mediated phosphorylation of residues S2461, S2462, and T2463.8 Mutation of these three residues resulted in decreased SERCA2a phosphorylation indicative of decreased SPEG-2 catalytic activity.13 However, mutation of these residues or knock-out of PKB did not result in complete loss of SERCA2a phosphorylation by SPEG, suggesting that PKB phosphorylation may positively regulate SPEG’s catalytic activity, but it is unlikely to be the sole regulator. Phosphoproteomic studies demonstrate phosphorylation by CAMKII (S2135).18 Additionally, tissue-specific mouse proteomics revealed two heart-specific phospho sites for SPEG.19 There are also several sites on SPEG that are hyper-phosphorylated with exercise, although the relevance of these phosphorylation events on protein function remains unknown.20,21
While other tandem kinase domains outside of the MLCK family such as ribosomal S6 kinase-1 have been shown to be important in activating each other,22 no such relationship has been shown for the tandem kinase domains in SPEG or other members of the OBSCN sub-family of the MLCK family. At least in vitro, the two catalytic domains seem to be able to act independently of one another in the phosphorylation of their substrates.11 Thus, further extensive work is required to dissect whether these tandem kinase domains have the ability to activate or inhibit one another in vivo.
3.2 Other structural domains in SPEG
Like most members of the MLCK family, SPEG contains several immunoglobulin-like and fibronectin III domains. These immunoglobulin-like and fibronectin III domains facilitate protein-protein interactions that allow MLCK family proteins to help form a cytoskeletal network and compartmentalize the cell.23 These binding domains can also localize these kinases to specific targets through direct binding events, unlike other kinases that rely on anchoring or scaffolding proteins to localize to their substrates.24 The crystal structure of APEG-1 lacking the 14 N-terminal amino acids revealed a single immunoglobulin domain containing an Arg-Gly-Asp (RGD) adhesion recognition motif that is crucial for interaction with extracellular proteins and cell adhesion.25 This functional domain also mediates a weak dimerization event with the smooth muscle APEG-1 protein isoform in vitro, although it has not been confirmed whether such an interaction occurs in vivo.25 The larger SPEG isoforms also contain multiple immunoglobulin-like and fibronectin III domains. In addition to facilitating protein-protein interactions, the repeat immunoglublin-like domains may provide SPEG with mechanosensitivity with a domain arrangement similar to that seen in the spring-like region of titin.21 However, further experimental work is necessary to identify the precise functions of these different domains.
A common feature of many members of the MLCK family is binding to myosin or actin within the muscle sarcomere.15 However, it is not definitively known whether SPEG is capable of binding either. SPEG localizes to the Z-disc on either side of DES in striated muscle and partially overlaps with α-actinin, but not myosin staining.9,26 SPEG also shows localization with the terminal cisternae, where it binds JPH2, RyR2, and SERCA2a.4,10,11 Recent studies have shown that SPEG also localizes to the intercalated disc and sarcolemma regions in adult cardiomyocytes in a manner similar to obscurin like-1 (OBSL1), a protein made from a gene physically linked to SPEG.27 Given that OBSL1 is almost completely composed of immunoglobulin-like domains, it is likely that SPEG also uses its immunoglobulin-like domains in a similar manner in order to localize within the cell. However, the functional significance of SPEG localizing to the intercalated disc or sarcolemma remains unknown.
4. SPEG as a calcium regulator
Thanks to the creation of various genetic mouse models of SPEG loss (Table 2), our knowledge about the roles in cardiac muscle of the SPEGα and SPEGβ isoforms has substantially improved within the last 10 years. Through these studies, we and others have found that the SPEGα and SPEGβ isoforms play critical roles in regulating cardiomyocyte contractility by means of phosphorylating several Ca2+-handling proteins (Figure 2).4,9,11,12 These phosphorylation events, in turn, directly modulate excitation–contraction coupling and contractility in striated muscle. In addition, SPEG has been shown to directly modulate transverse (T)-tubule stability, thereby contributing to the ultrastructural stability of key Ca2+ signalling domains within cardiomyocytes.4
