

SUMMARY
Tuberous sclerosis complex (TSC) is a neurodevelopmental disorder that often presents with psychiatric conditions, including autism spectrum disorder (ASD). ASD is characterized by restricted, repetitive, and inflexible behaviors, which may result from abnormal activity in striatal circuits that mediate motor learning and action selection. To test whether altered striatal activity contributes to aberrant motor behaviors in the context of TSC, we conditionally deleted Tsc1 from direct or indirect pathway striatal projection neurons (dSPNs or iSPNs, respectively). We find that dSPN-specific loss of Tsc1 impairs endocannabinoid-mediated long-term depression (eCB-LTD) at cortico-dSPN synapses and strongly enhances corticostriatal synaptic drive, which is not observed in iSPNs. dSPN-Tsc1 KO, but not iSPN-Tsc1 KO, mice show enhanced motor learning, a phenotype observed in several mouse models of ASD. These findings demonstrate that dSPNs are particularly sensitive to Tsc1 loss and suggest that enhanced corticostriatal activation may contribute to altered motor behaviors in TSC.
Graphical abstract
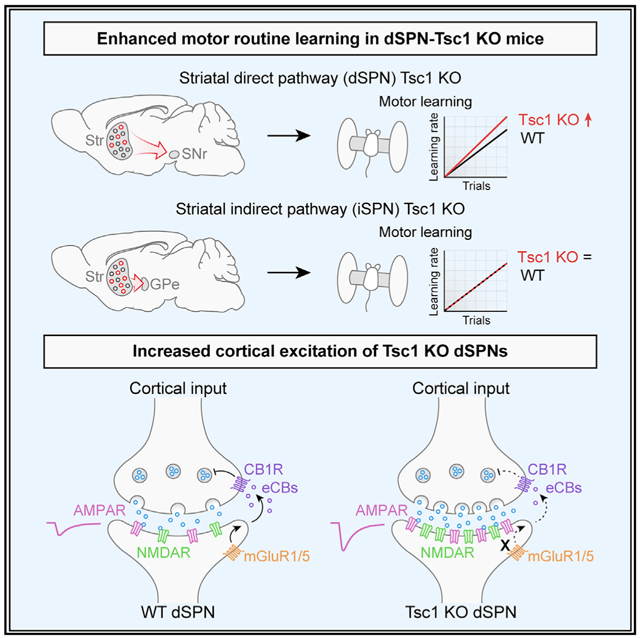
In brief
Benthall et al. show that loss of Tsc1 from striatal direct pathway neurons (dSPNs) impairs synaptic long-term depression, resulting in increased cortically driven firing. Enhanced synaptic transmission is associated with increased motor learning in dSPN-Tsc1 knockout mice. These findings have implications for altered motor behaviors in individuals with Tuberous Sclerosis Complex.
INTRODUCTION
Autism spectrum disorder (ASD) is characterized by social and communication deficits, as well as the presence of restricted and repetitive behaviors (RRBs). Over a hundred genes have been associated with ASD risk (Satterstrom et al., 2020), including genes that cause syndromic disorders, which are associated with a constellation of neurological, psychiatric, and medical conditions (Sztainberg and Zoghbi, 2016). One such disorder is Tuberous Sclerosis Complex (TSC), which is caused by mutations in either TSC1 or TSC2. Up to 50% of individuals with TSC are diagnosed with ASD and most have epilepsy and other behavioral or cognitive disorders (Curatolo et al., 2015; Davis et al., 2015). The TSC1 and TSC2 proteins form a complex that negatively regulates the mTORC1 signaling pathway, a central signaling hub controlling cellular metabolic processes such as protein and lipid synthesis and autophagy (Saxton and Sabatini, 2017). When the TSC1/2 complex is disrupted, mTORC1 signaling is constitutively active, leading to excessive cell growth and altered cellular metabolism (Huang and Manning, 2008). Dysregulation of mTORC1 signaling is not limited to TSC but may occur commonly in ASD (de Vries, 2010; Tang et al., 2014; Kelleher and Bear, 2008; Winden et al., 2018).
While epilepsy in TSC likely arises from altered excitability in forebrain circuits, the brain regions and cell types important for ASD-related behaviors are less well understood. We hypothesized that alterations in striatal circuits, which mediate motor learning, action selection, and habit formation, might contribute to the RRBs observed in TSC patients with ASD. Indeed, striatal alterations have been implicated in ASD by structural and functional MRI studies (Turner et al., 2006; Qiu et al., 2010; Di Martino et al., 2011). Further, in a study of TSC children with and without ASD, striatal metabolism was found to differ specifically in those children with ASD and was correlated with the presence of RRBs (Asano et al., 2001).
Mouse studies support the link between striatal alterations and ASD-related behaviors (Fuccillo, 2016; Li and Pozzo-Miller, 2020; Rothwell, 2016), showing that mutations in ASD-risk genes can endow the striatum with an enhanced ability to acquire fixed motor routines (Rothwell et al., 2014; Platt et al., 2017; Peñagarikano et al., 2011; Nakatani et al., 2009; Kwon et al., 2006; Hisaoka et al., 2018). Changes in striatal synaptic properties have been reported in multiple mouse models with mutations in ASD-risk genes (Fuccillo, 2016; Li and Pozzo-Miller, 2020; Peça et al., 2011; Peixoto et al., 2016). Our group has previously shown input- and cell-type-specific changes in synaptic transmission in striatal neurons with postnatal deletion of Tsc1 (Benthall et al., 2018). Therefore, the goal of this study was to determine whether loss of Tsc1 selectively in striatal neurons is sufficient to alter motor behaviors relevant to ASD.
Striatal function depends on the coordinated activity of two subpopulations of GABAergic principal cells: direct pathway spiny projection neurons (dSPNs) and indirect pathway spiny projection neurons (iSPNs). dSPNs preferentially express D1-type dopamine receptors, while iSPNs express D2-type dopamine receptors and A2A-type adenosine receptors (Gerfen and Surmeier, 2011). dSPNs and iSPNs are distinct in their projection targets, diverging as they leave the striatum to innervate the globus pallidus internal segment (GPi) and substantia nigra pars reticulata (SNr), or the globus pallidus external segment (GPe), respectively (Gerfen and Surmeier, 2011). At the simplest level, bulk activation of dSPNs increases locomotor behavior and action selection, while stimulation of iSPNs inhibits movement (Kravitz et al., 2010; Tai et al., 2012). Coordinated activity between dSPNs and iSPNs allows for the selection of appropriate actions to be performed in a given context, while inappropriate actions are suppressed. Cortical and thalamic inputs make the primary glutamatergic synapses onto dSPNs and iSPNs (Doig et al., 2010; Ding et al., 2008), and modification of the strength of this excitatory drive by long-term synaptic plasticity mediates motor learning and habit formation (Graybiel and Grafton, 2015; Gremel and Lovinger, 2017).
To determine how developmental loss of Tsc1 affects striatal synapses and whether this is sufficient to alter striatal-dependent motor behaviors, we generated mice in which Tsc1 was selectively deleted from either dSPNs or iSPNs. We find that motor routine learning is selectively enhanced in dSPN-Tsc1 KO mice, which also occurs in mice with global haploinsufficiency of Tsc2. Further, we find that Tsc1 deletion causes a non-cell autonomous enhancement of corticostriatal drive of dSPNs but not iSPNs, which is associated with loss of endocannabinoid-mediated long-term depression (eCB-LTD). Together, these findings demonstrate that increased cortical drive of dSPNs resulting from Tsc1 loss is sufficient to enhance the learning of a fixed motor routine.
RESULTS
Upregulation of mTORC1 and somatic hypertrophy in SPNs with Tsc1 deletion
To test whether striatal-specific disruption of the Tsc1/2 complex is sufficient to alter motor behaviors, we generated mice with conditional deletion of Tsc1 from dSPNs or iSPNs (Figures 1A and 1B). We used the EY217 Drd1-Cre founder line from GENSAT to delete Tsc1 from dSPNs as it has a more striatal-restricted expression profile than the commonly used EY262 line when bred to a Cre-dependent tdTomato reporter (Ai9) (Figures S1A and S1B). In preliminary studies, we found that Tsc1fl/fl;Drd1-Cre+(EY262) mice died prematurely, around postnatal day 15. The premature mortality may have been caused by seizures due to cortical Cre expression in this line (Figure S1B), as loss of Tsc1 from a relatively small percentage of cortical neurons is sufficient to induce seizures (Lim et al., 2017). Tsc1fl/fl;Drd1-Cre+(EY217);Ai9+/− mice (referred to as dSPN-Tsc1 KO) did not exhibit premature mortality and had only very sparse tdTomato expression in the cortex (Figures S1C and S1D), as well as in a small number of cells in the cerebellum (Figures S1E and S1F). Notably, Cre expression in Drd1-Cre(EY217);Ai9 mice was highest in the dorsal striatum, with 40%–45% of neurons exhibiting Cre-dependent recombination, whereas less than 8% of neurons in the ventral striatum exhibited Cre recombinase activity (Figures S2A-S2E).
Figure 1. Developmental loss of Tsc1 from striatal projection neurons induces mTORC1 activation.

(A and B) Images of sagittal brain sections showing the expression patterns of a tdTomato Cre reporter (red) in a Drd1-Cre+;Ai9+/− mouse (EY217 GENSAT founder line, A) and an Adora2a-Cre+;Ai9+/− mouse (KG139 GENSAT founder line, B). Right panels show higher magnification images of the dorsal striatum (boxed regions). Neurons are labeled in blue using an anti-NeuN antibody. Str, striatum; SNr, substantia nigra pars reticulata; GP, globus pallidus.
(C–E) Confocal images of dorsolateral striatum from a dSPN-Tsc1 WT (C), Tsc1 Het (D), and Tsc1 KO (E) mouse labeled with antibodies against phosphorylated S6 (p-S6, Ser240/244, green in the merged image) and NeuN (blue in the merged image). TdTomato (red in the merged imaged) is expressed in dSPNs.
(F) Cumulative distributions of dSPN p-S6 fluorescence intensity per cell, expressed as a percentage of wild type (WT). 1,800 cells from 6 dSPN-Tsc1 WT mice, 1,200 cells from 4 dSPN-Tsc1 Het mice, and 1,500 cells from 5 dSPN-Tsc1 KO mice were analyzed. Inset boxplots display the 25%–75%-ile p-S6 per cell by genotype (line at the median, whiskers = min to max). p < 0.0001, Kruskal-Wallis test; WT versus Het, ***p < 0.0001; WT versus KO, ***p < 0.0001, Dunn’s multiple comparisons test.
