

Abstract
The MolFind application has been developed as a nontargeted metabolomics chemometric tool to facilitate structure identification when HPLC biofluids analysis reveals a feature of interest. Here synthetic compounds are selected and measured to form the basis of a new, more accurate, HPLC retention index model for use with MolFind. We show that relatively inexpensive synthetic screening compounds with simple structures can be used to develop an artificial neural network model that is successful in making quality predictions for human metabolites. A total of 1955 compounds were obtained and measured for the model. A separate set of 202 human metabolites was used for independent validation. The new ANN model showed improved accuracy over previous models. The model, based on relatively simple compounds, was able to make quality predictions for complex compounds not similar to training data. Independent validation metabolites with feature combinations found in three or more training compounds were predicted with 97% sensitivity while metabolites with feature combinations found in less than three training compounds were predicted with >90% sensitivity. The study describes the method used to select synthetic compounds and new descriptors developed to encode the relationship between lipophilic molecular subgraphs and HPLC retention. Finally, we introduce the QRI (qualitative range of interest) modification of neural network backpropagation learning to generate models simultaneously based on quantitative and qualitative data.
Graphical Abstract

INTRODUCTION
Compared to targeted metabolomics where a predefined list of metabolites is examined, nontargeted metabolomics employs a less biased analysis of small-molecules present within a biological system.1 Recent advances in nontargeted metabolomics have led to the identification of disease biomarkers2–9 and generated optimism for gains in understanding disease mechanisms and associated druggable targets. Advances have arisen from parallel improvements in analytical and computational methodologies. High resolution high-performance liquid chromatography−mass spectrometry (HPLC-MS) has enabled high-throughput detection of metabolites in biological samples. Improvements in computational tools have enabled rapid discrimination of “features” that show significant variation across sample groups. A “feature” refers to an observed HPLC-MS peak corresponding to a specific metabolite. For non-targeted metabolomics, the ultimate goal is to match each feature with a verified molecular structure. As structures for the majority of human endogenous metabolites have not been enumerated, structure identification methodology remains a central focus of metabolomics research.
Currently, the typical structure identification process involves searching metabolite specific databases such as Metlin,10 HMDB,11 and KEGG12 using the experimental exact mass of the feature of interest. Even when exact mass matches are found, additional experimental data are necessary to confirm the match and such corollary metadata are typically absent. In many cases, an exact mass match is not found because metabolite specific databases are incomplete. For this reason, there are advantages in searching larger, more diverse, databases such as PubChem.13 Unfortunately, such searches return an excessive number of compounds, many of which are not biological. This is the principal dilemma when using databases for structure identification; small specific databases often do not contain the unknown, and large general databases contain too many false positives.
To address this dilemma, our group has developed MolFind14–17 to facilitate structure identification of features observed in HPLC-MS assays. In MolFind, a feature of interest is considered as a “target unknown”. The HPLC-MS exact mass of the target unknown is used to query large chemical databases and generate a candidate list of matches. The candidate list is filtered using multiple attributes predicted by computational models. The predicted attributes are retention index (RI)17–19 Ecom50,15,17 drift index (DI),14 whether a compound is biological or nonbiological (BioSM),20 and collision induced disassociation (CID) spectra (i.e., MS/MS).14,21 An experimental value for each attributes is measured for the target unknown and compared with predicted values for each candidate. Candidates whose predicted values most closely match the experimental values of the target unknown are returned as the most likely candidates. Comparisons are based on a “filter range” for each model derived from model validation statistics. Each filter range is centered on the experimental value of the target unknown and candidates whose predicted value falls outside the filter range are eliminated. The goal is to filter out false positives returned by the exact mass search thus reducing the candidate list to a manageable number to purchased or synthesize for final confirmation.
The effectiveness of the MolFind is dependent on the accuracy of its computational models and is thus also dependent on model training data. For optimal performance, training data needs sufficient structural diversity to cover the structure−property space where predictions are made. The width of the filter range is based on the standard error (SE) of prediction, thus the SE needs to be small to maximize the number of eliminated compounds. An ideal model will eliminate a large number of false positives while retaining the true positive (the match to the target unknown). Both high sensitivity and specificity are necessary for the models to provide meaningful enrichment in filtration.
HPLC retention index (RI) is an important attribute that has been modeled and used for filtration in MolFind.19 A major advantage of the HPLC-RI assay is the ease of transferability between laboratories since RI is based on relative (rather than absolute) retention time. Though RI has advantages as an attribute to measure, there are disadvantages in using RI as an attribute to model. Since every atom affects the RI value, it is optimal to have training set examples of every combination of chemotypes likely to be found in metabolites. Also, a wide range of RI values have been observed for metabolites and it is ideal to have data that uniformly covers the observed range. Thus, there is a large and diverse structure−property space to be populated to form the basis of a reasonable model. This leads to difficulty optimizing the model structure space using readily available compounds.
One goal of such a model is to predict RI for compounds in metabolomics specific databases. However, an ideal RI model must also generate reasonable RI predictions for metabolites that are not yet confirmed to exist in humans. Adequate modeling of currently unknown biochemical structure space presents significant challenges. In addition, it is impractical to obtain sufficient numbers of commercially available metabolites; both in terms of cost and the sheer numbers required for creating a model with adequate coverage of the applicability domain of human metabolites. Our previous RI model was based on 390 compounds and showed marginal performance on new data with 78% of independent validation (ival) compounds predicted within ±2 validation set SE.19 The lower than expected sensitivity was likely because of limitations in the measured RI range and the number of relevant chemotypes missing from the model data.
Here we have addressed these issues by developing a new artificial neural network (ANN) model based on 1955 synthetic compounds that were selected, purchased, and measured for the study. Compound selection was intended to project to both observed and likely human metabolite structure space. The experimental RI range was increased by 42% and an effort was made to minimize the occurrence of singletons or features of low population. New descriptors were also formulated to address observed deficiencies in QSRR structure description.
MATERIALS AND METHODS
Selection of Synthetic Compounds for the Model Data Set.
The goals of compound selection are similar to those of selecting a combinatorial library where an optimal data set has maximal diversity, uniform coverage of property space, and minimal redundancy.22 The challenge is to use inexpensive synthetic compounds to populate a human endogenous metabolite-like structure/property space, where much of that space is not fully enumerated. The resulting data must form the basis of a model that can make reasonable predictions for compounds whose composition is not known a priori with any certainty. In order to address this, an examination of the applicability domain is necessary. With our previous model, a bitkey analysis19 was used to define the applicability domain. A bitkey is a bit string where each bit encodes presence/absence of a structure features. The total string represents a predetermined set of features deemed to be important to the model target end point. The bitkey has one on-bit for each structure feature present such that compounds with the same bitkey value have the same combination of features from the predetermined set. For this study, each bit was set to encode a different heteroatomic structure feature. In the previous study, a 20% smaller SE was observed for predicted compounds that had three or more model training compounds with the same bitkey.19 This would suggest that a reasonable structure/property space could be created by generating bitkeys for known metabolites and obtaining at least three synthetic compounds to match each bitkey. An analysis of 3480 HPLC positive-ion MS detectable human metabolites showed 328 bitkeys, (328 unique combinations of heteroatomic features) ranging from 1 to 16 on-bits. For 198 bitkeys, there were 1 or 2 metabolites with that combination of features. With 328 bitkeys, it would be reasonable to consider creating a ∼2000 compound data set based on 3−8 compounds per bitkey but there are multiple problems with this approach.
Available compounds were not found to match every observed bitkey. Because there can be significant structure variation and a wide RI range among compounds with the same bitkey, it is unlikely that coverage could be obtained with 3−8 compounds. Since much of known human metabolite space is sparsely populated, use of known metabolites as a template for a model data set could result in isolated clusters separated by large, under-populated, interpolation regions.
For these reasons, a different approach was taken. We here hypothesize that a model for RI prediction in human endogenous metabolite-like space can be based on compounds with simple features and feature combinations, not unlike combinatorial chemistry building blocks.22,23 A triplet of three features can be described by three feature pairs, so if data is available for each possible pair, the ANN can make virtual combinations of higher order feature combinations and estimate their effect. We hypothesize that newly discovered metabolites will likely be composed of combinations of known structure features that have been observed in the nearly 8000 confirmed human metabolites. For this reason, the construction of the data set focused on obtaining compounds that sample single features and simple feature combinations found in confirmed human metabolites.
The first step was to enumerate classes of single heteroatomic functional groups found in HPLC-MS positiveion mode detectable metabolites. These compounds are comprised of carbon, hydrogen, and a single type of heteroatomic functional group including amine, pyridine, aniline, pyrrole, permanent charge nitrogen, amide-like (amide, urea, imide, etc.), thioamide-like (thioamide, thiourea, thioimide, etc.), and alpha-beta unsaturated ketone. Commercially available compounds were sorted into sets based on these types. Each set was sorted on the number of heteroatoms and the number of carbon atoms to roughly parallel the anticipated RI value. The compound with the largest number of heteroatoms and smallest number of carbon atoms was selected. Also, the compound with the largest number of carbon atoms and only one heteroatom was selected. Compounds were sampled from the space between to create a representative group from the set. An effort was made to select diversity in branching, ring structure, aromaticity, and shape in a process is similar to visual selection in a cluster-based design.22 A total of 414 compounds were selected to populate the single functional group sets.