Table 2.
Clinical characteristics of humans with SPEG mutations
| Patient | Gender/age | Genotype | Skeletal muscle findings | Cardiac findings | Ref |
|---|---|---|---|---|---|
| 1 | Female/died at 3 weeks |
Homozygous c.6697C>T p.G2233* Pathogenic Nonsense |
Severe hypotonia, respiratory insufficiency | No cardiac evaluation | 10 |
| 2 | Female/6 years |
Heterozygous c.3709_3715 + 29del36 p.T1237Sfs*46 Pathogenic Frameshift and c.4276C>T p.R1426* Pathogenic Nonsense |
Severe hypotonia, ophthalmoplegia, facial weakness, tracheostomy for respiratory insufficiency, sat unsupported at 2, unable to walk | Normal cardiac function at birth, at 2 months of age: dilated cardiomyopathy, severe depression of systolic function, left and right ventricular diastolic dysfunction. Drug treatments resulted in normal ventricular function by 1 year of age | 10 |
| 3 | Male/19 months |
Heterozygous c.2915_2916delCCinsA p.A972Dfs*79 Pathogenic Frameshift and c.8270G>T p.G2757V Likely pathogenic Missense |
Severe hypotonia, facial weakness, unsupported sitting at 18 months | Foetal bradycardia, dilated cardiomyopathy at 1 month of age, decreased left ventricular function, mitral insufficiency | 10 |
| 4 | Male/3 years |
Homozygous c.1626_1627insA p.T544Dfs*48 Pathogenic Frameshift |
Severe hypotonia, ophthalmoplegia with mild ptosis, polyphasic motor unit potentials on EMG, unsupported sitting at 12 months, unable to walk | Foetal bradycardia, no cardiomyopathy | 28 |
| 5 | Male/8 years |
Homozygous c.9586C>T p.R3196* Pathogenic Nonsense |
Severe hypotonia, facial weakness, walking with assistance at 3 years, walking independently at 4 years, normal eye movement | Foetal bradycardia, dilated cardiomyopathy, left ventricular ejection fraction 31% and worsening, mild mitral insufficiency | 28 |
| 6 | Female/10 years |
Heterozygous c.1071_1074dup p.K359Vfs*35 Pathogenic Frameshift and c.4399C>T p.R1467* Pathogenic Nonsense |
Hypotonia, positive Gower’s sign, walking independently at 30 months | Reduced myocardial contraction at 5 years that normalized with 1 year of drug treatment, no dilated cardiomyopathy | 29 |
| 7 | Male/died 19 weeks |
Homozygous c.7119C>A p.Y2373* Pathogenic Nonsense |
Floppy infant, no deep tendon reflexes, motor nerve conduction velocity amplitudes of median and peroneal nerve were below normal range (axonal neuropathy), polyphasic motor unit potentials | At 10 weeks fractional shortening of 30% and normal inner diameter of left ventricle, enlarged atria, abnormal trabeculation, intratrabecular recesses as pathognomonic of left ventricular non-compaction (LVNC) | 30 |
| 8 | Male/died 17 years |
Homozygous c.9185_9187delTGG p.V3062del Likely pathogenic In-frame deletion |
Proximal muscle weakness diagnosed at age 4, ophthalmoplegia at 12 | At age 6 biventricular hypertrophy, severe left ventricular dilation, poor muscle contractility, progressive dilated cardiomyopathy: fractional shortening went from 20% at age 10 to 9% at age 16, severe mitral valve insufficiency, died of cardiopulmonary insufficiency | 31 |
| 9 | Female/6.5 years |
Heterozygous c.2183delT p.L728Rfs*82 Pathogenic Frameshift and c.8962_8963insCGGG GCGAACGTTCGTG GCCAAGAT p.V2997Gfs*52 Likely pathogenic Frameshift |
Hypotonia, facial weakness, axial hypotonia, proximal muscle weakness, ophthalmoplegia, bilateral ptosis, intermittent strabismus, walking at 2 years | Sinus tachycardia, no signs of contractile dysfunction | 31 |
| 10 | Female/died 3 days (Twin of P11) |
Homozygous c.8710A>G p.T2904A Uncertain significance Missense |
Facial weakness, ptosis, respiratory insufficiency, hypotonia, axial muscle weakness | Sinus tachycardia, right atrium abnormality, dilated cardiomyopathy | 32 |
| 11 | Female/died 5 days (Twin of P10) |
Homozygous c.8710A>G p.T2904A Uncertain significance Missense |
Facial weakness, ptosis, respiratory insufficiency, hypotonia, axial muscle weakness | Sinus tachycardia, right atrium abnormality, dilated cardiomyopathy | 32 |
Figure 2.
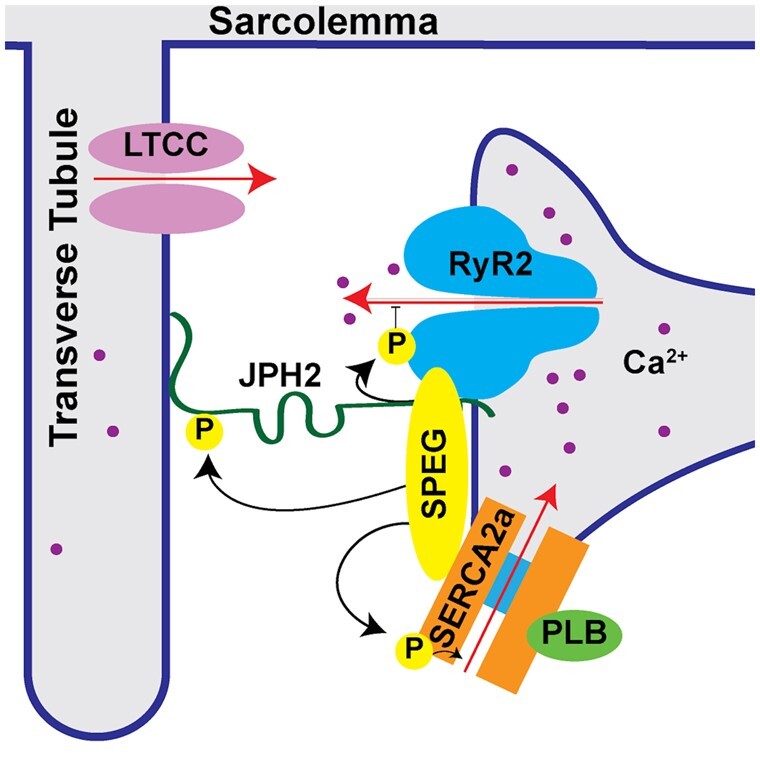
Subcellular organization of SPEG and its phosphorylation targets in cardiac muscle. SPEG phosphorylation of junctophilin-2 (JPH2) in cardiac muscle plays a role in T-tubule formation and stabilization. SPEG phosphorylation of RyR2 and SERCA2a in cardiac muscle modulates sarcoplasmic reticulum Ca2+ handling.