(G) Bar graphs display the mean ± SEM p-S6 level per mouse for each genotype (calculated from the data in F). Dots represent values for individual mice. p < 0.0001, F (2, 12) = 0.1553, one-way ANOVA; WT versus Het, p = 0.3701; WT versus KO, ***p < 0.0001, Holm-Sidak’s multiple comparisons test.
(H) Cumulative distributions of dSPN soma volume per cell, measured from the same cells as in (F), expressed as a percentage of WT. Inset boxplots display the 25%–75%-ile soma volume per cell by genotype (line at the median, whiskers = min to max). p < 0.0001, Kruskal-Wallis test; WT versus Het, **p = 0.0071; WT versus KO, ***p < 0.0001, Dunn’s multiple comparisons test.
(I) Bar graphs display the mean ± SEM soma volume per mouse for each genotype (calculated from the data in H). Dots represent values for individual mice. p = 0.1927, F (2,12) = 1.030, one-way ANOVA.
(J–L) Confocal images of dorsolateral striatum from an iSPN-Tsc1 WT (J), Tsc1 Het (K), and Tsc1 KO (L) mouse labeled with antibodies against phosphorylated S6 (p-S6, Ser240/244, green in the merged image) and NeuN (blue in the merged image). TdTomato (red in the merged imaged) is expressed in iSPNs.
(M) Cumulative distributions of iSPN p-S6 fluorescence intensity per cell, expressed as a percentage of WT. 1,500 cells from 5 iSPN-Tsc1 WT mice, 1,500 cells from 5 iSPN-Tsc1 Het mice, and 1,800 cells from 6 iSPN-Tsc1 KO mice were analyzed. Inset boxplots display the 25%–75%-ile p-S6 per cell by genotype (line at the median, whiskers = min to max). p < 0.0001, Kruskal-Wallis test; WT versus Het, ***p < 0.0001; WT versus KO, ***p < 0.0001, Dunn’s multiple comparisons test.
(N) Bar graphs display the mean ± SEM p-S6 level per mouse for each genotype (calculated from the data in M). Dots represent values for individual mice. p = 0.0481, F (2, 13) = 10.59, one-way ANOVA; WT versus Het, p = 0.4393; WT versus KO, *p = 0.0368, Holm-Sidak’s multiple comparisons test.
(O) Cumulative distributions of iSPN soma volume per cell, measured from the same cells as in (M), expressed as a percentage of WT. Inset boxplots display the 25%–75%-ile soma volume per cell by genotype (line at the median, whiskers = min to max). p < 0.0001, Kruskal-Wallis test; WT versus Het, *p = 0.0142; WT versus KO, ***p < 0.0001, Dunn’s multiple comparisons test.
(P) Bar graphs display the mean ± SEM soma volume per mouse for each genotype (calculated from the data in O). Dots represent values for individual mice. p = 0.0242, Kruskal-Wallis test; WT versus Het, p > 0.9999; WT versus KO, p = 0.0687, Dunn’s multiple comparisons test.
See also Figures S1 and S2.
To target iSPNs, we used the Adora2a-Cre (KG139) GENSAT mouse line as A2A receptors are enriched in iSPNs and exhibit selective expression in the striatum compared to other brain regions (Schiffmann and Vanderhaeghen, 1993). We found that Adora2a-Cre induced uniform tdTomato expression throughout the dorsal and ventral striatum in approximately 45% of neurons (Figures S2F-S2J). No seizures were observed in Tsc1fl/fl;Adora2a-Cre+;Ai9+/− mice (referred to as iSPN-Tsc1 KO).
To confirm that Tsc1 deletion upregulated mTORC1 signaling in dSPNs and iSPNs, we quantified the phosphorylation of ribosomal protein S6, which is a commonly used readout of mTORC1 activity. Loss of Tsc1 caused a gene dose-sensitive increase in p-S6 levels in dorsal striatal dSPNs and iSPNs, indicating mTORC1 pathway hyperactivity in both cell types (Figures 1C-1G and 1J-1N). Prior studies have demonstrated pronounced somatic hypertrophy in Tsc1 KO neurons in various brain regions, consistent with the known role of mTORC1 in regulating cell size (Bateupet al., 2011; Kosillo et al., 2019; Normand et al., 2013; Tsai et al., 2012; Feliciano et al., 2011; Malik et al., 2019; Tavazoie et al., 2005). However, in the striatum, we observed a relatively modest increase in soma volume in dSPN- and iSPN-Tsc1 KO mice (Figures 1H and 1I and 1O and 1P). dSPN-Tsc1 Het neurons had a small increase in soma volume compared to wild-type (WT) while iSPN-Tsc1 Het neurons had slightly reduced soma volume (Figures 1H and 1O). Together these results show that complete loss of Tsc1 from SPNs results in robust activation of mTORC1 signaling leading to moderate somatic hypertrophy. Heterozygous loss of Tsc1 mildly increases mTORC1 activity in dSPNs and iSPNs but does not strongly affect soma size.
Loss of Tsc1 from dSPNs, but not iSPNs, enhances motor routine learning
To determine whether SPN-specific loss of Tsc1 was sufficient to alter motor behaviors, we investigated general locomotor activity and self-grooming behavior in the open field. We found no significant differences in the total distance traveled, number of rears, or grooming bouts in dSPN- or iSPN-Tsc1 Het or KO mice compared to controls (Figures S3A-S3F). These results suggest that loss of Tsc1 from a single SPN subtype is not sufficient to alter gross motor behavior or induce spontaneous stereotypies. A summary of the behavior test results by genotype and sex is shown in Table S1.
The accelerating rotarod is a striatal-dependent motor learning assay (Yin et al., 2009) that is altered in multiple mouse models with mutations in ASD-risk genes (Fuccillo, 2016). In this test, mice learn to run on a rod revolving at increasing speed over four days of training with three trials performed each day (Rothwell et al., 2014). Over the course of training, mice develop a stereotyped motor routine to stay on the apparatus for increasing amounts of time and thus reach higher terminal velocities on later trials. We found that dSPN-Tsc1 Het and KO mice had similar initial rotarod performance as WT littermates on the first trial, reflecting normal baseline motor coordination (Figures 2A and 2B). However, dSPN-Tsc1 KO mice exhibited significantly enhanced motor learning measured by the slope of performance from the first to last trial for each mouse, compared to littermate controls (Figure 2C). dSPN-Tsc1 Het animals also displayed a mild enhancement of motor learning consistent with a gene dose-dependent effect (Figure 2A). The enhancement in rotarod performance was most pronounced for the more challenging 10–80 rpm acceleration trials. Across these trials (trials #7–12), 72% and 70% of dSPN-Tsc1 Het and KO mice, respectively, displayed continued improvement indicated by a positive slope of their learning curve (Figure 2D). This contrasted with the ∼50% of dSPN-Tsc1 WT mice that showed improvement across trials 7–12, reflecting a near ceiling level of performance by trial 7 (Figure 2D). These results were unlikely to be driven by changes in weight as this was not significantly different between genotypes (dSPN-Tsc1 WT male = 34.55 ± 2.42 g, Het male = 33.11 ± 1.65 g, KO male = 29.20 ± 1.35 g, p = 0.3453, Kruskal-Wallis test; dSPN-Tsc1 WT female = 31.30 ± 1.99 g, Het female = 26.50 ± 1.22 g, KO female = 25.44 ± 1.77 g; p = 0.0910, Kruskal-Wallis test). Further, we found that weight was not strongly correlated with rotarod learning rate for either sex (Figures S3G and S3H). We examined rotarod performance in mice with loss of Tsc1 in iSPNs, and found no difference in either initial motor performance, learning rate, or performance on the 10–80 rpm trials between genotypes (Figures 2E-2H). Together these results show that loss of Tsc1 from dSPNs, but not iSPNs, leads to enhanced motor routine learning.
Figure 2. Mutations in Tsc1 or Tsc2 enhance motor routine learning.

(A) Accelerating rotarod learning curve across 12 trials (4 days) in dSPN-Tsc1 WT (n = 30), Het (n = 25), and KO (n = 29) mice. Circles represent mean ± SEM. Repeated-measures two-way ANOVA p values are shown; geno F (4, 110) = 7.034, trial F (5.092, 560.1) = 105.8, g × t F (44, 120) = 1.987. RPM, revolutions per minute.
(B) Rotarod performance on trial 1 for dSPN-Tsc1 WT, Het, and KO mice quantified as terminal speed. Bars represent mean ± SEM. Dots represent values for individual mice (n is the same as in A). p = 0.7233, Kruskal-Wallis test.
(C) Overall learning rate of dSPN-Tsc1 WT, Het, and KO mice calculated as the slope of the line of performance on the first trial (1) to the last trial (12) for each mouse (RPM/day). Bars represent mean ± SEM. Dots represent values for individual mice (n is the same as in A). p = 0.0120, Kruskal-Wallis test; dSPN WT versus KO, **p = 0.0071; dSPN WT versus Het, p = 0.1170, Dunn’s multiple comparisons test.
(D) Percentage of dSPN-Tsc1 WT, Het, and KO mice with a positive learning curve (slope of performance) from trial 7 to trial 12 (10–80 RPM acceleration).
(E) Accelerating rotarod learning curve across 12 trials (4 days) in iSPN-Tsc1 WT (n = 34), Het (n = 13), and KO (n = 18) mice. Dots represent mean ± SEM. Repeated-measures two-way ANOVA p values are shown; geno F (2, 62) = 0.1839, trial F (4.935, 306) = 52.73, g × t F (22, 682) = 1.118.
(F) Rotarod performance on trial 1 for iSPN-Tsc1 WT, Het, and KO mice quantified as terminal speed. Bars represent mean ± SEM. Dots represent individual mice (n is the same as in E). p = 0.6737, Kruskal-Wallis test.
(G) Overall learning rate of iSPN-Tsc1 WT, Het, and KO mice calculated as the slope of the line of performance on the first trial to the last trial for individual mice (RPM/day). Bars represent mean ± SEM. Dots represent individual mice (n is the same as in E). p = 0.2761, Kruskal-Wallis test.
(H) Percentage of iSPN-Tsc1 WT, Het, and KO mice with a positive learning curve (slope of performance) from trial 7 to trial 12 (10–80 RPM acceleration).
(I) Accelerating rotarod learning curve across 12 trials (4 days) in global Tsc2 WT (n = 17) and Tsc2 Het (n = 31) mice. Circles represent mean ± SEM. Repeated-measures two-way ANOVA p values are shown; geno F (1, 46) = 2.910, trial F (5.997, 275.9) = 40.22, g × t F (11, 506) = 2.639.
(J) Rotarod performance on trial 1 for Tsc2 WT and Het mice quantified as terminal speed. Bars represent mean ± SEM. Dots represent individual mice (n is the same as in I). p = 0.2533, two-tailed unpaired t test.