Next, compounds with feature “pairs” were selected by taking the half triangle of the single functional group matrix (amine-pyridine, amine-aniline, etc.) and adding pairs with functional groups found in metabolites that cannot be detected on their own. Added groups included acid (carboxylate, phosphate, and sulfate), phenol, alcohol, ether, ester, ketone, furan, thiol, thioether, thioketone, and thiophene. The same sorting process was applied as was used for the single functional group sets and compounds were selected in the same manner. A total of 1290 compounds were selected to populate the “pairs” sets. Feature “triplets” were also examined, but examples of most combinations were not commercially available and only 197 compounds with three features were included. An additional 54 compounds with 4−6 features were also included. A total of 202 confirmed human endogenous metabolites were set aside as an independent validation set.
Bitkeys for the resulting data set ranged from 1 to 9 on-bits with 1866 compounds having 5 or fewer on-bits. This is in contrast to human metabolite data where 178 of the 328 observed bitkeys have 6−16 on-bits. For the 202 compound metabolite validation set, more than half have 5 or more on-bits with a maximum of 11. In general, human metabolites are more complex than the selected training data in terms of combinations of features and the number of features per molecule. This study will provide an indication concerning the plausibility of making predictions for complex structures using a model based on compounds with simple combinations of features from which the complex combinations are composed.
After the selected compounds were obtained, the decision was made to switch from modeling with structure descriptors based on the neutral form to the descriptors based on the calculated major microspecies and tautomer at the pH of the HPLC mobile phase. This resulted in changes to the structure space not accounted for during compound selection such as the internal rearrangement of some structures. Also, RI measurements were not obtained for every compound. These complications resulted in a model data space with less coverage and diversity than was intended. The structure and predicted major microspecies of all compounds used in the study are provided in Supporting Information files S1−S4.
Experimental Determination of Retention Index (RI) Values.
The determination of HPLC retention times of the model test compounds and the calculation of the corresponding nitro-n-alkane and n-amide retention indices has been described previously.19 Briefly, solutions of C1−C10 n-alkanes, C3−C14 n-amides, test compounds, internal control compounds, and external control compounds were prepared and analyzed by HPLC/MS using an OptiGuard (1 mm × 17 mm, 5 μm) guard column linked to a Zorbax SBC18 (1 mm × 150 mm, 3.5 μm) analytical column with a linear gradient of 0.01% heptafluorobutyric acid (HFBA)/water/acetonitrile with detection by positive ion electrospray mass spectrometric analysis. Specifically, the HPLC analysis was performed on an Agilent 1100 HPLC system using a solvent gradient of 100% 0.01% (v/v) HFBA/H2O to 100% 0.01% (v/v) HFBA/90% CH3CN/H2O at a flow rate of 75 μL/min. Using UV detection, a homologues series of nitro-n-alkanes (C1−C10) were analyzed on the same HPLC system at the beginning and end of each batch of sample analyses. The retention times of the sample compounds and the fourth-order polynomial relationship between the nitro-n-alkanes retention times and one hundred times the respective number of carbons in the nitro-n-alkane structure were used to calculate the retention index of each test and control compound. Since the homologues series of n-amides have the advantage of being detected by mass spectrometry, the nitro-n-alkane retention index values of each test and control compound were converted to a retention index value referenced to the C3−C14 homologous series of n-amides analyzed on the same HPLC system.
The positive ion electrospray detectable homologous series of n-alkylamide retention compounds were chosen such that they eluted within a similar retention time range as the non-MS detectable homologous series of nitro-n-alkanes that were previously used in our retention indices studies. The earliest eluting n-alkylamide, n-propanamide, has a retention time of approximately 2.3 min which compares with the 2.2 min retention time for the earliest eluting nitro-n-alkane, n-nitromethane, and the last eluting n-alkylamide, n-tetradecanamide, has a retention time of approximately 19.7 min which is similar to the 19.6 min retention time of the last eluting nitro-n-alkane, n-nitrodecane. The RI values of compounds that eluted before n-propanamide or after n-tetradecanamide were extrapolated, respectively, from the logarithmic linear regression equation for compounds eluting in the isocratic mobile phase or the fourth order polynomial equation for compounds eluting in the gradient portion of the mobile phase. Extrapolated values suffer a loss off accuracy since the nonlinearity of the system is unclear outside the range of standards. These values do, however, give a qualitative indication of the retention magnitude. In addition, compounds that elute substantially before n-propanamide elute very near the column void time and are not substantially retained. The RI variation for these compounds likely arises from different mechanisms than for compounds that are substantially retained. This creates three categories of measured values. Values > 1400 retention index units (RIU) suffer from extrapolation error. Values < 300 RIU suffer from extrapolation error and also likely arise from a different mechanism. Values between 300 and 1400 RIU are not entirely consistent with the other two categories of measurements. It is likely that the relationship between RI and descriptor values will also not be consistent across all three categories and this creates difficulties in using all of the available data for a model.
Despite measurement difficulties, a number of MS detectable human metabolites have been observed to elute outside the range of N-alkylamide standards. It is optimal for MolFind to be able to account for any MS detectable compound that appears as a feature of interest, even if it elutes outside the range of standards. This necessitates that the MolFind RI model account for measurements in all three categories. Our approach to this is discussed in the Materials and Methods section.
Structure Descriptors.
A total of 47 structure information representation (SIR) descriptors24–29 were used to build a series of ANN ensemble models. Descriptors values were calculated using winMolconn, v2.1.30 Descriptors were not selected by a statistically based selection algorithm. The descriptor set was chosen in an attempt to explicitly encode all molecular characteristics that influence reverse phase HPLC retention under known measurement conditions. Though the extent to which that goal has been met it is still being investigated, this description methodology has been used to create multiple successful models of HPLC data sets.14,15,17–19 A complete description of the descriptor set is given in the following sections.
Major Microspecies Calculation.
The pH of both HPLC mobile phase components was measured to be 2.5. There are multiple functional groups in model compounds whose protonation or ionization state is uncertain at this pH. Protonation adds a positive charge and a hydrogen bond donor, while occupying a hydrogen bond acceptor. Ionization has similar effects. Preliminary models suggested that these changes were too impactful to approximate with neutral structures. To address this, the Marvin Beans Calculator (cxcalc)31 was used to convert structures to the major macrospecies and tautomer at pH 2.5. All structure descriptors were calculated for the predicted major macrospecies.
Heteroatomic Feature Descriptors.
Most heteroatomic features were described using the Interaction Group (IGroup) E-State described previously.18,19 The IGroup indices were created for modeling solution properties and have been successfully used to model HPLC-RI of diverse data sets.18,19 IGroups explicitly encode information about every heteroatom by grouping Atom-level Electrotopological State (E-State)26 values for atoms that participate in similar noncovalent solution interactions. IGroups are created by assigning atoms from related functional groups to a unified set of descriptors. A list and description of the IGroup feature descriptors used for the study model is given in Table 1.
Table 1.
IGroup E-State Descriptors
| index | atom typesa | functional groupsb |
|---|---|---|
| pyridine_N | aNa | pyridine, diazole |
| pyridine_NH | −H | on protonated pyridine |
| alph_Nc | >N−, =N− | amine, guanidine, imine |
| alph_NH | −H | on any alph_N |
| aniline_N | >N−, =N− | aniline, benzamidine |
| anilne_NH | −H | on any aniline nitrogen |
| pyrrole_N | aNa− | pyrrole, indole, diphenylamine |
| pyrrole_NH | −H | on any pyrrole nitrogen |
| prot_N | >N+<, aaN+− | quaternary amine, pyridinium |
| amide_O | =O | amide like groups |
| amide_N | >N−, =N− | amide like groups |
| amide_NH | −H | on amide-like nitrogen |
| thioamide_dS | =S | thioamide-like groups |
| thioamide_N | >N−, =N− | thioamide-like groups |
| acid_dO | =O | acid-like groups (COOH, PO3) |
| acid_sO | −O | acid-like groups (COOH, PO3) |
| phenol_O | −OH | phenol |
| alph_ssOd | −O− | ether, ester |
| alph_dOe | =O | ester, ketone, aldehyde |
| alph_sOH | −O | alcohol |
| arom_O | aOa | furan, oxazole |
| alph_Sf | −S−, −SH, =S | thioether, thiol, thioketone |
| arom_S | aSa | thiophene, thiazole |
Valence state atom types included in the index.
Index is comprised of atoms from these functional groups.
Unassigned nitrogen atoms are added to this group by default.
Unassigned −O− oxygen atoms are added to this group by default.
Unassigned =O oxygen atoms are added to this group by default.
Unassigned sulfur atoms are added to this group by default.
Additionally, two E-State based internal hydrogen bonding indices were used to encode internal hydrogen bonding configurations. An integer count positive charge descriptor was used to encode the charge of protonated atoms and permanently charged nitrogen species (quaternary amine, pyridinium, etc.). An integer count negative charge descriptor was used to encode the charge of ionized atoms.
Lipophillic Features.