4.1 SPEG regulation of junctional membrane complex formation
In adult mice, induced cardiomyocyte-specific knockout of Speg results in a striking loss of T-tubules and JMCs.4 In cardiomyocytes, JMCs (dyads) are subcellular microdomains, in which voltage-gated L-type Ca2+ channels on the sarcolemma are held in close apposition to RyR2/SR Ca2+-release channels on the SR.33Cardiac-specific Speg knockout mice also exhibit decreased Ca2+-transient amplitudes indicative of reduced systolic SR Ca2+ release along with reduced SR Ca2+ load, both of which contribute to decreased contractility.4 Although cardiac-specific Speg knockout mice develop heart failure within 4 weeks of knockout induction, the JMC disruptions and Ca2+-handling alterations develop before any signs of cardiac failure manifest.4 These findings strongly suggest that loss of SPEG in adult cardiomyocytes is causally linked to the aforementioned JMC and Ca2+-handling changes, and are not secondary due to heart failure. Similar findings were reported in subsequent studies performed in skeletal muscle of mice with skeletal-muscle-specific SPEG knockout.34 SPEG knockout resulted in loss of JMCs (triads) in skeletal muscle as well as reduced SR Ca2+ release during contraction. Taken together, these results suggest that loss of SPEG is sufficient to cause disruption of striated muscle JMCs, thus leading to reduced SR Ca2+ release and impaired striated muscle contractility.
In cardiomyocytes, loss of JMCs and T-tubules has been possibly attributed to reduced SPEG-mediated phosphorylation of JPH2.4 JPH2 is a membrane-spanning structural protein that binds the sarcolemma at its N-terminus and the SR with its C-terminal transmembrane segment.35 Using cardiac-specific short hairpin RNA-mediated JPH2 knockdown in mice, we previously showed that loss of JPH2 during cardiac development prevents the development of mature T-tubules in cardiomyocytes.36 Subsequently, Quick et al.4 demonstrated that reduced phosphorylation of JPH2 in SPEG knockdown mice—despite unaltered levels of JPH2 protein—correlated with a loss of T-tubules and JMCs. Additional studies are needed to identify the exact residue(s) on JPH2 that are subject to SPEG phosphorylation and to determine whether phosphorylation of such residue(e) are essential for T-tubule/JMC stability. Interestingly, recent studies suggest that another type of post-translational modification (S-palmitoylation) of JPH2 is also critical for its role in tethering the SR to the plasma membrane.37 However, it is unknown whether S-palmitoylation of JPH2 affects binding to SPEG.
4.2 SPEG kinase regulation of SR Ca2+ handling proteins
In addition to affecting sarcomere and JMC structure, SPEG can directly affect excitation–contraction coupling by phosphorylating key Ca2+-handling proteins. Excitation–contraction coupling is the process by which an electrical, depolarizing signal propagates down T-tubules to activate L-type Ca2+-channels, whereby the subsequent Ca2+ influx activates SR Ca2+ release via RyR2, thereby causing contraction of the myofilaments.33 During relaxation of the cardiomyocyte, Ca2+ is pumped back into the SR via SERCA2a or extruded from the cell via the Na+/Ca2+-exchanger.
SPEG was identified as a major regulator of SR Ca2+ handling in unbiased proteomic analyses of JPH2 and RyR2 binding partners in adult mouse hearts.4 In this study, SPEG emerged as the only protein that binds to both JPH2 and RyR2 under stringent co-immunoprecipitation conditions. RyR2 is the main SR Ca2+-release channel responsible for excitation–contraction coupling.16,33 Interestingly, SPEG is capable of binding to RyR2 using the N-terminal portion of SPEG that is unique to SPEGβ.4 These findings suggest that SPEGα, which does not have the N-terminal region needed for RyR2 binding, probably does not regulate RyR2, although this has yet to be experimentally verified. Cardiomyocyte-specific SPEG conditional knockout mice develop increased Ca2+ spark frequency indicative of aberrant RyR2 activity.4 This increase in Ca2+ spark frequency was not seen by Quan et al.11, which may be caused by the fact that they measured Ca2+ fluxes in non-paced cardiomyocytes, which might interfere with normal SR Ca2+ homeostasis. An additional limitation to these studies is that non-spark mediated RyR2 leak was not measured through pharmacological tetracaine or dantrolene protocols.38
Recently, we identified serine 2367 (S2367) as the SPEG-phosphorylation site on RyR2.12 Genetic knockout of SPEG caused reduced S2367 phosphorylation on RyR2. Campbell et al.12 generated knock-in mice in which the S2367 site was genetically inactivated by mutating the residue to alanine. Complete inactivation of S2367 on RyR2 leads to enhanced RyR2 activity manifesting as an increase in SR Ca2+-spark frequency under diastolic conditions.12 These findings suggest that the S2367 phosphorylation site on RyR2 acts as an inhibitory site, which makes the SPEG-phosphorylation site unique among RyR2 site phosphorylated by other kinase (e.g. protein kinase A, Ca2+/calmodulin-dependent protein kinase II) that enhance RyR2 activity upon phosphorylation (Figure 3).39–47 Understanding how this phosphorylation site interacts with other binding partners and post-translational modifications on RyR2, as well as identifying the phosphatases with their corresponding regulatory subunits that regulate this site, will be important for establishing a more complete understanding of RyR2 dynamics.
Figure 3.
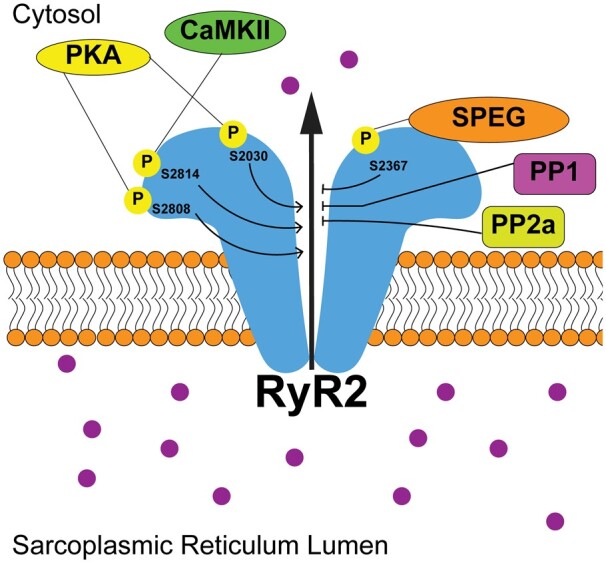
SPEG phosphorylation of RyR2-S2367 inhibits diastolic Ca2+ leak. While CaMKII phosphorylation (P) of S2814 and PKA phosphorylation of S2808 and S2030 have been shown to enhance RyR2 diastolic opening, enhanced targeting by protein phosphatase 1 and 2 (PP1 and PP2) along with phosphorylation of S2367 by SPEG inhibit diastolic Ca2+ leak.