(K) Overall learning rate of Tsc2 WT and Het mice calculated as the slope of the line of performance on the first trial to the last trial for individual mice (RPM/day). Bars represent mean ± SEM. Dots represent individual mice (n is the same as in I). **p = 0.0057, Mann-Whitney test.
(L) Percentage of Tsc2 WT and Het mice with a positive learning curve (slope of performance) from trial 7 to trial 12 (10–80 RPM acceleration).
To rule out that the motor learning phenotype in dSPN-Tsc1 KO mice was an anomaly due to cell-type-specific loss of Tsc1, we tested mice that were heterozygous for a germline loss-of-function mutation in Tsc2 (Onda et al., 1999). Heterozygous mice were used as germline homozygous deletion of Tsc2 is embryonic lethal (Onda et al., 1999). Similar to dSPN-Tsc1 KO mice, Tsc2+/− mice had normal initial motor performance but exhibited a significant increase in learning rate compared to WT littermates that became apparent in the more challenging trials of the test (Figures 2I-2L). Again, this was not likely due to a change in weight as this was not significantly different between genotypes (Tsc2 WT male = 27.41 ± 0.63 g versus Tsc2 Het male = 25.45 ± 0.47 g, p = 0.0637, Mann-Whitney test; Tsc2 WT female = 19.53 ± 0.55 g versus Tsc2 Het female = 19.53 ± 0.65 g; p = 0.9989, unpaired t test). These findings demonstrate that increased motor learning can be induced by even partial disruption of the Tsc1/2 complex and that loss of Tsc1 from dSPNs alone is sufficient to drive this change.
Loss of Tsc1 increases cortical drive of dSPNs
Motor learning relies on corticostriatal transmission; therefore, changes in SPN response to cortical activity could account for enhanced motor routine acquisition in dSPN-Tsc1 KO mice. To test this, we crossed the Tsc1;Drd1-Cre;Ai9 and Tsc1;Adora2a-Cre;Ai9 mice to the Thy1-ChR2-YFP mouse line, which expresses channelrhodopsin (ChR2-YFP) in a subset of cortical layer V pyramidal cells (Arenkiel et al., 2007; Figure 3A). To simulate a train of cortical inputs, we applied ten light pulses over the recording site in dorsolateral striatum and recorded responses in SPNs in the absence of any synaptic blockers (Figure 3B). By varying the intensity of cortical stimulation, either subthreshold depolarizations or action potentials (APs) could be elicited with a given probability. We quantified the percentage of stimuli that evoked APs at different light intensities and found that cortical terminal stimulation resulted in significantly increased spike probability in dSPN-Tsc1 KO cells compared to WT (Figures 3C and 3D; Figure S4). Notably, loss of one copy of Tsc1 in dSPNs was sufficient to increase corticostriatal drive as dSPN-Tsc1 Het neurons also showed enhanced cortically driven spiking (Figures 3C and 3D).
Figure 3. Tsc1 loss selectively increases cortico-dSPN synaptic drive.

(A) Confocal image of a sagittal brain section from a Tsc1wt/fl;Thy1-ChR2-EYFP+;Drd1-Cre+;Ai9+/− mouse. Right image shows cortical terminals expressing ChR2-EYFP (green) in the dorsal striatum. tdTomato (red) labels dSPNs. DAPI stained nuclei are in blue.
(B) Schematic of the experiment. Cortical terminals expressing ChR2 were stimulated with 10 pulses of blue light at 20 Hz and responses were recorded from dSPNs in dorsolateral striatum.
(C) Example traces of action potentials (AP) in dSPNs evoked by cortical terminal stimulation at 10% light intensity for the indicated genotypes.
(D) Quantification (mean ± SEM) of the percentage of cortical terminal stimuli that evoked APs in dSPNs. dSPN-Tsc1 WT n = 5 neurons from 2 mice, dSPN Het n = 7 neurons from 2 mice, dSPN KO n = 5 neurons from 2 mice. Mixed-effects model (REML) p values are shown; geno F (2, 14) = 12.25, stim F (2.125, 26.35) = 45.79, g × s (10, 62) = 2.164.
(E) Example traces of APs in iSPNs evoked by cortical terminal stimulation at 10% light intensity for the indicated genotypes.
(F) Quantification (mean ± SEM) of the percentage of cortical terminal stimuli that evoked APs in iSPNs. iSPN-Tsc1 WT n = 10 neurons from 3 mice, iSPN Het n = 10 neurons from 3 mice, iSPN KO n = 7 neurons from 3 mice. Repeated-measures two-way ANOVA p values are shown; geno F (2, 24) = 0.4633, stim F (2.464, 59.13) = 198.6, g × s F (12, 144) = 0.4655.
(G) Example traces of APs in dSPNs evoked by a 350 pA current step for the indicated genotypes.
(H) Quantification of the number of APs induced by depolarizing current steps in dSPN-Tsc1 WT, Het, and KO neurons. Circles represent mean ± SEM dSPN WT n = 13 neurons from 3 mice, dSPN Het n = 6 neurons from 2 mice, dSPN KO n = 11 neurons from 3 mice. Mixed-effects model (REML) p values are shown; geno F (2, 27) = 1.794, curr F (2.2127, 57.08) = 95.26, g × c F (24, 322) = 1.572.
(I) Example traces show EPSCs induced by optogenetic corticostriatal stimulation at different light intensities (5%–20%) in dSPN-Tsc1 WT, Het, and KO neurons.
(J) Quantification (mean ± SEM) of corticostriatal EPSC amplitude in dSPN-Tsc1 WT, Het, and KO neurons induced by different light intensities (0.5 ms pulse width). dSPN-Tsc1 WT n = 18 neurons from 8 mice, dSPN Het n = 7 neurons from 3 mice, dSPN KO n = 22 neurons from 10 mice. Repeated-measures two-way ANOVA p values are shown; geno F (2, 44) = 3.376, stim F (1.590, 69.98) = 119.4, g × s F (4, 88) = 4.597.
(K) Example traces show pairs of EPSCs evoked by optogenetic corticostriatal stimulation (5% light intensity) recorded at +40 mV (top traces) and −70 mV (bottom traces) from dSPN-Tsc1 WT, Het, and KO neurons.
(L) Quantification of AMPA:NMDA ratio per cell in dSPN-Tsc1 WT, Het, and KO neurons evoked by 5% light stimulation. Bars represent mean ± SEM. Dots represent values for individual neurons. dSPN-Tsc1 WT n = 14 neurons from 8 mice, dSPN Het n = 13 neurons from 5 mice, dSPN KO n = 12 neurons from 8 mice. p = 0.5765, F (2, 36) = 2.759, one-way ANOVA.
See also Figures S4 and S5.
We performed the cortical stimulation experiment in iSPNs and found that WT iSPNs were more readily driven to spike than dSPNs, consistent with their increased intrinsic excitability (Gertler et al., 2008; Benthall et al., 2018). Therefore, we reduced the length of the light pulse to avoid saturating the response. The number of cortically driven APs in iSPNs was not different between genotypes at any light intensity (Figures 3E and 3F). These results are consistent with the behavioral results and demonstrate that deletion of Tsc1 has a selective impact on dSPNs, while iSPNs are remarkably unaffected.
Increased cortico-dSPN excitability results from enhanced synaptic transmission
Given that dSPN-Tsc1 Het and KO neurons exhibited a clear enhancement of corticostriatal excitability that was not observed in iSPNs, we further analyzed dSPNs to investigate the potential mechanism for this change. Enhanced cortical drive of dSPN-Tsc1 Het and KO neurons could result from increased intrinsic membrane excitability, increased synaptic excitation, or both. To test whether changes in intrinsic excitability occurred in dSPN-Tsc1 KO neurons, we injected positive current and measured the number of APs fired as a function of current step amplitude. The input-output curve for dSPN-Tsc1 KO cells was shifted slightly to the right relative to dSPN-Tsc1 WT and Het neurons, indicating a small decrease in intrinsic membrane excitability (Figures 3G and 3H).
Intrinsic hypoexcitability has been observed in other neuron types with Tsc1 loss (Normand et al., 2013; Bateup et al., 2013; Kosillo et al., 2019; Tsai et al., 2012; Yang et al., 2012) and may result from changes in cell size, which impact passive membrane properties. However, we did not observe significant changes in membrane resistance or capacitance in dSPN-Tsc1 Het or KO cells (Figures S5A and S5B), which may reflect the relatively small changes in soma size in SPNs (see Figure 1). Since changes in intrinsic properties could not account for the enhanced corticostriatal excitability observed in dSPNs, we measured synaptic excitability by recording AMPA receptor (AMPAR)-mediated excitatory post-synaptic currents (EPSCs) evoked by optical stimulation of cortical terminals. We observed significantly larger EPSCs in dSPN-Tsc1 Het and KO cells compared to WT, particularly at higher light intensities (Figures 3I and 3J). When we measured the ratio of AMPAR currents recorded at −70 mV to NMDAR currents recorded at +40 mV (with 5% light power), we found no significant differences among genotypes (Figures 3K and 3L). These results suggest a general enhancement of excitatory synaptic transmission onto Tsc1 KO dSPNs rather than a selective potentiation of AMPAR-mediated responses.
To investigate the specific synaptic properties that were altered in dSPN-Tsc1 KO and Het neurons, we recorded miniature excitatory post-synaptic currents (mEPSCs) at 6 weeks of age and found a small increase in the amplitude and large increase in the frequency of mEPSCs onto dSPN-Tsc1 KO cells compared to WT (Figures 4A-4C). A significant increase in mEPSC frequency was also observed in dSPN-Tsc1 Het cells (Figure 4C). To determine the developmental timing of these changes, we recorded mEPSCs in dSPN-Tsc1 WT and KO neurons at 2, 3, and 4 weeks of age. We found that the increased mEPSC amplitude and frequency in dSPN-Tsc1 KO neurons did not emerge until 4 weeks of age, a time when corticostriatal circuits are maturing and becoming refined (Kuo and Liu, 2019; Peixoto et al., 2016; Figures S5C and S5D). Thus, loss of Tsc1 may not affect the initial development of excitatory synapses but could affect their activity-dependent refinement. Consistent with a lack of cortico-iSPN excitability change, no significant changes in mEPSC amplitude or frequency were observed in iSPN-Tsc1 Het or KO cells at 6 weeks of age (Figures 4D-4F).
Figure 4. Loss of Tsc1 increases excitatory synaptic transmission onto dSPNs.

(A) Example traces of miniature excitatory post-synaptic currents (mEPSCs) recorded from dSPN-Tsc1 WT, Het, and KO neurons.