One of the most difficult aspects of structure description as it relates to reverse phase RI is the description of the lipophilic parts of the molecule (lipophilic subgraphs). Lipophilic subgraphs interact with the HPLC stationary phase by Van der Waals dispersion and are responsible for retention. Many previously utilized description systems characterizing the potential for dispersion interactions are whole molecule approximations of molecular polarizability expressed as molar refraction (Rm).32,33 When examining a series of compounds with identical branching, heteroatom composition, and heteroatom position, Rm explains essentially all RI variance. For the series of amines, propylamine (Rm = 19.4, RI = 415), butylamine (Rm = 24.0, RI = 522), octylamine (Rm = 42.6, RI = 776), nonylamine (Rm = 47.6, RI = 785), undecylamine (Rm = 56.6, RI = 895), tridecylamine (Rm = 69.0, RI = 1014), and hexadecylamine (Rm = 81.6, RI = 1202), Rm correctly rank orders the series and accounts for 99% of RI variance.
Molar refraction, however, does not always correlate with RI over a series of structurally diverse compounds with similar Rm. Figure 1 shows nine C8H19N isomers with 33% variance in RI. Experimental Rm varies by only 3% and has a negligible correlation with RI.
Figure 1.

Retention index (RI), molar refraction (Rm), and lipophilic subgraph descriptor values (LpSgr1–3) for a series of C8H19N isomers. Rm does not parallel RI variation over a series of isomers. The Rm:RI correlation explains only 6% of RI variation. The value of the largest lipophilic subgraph descriptor for each isomer (LpSgr1) explains 91% of the RI variation and thus captures the structure property relationship to a greater extent.
An examination of the isomer series does not reveal any obvious descriptors (count of H-bond donors, count of methyl groups, etc.) that parallel RI. These data highlight the lack of an adequate system of description to relate the characteristics of lipophilic subgraphs to RI for molecules with similar Rm.
When polarizability is similar across a series of molecules, other factors must be examined to explain RI variance. It is not only dispersion potential that influences stationary phase interactions, but also interaction efficiency. An examination of Figure 1 suggests structural characteristics that influence the efficiency of lipophilic subgraph stationary phase interactions. A compound is more efficiently retained when all of the lipophilic atoms are contained in a single continuous subgraph. For isomers with the same number of subgraphs, increased branching leads to shorter retention times. Based on such observations, we here introduce the lipophilic molecular subgraph descriptors (LpSgrN, where N is the subgraph number). The LpSgrN indices are a new system of structure description to encode the size, dispersion potential, distribution of hydrophilic centers, and branching of individual lipophilic subgraphs in molecules. The LpSgrN indices were calculated as follows.
A graph search algorithm was used to enumerate each lipophilic subgraph using a list of hydrophilic atom types as search stop atoms. Any protonated or ionized atom was considered a stop, along with phosphorus, oxygen (excepting −O− in ether or ester), and nitrogen (excepting pyrrole and nonprotonated pyridine). Carbon and sulfur were assigned as lipophilic. Once the atom list for each separate subgraph was identified, an atom-level refraction (AtR) value was assigned based on hydrophobic constants suggested by Ghouse and Crippen.32 Next, each AtR was adjusted to account for the influence of nearby electronegative atoms. Strongly electro-negative atoms reduce the polarizability of neighbors by withdrawing electron density. To take this into account, each AtR was modified according to eqs 1a and 1b.
| (1a) |
∑KHEΔ(i) = sum of Kier−Hall electronegativity difference adjustments for atom i; KHE(i) = Kier−Hall electronegativity for atom i; KHE(j) = Kier−Hall electronegativity for atom j; dij2 = square of path distance between atoms i and j
| (1b) |
KHEΔ_AtR(i) = KHE difference adjusted atom-level refraction for atom i; scale = scale factor = 0.1; AtR(i) = Ghouse Crippen atom-level refraction for atom i.
Using eq 1a, for each pair of atoms i,j, the Kier−Hall electronegativity difference (KHEΔ) was taken and divided by the square of the i,j path distance so the effect is more pronounced for close neighbors. For each subgraph atom i, the absolute value of KHEΔ for every atom pair is accumulated in a sum ∑KHEΔ(i). The absolute value was added to ∑KHEΔ(i) where KHE(i) < KHE(j), meaning that i is less electronegative than j and is losing electron density to j. Where KHE(i) > KHE(j), the absolute value was subtracted because i is gaining electron density from j. The larger the accumulated sum, the more electron density is lost resulting in loss of polarizability. AtR for each atom was modified by ΣKHEΔ according to eq 1b where AtR for each atom i was multiplied by the product of ∑KHEΔ(i) and a scaling factor. The result is the Kier−Hall electronegativity difference adjusted atom-level refraction (KHEΔ_AtR).
Next, steric interference from hydrophilic atoms was addressed by reducing KHEΔ_AtR(i) for lipophilic subgraph atoms with at least one hydrophilic neighbor. KHEΔ_AtR(i) was multiplied by an interference coefficient based on the count of hydrophilic atoms alpha and beta to i. The interference coefficient was 1.0 if there were no alpha or beta hydrophilic neighbors. KHEΔ_AtR for each atom was multiplied by its interference coefficient and the resulting values were summed across atoms in the subgraph to create the Kier-Hall electronegativity difference adjusted, hydrophilic steric interference adjusted, subgraph refraction (KHEΔ_HSI_SubGrR). The steps for applying the interference coefficients to the lipophilic subgraph of three phenylenediamine isomers are illustrated in Figure 2. The full table of interference coefficients is included in Supporting Information file S5.
Figure 2.
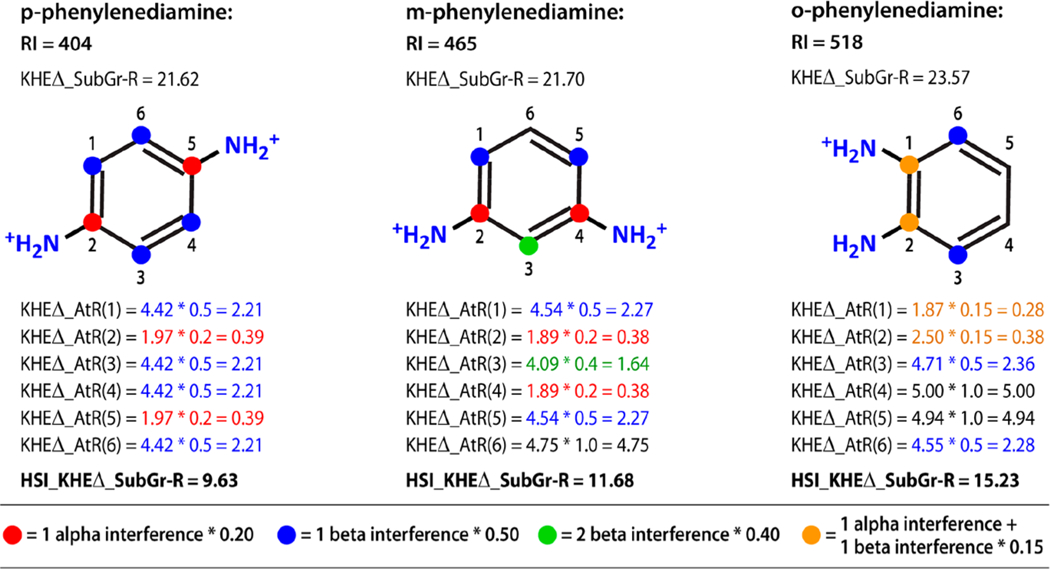
Calculation of hydrophilic steric interference adjustment for the lipophilic subgraph in three phenylenediamine isomers. The KHEΔ_AtR values are adjusted to take into account the steric interference of dispersion interactions caused by hydrophilic atoms associated with solvent. The interference coefficients are given in the color coded key and are based on the count of alpha and beta hydrophilic atoms. The KHEΔ_AtR values do not vary nearly as much as RI and explain only 76% of RI variance. Following the steric interference adjustment, the HSI_KHEΔ_SubGrAtR values explain 96% of RI variance. This ortho, meta, and para series of isomers illustrate that when hydrophilic atoms are close together relative to the lipophilic subgraph, the lipophilic subgraph is more exposed resulting in a longer retention time. The o-phenylenediamine isomer has a longer retention not only because of greater lipophilic subgraph exposure but also because an internal hydrogen bonding configuration has been introduced and because the major microspecies is only +1.
Finally, each KHEΔ_HSI_SubGrR was adjusted according to eq 2 to take branching and rings into account. A “branch degree” term was created from the quotient of the simple chi path 2 value for an unbranched structure with the same number of atoms as the subgraph (2χn) and the actual simple chi path 2 (2χs) value of the subgraph. Since chi path 2 increases with branching, the branch degree is 1 where the lipophilic subgraph is unbranched and gets smaller with each branch point.
| (2) |
KHEΔ_HSI_SubGrRs = sum of Ghouse Crippen atom level refraction values for subgraph s after Kier−Hall electro-negativity difference and steric adjustments have been applied; 2χn = simple chi path 2 index for straight chain structure with the same number of atoms as s; 2χs = simple chi path 2 index for lipophilic subgraph s; LpSgr-s = final lipophilic subgraph index value for subgraph s.
The resulting product gives the final descriptor value for each lipophilic subgraph (LpSgr) where the sum of the Ghouse and Crippen hydrophobic constants for the subgraph atom has been reduced by the presence of local electronegative atoms, steric interference from hydrophilic centers, and the presence of rings and branching. The LpSgr-s index values for each molecule were rank ordered and each value was assigned to a distinct descriptor where LpSgr1 is the lipophilic subraph with the largest value, LpSgr2 is the second largest value, etc. Looking at Figure 1, LpSgr1 explains 91% of RI variance and LpSgr1 and LpSgr2 together explain 99% of RI variance where Rm explains only 6%. The first 7 lipophilic subgraph index values (LpSgr1−LpSgr7) were used in the model. An example of the calculation steps of LpSgr1 and LpSgr2 for dibutylamine and di-sec-butylamine is given in Figure 3.
