

ABSTRACT
The opportunistic pathogen Staphylococcus aureus is protected by a cell envelope that is crucial for viability. In addition to peptidoglycan, lipoteichoic acid (LTA) is an especially important component of the S. aureus cell envelope. LTA is an anionic polymer anchored to a glycolipid in the outer leaflet of the cell membrane. It was known that deleting the gene for UgtP, the enzyme that makes this glycolipid anchor, causes cell growth and division defects. In Bacillus subtilis, growth abnormalities from the loss of ugtP have been attributed to both the absence of the encoded protein and the loss of its products. Here, we show that growth defects in S. aureus ugtP deletion mutants are due to the long, abnormal LTA polymer that is produced when the glycolipid anchor is missing from the outer leaflet of the membrane. Dysregulated cell growth leads to defective cell division, and these phenotypes are corrected by mutations in the LTA polymerase gene, ltaS, that reduce polymer length. We also show that S. aureus mutants with long LTA are sensitized to cell wall hydrolases, beta-lactam antibiotics, and compounds that target other cell envelope pathways. We conclude that control of LTA polymer length is important for S. aureus physiology and promotes survival under stressful conditions, including antibiotic stress.
IMPORTANCE Methicillin-resistant Staphylococcus aureus (MRSA) is a common cause of community- and hospital-acquired infections and is responsible for a large fraction of deaths caused by antibiotic-resistant bacteria. S. aureus is surrounded by a complex cell envelope that protects it from antimicrobial compounds and other stresses. Here, we show that controlling the length of an essential cell envelope polymer, lipoteichoic acid, is critical for controlling S. aureus cell size and cell envelope integrity. We also show that genes involved in LTA length regulation are required for resistance to beta-lactam antibiotics in MRSA. The proteins encoded by these genes may be targets for combination therapy with an appropriate beta-lactam.
KEYWORDS: beta-lactams, cell division, cell envelope, cell wall, Gram-positive bacteria, lipoteichoic acid, peptidoglycan, peptidoglycan hydrolases, teichoic acids
INTRODUCTION
The bacterial cell envelope is a barrier that protects bacteria from unpredictable and often hostile environments. In Gram-positive bacteria, such as Staphylococcus aureus, the cell envelope comprises the cell membrane and a thick peptidoglycan (PG) layer that is decorated with a variety of proteins and polymers important for viability and virulence. Among these polymers are teichoic acids, which are negatively charged and divided into two classes based on their subcellular localization. One class, wall teichoic acids (WTA), are covalently linked to PG; the other class, lipoteichoic acids (LTA), are associated with the cell membrane through a glycolipid anchor (1). WTA and LTA play partially redundant roles in cell envelope integrity and cannot be deleted simultaneously (2–4).
In S. aureus, both WTA and LTA have been implicated in the control of cell morphology and division (3, 5, 6), virulence (7–14), osmoregulation (15–18), antimicrobial resistance (6, 19–22), and spatiotemporal regulation of cell wall enzymes (23–27). These functions may be accomplished through organization and regulation of cell envelope enzymes, binding of protons and divalent cations, and alteration of the physicochemical properties of the cell envelope. However, LTA is more important than WTA for cell viability. S. aureus can grow under standard laboratory conditions without WTA (28), but cells lacking lipoteichoic acid synthase (LtaS), the enzyme that assembles LTA on the cell surface, are not viable and rapidly acquire suppressor mutations (3, 5, 16, 29, 30).
The usual glycolipid anchor and the starting unit for LTA is diglucosyl-diacylglycerol (Glc2DAG), which is synthesized from UDP-glucose and diacylglycerol (DAG) by the glycosyltransferase UgtP (also called YpfP) (Fig. 1A and B) (31, 32). Glc2DAG is exported to the cell surface by LtaA (10). The lipoteichoic acid polymerase is LtaS, a polytopic membrane protein with an extracellular domain that contains the active site (4, 33). LtaS transfers phosphoglycerol units derived from phosphatidylglycerol (Ptd-Gro) to the Glc2DAG starter unit, producing DAG as a by-product (5, 34, 35). DAG is recycled to Ptd-Gro by a salvage pathway (36). LTA is heavily decorated with d-alanyl residues, a modification of teichoic acids that is important in autolysin regulation and has also been implicated in S. aureus virulence (8, 12, 13, 19). Notably, deleting the glycosyltransferase gene ugtP or the genes encoding the enzymes that produce UDP-glucose, its substrate, does not result in the loss of LTA, although such mutations result in morphological and fitness defects (10, 11, 32). Instead, LtaS uses Ptd-Gro rather than Glc2DAG as the lipid starter unit for LTA assembly.