(B and C) Mean ± SEM mEPSC amplitude (B) and frequency (C) recorded from dSPN-Tsc1 WT, Het, and KO neurons. Dots represent individual neurons. For (B), p = 0.0337, F (2, 38) = 3.712, one-way ANOVA; p = 0.0766 (dSPN WT versus Het), *p = 0.0217 (dSPN WT versus KO), Holm-Sidak’s multiple comparisons tests. For (C), p = 0.0014, F (2, 38) = 7.822, one-way ANOVA; **p = 0.0049 (dSPN WT versus Het), **p = 0.0011 (dSPN WT versus KO), Holm-Sidak’s multiple comparisons tests. For both (B) and (C), dSPN WT n = 13 neurons from 3 mice, dSPN Het n = 15 neurons from 3 mice, dSPN KO n = 13 neurons from 3 mice.
(D) Example traces of mEPSCs recorded from iSPN-Tsc1 WT, Het, and KO neurons.
(E and F) Mean ± SEM mEPSC amplitude (E) and frequency (F) recorded from iSPN-Tsc1 WT, Het, and KO neurons. Dots represent individual neurons. For (E), p = 0.2942, F (2, 42) = 1.260, one-way ANOVA. For (F), p = 0.4083, F (2, 42) = 0.9151, one-way ANOVA. For both (E) and (F), iSPN WT n = 16 neurons from 3 mice, iSPN Het n = 15 neurons from 3 mice, iSPN KO n = 14 neurons from 3 mice.
(G and H) Confocal images of individual Tsc1 WT (G) and KO (H) dSPNs labeled with tdTomato.
(I and J) Representative images of dendritic spines from a Tsc1 WT (I) and KO (J) dSPN.
(K) Quantification (mean ± SEM) of dendritic spine density per 10 μm of dendrite in Tsc1 WT and KO dSPNs. Dots represent the average spine density for individual neurons. dSPN WT n = 6 neurons (12 dendrites) from 3 mice, dSPN KO n = 6 neurons (13 dendrites) from 3 mice. p = 0.8182, Mann-Whitney test.
(L) Electrical stimulation (50 ms ISI) was used to evoke pairs of excitatory synaptic currents in Tsc1 WT and KO dSPNs. Paired pulse ratio (PPR) was measured as the amplitude of the second response divided by the first response. Bars represent mean ± SEM, dots represent individual neurons. dSPN WT n = 28 neurons from 10 mice, dSPN KO n = 24 neurons from 9 mice. **p = 0.0038, two-tailed unpaired t test.
See also Figure S5.
While changes in mEPSC amplitude usually reflect increased post-synaptic AMPAR content, increased mEPSC frequency can result from a greater number of synaptic contacts or a change in presynaptic release probability. To measure synapse number, we sparsely labeled dSPNs in the dorsal striatum using an AAV expressing Cre-dependent tdTomato. We imaged and reconstructed individual dSPNs and quantified the density of dendritic spines, which are the sites of corticostriatal synapses onto SPNs. We found that dSPN-Tsc1 KO neurons had equivalent spine density to dSPN-Tsc1 WT cells (Figures 4G-4K), suggesting that the increased mEPSC frequency was due to a change in presynaptic release probability. To investigate this, we measured the paired pulse ratio (PPR) of AMPAR currents evoked by two electrical stimuli delivered 50 ms apart. The stimulating electrode was placed in or just above the corpus callosum to preferentially activate cortical inputs (Ding et al., 2008; Assous et al., 2019). We found a significant decrease in PPR in dSPN-Tsc1 KO cells compared to WT, consistent with increased presynaptic release probability of cortical inputs onto dSPNs (Figure 4L).
Endocannabinoid-mediated long-term depression is impaired in dSPN-Tsc1 KO neurons
One mechanism that could explain enhanced presynaptic corticostriatal transmission onto dSPN-Tsc1 KO neurons is a loss of long-term depression (LTD), which would render cells unable to depress excitatory inputs. Corticostriatal terminals express CB1 receptors that mediate endocannabinoid-LTD (eCB-LTD), a prominent form of striatal synaptic depression (Kreitzer and Malenka, 2008; Lovinger, 2010). Upon coincident activation of group 1 metabotropic glutamate receptors (mGluR1/5) and L-type calcium channels, SPNs release the endocannabinoids anandamide (AEA) or 2-arachidonoylglycerol (2-AG), which act as retrograde signals that activate presynaptic CB1Rs to decrease corticostriatal release probability (Lüscher and Huber, 2010). It was previously thought that eCB-LTD occurs primarily in iSPNs (Kreitzer and Malenka, 2007), but recent studies using selective stimulation of corticostriatal terminals have revealed eCB-LTD in both SPN subtypes (Wu et al., 2015). Since another form of mGluR-dependent LTD expressed in the hippocampus is impaired in multiple TSC mouse models (Bateup et al., 2011; Auerbach et al., 2011; Chévere-Torres et al., 2012; Potter et al., 2013), and selective disruption of 2-AG release from dSPNs causes increased glutamatergic transmission (Shonesy et al., 2018), we reasoned that loss of eCB-LTD could occur in dSPNs with Tsc1 deletion.
We induced eCB-LTD in dSPNs with wash-on of the group 1 mGluR agonist DHPG and monitored the amplitude of EPSCs in response to optogenetic stimulation of cortical terminals in the dorsolateral striatum. Stimulation strength was adjusted for each cell to evoke 500–700 pA currents during the baseline period. While dSPN-Tsc1 WT neurons showed a long-lasting synaptic depression to ∼79% of baseline levels, eCB-LTD did not consistently occur in dSPN-Tsc1 KO neurons, which exhibited only a small initial reduction in EPSC amplitude during DHPG application that was not maintained (Figure 5A). dSPN-Tsc1 Het cells exhibited an intermediate level of eCB-LTD (Figure S6A), consistent with their intermediate increase in mEPSC frequency and corticostriatal drive (see Figures 3 and 4). To ensure that the protocol was inducing eCB-dependent LTD, we washed on DHPG in the presence of AM-251, a CB1R antagonist, and found that LTD was blocked in control dSPNs (Figure S6B). The deficit in eCB-LTD in Tsc1-KO dSPNs could not be explained by a difference in presynaptic CB1R function, as direct activation of CB1Rs with the agonist WIN-2 depressed corticostriatal EPSCs to a similar extent in Tsc1 KO and WT dSPNs (Figure 5B).
Figure 5. dSPN-Tsc1 KO neurons have impaired eCB-LTD.

(A) Corticostriatal terminals in the dorsolateral striatum were stimulated with blue light to evoke EPSCs in dSPN-Tsc1 WT and KO neurons. DHPG (100 μM) was washed on for 10 min to induce eCB-LTD. Data are presented as mean ± SEM percentage of baseline (dashed line). Repeated-measures two-way ANOVA p values are shown; geno F (1, 10) = 15.93, time F (3.479, 34.79) = 7.993, g × t F (25, 250) = 2.563. dSPN WT n = 7 neurons from 3 mice, dSPN KO n = 5 neurons from 4 mice. Example traces on the right show the average EPSCs from the baseline period (“1”) and 35–40 min after DHPG application (“2”) for each genotype.
(B) Corticostriatal terminals were stimulated with blue light to evoke EPSCs in dSPN-Tsc1 WT and KO neurons. WIN-2 (2 μM) was applied at time 0 to activate CB1 receptors. Data are presented as mean ± SEM percentage of baseline (dashed line). Mixed-effects model (REML) p values are shown; geno F (1, 4) = 0.2433, time F (2.410, 9.641) = 29.36, g × t F (1.551, 4.329) = 0.5214. dSPN WT n = 5 neurons from 4 mice, dSPN KO n = 4 neurons from 3 mice. Example traces on the right show the average EPSC from the last 5 min of the baseline period (“1”) and 19–24 min after WIN-2 application (“2”).
See also Figure S6.
To investigate a potential molecular basis for the loss of eCB-LTD in Tsc1 KO dSPNs, we performed translating ribosome affinity purification (TRAP) to assess the translational status of mRNAs encoding key proteins involved in eCB-LTD. To do this, we engineered an AAV to express a Cre-dependent GFP-tagged ribosomal protein (EGFP-L10a) and injected this into the dorsal striatum of Drd1-Cre+ mice that were WT or homozygous floxed for Tsc1 (Figures 6A and 6B). This approach enabled selective expression of EGFP-L10a in dSPNs (Figure 6C) and isolation of ribosome-bound mRNAs from dSPNs using TRAP (Heiman et al., 2008, 2014). We verified the specificity of this approach using qPCR to quantify the relative levels of Drd1 (D1 receptor) and Drd2 (D2 receptor) mRNA in the TRAP samples compared to the unbound samples, which include RNA from all striatal cell types. As expected, Drd1 mRNA was significantly enriched in the TRAP samples from both dSPN-Tsc1 WT and dSPN-Tsc1 KO mice compared to the unbound samples (Figure 6D). Accordingly, Drd2 mRNA was depleted from the TRAP samples, demonstrating preferential isolation of mRNA from dSPNs (Figure 6E).
Figure 6. dSPN Tsc1 KO cells exhibit reduced ribosome engagement of mRNAs involved in mGluR-eCB signaling.

(A) Schematic of the design of AAV-FLEX-TRAP, which allows Cre-dependent expression of a GFP-tagged ribosomal subunit (EGFP-L10a).
(B) Schematic of the workflow for the Translational Ribosome Affinity Purification (TRAP) experiment.
(C) Images of EGFP-L10a expression from an AAV5-hSyn-DIO-EGFP-L10a injection into the dorsolateral striatum of a Tsc1wt/wt;Drd1-Cre+;Ai9+/− mouse showing expression in tdTomato+ dSPNs. In the merged image, EGFP-L10 is in green, tdTomato is in red, and NeuN immunostaining is in blue to label neurons.
(D and E) Quantification of relative Drd1 (D) and Drd2 (E) mRNA levels in TRAP samples (ribosome bound mRNA isolated from dSPNs) versus the unbound (“UN,” all striatal tissue) from the same mouse measured by qPCR. Bars represent mean ± SEM. Dots represent individual samples, taken from one mouse. For (D), ***p = 0.0009 (dSPN WT); **p = 0.0053 (dSPN KO), paired t tests. For (E), ***p = 0.0003 (dSPN WT); ***p = 0.0007 (dSPN KO), paired t tests. For (D) and (E), dSPN-Tsc1 WT n = 10 and dSPN-Tsc1 KO n = 7 mice.
(F–J) Quantification of Grm1 (F), Grm5 (G), Homer1 (H), Plcb1 (I), and Drd1 (J) mRNA levels in TRAP samples (ribosome bound mRNA isolated from dSPNs) relative to Actb from dSPN-Tsc1 WT and KO mice measured by qPCR. Bars represent mean ± SEM and dots represent individual mice. For (F), **p = 0.0022; (G), **p = 0.0018; (H), *p = 0.0346; (I), **p = 0.0025; (J), p = 0.9702; two-tailed unpaired t tests. For all panels, dSPN-Tsc1 WT n = 10 and dSPN-Tsc1 KO n = 8 mice (except for Drd1, dSPN KO n = 7 mice).