Figure 3.
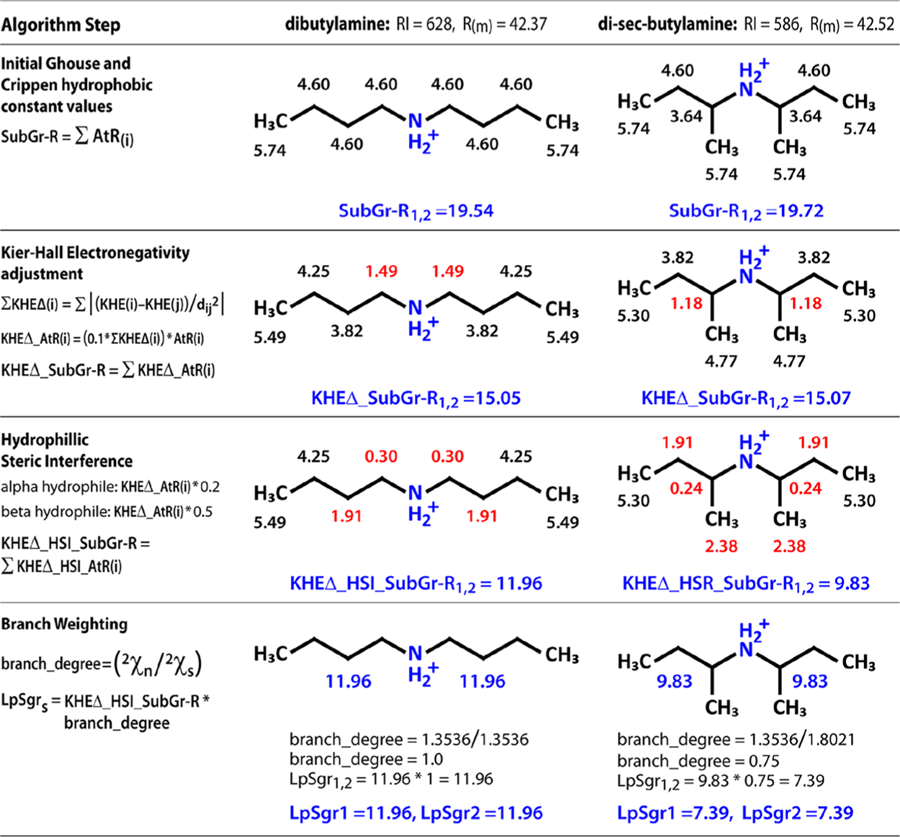
Calculating lipophilic subgraph descriptors for dibutylamine and di-sec-butylamine. Step 1 assigns atom-level refraction (AtR) values based on the Gouse Crippen approximation. Step 2 adjusts AtR by summing the Kier−Hall electronegativity difference against all other atoms in the molecule and multiplying AtR by a scaling factor to create KHEΔ_AtR(i) for each atom. The third step adjusts KHEΔ_AtR by multiplying an interference coefficient based on the count of alpha and beta hydrophilic atoms to create KHEΔ_HSI_AtR(i). Finally, KHEΔ_HSI_AtR(i) for each atom in the subgraph is summed and multiplied by a branch degree coefficient. The series of four steps results in descriptor values that reflect the difference in RI value where molar refraction does not. Intermediate subgraph sum values are given for each step to illustrate.
Graph Based Feature Descriptors.
Graph based feature descriptors used in the model included the count of rings, circuits, and rotatable bonds. Molecular connectivity indices were also used including 4χpc (chi simple path-cluster 4), 3χp (chi simple path-3), and 10χp (chi simple path-10) indices.34,35
Global Descriptors.
Global descriptors encode structure characteristics common to every compound and seven were used for this study. The molecular connectivity 0χv index (chi valence 0)35 was used to approximate molecular volume. A pair of global indices (rvalHyd and sumLpSubGr) was used to characterize hydrophilicity and lipophilicity. The rvalHyd index (ratio valence hydrophilic index)19 gives the sum of atom level E-State values of hydrophilic atoms divided by the total atom level E-State for the molecule. This index quantifies the proportion of valence electron density associated with hydrophilic atoms. A new global index of lipophilicity, sum of lipophilic subgraph refraction (sumLpSubGr) was created for this study as the sum of the LpSgr1−LpSgr7 lipophilic subgraph indices described above. The global shape descriptors 2κα (kappa alpha 2),27 flatness, and inv_dx2 (inverted difference simple chi 2) described in previous HPLC RI models19 were also used.
A list of descriptors and their definitions is given in Supporting Information file S6, and the descriptor values for all study compounds is given in Supporting Information files S7 and S8.
Input Normalization and Scaling.
When using the RPROP36 learning algorithm, the fann library,37 on which the ANN model software is based, restricts descriptor and target input values to a range of 0.0−1.0. To accommodate this requirement, descriptor values were normalized by Z-score and scaled. Feature descriptors were normalized using the mean and standard deviation of rows with nonzero values. Feature descriptors values of 0.0 were not normalized so as to retain their null set information. Global descriptors were normalized using the mean and standard deviation for all rows. Following normalization, all global descriptors and feature descriptors with nonzero normal values were scaled from 0.1 to 0.7. The use of 0.7 as a maximum leaves “headroom” for any compound predicted by the model that has a larger input value than the data set maximum. Feature descriptor normal values of 0.0 were not scaled. The flatness and inv_dx2 global descriptors were scaled from 0 to 1 since both have a theoretical minimum and maximum that cannot be surpassed.
Descriptor Reduction Tests.
Though a statistically based descriptor selection algorithm was not employed in this study, the set of 47 descriptors was subjected to a reduction test. An ANN model was built for each n − 1 subset (all possible models with 46 of the 47 descriptors) and analyzed by the statistical criteria used for the n = 47 model. The top 10 models were kept and models were built for n − 2 subsets (models with 45 of the 46 model descriptors). The procedure was implemented up to n-3 but no model was found with validation statistics superior to the n = 47 model.
Descriptor Correlations.
The linear correlation of input descriptors is of less interest when a nonlinear modeling method is used but a correlation analysis is necessary to complete the descriptor profile. A total of four pairs of model descriptors have a linear correlation >0.90. The alaphatic nitrogen (alph_N) and hydrogen on alaphatic nitrogen (alph_NH) descriptors are correlated at 0.95. The thioamide-like double bond sulfur (tamide_dS) and thioamide-like single bond nitrogen (tamide_N) descriptors are also correlated at 0.95. The acid-like double bond oxygen (acid_dO) and acid-like single bond oxygen descriptors (acid_sO) are correlated at 0.93. The chi valence 0 and chi simple path 3 descriptors are correlated at 0.92.
The intercorrelation of the amine descriptors is likely because nearly all amines are protonated by Marvin cxcalc at the pH of the mobile phase. The two descriptors run largely in parallel because every amine has at least one associated hydrogen atom. The intercorrelation of the thioamide descriptors likely results from the over representation of secondary thioamides in the data. The intercorrelation of the acid-like descriptors likely results from too many of the acid groups being carboxylates, each with one = O and one −OH. In these cases, nearly 90% of the information from the descriptor pair is redundant. However, the descriptor reduction tests demonstrate that the model is not improved by the removal of either of the descriptors so it is likely that the independent 10% of information is useful. A correlation matrix is given in Supporting Information file S9.
MODELING METHODS
Data Set Partitioning.
The 1955 compound data set was partitioned into a 4 × 10 × 10 ensemble described previously.19 To create the ensemble, the 23 IGroup feature descriptors were used to generate a bitkey for each row. Compounds were sorted by bitkey to generate 278 classes of compounds where all class members have the same heteroatomic features. Classes were organized from simple to complex as defined by the count of on-bits. Within each class, compounds were rank ordered on experimental RI. Classes with the same count of on bits were then ordered based on the number of row members. The result was an organization of the data such that the first compounds were the 1-on-bit class with the largest number of members and the last compounds were the 9-on-bit class with the fewest members. No compounds in the model data set had more than 9-on-bits, meaning nonzero descriptor values for more than 9 of the 23 IGroup descriptors.
After ordering, rows were divided into four fit/validate splits (A, B, C, D). Each split was constructed by iterating through the bitkey order and assigning every fourth compound to validate. Assignment to validation started with the first row for split A, the second row for split B, the third row for split C, and the fourth row for split D. The result was ∼25% of data rows assigned to the validate set of each split with the remaining ∼75% in the fit set. Assignment to validation was mutually exclusive where each compound was assigned to the validate set of exactly one of the four splits. The fit set of each split was further subdivided into 10 folds by assigning every 10th compound to test. Test set assignment was similar to validation assignment in that for the first fold, test set assignment began with the first row and every 10th compound thereafter. For the second fold, test set assignment began with the second row, etc. This resulted in ∼90% of the data for each fold in a training set and ∼10% as a test set for cross validation. Test set assignment is also mutually exclusive where each fit set compound was in the test set of exactly one fold.