SPEG also phosphorylates SERCA2a. Quan et al.11 reported that SPEG phosphorylation of SERCA2a enhances SR Ca2+-reuptake activity. SPEG knockout in adult mouse cardiomyocytes resulted in a significant decrease in SERCA2a activity.11In vitro studies demonstrated that the SPEG-2 kinase domain was sufficient to phosphorylate a peptide containing SERCA2a-T484. Additionally, SPEG-knockout mice had decreased SERCA2a-T484 phosphorylation. In silico studies also demonstrated that SPEG was able to increase oligomerization of SERCA2a and that this was mediated through SPEG phosphorylation of SERCA2a-T484.11 Loss of SPEG levels resulting in enhanced SERCA2a activity was not seen in SPEG cKO mice by Quick et al.4 However, this could be potentially explained by the smaller sample sizes that might fail to detect existent differences. Differences in both genetic background and tamoxifen dosing also resulted in mice having a more rapid onset of heart failure in Quick et al.4 compared to Quan et al.11 Future studies in SERCA-T484 phospho-resistant or phospho-mimetic mouse lines are required to definitely validate the precise role of SERCA2a-T484 phosphorylation by SPEG in the regulation of ventricular cardiomyocyte Ca2+ homeostasis.
Interestingly, Alsina et al.39 recently reported that the RyR2/SR Ca2+-release channel and the SERCA2a/phospholamban/Ca2+ reuptake transporter are part of a macromolecular protein super-complex. It was shown that a protein phosphatase type-1 regulatory subunit type-3A plays a key role in organizing these SR macromolecular complexes.39 Given that SPEG binds to both RyR2 and SERCA2a,11,12 it may also play a role in the structural organization and regulation of SR Ca2+-handling protein complexes.
5. Human diseases caused by SPEG mutations
Inherited compound heterozygous or homozygous loss-of-function mutations in SPEG have been shown to cause centronuclear myopathy (CNM), a form of congenital myopathy marked by muscle weakness and centralized skeletal muscle nuclei on histological examination.48 In addition, many affected patients may also have concomitant dilated cardiomyopathy or other cardiac components.48 CNM can also be caused by mutations in other genes such as myotubularin (MTM1), bridging integrator-1, or dynamin-2, genes that all encode proteins with important roles in T-tubule formation.49
The first three human SPEG mutations associated with CNM and dilated cardiomyopathy were reported by Agrawal et al.10 in 2014. Since then, a total of 11 patients with disease-causing SPEG mutations have been reported (Figure 4).32 Patient demographics, family history, clinical symptoms, and cellular/molecular findings in patient muscle biopsies are summarized in Table 2. Most patients with SPEG mutations suffered from hypotonia and skeletal muscle weakness, as expected for those afflicted by CNM as well as from cardiac abnormalities, including dilated cardiomyopathy, bradycardia, and eventually heart failure (Table 2). While seven mutations suffered from homozygous autosomal-recessive SPEG mutations, four patients exhibited compound heterozygous mutations. Three of the 15 mutations are likely pathogenic with 11 being considered pathogenic variants. One mutation in patients 10 and 11- both twins homozygous for the same mutation - is classified as a variant of unknown significance.32 However, given the close approximation of the mutation to a critical functional domain and the fact that both twins had severe disease consistent with CNM, it is still possible that the mutation is disease causing. Interestingly, 11 out of 15 mutations cause truncated mutations through either nonsense or frameshift mutations. Therefore, it is believed that the striated muscle phenotype is generally caused by a loss-of-function mutation phenotype.
Figure 4.

Localization of SPEG mutation associated with human disease. Diagram showing SPEG mutations identified in patients (P) 1-11 reported to date (see Table 2) relative to the functional protein domains. All protein domains are drawn to scale. *Nonsense mutation. Fs, frameshift.
Patients with variants in the N-terminal part of the SPEG locus that is unique to SPEGβ generally did not develop a cardiomyopathy.28,29,31 As the SPEGβ isoform is driven by an alternative promoter, it is predicted that these patients will at least have some amount of SPEGα. Thus, it appears that SPEGβ and SPEGα may have a great deal of redundancy in cardiac muscle function, although this needs to be experimentally validated. Additionally, some patients had mutations within the SPEG-2 kinase domain.32 With the exception of one patient, who was compound heterozygous for another N-terminal mutation that only affects SPEGβ, these patients had primarily a cardiomyopathic phenotype.32 However, these patients had no or a relatively mild skeletal myopathy, suggesting that proteins phosphorylated by kinase domain SPEG-2 play a more important role in cardiac muscle cells.
The causality of SPEG loss in the pathogenesis of myopathy and dilated cardiomyopathy has been established using various SPEG mouse models. Germline Speg knockout mice exhibit increased skeletal myocyte central nuclei reflective of findings in CNM-affected individuals.10 Moreover, germline Speg knockout mice also develop severe neonatal dilated cardiomyopathy with a nearly 100% fatality rate in pups.9 While it is likely that frameshift mutations cause reduced SPEG levels and a loss-of-function phenotype,10 it is still unclear how SPEG missense mutations lead to human disease.