We compared the relative expression levels of mRNAs encoding proteins required for eCB-LTD including the two types of group 1 mGluRs (Grm1 and Grm5), the requisite mGluR scaffold protein Homer 1 (Homer1), and phospholipase C beta (Plcb1), which promotes the formation of diacylglycerol that is subsequently converted to 2-AG by diacylglycerol lipase (Ohno-Shosaku and Kano, 2014). We also compared the levels of ribosome-bound Drd1 as a control. We found that dSPN-Tsc1 KO mice had increased amounts of total ribosome-bound mRNA (0.0518 ± 0.0077 versus 0.0313 ± 0.0031 ratio of TRAP-isolated mRNA to unbound RNA in dSPN KO versus WT mice, p = 0.0138, unpaired t test), consistent with a global increase in protein synthesis resulting from high mTORC1 activity. However, all four components of the eCB-LTD pathway showed a relative reduction in ribosome-bound mRNA levels in dSPN-Tsc1 KO mice compared to WT when normalized to Actb (B-Actin) (Figures 6F-6I). Notably, levels of Drd1 mRNA were not significantly different between genotypes (Figure 6J). The amount of ribosome-bound mRNA is generally thought to reflect translation efficiency (Gobet and Naef, 2017); therefore, these results suggest a relative downregulation in the translation of multiple mRNAs encoding proteins required for eCB-LTD. Such decreased expression may contribute to the lack of functional eCB-LTD in Tsc1 KO dSPNs.
DISCUSSION
In this study, we tested whether cell-type-specific deletion of Tsc1 from striatal neurons was sufficient to alter synaptic function and motor behaviors. We found that in both direct and indirect pathway SPNs, developmental loss of Tsc1 upregulated mTORC1 signaling and modestly increased soma size. However, we found that mTORC1 activation in dSPNs, but not iSPNs, enhanced motor routine learning in the absence of spontaneous stereotypies or locomotor hyperactivity. Further, we found that loss of Tsc1 from dSPNs was associated with increased corticostriatal synaptic excitability and an impairment in eCB-LTD. Notably, loss of one copy of Tsc1 was sufficient to increase cortical drive of dSPNs and enhance motor learning. These findings implicate the striatal direct pathway as a possible driver of altered motor behaviors in TSC.
It has been shown that loss of Tsc1 in either the cerebellum or thalamus is sufficient to cause social behavior deficits and spontaneous RRBs including repetitive self-grooming (Tsai et al., 2012; Normand et al., 2013). Given the central role of the striatum in action selection and motor learning, we hypothesized that alterations in striatal circuits might also contribute to altered motor behaviors in mouse models of TSC. Indeed, siRNA-mediated knockdown of Tsc1 in the dorsal striatum has been shown to induce behavioral changes (Lee et al., 2018). Here, we found that mice with loss of Tsc1 in dorsal striatal dSPNs, but not iSPNs, had enhanced performance on the accelerating rotarod, a motor learning task that relies on corticostriatal transmission (Yin et al., 2009; Kupferschmidt et al., 2019). Previous work has shown that striatal mTORC1 signaling is required for motor learning on the accelerating rotarod, as both pharmacological and genetic intrastriatal inhibition of mTOR signaling impairs rotarod learning (Bergeron et al., 2014). Our results are congruent with these findings and show that increasing mTORC1 activity in dSPNs enhances rotarod performance. Notably, an extended accelerating rotarod task may be necessary to reveal motor learning phenotypes, as Tsc1 and Tsc2 heterozygous mice were reported to have no changes in rotarod performance when an abbreviated version of the task was used (Sato et al., 2012).
Performance in the accelerating rotarod is commonly affected in mice with mutations in ASD-risk genes, as several other genetic ASD mouse models exhibit enhanced rotarod performance (Kwon et al., 2006; Rothwell et al., 2014; Nakatani et al., 2009; Peñagarikano et al., 2011; Platt et al., 2017; Chadman et al., 2008; Hisaoka et al., 2018). However, this is not the case for all ASD-risk genes (Wang et al., 2016; Portmann et al., 2014; Yin et al., 2021). Most of the aforementioned mouse models have global gene deletions in which brain regions and circuits outside of the striatum could contribute to the phenotype. However, two studies showed that selectively disrupting Nlgn3 or Chd8 in striatal neurons was sufficient to increase rotarod learning (Rothwell et al., 2014; Platt et al., 2017). Moreover, similar to what was observed here, loss of Nlgn3 from dSPNs alone led to enhanced motor learning, implicating the direct pathway as a key driver of this phenotype (Rothwell et al., 2014). Interestingly, in these studies, ventral striatal disruption of the gene was responsible for the motor learning improvement. Here, deletion of Tsc1 from dSPNs was largely restricted to the dorsal striatum, and our findings are consistent with literature establishing the dorsal striatum’s role in motor learning (Yin et al., 2009). Further studies will be needed to define the contributions of dorsal versus ventral striatal circuits to motor learning and elucidate the contributions of specific striatal subregions to behavioral changes in TSC mouse models.
While enhanced motor routine learning may be a shared phenotype across multiple mouse models of ASD, the synaptic and cellular mechanisms driving this phenotype may be distinct depending on the specific gene that is altered. dSPN-Nlgn3 KO mice displayed normal excitatory synaptic transmission and eCB-LTD but had a deficit in inhibition (Rothwell et al., 2014). In Chd8+/− mice, there was increased spontaneous excitatory transmission onto SPNs; however, the SPN sub-type was not defined (Platt et al., 2017). We found that dSPN-Tsc1 Het and KO cells had strongly enhanced corticostriatal excitation that was due to increased synaptic, but not intrinsic, excitability. The enhanced cortical drive was likely driven by changes in presynaptic function as we found increased release probability and mEPSC frequency in dSPN-Tsc1 KO neurons, consistent with our prior study (Benthall et al., 2018). Given that the cortical inputs were WT in our model, a potential explanation for increased release probability is a change in retrograde signaling from dSPNs to cortical terminals. Indeed, we found that eCB-LTD was disrupted in Tsc1 KO dSPNs despite normal presynaptic CB1 receptor function. This suggests that loss of Tsc1 and upregulation of mTORC1 signaling in dSPNs interferes with one or more post-synaptic processes required for this form of plasticity: (1) expression of group 1 mGluRs, (2) signaling downstream of mGluRs, and/or (3) synthesis and release of eCBs. We found that multiple mRNAs encoding proteins required for eCB-LTD exhibited relatively reduced ribosome engagement in dSPN-Tsc1 KO cells compared to WT. Together, this suggests that loss of Tsc1 and deregulation of mTORC1 signaling in dSPNs impairs post-synaptic mGluR signaling via altered expression of key components of this pathway. Notably, striatal eCB-LTD is also disrupted in a mouse model of the neurodevelopmental disorder Fragile X Syndrome and in mice with an ASD-linked R451C Nlgn3 mutation (Jung et al., 2012; Martella et al., 2018). In both of these mouse models, the phenotypes could be improved by pharmacologically enhancing cannabinoid signaling.
A chronic impairment of eCB-LTD in Tsc1 KO dSPNs may lead to increased glutamatergic transmission over time. In support of this idea, disruption of endocannabinoid synthesis in dSPNs via cell-type-specific loss of DAGLα, the enzyme required for 2-AG production, is sufficient to cause glutamatergic hyperactivity (Shonesy et al., 2018). Therefore, loss of eCB-LTD may be the primary driving mechanism that leads to increased cortical activation of Tsc1 KO dSPNs. This is consistent with our observation that changes in mEPSC properties in dSPN-Tsc1 KO neurons do not arise until 4 weeks of age and later. While initial synapse formation may occur normally in Tsc1 KO dSPNs, activity-dependent refinement of cortical synapses, which occurs over the first few weeks of postnatal life (Kuo and Liu, 2019), may be impaired by an inability to depress cortical inputs. Interestingly, the study by Shonesy et al. showed that, while disruption of 2-AG signaling in dSPNs led to synaptic and behavioral consequences, loss of DAGLα from iSPNs had no measurable effects on behavior (Shonesy et al., 2018), consistent with the lack of behavior changes reported here for iSPN-Tsc1 KO mice.
Taken together, our results support a model whereby developmental loss of Tsc1 from dSPNs impairs eCB-LTD resulting in unchecked corticostriatal drive. While further work will be needed to establish a causal link between enhanced cortico-dSPN activity and increased rotarod learning, it is possible that perturbed presynaptic plasticity in Tsc1 KO dSPNs may alter corticostriatal coupling during rotarod training, leading to atypical learning in this paradigm (Kupferschmidt et al., 2019). In terms of autistic behaviors in individuals with TSC, our findings suggest that striatal synaptic and circuit changes, which in mice increases the ability to learn a stereotyped motor routine, could be a contributor to the emergence of restricted, repetitive patterns of behavior.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Helen S. Bateup (bateup@berkeley.edu).
Materials availability
The AAV-hSyn-DIO-EGFP-L10a-WPRE-hGH and AAV-Ef1a-DIO-EGFP-L10a-WPRE-hGH plasmids and corresponding maps generated in this study are available from the corresponding author upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
To generate conditional deletion of Tsc1 in dSPNs, Tsc1fl/fl mice (Jackson Laboratory strain #005680; Kwiatkowski et al., 2002) of mixed background were crossed with Drd1a-Cre (EY217) mice (GENSAT [MMRRC #030778-UCD] (Gong et al., 2007). To delete Tsc1 from iSPNs, Tsc1fl/fl mice were crossed with Adora2a-Cre (KG139) mice (GENSAT (MMRRC #031168-UCD) (Gong et al., 2007)). To identify Cre-expressing neurons, dSPN-Tsc1 KO and iSPN-Tsc1 KO mice were bred to the tdTomato Cre reporter Ai9 mouse line (Jackson Laboratory strain #007909 (Madisen et al., 2010)). To express YFP-tagged ChR2 in a subset of Layer V cortical pyramidal cells, the Thy1-ChR2-YFP mouse line (Jackson Laboratories strain #007612; Arenkiel et al., 2007) was bred into each line. Tsc2+/− mice (Jackson Laboratory strain #004686; Onda et al., 1999) were used for the rotarod experiments.
Mice were group housed on a 12 h light/dark cycle and given ad libitum access to standard rodent chow and water. Both male and female animals were used for experimentation. The ages, sexes, and numbers of mice used for each experiment are indicated in the respective method details and figure legends. All mice used for experiments were heterozygous or hemizygous for the Ai9, Drd1-Cre, Adora2a-Cre, or Thy1-ChR2-YFP transgenes to avoid potential physiological or behavioral alterations.