FIG 1.
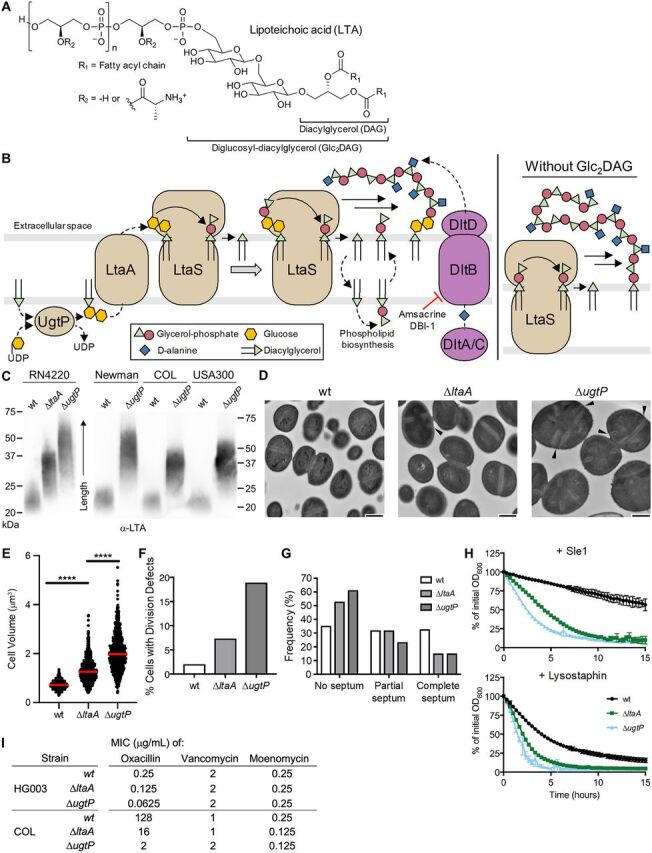
S. aureus mutants unable to synthesize LTA on Glc2DAG produce long LTA polymers anchored on Ptd-Gro, have cell size and division defects, and are susceptible to PG hydrolases and beta-lactam antibiotics. (A) LTA is a polymer of 30 to 50 phosphoglycerol repeats linked to a Glc2DAG lipid anchor. Repeat units are heavily modified with d-alanyl esters. (B) UgtP uses DAG and UDP-glucose to synthesize Glc2DAG, which is then exported by LtaA to the cell surface. LtaS polymerizes phosphoglycerol units derived from Ptd-Gro on the lipid anchor. Each elongation cycle generates DAG. The dlt pathway adds d-alanyl esters to LTA. In the absence of Glc2DAG, LtaS uses Ptd-Gro as an alternative lipid anchor, resulting in abnormally long LTA. (C) Anti-LTA Western blot of exponential-phase S. aureus lysates. RN4220 and Newman are methicillin-sensitive S. aureus (MSSA) strains, while COL and USA300 are MRSA strains. (D) TEM of RN4220 strains. Arrowheads indicate septal defects. Bars, 500 nm. (E) RN4220 cell volumes were calculated from cells lacking visible septa. The median is shown with a red bar. ****, P < 0.0001. (F) RN4220 cells were classified based on the cell cycle stage and presence of defects (see Fig. S1C for the classification scheme). Cells containing misplaced and/or multiple septa (classes C and E) were totalled. (G) Cell cycle stage frequency plot of classified cells without visible defects. (H) RN4220 strains were suspended in phosphate-buffered saline and treated with lytic enzymes. The decrease in OD600 was tracked over time. For all plots, the averages and standard deviations from 3 technical replicates are shown. (I) MIC table of oxacillin, vancomycin, and moenomycin against select S. aureus strains. HG003 is an MSSA strain, and COL is an MRSA strain.