6. Altered SPEG expression in heart failure
Heart failure is a clinical syndrome characterized by symptoms and signs resulting from the inability of the heart to meet the metabolic demands of the body.50 It affects over 6.5 million people older than 20 years of age in the USA alone.51 Current treatment methods are inadequate with a 5-year survival of only 42.3%, which in part reflects a poor understanding of the molecular origin of this disease.52
In heart tissue samples from patients with heart failure with a reduced ejection fraction, SPEG mRNA levels were greatly decreased compared in comparison to patients with non-failing hearts.4 In experimental mouse models of heart failure induced by isoproterenol or transverse aortic constriction, SPEG mRNA levels were also reduced.11 A reduction in cardiac SPEG levels appears to be causally linked to the development of heart failure. In studies by two separate labs, tamoxifen-inducible cardiomyocyte-specific SPEG knockout in mice induced the development of severe dilated cardiomyopathy within weeks resulting in significantly decreased survival.4,11 The loss of SPEG within cardiomyocytes causes a major loss of T-tubules and JMCs, possibly in part due to hypo-phosphorylation of JPH2.4 In addition, enhanced RyR2-mediated SR Ca2+ leak and reduced SERCA2a activity lead to aberrant SR Ca2+ handling and reduced myocyte contractility further contributing to the development of heart failure.4,11
Finally, studies in heterozygous SPEG knockout mice revealed the absence of a baseline cardiac phenotype suggesting that reductions in SPEG levels up to at least 50% are tolerated.53 However, following the induction of pressure overload induced by transverse aortic constriction, heterozygous SPEG mice exhibited a significant increase in fibrosis and chamber dilation as well as a decrease in ejection fraction in comparison to wild-type control mice. Heterozygous SPEG knockout mice also exhibited more profound T-tubule and JMC disruption after transverse aortic constriction relative to controls.53 Thus, mildly reduced SPEG levels may predispose to exacerbated heart failure development, while severe reductions in SPEG levels may be sufficient to cause heart failure without additional stressors. Taken together, these studies suggest a causative role for decreased SPEG levels or activity in human heart failure.
7. Altered SPEG function in atrial fibrillation
Atrial fibrillation is the most frequently diagnosed cardiac arrhythmia affecting between 3 and 6 million people in the USA alone.54 Because the number of atrial fibrillation cases dramatically increase with age and women generally live longer than men, more women than men suffer from atrial fibrillation. Atrial fibrillation is more common among patients suffering from obesity, hypertension, diabetes heart failure, and chronic kidney disease.55,56 Several mechanisms contribute to the development of atrial fibrillation, including electrical remodelling, structural remodelling, and inflammatory signalling.57–59 Ca2+-handling abnormalities have been identified in patients with either paroxysmal (early stage) or persistent (more advanced) atrial fibrillation, although the underlying molecular mechanisms are quite distinct.60–62
Recently, Campbell et al.12 reported than reduced SPEG levels may play a role in atrial fibrillation pathogenesis. Patients with paroxysmal atrial fibrillation exhibited reduced SPEG protein levels in right atrial biopsies compared to patients in sinus rhythm.12 Interestingly, SPEG levels were unchanged in chronic atrial fibrillation, indicating that it is unlikely that this is secondary to atrial fibrillation induced atrial remodelling changes.12 To determine whether decreased SPEG levels could be sufficient to enhance the susceptibility to atrial fibrillation, atrial SPEG knockout mice were developed using an atrial-specific adeno-associated virus 9 used to deliver Cre to SPEG-floxed mice (Table 1).12,63 Atrial SPEG knockout mice were more susceptible to atrial fibrillation induction using programmed electrical stimulation, suggesting that reduced SPEG levels generate a substrate for atrial arrhythmia formation.
Previous studies revealed evidence for increased SR Ca2+ leak and SR Ca2+ load in patients with paroxysmal atrial fibrillation.64 Abnormal SR Ca2+ leak has been attributed to enhanced RyR2 protein expression and reduced protein phosphatase-1 regulatory subunit levels, independent of changes in protein kinase A or Ca2+/calmodulin-dependent protein kinase II phosphorylation of RyR2 at S2808 and S2814, respectively.39,61,64 Atrial cardiomyocytes from atrial SPEG-knockout mice exhibited an increase in SR Ca2+ leak evidenced by an enhanced Ca2+-spark frequency. Moreover, reduced SPEG protein levels were associated with reduced RyR2-S2367 phosphorylation levels in patients with paroxysmal atrial fibrillation.12 In contrast, phosphorylation levels of other, well-characterized RyR2 phosphorylation sites S2808 and S2814 were not altered, consistent with previous publications.60,61 Thus, a reduction in S2367 phosphorylation might be a unique marker of RyR2 dysfunction in paroxysmal atrial fibrillation patients.
Studies in knock-in mouse models, in which the S2367 phosphorylation site was genetically ablated, revealed that reduced SPEG phosphorylation of RyR2 predisposes mice to atrial fibrillation induction.12 Conversely, knockin mice with a constitutively phosphorylated S2367 residue due to mutation S2367D were protected against atrial fibrillation induction after a carbachol challenge.61Hypo-phosphorylation of S2367 in atrial cardiomyocytes from RyR2-S2367A mice led to an increased number of spontaneous SR Ca2+ sparks and Ca2+ waves, consistent with enhanced cellular triggered activity. Confocal imaging studies of Ca2+ spark latency times65 revealed a leftward shift in spark-to-spark delay time in atrial cardiomyocytes from S2367A mice, validating that RyR2 single-channel activity is enhanced due to a loss of S2367 phosphorylation.12 While these studies provide strong evidence that reduced SPEG levels and reduced SPEG phosphorylation of RyR2 contribute to aberrant SR Ca2+ handling and the evolution of an atrial fibrillation-promoting substrate, extensive additional work is needed to assess the impact on other SPEG targets (i.e. JPH2, SERCA2a) and their potential roles in atrial fibrillation pathogenesis.