All animal procedures were conducted in accordance with protocols approved by the University of California, Berkeley Institutional Animal Care and Use Committee (IACUC) and Office of Laboratory Animal Care (OLAC).
METHOD DETAILS
Brain sectioning and immunohistochemistry
Adult mice were anesthetized with isoflurane and transcardial perfusion was performed with 10 mL of 1x PBS followed by 10 mL of ice cold 4% PFA in 1x PBS (EMS, 15710-S) and post-fixed in 4% PFA in 1x PBS overnight at 4°C. 30 μm coronal sections were made using a freezing microtome (American Optical, AO 860) and stored in 1x PBS at 4°C.
For immunohistochemistry, individual wells of sections were washed with 1x PBS, then blocked for 1 hour at RT with BlockAid blocking solution (Life Tech, B10710). Primary antibodies diluted in PBS-Tx (1x PBS with 0.25% Triton X-100 (Sigma, T8787)) were added and tissue was incubated for 48 h with gentle shaking at 4°C. Sections were then washed 3 × 10 min with PBS-Tx. Secondary antibodies diluted 1:500 in PBS-Tx were added and incubated with shaking for 1 h at room temperature. Sections were washed 3 × 10 min in 1x PBS and then rinsed once in 0.1M PB. Sections were mounted onto SuperFrost slides (VWR, 48311-703) and coverslipped with VECTASHIELD HardSet with DAPI (VWR, 101098-050). The following antibodies were used: anti-phosphorylated S6 ribosomal protein (Ser240/244, 1:800, Cell Signaling Technology, 5364S), anti-GFP (1:1000, Abcam, ab13970), anti-NeuN (1:800, Millipore, MAB377), Alexa Fluor 488 and Alexa Fluor 633 conjugated secondary antibodies (1:500, Invitrogen, A-21070 and A-31553).
Confocal microscopy and image analysis
To analyze p-S6 levels and soma volume, Z stack images of striatal sections were taken on a confocal microscope (Zeiss LSM 780 AxioExaminer or Olympus FLUOVIEW FV3000) with a 20x objective using the same acquisition settings for each section. For quantification, cellular regions of interest (ROIs) were automatically created based on the tdTomato signal with the Surfaces module in Imaris software (Oxford Instruments). The mean p-S6 fluorescence intensity per ROI and average soma volume were calculated using Imaris. Values for Tsc1 Het and KO cells were normalized to the average of all wild-type cells imaged in the same batch. To generate cumulative probability plots, 300 cells from each mouse were used (100 or 150 cells per section, 2-3 sections per mouse).
To analyze Cre-mediated recombination patterns in Drd1- and Adora2a-Cre;Ai9 mice, Z stack images of cortex, cerebellum, dorsolateral striatum, dorsomedial striatum, nucleus accumbens lateral shell, and nucleus accumbens medial shell were taken on a confocal microscope (Olympus FLUOVIEW FV3000) with a 20x objective. For quantification, ROIs were manually defined in ImageJ for all NeuN positive cells (for striatum and nucleus accumbens) or automatically created based on the DAPI signal with the Surfaces module in Imaris software (for cortex) and used to determine co-localization with the Cre-dependent tdTomato signal.
To analyze EGFP-L10a expression, Z stack images of dorsal striatum were taken on a confocal microscope (Olympus FLUOVIEW FV3000) with a 20x objective. For quantification, ROIs were manually defined in ImageJ for all GFP positive cells and used to verify co-localization with the Cre-dependent tdTomato signal. 85%–93% of EGFP-L10a positive cells in the dorsal striatum were tdTomato positive.
Dendritic imaging and spine analysis
Neonatal (P1-4) dSPN-Tsc1 WT and KO mice were cryoanesthetized and injected bilaterally with 200 nL AAV9.CAG.Flex.tdTomato.WPRE.bGH (Penn Vector Core, AllenInstitute864), diluted 1:500 to achieve sparse transduction. Injections were targeted to the dorsal striatum, with coordinates approximately 1.2 mm lateral to midline, 2.0 mm posterior to bregma, and 1.5 mm ventral to the head surface. At age P40-50, mice were perfused and brains were post-fixed with 4% paraformaldehyde, then sectioned at 80 μm. Sections were blocked for 1 hr at RT in BlockAid (ThermoFisher, B10710) and incubated for 48 hr at 4°C with an antibody against RFP (1:1000, Rockland (VWR), RL600-401-379). Sections were washed 3 × 10 min in PBS-Tx and incubated for 1 hr at RT with Alexa Fluor 546 secondary antibody (1:500, Invitrogen, A-11035). Sections were washed 3 × 10 min in 1x PBS and mounted onto slides using VECTASHIELD HardSet with DAPI (VWR, 101098-050). Z stack images of individual dendrites were taken on a confocal microscope (Zeiss LSM 880 NLO AxioExaminer) with a 63x objective using Airyscan. To quantify spine density, dendrites and spines were reconstructed using the FilamentTracer module in Imaris software (Oxford Instruments). The spine density of each dendrite was calculated using Imaris. Spine density analysis was initially separated into the first 40 μm of the dendrite (proximal) and the final 40 μm of the dendrite (distal). There was no significant difference between proximal and distal spine density within cells or across genotypes. These values were therefore combined and spine density of the 80 μm length of dendrite is reported.
Behavioral analysis
Behavior studies were carried out in the dark phase of the light cycle under red lights (open field) or white lights (rotarod). Mice were habituated to the behavior testing room for at least 30 min prior to testing and covered by a black-out curtain. Mice were given at least one day between different tests. All behavior equipment was cleaned between each trial and mouse with 70% ethanol, and additionally rinsed in diluted soap followed by water at the end of the day. If male and female mice were to be tested on the same day, male mice were run first then returned to the husbandry room, after which all equipment was thoroughly cleaned prior to bringing in female mice for habituation. All animals to be tested from a given cage were run in each behavior test in the same day. Behavioral tests were performed with young adult male and female mice (6-10 weeks old). Mice had access to a running wheel in their home cage. The experimenter was blind to genotype throughout the testing and scoring procedures.
Open field
Exploratory behavior in a novel environment and general locomotor activity were assessed by a 60 min session in an open field chamber (40 cm L x 40 cm W x 34 cm H) made of transparent plexiglass. Horizontal infrared photobeams were positioned to detect rearing. The mouse was placed in the bottom right hand corner of the arena and behavior was recorded using an overhead camera and analyzed using the ANY-maze (Stoelting Co.) behavior tracking software. An observer manually scored self-grooming behavior during the first 20 minutes of the test. A grooming bout was defined as an unbroken series of grooming movements, including licking of body, paws, or tail, as well as licking of forepaws followed by rubbing of face with paws.
Rotarod
The accelerating rotarod test was used to examine motor learning. Mice were trained on a rotarod apparatus (Ugo Basile, 47650) for four consecutive days. Three trials were completed per day with a 5 min break between trials. The rotarod was accelerated from 5-40 revolutions per minute (rpm) over 300 s for trials 1-6 (days 1 and 2), and from 10-80 rpm over 300 s for trials 7-12 (days 3 and 4). On the first testing day, mice were first acclimated to the apparatus by being placed on the rotarod rotating at a constant 5 rpm for 60 s and returned to their home cage for 5 minutes prior to starting trial 1. Latency to fall, or to rotate off the top of the rotarod barrel, was measured by the rotarod stop-trigger timer.
Electrophysiology
Mice (P40-50) were perfused transcardially with ice-cold ACSF (pH = 7.4) containing (in mM): 127 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 25 glucose, bubbled continuously with carbogen (95% O2 and 5% CO2). Brains were rapidly removed and coronal slices (275 μm) were cut on a VT1000S vibratome (Leica) in oxygenated ice-cold choline-based external solution (pH = 7.8) containing (in mM): 110 choline chloride, 25 NaHCO3, 1.25 NaHPO4, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 25 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. Slices were recovered in ACSF at 34°C for 15 min and then kept at RT before recording.
Recordings were made with a MultiClamp 700B amplifier (Molecular Devices) at RT using 3-5 MOhm glass patch electrodes (Sutter, BF150-86-7.5). Data were acquired using ScanImage software, written and maintained by Dr. Bernardo Sabatini (https://github.com/bernardosabatini/ SabalabAcq). Traces were analyzed in Igor Pro (Wavemetrics). Recordings with a series resistance > 25 MOhms or holding current above −200 pA were rejected.
Current-clamp recordings
Current clamp recordings were made using a potassium-based internal solution (pH = 7.4) containing (in mM): 135 KMeSO4, 5 KCl, 5 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 phosphocreatine, and 1 ETGA. For corticostriatal excitability experiments, optogenetic stimulation consisted of a full-field pulse of blue light (470 nm, 0.05 ms for iSPNs or 0.5 ms for dSPNs, CoolLED) through a 63x objective. Light power was linear over the range of intensities tested (see Figure S4). No synaptic blockers were included. For intrinsic excitability experiments, NBQX (10 μM, Tocris, 1044), CPP (10 μM, Tocris, 0247) and picrotoxin (50 μM, Abcam, 120315) were added to the external solution to block synaptic transmission. 500 ms depolarizing current steps were applied to induce action potentials. No holding current was applied to the membrane.
Voltage-clamp recordings
Voltage-clamp recordings were made using a cesium-based internal solution (pH = 7.4) containing (in mM): 120 CsMeSO4, 15 CsCl, 10 TEA-Cl, 8 NaCl, 10 HEPES, 1 EGTA, 5 QX-314, 4 Mg-ATP, and 0.3 Na-GTP. Recordings were acquired with the amplifier Bessel filter set at 3 kHz. Miniature excitatory synaptic currents (mEPSCs) were recorded in the presence of TTX (1 μM, Abcam, 120055) to prevent action potential-mediated release. Picrotoxin (50 μM) and CPP (10 μM) were included for mEPSC experiments to isolate AMPAR-mediated events. Corticostriatal synaptic stimulation experiments to measure evoked AMPA-mediated EPSCs were performed in picrotoxin (50 μM) and CPP (10 μM), and optogenetic stimulation consisted of a full-field pulse of blue light (470 nm, 0.15 ms) through a 63x objective. To measure AMPA/NMDA ratio, experiments were performed in 50 μM picrotoxin and the membrane was held at different potentials to isolate primarily AMPAR (−70 mV) or compound AMPAR and NMDAR (+40 mV) currents. The current amplitude at +40 mV was measured 50 ms after stimulation, by which time the AMPAR-mediated current has decayed. To measure paired pulse ratio, experiments were performed in 50 μM picrotoxin and the membrane was held at −70 mV. To preferentially excite cortical inputs, a concentric bipolar stimulating electrode (FHC) was placed in the corpus callosum dorsomedial to the recording site in dorsolateral striatum. A 0.2 ms stimulus was applied with a 50 ms interstimulus-interval.
eCB-LTD
Endocannabinoid-mediated long-term depression (eCB-LTD) was induced in Tsc1;Drd1-Cre;Ai9;Thy1-ChR2-YFP mice by bath application of the group 1 mGluR agonist DHPG (100 μM, Sigma, D3689) for 10 min, following a 10 min baseline measurement of EPSC amplitude with single full field light pulses (3%–15% light intensity, 0.15 ms) delivered every 30 s to stimulate corticostriatal terminals. Light intensity was adjusted for each cell to evoke 500-700 pA currents during the baseline period. DHPG was subsequently washed off and EPSC amplitude was monitored every 30 s for an additional 40 min. Picrotoxin (50 μM) was added to the bath during eCB-LTD experiments to isolate excitatory events, and perfusion flow rate was set to 5 mL/min. Cells were held at −50 mV to facilitate opening of L-type calcium channels. The CB1R antagonist AM 251 (10 μM, Tocris, 1117) was added to the bath during a subset of eCB-LTD experiments with dSPN-Tsc1 WT cells to verify that the LTD observed during these experiments was dependent upon CB1R activation. For CB1R agonism experiments, WIN-2 (2 μM, EMD Millipore, 504344) was applied to the bath throughout the recording.