In Bacillus subtilis, deleting ugtP or the genes for the enzymes that produce UDP-glucose also results in morphological defects (37–40). In one study, ugtP mutant cells were shown to be shorter than wild-type cells, and it was proposed that UgtP is a nutrient sensor that negatively regulates cytokinesis in a manner that depends on UDP-glucose levels (38). In this model, under nutrient-rich conditions, high levels of UDP-glucose localize UgtP to the cytokinetic ring and inhibit FtsZ polymerization or constriction, which delays cell division to provide cells time to grow to a larger size (38, 41). ΔugtP cells are therefore small, because UgtP is not present to slow cell division. Other studies in B. subtilis are not consistent with a role for UgtP in nutrient sensing, because mutant cells were found to be enlarged in its absence or to have shape rather than size alterations (39, 40). Instead, loss of the glycolipids produced by UgtP was proposed to be responsible for the defects of ΔugtP in this organism (42).
UgtP evidently does not act to increase cell size in S. aureus, because ΔugtP mutant cells are larger, rather than smaller, than those of the wild type (32). Like other S. aureus cells that grow too large (6, 43–46), the mutant cells have cell division defects, such as multiple and misplaced septa (32). While these defects may result directly from the loss of the ugtP gene product, the ΔugtP deletion is pleiotropic and causes multiple effects, including the absence of the disaccharide anchor Glc2DAG and a reported abnormal lengthening of LTA polymers (10). Whether increased cell size and dysregulated cell division directly result from the loss of the ugtP gene product, from the loss of intracellular Glc2DAG, or from the abnormally long LTA polymers has been unclear.
Mutants lacking ltaA, which encodes the flippase that exports Glc2DAG to the cell surface, provide a means to distinguish among these possibilities. ΔltaA mutants express UgtP and synthesize Glc2DAG but are unable to export it efficiently; they may have higher intracellular concentrations of Glc2DAG than wild-type cells (10). The LTA polymers produced by ΔltaA mutants are longer than those of the wild type but shorter than those of ΔugtP mutant cells, being a heterogeneous mixture in which some polymers are assembled on Ptd-Gro and some on Glc2DAG (which is exported to the cell surface by an alternative, unknown mechanism) (10). Although ΔltaA cells have an altered distribution of glycolipids in the membrane leaflets, and ΔugtP cells lack glycolipids entirely, we hypothesized that if the defects observed in ΔugtP cells were caused by the abnormally long LTA polymers, we should also observe cell size and division defects in the ΔltaA mutant. Under this assumption, we predicted that the ΔltaA mutant would have less pronounced phenotypes than the ΔugtP mutant due to the intermediate polymer length in the ΔltaA mutant.
Here, we show that the production of long, abnormal LTA is sufficient to alter cell size and lead to cell division defects. We also report that LTA pathway mutants with these morphological defects are highly susceptible to beta-lactam antibiotics and PG hydrolases and are dependent on other cell envelope pathways that are dispensable in wild-type strains. We used an inhibitor of one of these pathways to select for suppressor mutations in ΔugtP strains and found that most of the suppressor mutations were located in the LTA polymerase gene, ltaS, and caused a reduction in LTA polymer length. Polymer abundance was frequently decreased as well, in some cases to almost undetectable levels. The ltaS suppressor mutations partially reversed the cell size and division abnormalities caused by the ΔugtP mutation. Taken together, these studies indicate that LTA length and abundance play a crucial role in controlling cell size and cell envelope integrity in S. aureus.
RESULTS
ΔugtP and ΔltaA mutants are larger than wild-type cells and have cell division defects.