8. Conclusions
SPEG is a member of the myosin light chain kinase family critical for cardiac muscle Ca2+ handling.12,32,34 Genetic mutations in SPEG have been linked to a clinically heterogeneous condition known as CNM that can include congenital myopathy and dilated cardiomyopathy, depending on the mutation.10,28,29,31,32 Reduced protein levels of SPEG have also been found as potential contributors to the development of heart failure and atrial fibrillation.4,12
Disease modelling of SPEG alterations in mice has provided deep insights into the mechanisms by which SPEG alterations could cause cardiac pathology. Cardiac-restricted SPEG knockout mice develop dilated cardiomyopathy and premature death, suggesting a causal relationship between low SPEG levels and heart failure.4 In addition, atrial-specific SPEG knockdown caused an increased susceptibility to atrial fibrillation, suggesting that loss of SPEG could create a pro-arrhythmogenic substrate.12 SPEG has several functions in cardiac muscle, including stabilization of T-tubules and JMCs, regulation of SR Ca2+ release via RyR2, and modulation of SR Ca2+ reuptake via SERCA2a.4,11,12 Additionally, given SPEG's localization to the sarcolemma outside of the JMC,12 it will be important to examine whether SPEG has additional targets in these regions of the cardiomyocyte. It is not yet known what the consensus substrate recognition motif is for either of the SPEG kinase domains. However, identification of further substrates will aid in this endeavour.
Protein kinases serve as promising pharmacological targets for small molecule inhibitors or activators.66 Thus, a further refinement of our understanding of SPEG function is expected to foster the development of novel therapeutic strategies for frequent but currently sub-optimally treated diseases like heart failure and atrial fibrillation. Future work will also need to elucidate whether there are differences in the precise mechanisms by which SPEG causes atrial fibrillation vs. heart failure. It may be that SPEG is more important as a T-tubule regulator in the ventricle while SPEG phosphorylation of RyR2 may be more critical in the atria. The mechanisms of SPEG down-regulation may also be different between the two disease processes.
SPEGβ and SPEGα are both expressed in cardiac muscle. However, future work will be needed to test whether there are roles unique to these different isoforms. Patients with human mutations that only affect the region unique to SPEGβ with SPEGα function preserved appear to have a milder cardiomyopathy phenotype suggesting some overlap in function.32 However, immunoprecipitation data to date suggests that it is only the SPEGβ isoform that binds RyR2,4 but it is not yet known whether SPEGα can still complex or phosphorylate RyR2 without directly binding the channel.
Over the last 5 years, SPEG has emerged as a key regulator of cardiac Ca2+ homeostasis. It will be important to examine how SPEG interacts with other novel regulators of sarcoplasmic reticulum Ca2+ release as the field of excitation–contraction coupling continues to progress. Additionally, SPEG may have additional kinase targets as well as structural roles that will need to be better understood in order to translate our knowledge into treatment for patients with cardiovascular disease.
Acknowledgements
We would like to acknowledge Brian Martin for useful revisions of this manuscript. We also wish to acknowledge the Medical Scientist Training Program at Baylor College of Medicine, Houston, TX.
Conflict of interest: X.H.T.W. is a founding partner of Elex Biotech, a start-up company that developed drug molecules to target ryanodine receptors for treatment of cardiac arrhythmias. Other authors have no conflicts related to this study.
Funding
H.C. was funded by the National Institutes of Health (NIH) F30 fellowship HL140782. A.P.Q. was funded by the American Heart Association predoctoral fellowship 14PRE20490083, and NIH T32 training grant HL007676. D.D. was funded by the NIH grants R01-HL131517, R01-HL136389, and R01-HL089598 and the German Research Foundation (DFG) grant Do 769/4-1. X.W. was funded through NIH grants HL089598, HL091947, HL117641, and HL147108.
Data availability
No new data was generated or analyzed in support of this article.
References
- 1.Heijman J, Dewenter M, El-Armouche A, Dobrev D.. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol 2013;64:90–98. [DOI] [PubMed] [Google Scholar]
- 2.Vlahos CJ, McDowell SA, Clerk A.. Kinases as therapeutic targets for heart failure. Nat Rev Drug Discov 2003;2:99–113. [DOI] [PubMed] [Google Scholar]
- 3.Goette A, Lendeckel U, Klein HU.. Signal transduction systems and atrial fibrillation. Cardiovasc Res 2002;54:247–258. [DOI] [PubMed] [Google Scholar]
- 4.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A, Wehrens XH.. SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res 2017;120:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh CM, Yoshizumi M, Endege WO, Kho CJ, Jain MK, Kashiki S, de los Santos R, Lee WS, Perrella MA, Lee ME.. APEG-1, a novel gene preferentially expressed in aortic smooth muscle cells, is down-regulated by vascular injury. J Biol Chem 1996;271:17354–17359. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh CM, Fukumoto S, Layne MD, Maemura K, Charles H, Patel A, Perrella MA, Lee ME.. Striated muscle preferentially expressed genes alpha and beta are two serine/threonine protein kinases derived from the same gene as the aortic preferentially expressed gene-1. J Biol Chem 2000;275:36966–36973. [DOI] [PubMed] [Google Scholar]
- 7.Tam JL, Triantaphyllopoulos K, Todd H, Raguz S, de Wit T, Morgan JE, Partridge TA, Makrinou E, Grosveld F, Antoniou M.. The human desmin locus: gene organization and LCR-mediated transcriptional control. Genomics 2006;87:733–746. [DOI] [PubMed] [Google Scholar]