In each of the four fit/validate splits, ∼75% of the overall data resides in 10 folds of training data and each fold has its own leave-10%-out test set. Additionally, ∼25% of the data is reserved for validation. Since the ensemble contains four fit/validate splits, a validation prediction was available for every data set compound and every compound was used for training in three splits. Because the data rows were ordered in both structure and property space using the bitkey and RI values, a relatively equivalent number of structures from each structural class was assigned to train, test, and validate for each fold in the data split. The ordering of each bitkey class on RI helps to ensure an even distribution in property space.15,17,18
Additionally, 202 confirmed human endogenous metabolites were set aside as an independent validation set. Since MolFind is intended to identify metabolites from a candidate set, it was decided to set aside all metabolites in a second validation set. Since all compounds in the model data set were synthetic, this method will provide information as to the effectiveness of using synthetic compounds as the basis of a model to predict human endogenous biochemicals.
ANN Learning Method.
A separate 10-fold ensemble model was built for each of the four fit/validate splits. Each model employed an architecture of an input layer with 47 input neurons, 1 hidden layer with 23 hidden neurons, and an output layer with 1 output neuron. The architecture was based on the previous model where the optimal architecture was found to have a single hidden layer with half the number of hidden neurons as model inputs.19 Each of the 10 folds of each split was trained separately by generating 50 models for the fold, each trained from a different set of random starting weights. Model training utilized the RPROP36 algorithm implemented in a proprietary application built with the fann library.37 A previous study showed that the use of validate statistics for the stopping and model selection criteria produced a more effective model for predicting new data than a model developed with test set stopping and selection.19 For this reason, each model was trained on the fold training set rows using the validation set mean absolute error (MAE) as the stopping criteria (training continued until the validation MAE minimized). Validation MAE was also used as the model selection criteria meaning that the validation MAE was evaluated for all 50 fold models and the model with the lowest validation MAE was selected as the final fold model. Test set predictions were also made for the purpose of statistical analysis. This process was repeated for all 10 folds resulting in 9 train predictions and 1 test prediction for fit set rows and 10 predictions for validate rows.
The above process was repeated for each of the four fit/validate sets resulting in 40 final fold models selected from 2000 total models. To create final ensemble statistics, averages were taken over all 40 models. Each compound was assigned to the training set in 36 of the 40 models, the test set in three models, and the validation set in 10 models. Final train, test, and validation prediction values were created by averaging the predictions from each set. Finally, each of the 40 models in the ensemble was used to make a prediction for the 202 independent validation compounds and the 40 predicted values were averaged to create an independent validation prediction. Statistics were calculated for the train, test, validate, and independent validate sets of the data based on the averages.
Since test set stopping is the more common procedure, a second 4 × 10 × 10 ensemble model was created using test set stopping and model selection. This model was developed in identical fashion to the first ensemble except that models were trained until the test set MAE minimized and the fold model with the best test MAE was selected as the final fold model. Model statistics were calculated in the same way as for the model built on validate set statistics.
After the final ensemble models had been generated, network architecture was optimized by creating new models based on networks with a single hidden layer of 5−55 hidden neurons in increments of 5. The stopping MAE was plotted against the number of hidden neurons and a polynomial curve fit was applied to interpolate the number of hidden neutrons where the polynomial minimized. A new model was created using the architecture suggested by the polynomial interpolation as well as models for n − 1, n − 2, n + 1, and n + 2 of the suggested number of hidden neurons. The best model was found to have a network architecture of a single hidden layer with 22 hidden neurons which is similar to that found in the previous study.
Quantitative Range of Interest (QRI) Adjustment.
Here we introduce a novel adjustment to the RPROP method implemented to address the issue of measured RI values that fall outside the range of N-amide standards. This adjustment is called quantitative range of interest (QRI). As mentioned in the Materials and Methods section, compounds that elute before the first standard (n-propanamide) elute very near to the column void time. These compounds are not substantially retained by the column. The RI variation of these compounds likely arises from fundamentally different mechanisms than compounds that are substantially retained. Because confirmed endogenous metabolites have been observed to elute in this same region, it was deemed necessary to include this data as a means to facilitate predictions for endogenous compounds that also elute before the first standard. Also, it is unknown how accurate extrapolation is for compounds that elute after the last standard. The QRI adjustment specifies a “quantitative range of interest” in the target end point where the data are reliable and it is important for predictions to be as accurate as possible. Data points that fall outside the QRI are still utilized during learning, but qualitative predictions are considered sufficient. The QRI adjustment modifies the ANN cost function for rows outside the QRI to zero the row error as long as the prediction is qualitatively correct. For compounds with measured RI values below 300 RIU, the contribution to the cost function for that row will be 0.0 as long as the prediction is also below 300 RIU. These qualitatively correct predictions are considered true positives regardless of the magnitude of the residual. If the prediction is above 300 RIU, the prediction is qualitatively incorrect and the cost was assessed as predicted−observed. The result is that once a prediction for a data row outside the QRI is pushed past the QRI boundary, the model is no longer aware of any error for that row and no additional weight adjustments are made regarding that row.
It was necessary to make a parallel modification to the program that was used to calculate model statistics. Predictions for compounds with RI values outside the QRI that classify as true positive predictions were not included in the MAE, meaning that the MAE was calculated only on rows in the QRI, plus rows outside the QRI that were false negatives.
RESULTS
Validation set and I-val set statistical results for the ANN-t (test set stopping) and ANN-v (validation set stopping) models are given in Table 2 along with the parallel statistics from our previous ANN RI model.19
Table 2.
Statistics Results of Modeling Methods Compared to Previous Model Results
| training set |
test set |
validate set |
i-val set |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| model | r 2a | MAEb | SEc | r 2 | MAE | SE | r 2 | MAE | SE | ±2SE | % TPe |
| previous ANNf | 0.98 | 25.3 | 32.6 | 0.87 | 51.4 | 65.6 | 0.91 | 42.7 | 54.8 | 219 | 78% g |
| ANN-t test optimizedh | 0.97 | 27.7 | 36.1 | 0.95 | 38.8 | 50.0 | 0.94 | 42.1 | 54.1 | 216 | 93% |
| ANN-v validate optimizedi | 0.98 | 29.8 | 38.6 | 0.94 | 47.0 | 60.6 | 0.95 | 39.3 | 50.7 | 203 | 94% |
Square of the Pearson correlation coefficient.
Mean absolute error.
Standard error.
±2 standard error filter range from validate SE.
Percent true positive predictions of independent validate set.
Results from previous study19 based on 390 compounds.
1492 compound i-val set predicted by the previous model.
ANN model with test set stopping and selection.
ANN model with validate set stopping and selection.
As can be seen from Table 2, the results from the ANN-t and ANN-v methods are similar. Results from the ANN-v model will be discussed in full because the validate MAE, and SE, and i-val set sensitivity were slightly better than for the ANN-t model. Results and discussion about the ANN-t model can be found in Supporting Information file SI10.
The ANN-v model showed a train r2 (square of the Pearson correlation coefficient) of 0.98, test q2 of 0.94, and validation r2 of 0.95. The ANN-v MAE was 28.8 RIU for train, 46.9 RIU for test, and 39.3 RIU for validate. The SE was 38.6 RIU for train, 60.5 RIU for test, and 50.7 RIU for validate. The ±2SE filter range based on the ANN-v validation statistics is 203 RIU which is 14.7% of the total data range of 1381 RIU.
Recalling the QRI adjustment, it was necessary to statistically account for predictions made for these compounds. These predictions are qualitative and were not included in the r2, MAE, and SE cited above. In Figure 4, QRI predictions are shown as black diamonds. Predictions for compounds with RI values outside the upper and lower QRI boundaries are shown as white circles at either end of the plot. The range of predictions made for these compounds is shown on the y-axis and the same x-axis value is used for every data point. In order to use this model to make predictions for a target unknown that elutes outside the QRI, it is necessary to apply a filter similar to the ±2SE filter range used for the QRI compounds. SE is the standard deviation of the error such that 95% of residuals are smaller than ±2 SE. To create a value similar to the SE to apply to the qualitative predictions outside the range of standards, we evaluated the magnitude of the cutoff value necessary to capture 95% of the validation predictions made for compounds outside the QRI.
Figure 4.

ANN-v train and validate results for 1955 compound data set. Round data points on each end represent compounds with experimental values outside the QRI (outside the range of N-amide standards, RIU < 300, RIU > 1400). On the validate data plot, the gray boxes at each end indicate the prediction cutoff for ∼95% true positive predictions. Data points within each box are true positive predictions while those outside are false negatives. These cutoff values approximate the ±2SE filter range for compounds with predicted values outside the range of standards. The center gray box illustrates the y-range of the ±2 SE filter range which constitutes 14.7% of the overall data range of 1381 RIU.
For the ANN-v model, a cutoff of 427 RIU captures 94% of predictions for compounds with RI values below the lower boundary. Similarly, a cutoff of 1278 RIU captures 94% of predictions for compounds with RI values above the upper boundary. If a target unknown elutes before the first standard, predictions will be made for compounds in the candidate set and compounds with predicted values >427 RIU will be excluded. If a target unknown elutes after the last standard, candidate set compounds with predicted RI < 1278 will be excluded. These cutoff values are shown in Figure 4 as the shaded gray boxes at each end of the validate plot. Plots of the train and validation predictions for the ANN-v model are given in Figure 4.
Comparison of Statistics for Predictions Made in Retention Time Units.