AAV-FLEX-TRAP plasmid and virus construction
The AAV-hSyn-DIO-EGFP-L10a-WPRE-hGH and AAV-Ef1a-DIO-EGFP-L10a-WPRE-hGH plasmids were assembled from pAAV-hSyn-DIO-mCherry (Addgene plasmid #50459) and pAAV-EF1A-DIO-mCherry (Addgene plasmid #50462), which were gifts from Dr. Bryan Roth, and an EGFP-L10a construct, which was a gift from Dr. Anne Schaefer. AAV serotype 5 viruses were prepared by the University of Pennsylvania Vector Core with a titer of 5.97 × 1012 for AAV5-hSyn-DIO-EGFP-L10a-WPRE-hGH and 3.22 × 1012 for AAV5-Ef1a-DIO-EGFP-L10a-WPRE-hGH.
Stereotaxic intracranial injections
Mice were anesthetized with isoflurane and mounted on a stereotaxic frame equipped with ear cups. 800 nL of an AAV serotype 5 Ef1a or hSyn promoter-driven DIO-EGFP-L10a virus (AAV-FLEX-TRAP) was bilaterally injected into the dorsal striatum of 3-8 week old Tsc1;Drd1-Cre mice of both sexes. Coordinates for injection were ± 1.6 M/L, +0.6 A/P, −1.3 D/V. Mice were used for experiments 11-14 days after AAV-FLEX-TRAP virus injection.
Translating ribosome affinity purification (TRAP)
Anti-GFP magnetic bead preparation
Each TRAP experiment was performed on 6 samples in parallel, with dSPN Tsc1 WT and KO mice processed together. For 6 mice, two batches of beads were prepped in parallel in separate tubes. TRAP was performed according to published methods (Heiman et al., 2014, Heiman et al., 2008). All steps were performed on ice unless otherwise noted. 450 μL of Dynabeads MyOne Streptavidin T1 (ThermoFisher, 65601) were washed using a magnetic tube rack in RNase-free 1x PBS and then incubated in Protein L solution (850 μL PBS + 150 μg Protein L, ThermoFisher, 29997) for 35 min at RT. Beads were washed 5x with 3% IgG Protease-free BSA to block, then incubated with 150 μg of two different anti-GFP antibodies (19C8 and 19F7, Memorial Sloan Kettering Antibody and Bioresource Core), diluted in 900 μL PBS, for 1 hr at RT. Beads were then washed 3x in 0.15 M KCl buffer without cyclohexamide (−CHX), then resuspended in 630 μL of 0.15 M KCl (+CHX).
Immunoprecipitation
Mice were anesthetized, and brains were dissected and blocked to contain mainly striatum. Bilateral striata from each animal were placed into glass homogenization tubes on ice, and pestles were inserted. Homogenization took place at 4°C (3 strokes at 300 RPM, 12 strokes at 900 RPM, Yamato Lab Stirrer, LT400), care was taken to avoid generating bubbles. Lysates were poured into pre-chilled Eppendorf tubes and centrifuged for 10 min at 2,000 x g at 4°C to precipitate large organelles. Samples were then transferred to a new pre-chilled tube and volumes were measured. 10% NP-40 (1/9 sample volume, ∼70-80 μL) was added, then DHPC (1/9 new sample volume) was added, and samples were incubated on ice for 5 min. 200 mg DHPC stock (Avanti Polar Lipids, 850306P) was dissolved in 1.38 mL ddH2O. Samples were then centrifuged for 10 min at 16,000 x g at 4°C to precipitate mitochondria. Antibody-bound beads were resuspended by inversion, and 200 μL of beads were added to 6 separate tubes. Supernatants from samples were then transferred into tubes with beads and incubated on rotators at 4°C overnight.
Isolation and purification of RNA
Samples were spun down briefly and placed on magnets pre-chilled on ice. Supernatants were collected and transferred to pre-chilled “unbound” tubes. Beads were washed 4x with 0.35 M KCl buffer, with samples sitting on ice for 1 min between washes to reduce background binding. Beads were resuspended in 350 μL RLT-beta-ME from an RNeasy kit (QIAGEN, 74004). 100 μL of unbound samples were added separately to 350 μL RLT-beta-ME. Samples (bound and unbound) were then rotated for 10 min at RT. Samples were placed on the magnet and supernatants were removed and added into fresh tubes containing 350 μL of 80% EtOH, mixed, and then all 700 μL of sample + EtOH was added to an RNeasy kit pre-chilled column. 350 μL of unbound sample was also mixed with 350 μL of EtOH and added to an RNeasy column. At this point there were 12 columns, one bound and one unbound sample for each mouse. Samples were centrifuged for 30 s at 8000 x g at RT. Flow-through was passed through the column twice more to repeat binding. Flow-through was then discarded and 500 μL of RPE buffer was added to each column and spun for 30 s at 8000 x g. Flow-through was discarded and 500 μL of 80% EtOH was added to the column. Columns were spun for 2 min at 8000 x g at RT. Flow-through was discarded, and columns were dried by spinning for 5 min at full speed with the cap open. Dried columns were placed into new collection tubes (not pre-chilled) and 28 μL RNase-free water was added directly to the column membrane. Columns were incubated for 5 min at RT with the caps closed, then spun for 1 min at max speed at RT. RNA concentration and quality was determined by NanoDrop and Bioanalyzer at the UC Berkeley Functional Genomics Laboratory core facility.
Quantitative PCR
Reverse transcription was performed using random hexamer primers and Superscript III reverse transcriptase (ThermoFisher, 18080051). Real-time PCR was performed in triplicate with 1 uL cDNA using a Bio-Rad CFX384 thermal cycler with TaqMan Universal PCR Master Mix, no AmpErase UNG (Lifetech, 4324018) and TaqMan probes. The following TaqMan probes were used: Grm1 (Mm01187086_m1), Grm5 (Mm00690332_m1), Homer1 (Mm00516275), Plcb1 (Mm00479998), Drd1 (Mm01353211_m1), Drd2 (Mm00438545_m1), and Actb (Mm02619580_g1). Values for all mRNAs were normalized to Actb for each sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments were designed to compare the main effect of genotype within each mouse line. The sample sizes were based on prior studies and are indicated in the figure legend for each experiment. Whenever possible, quantification and analyses were performed blind to genotype. GraphPad Prism version 9 was used to perform statistical analyses. The statistical tests and outcomes for each experiment are indicated in the respective figure legend. Two-tailed paired or unpaired t tests were used for comparisons between two groups. For data that did not pass the D’Agostino & Pearson normality test, a Mann-Whitney test was used. A one-way ANOVA with Holm-Sidak’s post hoc tests was used to compare the means of three or more groups. For data that did not pass the D’Agostino and Pearson normality test, a Kruskal-Wallis test with Dunn’s post hoc tests was used. Repeated-measures (RM) two-way ANOVAs were used to compare differences between groups for experiments with two independent variables. For datasets that had values randomly missing, mixed-effects model analysis was performed in place of a RM two-way ANOVA. P values were corrected for multiple comparisons. Statistical significance was defined in the figure panels as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Phospho-S6 Ribosomal Protein (Ser240/244) (D68F8) XP | Cell Signaling Technology | Cat # 5364S; RRID:AB_10694233 |
| Chicken polyclonal anti-GFP | Abcam | Cat # ab13970; RRID:AB_300798 |
| Mouse monoclonal anti-NeuN | Millipore Sigma | Cat # MAB377; RRID:AB_2298772 |
| Monoclonal anti-GFP | Memorial Sloan Kettering Antibody and Bioresource Core | 19C8 & 19F7 |
| Alexa Fluor 633 goat anti-rabbit secondary | Thermo Fisher Scientific | Cat # A-21070; RRID:AB_2535731 |
| Alexa Fluor 488 goat anti-mouse secondary | Thermo Fisher Scientific | Cat # A-31553; RRID:AB_221604 |
| Bacterial and virus strains | ||
| AAV5-hSyn-DIO-EGFP-L10a-WPRE-hGH | Custom made by Penn Vector Core for this paper | N/A |
| AAV5-Ef1a-DIO-EGFP-L10a-WPRE-hGH | Custom made by Penn Vector Core for this paper | N/A |
| AAV9-CAG-Flex-tdTomato-WPRE-bGH | Penn Vector Core | AllenInstitute864 |
| Chemicals, peptides, and recombinant proteins | ||
| Tetrodotoxin | Abcam | Cat # 120055 |
| Picrotoxin | Abcam | Cat # 120315 |
| CPP | Tocris | Cat # 0247 |
| NBQX | Tocris | Cat # 1044 |
| DHPG | Sigma | Cat # D3689 |
| AM 251 | Tocris | Cat # 1117 |
| WIN-2 | EMD Millipore | Cat # 504344 |
| Dynabeads MyOne Streptavidin T1 | ThermoFisher | Cat # 65601 |
| Grm1 TaqMan probe | ThermoFisher | Mm01187086_m1 |
| Grm5 TaqMan probe | ThermoFisher | Mm00690332_m1 |
| Homer1 TaqMan probe | ThermoFisher | Mm00516275 |
| Plcb1 TaqMan probe | ThermoFisher | Mm00479998 |
| Drd1 TaqMan probe | ThermoFisher | Mm01353211_m1 |
| Drd2 TaqMan probe | ThermoFisher | Mm00438545_m1 |
| Actb TaqMan probe | ThermoFisher | Mm02619580_g1 |
| Critical commercial assays | ||
| RNeasy Kit | QIAGEN | Cat # 74004 |
| SuperScript III First-Strand Synthesis System | ThermoFisher | Cat # 18080051 |
| TaqMan Universal PCR Master Mix, no AmpErase UNG | ThermoFisher | Cat # 4324018 |
| Experimental models: Organisms/strains | ||
| Mouse: Tsc1fl/fl | Jackson Laboratory | JAX stock #005680 |
| Mouse: Tsc2+/− | Jackson Laboratory | JAX stock #004686 |
| Mouse: Drd1a-Cre (EY217) | GENSAT | MMRRC #030778-UCD |
| Mouse: Adora2a-Cre (KG139) | GENSAT | MMRRC #031168-UCD |
| Mouse: Ai9 | Jackson Laboratory | JAX stock #007909 |
| Mouse: Thy1-ChR2-YFP | Jackson Laboratory | JAX stock #007612 |
| Software and algorithms | ||
| ImageJ | NIH | N/A |
| GraphPad Prism version 9 | GraphPad Software | N/A |
| ANY-maze | Stoelting Co | N/A |
| ScanImage | Dr. Bernardo Sabatini | https://github.com/bernardosabatini/ SabalabAcq |
Highlights.