- 8.Kusić D, Connolly J, Kainulainen H, Semenova EA, Borisov OV, Larin AK, Popov DV, Generozov EV, Ahmetov II, Britton SL, Koch LG, Burniston JG.. Striated muscle-specific serine/threonine-protein kinase beta segregates with high versus low responsiveness to endurance exercise training. Physiol Genomics 2020;52:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Ramjiganesh T, Chen YH, Chung SW, Hall SR, Schissel SL, Padera RF, Liao R, Ackerman KG, Kajstura J, Leri A, Anversa P, Yet SF, Layne MD, Perrella MA.. Disruption of striated preferentially expressed gene locus leads to dilated cardiomyopathy in mice. Circulation 2009;119:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloğlu G, Talim B, Yau KS, Allcock RJ, Laing NG, Perrella MA, Beggs AH.. SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet 2014;95:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY, Chen S.. SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ Res 2019;124:712–726. [DOI] [PubMed] [Google Scholar]
- 12.Campbell H, Quick AP, Abu-Taha I, Chiang DY, Kramm CF, Word TA, Brandenburg S, Hulsurkar M, Alsina KM, Liu H, Martin B, Uhlenkamp D, Moore OM, Lahiri SK, Corradini E, Kamler M, Heck AJR, Lehnart SE, Dobrev D, Wehrens XHT.. Loss of SPEG inhibitory phosphorylation of RyR2 promotes atrial fibrillation. Circulation 2020;142:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quan C, Du Q, Li M, Wang R, Ouyang Q, Su S, Zhu S, Chen Q, Sheng Y, Chen L, Wang H, Campbell DG, MacKintosh C, Yang Z, Ouyang K, Wang HY, Chen S.. A PKB-SPEG signaling nexus links insulin resistance with diabetic cardiomyopathy by regulating calcium homeostasis. Nat Commun 2020;11:2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temmerman K, Simon B, Wilmanns M.. Structural and functional diversity in the activity and regulation of DAPK-related protein kinases. FEBS J 2013;280:5533–5550. [DOI] [PubMed] [Google Scholar]
- 15.Gautel M.Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch 2011;462:119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bers DM.Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 2008;70:23–49. [DOI] [PubMed] [Google Scholar]
- 17.Puchner EM, Alexandrovich A, Kho AL, Hensen U, Schäfer LV, Brandmeier B, Gräter F, Grubmüller H, Gaub HE, Gautel M.. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci USA 2008;105:13385–13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scholten A, Preisinger C, Corradini E, Bourgonje VJ, Hennrich ML, van Veen TA, Swaminathan PD, Joiner ML, Vos MA, Anderson ME, Heck AJ.. Phosphoproteomics study based on in vivo inhibition reveals sites of calmodulin-dependent protein kinase II regulation in the heart. J Am Heart Assoc 2013;2:e000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP.. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010;143:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo H, Isserlin R, Emili A, Burniston JG.. Exercise-responsive phosphoproteins in the heart. J Mol Cell Cardiol 2017;111:61–68. [DOI] [PubMed] [Google Scholar]
- 21.Potts GK, McNally RM, Blanco R, You JS, Hebert AS, Westphall MS, Coon JJ, Hornberger TA.. A map of the phosphoproteomic alterations that occur after a bout of maximal-intensity contractions. J Physiol 2017;595:5209–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards SA, Fu J, Romanelli A, Shimamura A, Blenis J.. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr Biol 1999;9:810–820. [DOI] [PubMed] [Google Scholar]
- 23.Barclay AN.Membrane proteins with immunoglobulin-like domains—a master superfamily of interaction molecules. Semin Immunol 2003;15:215–223. [DOI] [PubMed] [Google Scholar]
- 24.Lester LB, Scott JD.. Anchoring and scaffold proteins for kinases and phosphatases. Recent Prog Horm Res 1997;52:409–429; discussion 429–430. [PubMed] [Google Scholar]
- 25.Manjasetty BA, Niesen FH, Scheich C, Roske Y, Goetz F, Behlke J, Sievert V, Heinemann U, Büssow K.. X-ray structure of engineered human Aortic Preferentially Expressed Protein-1 (APEG-1). BMC Struct Biol 2005;5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huxley AF.Cross-bridge action: present views, prospects, and unknowns. J Biomech 2000;33:1189–1195. [DOI] [PubMed] [Google Scholar]
- 27.Geisler SB, Robinson D, Hauringa M, Raeker MO, Borisov AB, Westfall MV, Russell MW.. Obscurin-like 1, OBSL1, is a novel cytoskeletal protein related to obscurin. Genomics 2007;89:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Castiglioni C, Kaçar Bayram A, Fattori F, Pekuz S, Araneda D, Per H, Erazo R, Gümüş H, Zorludemir S, Becker K, Ortega X, Bevilacqua JA, Bertini E, Cirak S.. Insights from genotype-phenotype correlations by novel SPEG mutations causing centronuclear myopathy. Neuromuscul Disord 2017; 27:836–842. [DOI] [PubMed] [Google Scholar]
- 29.Lornage X, Sabouraud P, Lannes B, Gaillard D, Schneider R, Deleuze JF, Boland A, Thompson J, Böhm J, Biancalana V, Laporte J.. Novel SPEG mutations in congenital myopathy without centralized nuclei. J Neuromuscul Dis 2018;5:257–260. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Schänzer A, Kampschulte B, Daimagüler HS, Logeswaran T, Schlierbach H, Petzinger J, Ehrhardt H, Hahn A, Cirak S.. A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol Commun 2018;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qualls AE, Donkervoort S, Herkert JC, D'gama AM, Bharucha-Goebel D, Collins J, Chao KR, Foley AR, Schoots MH, Jongbloed JDH, Bönnemann CG, Agrawal PB.. Novel SPEG mutations in congenital myopathies: genotype-phenotype correlations. Muscle Nerve 2019;59:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang J, Ma W, Chen Y, Jiang R, Zeng Q, Tan J, Jiang H, Li Q, Zhang VW, Wang J, Tang H, Luo L.. Novel SPEG variant cause centronuclear myopathy in China. J Clin Lab Anal 2020;34:e23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landstrom AP, Dobrev D, Wehrens XHT.. Calcium signaling and cardiac arrhythmias. Circ Res 2017;120:1969–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huntoon V, Widrick JJ, Sanchez C, Rosen SM, Kutchukian C, Cao S, Pierson CR, Liu X, Perrella MA, Beggs AH, Jacquemond V, Agrawal PB.. SPEG-deficient skeletal muscles exhibit abnormal triad and defective calcium handling. Hum Mol Genet 2018;27:1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeshima H, Hoshijima M, Song LS.. Ca2+ microdomains organized by junctophilins. Cell Calcium 2015;58:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, Landstrom AP, Song LS, Ackerman MJ, Wehrens XH.. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res 2013;100:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang M, Hu J, White FKH, Williamson J, Klymchenko AS, Murthy A, Workman SW, Tseng GN.. Palmitoylation of junctophilin-2 is critical for its role in tethering the sarcoplasmic reticulum to the plasma membrane. J Biol Chem 2019;294:13487–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Ríos E, Shannon TR.. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J 2010;98:2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alsina KM, Hulsurkar M, Brandenburg S, Kownatzki-Danger D, Lenz C, Urlaub H, Abu-Taha I, Kamler M, Chiang DY, Lahiri SK, Reynolds JO, Quick AP, Scott L, Word TA, Gelves MD, Heck AJR, Li N, Dobrev D, Lehnart SE, Wehrens XHT.. Loss of protein phosphatase 1 regulatory subunit PPP1R3A promotes atrial fibrillation. Circulation 2019; 140:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR.. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003;113:829–840. [DOI] [PubMed] [Google Scholar]