HPLC RI measurements are made as retention time (tR) and converted to retention index (RI) based on the measured tR values of the 11 N-amide standards. The distribution median of tR for the 1955 compound model data set is 8.9385 min with a first quartile (Q1) of 7.1478, third quartile (Q3) of 11.8675, mean of 9.5440, and standard deviation of 3.8401. The distribution median of RI is 652 RIU with Q1 of 541, Q3 of 858, mean of 708, and standard deviation of 251. Where the distributions are so different, it is instructive to report the validation set statistics calculated for predicted tR along with predicted RI. The validation set r2 for predicted versus experimental tR is 0.942 which is slightly lower than the validation set r2 for predicted versus experimental RI of 0.949. This is a small difference considering that the tR range is much smaller than the RI range. The validate MAE values cannot be compared directly because of the difference in units but the relative average error for the tR predictions is 3.59%, which again is slightly larger than the relative average error for the RI predictions of 3.57%.
Independent Validation.
The ANN-v model was used to make predictions for the 202 compound independent validation (i-val) set. These predictions simulate a match to a target unknown present on a candidate list. Each compound on the candidate list is considered a “positive” because their exact mass matches the observed exact mass of the target unknown. The role of the RI model in MolFind is to filter out false positives while leaving the true positive (actual structural match) on the list. In this context, each i-val compound can be considered a match where a true positive prediction is expected. To be a true positive, a prediction must fall within the ±2SE filter range for QRI compounds, or within the boundary cutoff for compounds outside the QRI.
Figure 5 shows the ANN-v i-val predictions. The gray boxes at the far left indicate the prediction cutoff for 95% true positive predictions of compounds that elute before the first standard. White circle points within the box are true positive predictions while gray diamonds outside are false negatives. The predictions for compounds eluting before the first standard were very good with only one false negative prediction. Overall, the model achieved 94% sensitivity with the i-val data.
Figure 5.
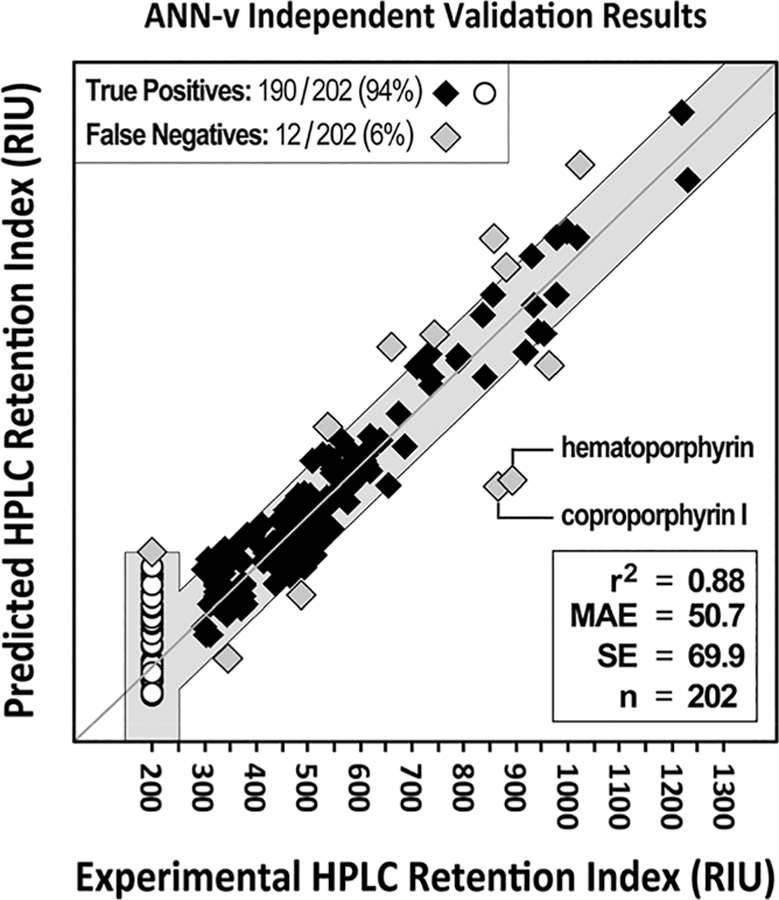
ANN-v independent validation set prediction results for 202 human endogenous compounds. The shaded gray area represents the region of true positive predictions based on the ±2SE filter window of 203 RIU from the ANN-v validation set statistics and the lower QRI cutoff. Points inside the gray area are the true positive predictions (black diamond and white circles). Points outside the gray area are the false negative predictions (gray diamonds). The worst predictions are for two porphyrins; coproporphyrin I AE = 305 RIU, and hematoporphyrin AE = 320 RIU. The MAE and SE are significantly worse than for the validation set data. Much of this difference is due to the two poorly predicted porphyrins. Sensitivity is close to the 95% that would be expected with the ±2SE filter range.
DISCUSSION
The aim of this study was to develop an improved ANN RI model for use with MolFind. Improvements included expanding the structure space of the previous model. Because it was not possible to obtain examples of every observed chemotype and likely combination of chemotypes, a method was employed to populate the space of single features and feature pairs as completely as possible. This design was based on the hypothesis that a neural network can infer reasonable predictions for higher order feature combinations based on the information contained in relatively simple feature combinations. Feature triplets were populated to the extent possible given compound availability and the data included a small number of compounds with up to 6 features available from previous studies. With this method, we were able to use simple, relatively inexpensive, synthetic compounds to generate of a model designed to predict RI values for endogenous metabolites.
It was also necessary to expand the RI data range to encompass that observed in human metabolites. Data range expansion included the introduction of the QRI methodology to allow for the inclusion of compounds with measured values outside the range of RI standards. Finally, structure description was addressed with the introduction of the lipophilic molecular subgraph descriptors. A 202 compound independent validation set of confirmed human metabolites was used to evaluate the relative success of the methodology.
The ANN-v model exceeded the performance of the previous model in the absolute magnitude of the ±2SE filter range. The performance increase is more substantial when comparing the magnitude of the filter range to the overall data range. Figure 6 gives the magnitude of the filter range as a percentage of the RI data range for the previous model and the study models.
Figure 6.
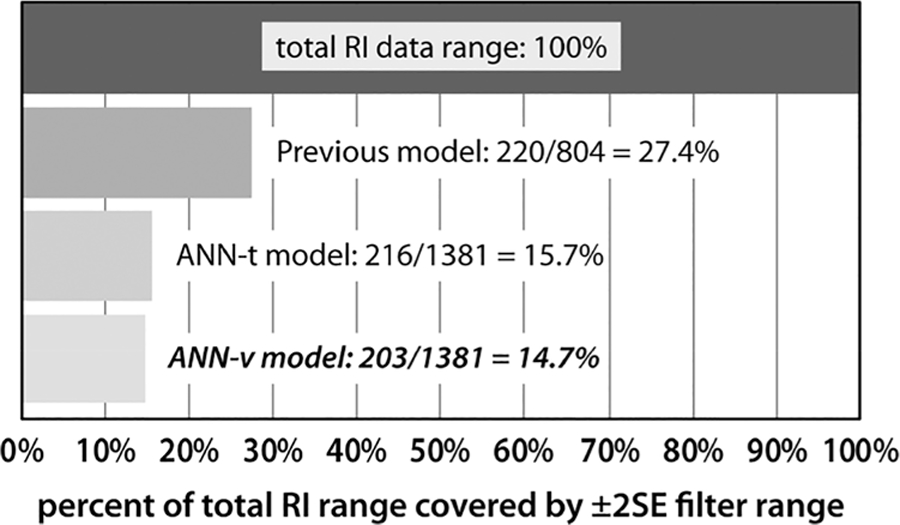
Percent of total RI data range covered by ±2SE filter range for study models and the previous model.19 The absolute magnitude of each data range and filter range are given along with the percent coverage. The bar graph is rendered using percent coverage because the data range varies across the models shown. The top bar illustrates the 100% data range. Each bar underneath represents the percent covered by the filter range of the listed model. The best result is for the ANN-v model where the filter range covers 14.7% of the data range and is 47% smaller than the coverage of the previous model. The percent coverage is an estimate of the effectiveness of each model in filtering out false positives from the exact mass candidate list.
Because the chemical composition of human metabolites is not fully enumerated, an important goal of this investigation was to create a model that could make reasonable predictions for compounds with metabolite-like chemotype combinations not found in the training data. Predictions for the 202 compound i-val set were used to evaluate success in this regard. The measure of effectiveness is the ability of the model to predict compounds as true positives even where the training data has no compounds with a bitkey match. Figure 7 gives the results of this sensitivity and applicability domain analysis for the ANN-v model.
Figure 7.