Selective loss of Tsc1 from striatal direct pathway neurons enhances motor learning
Tsc1 KO direct pathway neurons exhibit increased corticostriatal synaptic drive
Tsc1 KO direct pathway neurons have impaired endocannabinoid-mediated LTD
Tsc1 KO indirect pathway neurons do not show major changes in synaptic function
ACKNOWLEDGMENTS
This work was supported by a SFARI Pilot Award (# 307866), R21 NS096415, R56 MH111821, and R01 NS105634 grants to H.S.B. H.S.B. is a Chan Zuckerberg Biohub Investigator and was supported by an Alfred P. Sloan Foundation fellowship. K.N.B. was supported by an NSF graduate research fellowship (DGE 1106400). We thank Caroline Keeshen for assistance with mouse colony maintenance and initial pilot experiments. We thank Dr. John Blair for assistance with cloning the AAV-FLEX-TRAP construct. We thank Dr. Anne Schaefer for advice on the TRAP protocol and for providing the EGFP-L10a construct. We thank Kamran Ahmed for generating the figure schematics and graphical abstract. Confocal imaging was conducted, in part, at the CRL Molecular Imaging Center, supported by the Helen Wills Neuroscience Institute and NSF DBI-1041078. We thank Holly Aaron and Feather Ives for their microscopy training and assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109511.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, and Feng G (2007). In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 54, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, Mangner TJ, Chakraborty PK, and Chugani HT (2001). Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology 57, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Assous M, Martinez E, Eisenberg C, Shah F, Kosc A, Varghese K, Espinoza D, Bhimani S, Tepper JM, Shiflett MW, and Tran TS (2019). Neuropilin 2 Signaling Mediates Corticostriatal Transmission, Spine Maintenance, and Goal-Directed Learning in Mice. J. Neurosci 39, 8845–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, and Bear MF (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, and Sabatini BL (2011). Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci 31, 8862–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, and Sabatini BL (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benthall KN, Ong SL, and Bateup HS (2018). Corticostriatal Transmission Is Selectively Enhanced in Striatonigral Neurons with Postnatal Loss of Tsc1. Cell Rep. 23, 3197–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron Y, Chagniel L, Bureau G, Massicotte G, and Cyr M (2014). mTOR signaling contributes to motor skill learning in mice. Front. Mol. Neurosci 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadman KK, Gong S, Scattoni ML, Boltuck SE, Gandhy SU, Heintz N, and Crawley JN (2008). Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 1, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chévere-Torres I, Kaphzan H, Bhattacharya A, Kang A, Maki JM, Gambello MJ, Arbiser JL, Santini E, and Klann E (2012). Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the ΔRG mouse model of tuberous sclerosis complex. Neurobiol. Dis 45, 1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatolo P, Moavero R, and de Vries PJ (2015). Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 14, 733–745. [DOI] [PubMed] [Google Scholar]
- Davis PE, Peters JM, Krueger DA, and Sahin M (2015). Tuberous Sclerosis: A New Frontier in Targeted Treatment of Autism. Neurotherapeutics 12, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries PJ (2010). Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics 7, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino A, Kelly C, Grzadzinski R, Zuo XN, Mennes M, Mairena MA, Lord C, Castellanos FX, and Milham MP (2011). Aberrant striatal functional connectivity in children with autism. Biol. Psychiatry 69, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Peterson JD, and Surmeier DJ (2008). Corticostriatal and thalamostriatal synapses have distinctive properties. J. Neurosci 28, 6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Moss J, and Bolam JP (2010). Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J. Neurosci 30, 14610–14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano DM, Su T, Lopez J, Platel JC, and Bordey A (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest 121, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo MV (2016). Striatal Circuits as a Common Node for Autism Pathophysiology. Front. Neurosci 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, and Surmeier DJ (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci 28, 10814–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobet C, and Naef F (2017). Ribosome profiling and dynamic regulation of translation in mammals. Curr. Opin. Genet. Dev 43, 120–127. [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, and Gerfen CR (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Grafton ST (2015). The striatum: where skills and habits meet. Cold Spring Harb. Perspect. Biol 7, a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, and Lovinger DM (2017). Associative and sensorimotor cortico-basal ganglia circuit roles in effects of abused drugs. Genes Brain Behav. 16, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, and Heintz N (2014). Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat. Protoc 9, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaoka T, Komori T, Kitamura T, and Morikawa Y (2018). Abnormal behaviours relevant to neurodevelopmental disorders in Kirrel3-knockout mice. Sci. Rep 8, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, and Manning BD (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J 412, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, Ginger M, Frick A, DiPatrizio NV, Mackie K, et al. (2012). Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun 3, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, and Bear MF (2008). The autistic neuron: troubled translation? Cell 135, 401–406. [DOI] [PubMed] [Google Scholar]
- Kosillo P, Doig NM, Ahmed KM, Agopyan-Miu AHCW, Wong CD, Conyers L, Threlfell S, Magill PJ, and Bateup HS (2019). Tsc1-mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat. Commun 10, 5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, and Kreitzer AC (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo HY, and Liu FC (2019). Synaptic Wiring of Corticostriatal Circuits in Basal Ganglia: Insights into the Pathogenesis of Neuropsychiatric Disorders. eNeuro 6, ENEURO.0076-19.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt DA, Augustin SM, Johnson KA, and Lovinger DM (2019). Active Zone Proteins RIM1αβ Are Required for Normal Corticostriatal Transmission and Action Control. J. Neurosci. 39, 1457–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, and Onda H (2002). A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11, 525–534. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, and Parada LF (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim H, and Han PL (2018). Striatal Inhibition of MeCP2 or TSC1 Produces Sociability Deficits and Repetitive Behaviors. Exp. Neurobiol 27, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, and Pozzo-Miller L (2020). Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res 98, 2130–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JS, Gopalappa R, Kim SH, Ramakrishna S, Lee M, Kim WI, Kim J, Park SM, Lee J, Oh JH, et al. (2017). Somatic Mutations in TSC1 and TSC2 Cause Focal Cortical Dysplasia. Am. J. Hum. Genet 100, 454–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM (2010). Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 58, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, and Huber KM (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Pai EL, Rubin AN, Stafford AM, Angara K, Minasi P, Rubenstein JL, Sohal VS, and Vogt D (2019). Tsc1 represses parvalbumin expression and fast-spiking properties in somatostatin lineage cortical interneurons. Nat. Commun 10, 4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Meringolo M, Trobiani L, De Jaco A, Pisani A, and Bonsi P (2018). The neurobiological bases of autism spectrum disorders: the R451C-neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur. J. Neurosci 47, 701–708. [DOI] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, Machan JT, Moore CI, Connors BW, and Zervas M (2013). Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron 78, 895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, and Kano M (2014). Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol 29, 1–8. [DOI] [PubMed] [Google Scholar]
- Onda H, Lueck A, Marks PW, Warren HB, and Kwiatkowski DJ (1999). Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J. Clin. Invest 104, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, and Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat. Neurosci 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Zhou Y, Slaymaker IM, Shetty AS, Weisbach NR, Kim JA, Sharma J, Desai M, Sood S, Kempton HR, et al. (2017). Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 19, 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G, Bader PL, Grueter BA, Goold C, Fisher E, et al. (2014). Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. 7, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter WB, Basu T, O’Riordan KJ, Kirchner A, Rutecki P, Burger C, and Roopra A (2013). Reduced juvenile long-term depression in tuberous sclerosis complex is mitigated in adults by compensatory recruitment of mGluR5 and Erk signaling. PLoS Biol. 11, e1001627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu A, Adler M, Crocetti D, Miller MI, and Mostofsky SH (2010). Basal ganglia shapes predict social, communication, and motor dysfunctions in boys with autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 539–551. [DOI] [PubMed] [Google Scholar]
- Rothwell PE (2016). Autism Spectrum Disorders and Drug Addiction: Common Pathways, Common Molecules, Distinct Disorders? Front. Neurosci. 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, and Südhof TC (2014). Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, and Mizuguchi M (2012). Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun 3, 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann SN, and Vanderhaeghen JJ (1993). Adenosine A2 receptors regulate the gene expression of striatopallidal and striatonigral neurons. J. Neurosci 13, 1080–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Parrish WP, Haddad HK, Stephenson JR, Báldi R, Bluett RJ, Marks CR, Centanni SW, Folkes OM, Spiess K, et al. (2018). Role of Striatal Direct Pathway 2-Arachidonoylglycerol Signaling in Sociability and Repetitive Behavior. Biol. Psychiatry 84, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y, and Zoghbi HY (2016). Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci 19, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Tai LH, Lee AM, Benavidez N, Bonci A, and Wilbrecht L (2012). Transient stimulation of distinct subpopulations of striatal neurons mimics changes in action value. Nat. Neurosci 15, 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, and Sabatini BL (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci 8, 1727–1734. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, and Sahin M (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KC, Frost L, Linsenbardt D, McIlroy JR, and Müller RA (2006). Atypically diffuse functional connectivity between caudate nuclei and cerebral cortex in autism. Behav. Brain Funct 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winden KD, Ebrahimi-Fakhari D, and Sahin M (2018). Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 41, 1–23. [DOI] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, and Ding JB (2015). Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 10, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, and Jan LY (2012). Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 75, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, and Costa RM (2009). Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat. Neurosci 12, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Jones N, Yang J, Asraoui N, Mathieu ME, Cai L, and Chen SX (2021). Delayed motor learning in a 16p11.2 deletion mouse model of autism is rescued by locus coeruleus activation. Nat. Neurosci 24, 646–657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.