- 41.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR.. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA 2006;103:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wehrens XH, Lehnart SE, Reiken SR, Marks AR.. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004;94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 43.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH.. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 2009;119:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH.. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 2010;122:2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Oort RJ, Respress JL, Li N, Reynolds C, De Almeida AC, Skapura DG, De Windt LJ, Wehrens XH.. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension 2010;55:932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR.. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000;101:365–376. [DOI] [PubMed] [Google Scholar]
- 47.Potenza DM, Janicek R, Fernandez-Tenorio M, Camors E, Ramos-Mondragón R, Valdivia HH, Niggli E.. Phosphorylation of the ryanodine receptor 2 at serine 2030 is required for a complete β-adrenergic response. J Gen Physiol 2019;151:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tasfaout H, Cowling BS, Laporte J.. Centronuclear myopathies under attack: a plethora of therapeutic targets. J Neuromuscul Dis 2018;5:387–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cassandrini D, Trovato R, Rubegni A, Lenzi S, Fiorillo C, Baldacci J, Minetti C, Astrea G, Bruno C, Santorelli FM; Italian Network on Congenital Myopathies. Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr 2017;43:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; ESC Scientific Document Group. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 51.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2018 update: a report from the American heart association. Circulation 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 52.Connell P, Word TA, Wehrens XHT.. Targeting pathological leak of ryanodine receptors: preclinical progress and the potential impact on treatments for cardiac arrhythmias and heart failure. Expert Opin Ther Targets 2020;24:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shu C, Huang H, Xu Y, Rota M, Sorrentino A, Peng Y, Padera RF, Huntoon V, Agrawal PB, Liu X, Perrella MA.. Pressure overload in mice with haploinsufficiency of striated preferentially expressed gene leads to decompensated heart failure. Front Physiol 2018;9:863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ.. Worldwide epidemiology of atrial fibrillation: a global burden of disease 2010 study. Circulation 2014;129:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heijman J, Voigt N, Nattel S, Dobrev D.. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 2014;114:1483–1499. [DOI] [PubMed] [Google Scholar]
- 56.Molina CE, Abu-Taha IH, Wang Q, Roselló-Díez E, Kamler M, Nattel S, Ravens U, Wehrens XHT, Hove-Madsen L, Heijman J, Dobrev D.. Profibrotic, electrical, and calcium-handling remodeling of the atria in heart failure patients with and without atrial fibrillation. Front Physiol 2018;9:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nattel S, Dobrev D.. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol 2016;13:575–590. [DOI] [PubMed] [Google Scholar]
- 58.Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S.. The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 2016;109:467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha I, Ghezelbash S, Reynolds CL, Shen YH, LeMaire SA, Schmitz W, Müller FU, El-Armouche A, Tony Eissa N, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N.. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 2018;138:2227–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D.. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014;129:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiang DY, Zhang M, Voigt N, Alsina KM, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N.. Identification of microRNA-mRNA dysregulations in paroxysmal atrial fibrillation. Int J Cardiol 2015;184:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR.. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005;111:2025–2032. [DOI] [PubMed] [Google Scholar]
- 63.Ni L, Scott L, Campbell HM, Pan X, Alsina KM, Reynolds J, Philippen LE, Hulsurkar M, Lagor WR, Li N, Wehrens XHT.. Atrial-specific gene delivery using an adeno-associated viral vector. Circ Res 2019;124:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D.. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012;125:2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramay HR, Liu OZ, Sobie EA.. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res 2011;91:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C, Zecha J, Reiter K, Qiao H, Helm D, Koch H, Schoof M, Canevari G, Casale E, Depaolini SR, Feuchtinger A, Wu Z, Schmidt T, Rueckert L, Becker W, Huenges J, Garz AK, Gohlke BO, Zolg DP, Kayser G, Vooder T, Preissner R, Hahne H, Tõnisson N, Kramer K, Götze K, Bassermann F, Schlegl J, Ehrlich HC, Aiche S, Walch A, Greif PA, Schneider S, Felder ER, Ruland J, Médard G, Jeremias I, Spiekermann K, Kuster B.. The target landscape of clinical kinase drugs. Science 2017;358:eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data was generated or analyzed in support of this article.