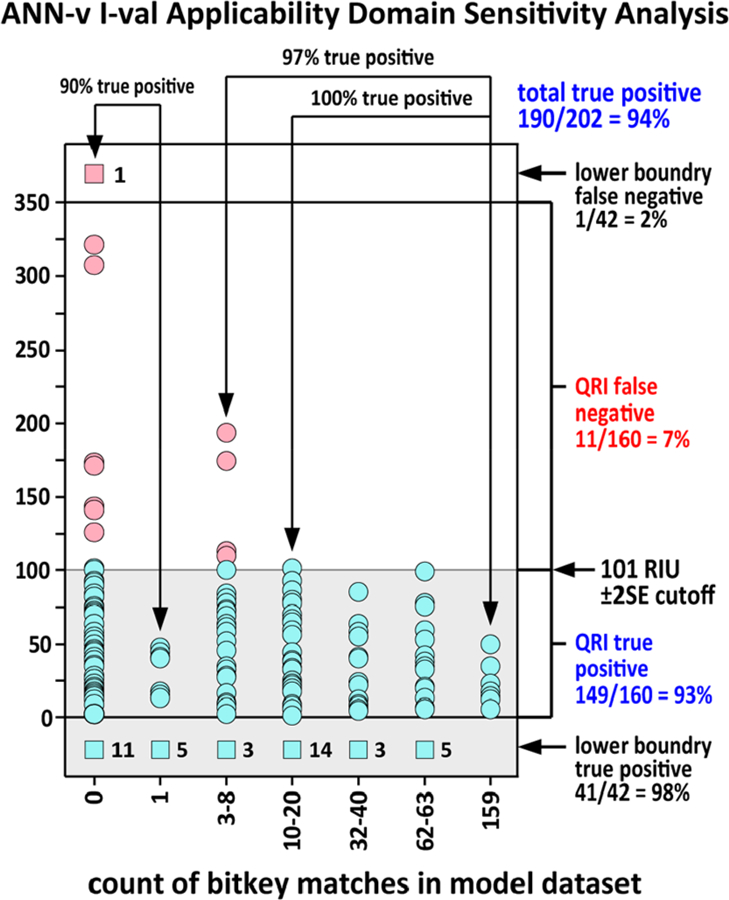
ANN-v bitkey applicability domain analysis for the 202 compound human endogenous metabolite i-val set. The x-axis groups compounds by the number of bitkey matches in the model data set. Prediction absolute error is shown on the y-axis. Green dots in the shaded area are true positive predictions based on the ±2SE filter range. Red dots above the sensitivity cutoff are false negative predictions. The ±2SE sensitivity cutoff is marked on the right. Compounds represented by green boxes at the bottom are true positive predictions for compounds below the QRI lower boundary. The number of true positive predictions is given in the number to the right of the box. The red box at the top of the plot, column 0, represents a false negative prediction for a compound outside the QRI lower boundary. The ANN-v model showed an overall sensitivity of 94%. Sensitivity was 97% when there were three or more compounds in the model data with a bitkey matching the predicted compound and 90% where there were two or fewer compounds in the model data with a matching bitkey.
In Figure 7, true positive predictions are shown in green while false negative predictions are shown in red. Since MolFind uses a ± 2SE filter range, to count as a true positive, the predicted value must fall within ±2SE of the measured value. The ±2SE cutoff is marked as the gray shaded area on the figure. The column on the left shows predictions for 69 compounds with no data set bitkey matches. False negatives are only observed for compounds with 8 or fewer bitkey matches. Sensitivity was 97% for compounds with 3 or more matches. Sensitivity is lowest for compounds with 0 matching bitkeys but is still reasonable at 90%. For the ANN-v model, lower boundary QRI predictions are true positive if the predicted value is <427 RIU. Forty one of the lower boundary QRI predictions were true positive and one was a false negative.
The ANN-v model was able to make reasonable predictions for the majority of compounds that were more complex than those found in the model data. The maximum number of on-bits in the model data was 9. For the ANN-v model, 9 of 10 compounds with 10 or 11 on-bits were predicted as a true positive. Of the 12 total false negatives, only one has 10 on-bits and the rest range from 1 to 8 on-bits with an average of 5. Though the model data consisted of compounds with an average of 3.5 on-bits, the model was able to make useful predictions for compounds with up to 11 on-bits. Though the number of compounds in this category is quite small, 90% sensitivity for compounds more complex than the training data suggests that the method of basing the model on relatively simple compounds has yielded interesting results and did not greatly harm predictions made from more complicated structures.
An analysis of the data displayed in Figure 7 suggests that it would be necessary to use a filter range closer to ±3SE in order to achieve 95% sensitivity for compounds with <2 bitkey matches. Since the bitkey values can be generated for all compounds being screened, it may be helpful in practice to use the ±3SE filter range when making predictions for candidate list compounds with <2 bitkey matches in the model data.
Though the performance of the ANN-v model is encouraging, there are several limitations of this work. The overall model data are skewed toward the bottom end of the RI range with approximately three compounds <750 RIU for every compound >750 RIU. This is in part because a number of purchased compounds anticipated to have large RI values were insufficiently soluble to obtain RI measurements. As a result, the validation SE is higher for compounds in the upper RI range. The existence of an improved model should allow for the identification of additional compounds that can be added to even out the RI distribution.
Because model descriptors were calculated based on the major microspecies and tautomer at pH 2.5 as calculated by Marvin cxcalc, it is likely that some RI predictions were compromised by inaccurate pKa predictions used to generate the major microspecies. Since models based on the neutral form were not nearly as successful, there does not appear to be any simple remedy for this issue other than improvements to the cxcalc software or the identification of a more accurate pKa predictor.
Concerning the model data space, there are a number of observed human metabolites with bitkey values that are absent from the current model data. Though the bitkey coverage of known metabolites is much greater with the models from this study than in the previous model, it would undoubtedly be helpful to obtain compounds to represent observed bitkeys for all known metabolites. Based on the i-val data, 100% sensitivity was only achieved when there were at least 10 bitkey matches in the model data. While good sensitivity was seen with three bitkey matches, the additional population of all bitkeys with <10 examples would likely be helpful.
CONCLUSIONS
The use of a human endogenous metabolite data for independent validation allowed for evaluating the sensitivity of the ANN-v model. The model showed improved specificity when predicting new data as compared to the previous model.19 This would suggest that the combination of the selection process for compounds to expand the model data space, the addition of the lipophilic subgraph descriptors, and the implementation of the QRI ANN learning methodology were collectively useful in advancing one of the major goals of the study. The data generated by this study does not provide insight into the specif icity of the ANN-v model. This is the true negative prediction rate, or in this case, an indication of how effective the model would be in filtering out false positives from the candidate list. The fact that the ±2SE filter range for the best ANN model is 47% smaller than the filter range for the previous model suggests that the new model will be a more effective filter, but this must be confirmed by additional work.
Given the chemical diversity of known and likely metabolites, the reasonable predictions made for compounds with no bitkey matches and for compounds more complex than training data suggest that a model can be built using compounds with simple features and feature combinations and still make predictions for more complex feature combinations that are not represented in the model data. Additional confirmation of this methodology would be significant in the development of global models in general.
It is likely that the model could be further improved through the addition of compounds that satisfy bitkey values from observed human metabolites currently missing from model data. Since there are approximately 3 to 1 compounds in the lower half of the RI data range, it is also likely that improvements could be made by adding compounds to the upper half of the RI range in both the model data and the human endogenous metabolite validation set. Since the model is somewhat dependent on the prediction of the correct major microspecies and tautomer at the mobile phase pH, new pKa prediction software should also be evaluated on an ongoing basis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ritvik Dubey and Janine Johnson for help with data collection.
Funding
This work was funded by The National Institutes of Health Grant GM087714.
ABBREVIATIONS
- HPLC
high performance liquid chromatography
- Tof-MS
time-of-flight mass spectroscopy
- MLR
multiple linear regression
- QRI
quantitative range of interest
- i-val
independent validation set
- MAE
mean absolute error
- SE
standard error
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00496.
S1: sdf file for 1955 ANN model compounds (neutral form). S2: sdf file for 1955 ANN model compounds (major microspecies, tautomer, at pH 2.5). S3: sdf file for 202 compound human endogenous metabolite i-val data set (neutral form). S4: sdf file for 202 compound i-val data set (major microspecies, tautomer, at pH 2.5). S5: text file with table of hydrophilic atom interference coefficients. S6: text file with column header definitions for subsequent data files S7 and S8 (includes descriptor definitions for model indices). S7: text file with ANN-v model data for 1955 compounds (includes compound identification data, experimental values, model predicted values, and model inputs). S8: text file with ANN-v model data for 202 i-val compounds (includes compound identification data, experimental values, model predicted values, and model inputs). S9: excel spreadsheet with ANN model input correlation matrix. S10: results and discussion for of ANN-t (test set optimized) model (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).Naz S; Vallejo M; García A; Barbas C Method Validation Strategies Involved in Non-Targeted Metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [DOI] [PubMed] [Google Scholar]
- (2).Brindle JT; Antti H; Holmes E; Tranter G; Nicholson JK; Bethell HWL; Clarke S; Schofield PM; McKilligin E; Mosedale DE; Grainger DJ Rapid and Noninvasive Diagnosis of the Presence and Severity of Coronary Heart Disease Using 1H-NMR-Based Metabolomics. Nat. Med 2002, 8, 1439–1444. [DOI] [PubMed] [Google Scholar]
- (3).Sreekumar A; Poisson LM; Rajendiran TM; Khan AP; Cao Q; Yu J; Laxman B; Mehra R; Lonigro RJ; Li Y; Nyati MK; Ahsan A; Kalyana-Sundaram S; Han B; Cao X; Byun J; Omenn GS; Ghosh D; Pennathur S; Alexander DC; Berger A; Shuster JR; Wei JT; Varambally S; Beecher C; Chinnaiyan AM Metabolomic Profiles Delineate Potential Role for Sarcosine in Prostate Cancer Progression. Nature 2009, 457, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (4).Tsang TM; Huang JT; Holmes E; Bahn S Metabolic Profiling of Plasma from Discordant Schizophrenia Twins: Correlation Between Lipid Signals and Global Functioning in Female Schizophrenia Patients. J. Proteome Res 2006, 5, 756–760. [DOI] [PubMed] [Google Scholar]
- (5).Woolas RP; Conaway MR; Xu F; Jacobs IJ; Yu Y; Daly L; Davies AP; O’Briant K; Berchuck A; Soper JT; Clarke-Pearson DL; Rodriguez G; Oram DH; Bast RC Jr. Combinations of Multiple Serum Markers are Superior to Individual Assays for Discriminating Malignant from Benign Pelvic Masses. Gynecol. Oncol 1995, 59, 111–116. [DOI] [PubMed] [Google Scholar]
- (6).Yin P; Zhao X; Li Q; Wang J; Li J; Xu G Metabolomics Study of Intestinal Fistulas Based on Ultraperformance Liquid Chromatography Coupled with Q-TOF Mass Spectrometry (UPLC/Q-TOF MS). J. Proteome Res 2006, 5, 2135–2143. [DOI] [PubMed] [Google Scholar]
- (7).Zhang S; Nagana Gowda GA; Asiago V; Shanaiah N; Barbas C; Raftery D Correlative and Quantitative 1H NMR-Based Metabolomics Reveals Specific Metabolic Pathway Disturbances in Diabetic Rats. Anal. Biochem 2008, 383, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang Z; Barnhill SD; Zhang H; Xu F; Yu Y; Jacobs I; Woolas RP; Berchuck A; Madyastha KR; Bast RC Jr. Combination of Multiple Serum Markers Using an Artificial Neural Network to Improve Specificity in Discriminating Malignant from Benign Pelvic Masses. Gynecol. Oncol 1999, 73, 56–61. [DOI] [PubMed] [Google Scholar]
- (9).Zhao X; Wang W; Wang J; Yang J; Xu G Urinary Profiling Investigation of Metabolites with Cis-Diol Structure from Cancer Patients Based on UPLC-MS and HPLC-MS as Well as Multivariate Statistical Analysis. J. Sep. Sci 2006, 29, 2444–2451. [DOI] [PubMed] [Google Scholar]
- (10).Smith CA; Maille G; Want EJ; Qin C; Trauger SA; Brandon TR; Custodio DE; Abagyan R; Siuzdak G METLIN: a Metabolite Mass Spectral Database. Ther. Drug Monit 2005, 27, 747–751. [DOI] [PubMed] [Google Scholar]
- (11).Wishart DS; Tzur D; Knox C; Eisner R; Guo AC; Young N; Cheng D; Jewell K; Arndt D; Sawhney S; Fung C; Nikolai L; Lewis M; Coutouly MA; Forsythe I; Tang P; Shrivastava S; Jeroncic K; Stothard P; Amegbey G; Block D; Hau DD; Wagner J; Miniaci J; Clements M; Gebremedhin M; Guo N; Zhang Y; Duggan GE; MacInnis GD; Weljie AM; Dowlatabadi R; Bamforth F; Clive D; Greiner R; Li L; Marrie T; Sykes BD; Vogel HJ; Querengesser L HMDB: the Human Metabolome Database. Nucleic Acids Res 2007, 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kanehisa M; Goto S KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang Y; Xiao J; Suzek TO; Zhang J; Wang J; Bryant SH PubChem: a Public Information System for Analyzing Bioactivities of Small Molecules. Nucleic Acids Res 2009, 37, W623–W633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Menikarachchi LC; Cawley S; Hill DW; Hall LM; Hall LH; Lai S; Wilder J; Grant DF MolFind: A Software Package Enabling HPLC/MS-Based Identification of Unknown Chemical Structures. Anal. Chem 2012, 84, 9388–9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kertesz TM; Hill DW; Albaugh DR; Hall LH; Hall LM; Grant DF Database Searching for Structural Identification of Metabolites in Complex Biofluids for Mass Spectrometry-Based Metabonomics. Bioanalysis 2009, 1, 1627–1643. [DOI] [PubMed] [Google Scholar]
- (16).Hill DW; Kertesz TM; Fontaine D; Friedman R; Grant DF Mass Spectral Metabonomics Beyond Elemental Formula: Chemical Database Querying by Matching Experimental With Computational Fragmentation Spectra. Anal. Chem 2008, 80, 5574–5582. [DOI] [PubMed] [Google Scholar]
- (17).Hall LM; Hall LH; Kertesz TM; Hill DW; Sharp TR; Oblak EZ; Dong YW; Wishart DS; Chen MH; Grant DF Development of Ecom50 and Retention Index Models for Non-targeted Metabolomics: Identification of 1,3-Dicyclohexylurea in Human Serum by HPLC/Mass Spectrometry. J. Chem. Inf. Model 2012, 52, 1222–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Albaugh DR; Hall LM; Hill DW; Kertesz TM; Parham M; Hall LH; Grant DF Prediction of HPLC Retention Index Using Artificial Neural Networks and IGroup E-State Indices. J. Chem. Inf. Model 2009, 49, 788–799. [DOI] [PubMed] [Google Scholar]
- (19).Hall LM; Hill DW; Menikarachchi LC; Chen MH; Hall LH; Grant DF Optimizing Artificial Neural Network Models for Metabolomics and Systems Biology: an Example Using HPLC Retention Index Data. Bioanalysis 2015, 7, 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hamdalla MA; Mandoiu II; Hill DW; Rajasekaran S; Grant DF BioSM: A Metabolomics Tool for Identifying Endogenous Mammalian Biochemical Structures in Chemical Structure Space. J. Chem. Inf. Model 2013, 53, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hill DW; Baveghems CL; Albaugh DR; Kormos TM; Lai S; Ng HK; Grant DF Correlation of Ecom50 Values Between Mass Spectrometers: Effect of Collision Cell Radiofrequency Voltage on Calculated Survival Yield. Rapid Commun. Mass Spectrom 2012, 26, 2303–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Linusson A; Gottfries J; Lindgren F; Wold S Statistical Molecular Design of Building Blocks for Combinatorial Chemistry. J. Med. Chem 2000, 43, 1320–1328. [DOI] [PubMed] [Google Scholar]
- (23).Linusson A; Elofsson M; Andersson IE; Dahlgren MK Statistical Molecular Design of Balanced Compound Libraries for QSAR. Curr. Med. Chem 2010, 17, 2001–2016. [DOI] [PubMed] [Google Scholar]
- (24).Hall LH; Kier LB; Hall LM ADME-Tox Approaches, Electrotopological State Indices to Assess Molecular Absorption, Distribution, Metabolism, Excretion, and Toxicity. In Comprehensive Medicinal Chemistry II; Taylor JB, Triggle DJ, Eds.; Elsevier: Oxford, 2007; Vol. 5, pp 555–576. [Google Scholar]
- (25).Hall LH; Kier LB; Hall LM Computer Assisted Drug Design, Topological Quantitative Structure-Activity Relationship Applications: Structure Information in Drug Discovery. In Comprehensive Medicinal Chemistry II; Taylor JB, Triggle DJ, Eds.; Elsevier: Oxford, UK, 2007; Vol. 4, pp 555–576. [Google Scholar]
- (26).Hall LH; Kier LB Molecular Structure Description: The Electrotopological State; Academic Press: San Diego, CA, 1999. [Google Scholar]
- (27).Kier LB; Hall LH The Kappa Indices for Modeling Molecular Shape and Flexibility. In Topological Indices and Related Descriptors in QSAR and QSPR; Balaban AT, Devillers J, Eds.; Gordon and Breach: Reading, UK, 1999; pp 455–489. [Google Scholar]
- (28).Hall LH Structure-Information, an Approach to Prediction of Biological Activities and Properties. Chem. Biodiversity 2004, 1, 183–201. [DOI] [PubMed] [Google Scholar]
- (29).Hall LH; Hall LM; Kier LB; Parham ME; Votano JR Interpretation of the Role of the Electrotopological State and Molecular Connectivity Indices in the Prediction of Physical Properties and ADME-Tox Behavior. Case study: Human Plasma Protein Binding. Proceedings of the Solvay Conference, Virtual ADMET Assessment in Target Selection and Maturation; IOS Press, 2006; pp 67–99. [Google Scholar]
- (30).winMolconn, version 1.2.2.1; Hall Associates Consulting: Quincy, MA, 2011. [Google Scholar]
- (31).Marvin Beans Calculator, version 15.8.3.0; ChemAxon: Budapest, Hungary, 2015. [Google Scholar]
- (32).Ghose AK; Crippen GM Atomic Physicochemical Parameters for Three-Dimensional Structure-Assisted Quantitative Structure-Activity Relationships 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Model 1987, 27, 21–35. [DOI] [PubMed] [Google Scholar]
- (33).Hansch C; Leo A; Hoekman DH Exploring QSAR.; Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington D.C., 1995. [Google Scholar]
- (34).Kier LB; Hall LH Molecular Connectivity in Chemistry and Drug Research; Academic Press: New York, 1976. [Google Scholar]
- (35).Kier LB; Hall LH Molecular Connectivity in Structure Activity Analysis; John Wiley: New York, 1986. [Google Scholar]
- (36).Riedmiller M; Braun H A Direct Adaptive Method for Faster Backpropagation Learning: The RPROP Algorithm Proceedings of 1993 IEEE International Conference on Neural Networks (ICNN ‘93), 1993; pp 586–591. [Google Scholar]
- (37).Nissen S Implementation of a fast artificial neural network library (fann); University of Copenhagen (DIKU): Copenhagen, 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.