Earlier studies of this pathway were performed in mutagenized laboratory strains derived from NCTC 8325 and reached different conclusions on the impact of deleting ugtP on LTA length, with one study reporting increased length and the other reporting decreased length (10, 24). Because both results were in mutagenized strains, we examined LTA from the wild type and ΔugtP mutants in several other strain backgrounds and found that in all cases, LTA length is increased in ΔugtP mutants that do not produce Glc2DAG (Fig. 1C; also, see Fig. S1A in the supplemental material). A previous study in S. aureus reported that ΔugtP cells are larger than wild-type cells and have other morphological defects (32), but ΔltaA mutant cells have not been examined in detail. We compared the morphology of otherwise isogenic wild-type, ΔltaA, and ΔugtP strains by transmission electron microscopy (TEM) and quantified cell size using bright-field and epifluorescence microscopy after staining with a membrane dye. Compared with the wild type, ΔltaA cells appeared larger by TEM and ΔugtP cells were clearly larger (Fig. 1D). Quantification of cell size confirmed these observations: cell volume increased from 0.72 ± 0.14 μm3 for the wild type to 1.26 ± 0.38 μm3 for ΔltaA cells and 1.98 ± 0.59 μm3 for ΔugtP cells (Fig. 1E; Fig. S1B). ΔltaA and ΔugtP cells also displayed cell division defects, including multiple and misplaced septa (Fig. S1C and D). Only 2.1% (n = 536) of wild-type cells had these defects, compared with 7.3% (n = 518) for ΔltaA cells and 18.9% (n = 555) for ΔugtP cells (Fig. 1F). Furthermore, ΔltaA and ΔugtP cells were more commonly observed without partial or complete septa, indicating that they spend a longer time growing prior to initiating septal synthesis (Fig. 1G; Fig. S1E). This enrichment of cells without septa implies a delay in cell division in mutants that produce longer LTA polymers, which likely explains their large cell size. An important challenge for the future will be to elucidate the mechanism by which LTA polymer length influences cell size and division. Together, the shared phenotypes of ΔltaA and ΔugtP mutants indicate that control over cell growth and division is adversely affected when LTA is abnormally long and assembled on a Ptd-Gro, rather than on a Glc2DAG, membrane anchor.
ΔugtP and ΔltaA mutants are more susceptible to lytic enzymes than the wild type.
Given the established connection between teichoic acids and enzymes that act on the cell wall, we used ΔltaA and ΔugtP mutants to probe whether production of abnormal LTA causes susceptibility to lytic enzymes that degrade peptidoglycan. We employed two enzymes that act on distinct sites on peptidoglycan: lysostaphin, an endopeptidase produced in other Staphylococcus spp. that hydrolyzes the peptide cross bridges between PG strands (47, 48), and a maltose-binding protein (MBP) fusion of Sle1, an amidase native to S. aureus that hydrolyzes the amide bond between N-acetylmuramic acid and the stem peptide of PG (49). We found that ΔltaA and ΔugtP mutants were substantially more susceptible to these enzymes than were wild-type cells (Fig. 1H; Fig. S1F), indicating that cells that make long, abnormal LTA have alterations in their cell envelopes that cause increased susceptibility to enzymes that hydrolyze PG.
ΔugtP and ΔltaA MRSA mutants are sensitized to beta-lactam antibiotics.
Previous work has shown that when ltaS is depleted or when WTA is removed, methicillin-resistant S. aureus (MRSA) becomes susceptible to some beta-lactam antibiotics, including oxacillin (6, 50). Cells depleted of LTA and WTA-null cells also have cell size and division defects that resemble those that we observed for ΔltaA and ΔugtP mutants (5, 6). These observations prompted us to test whether deleting ltaA or ugtP in a MRSA background also affected beta-lactam susceptibility. Although the MIC for oxacillin was only modestly decreased when ltaA or ugtP was deleted in a methicillin-sensitive background, it decreased 8-fold and 64-fold for ΔltaA and ΔugtP mutants, respectively, in MRSA (Fig. 1I; Fig. S1G). In contrast, there was little or no change in the MICs of antibiotics that primarily affect peptidoglycan polymerization (moenomycin and vancomycin) rather than cross-linking. Unlike vancomycin and moenomycin, the mechanism of action of beta-lactams generates a large number of un-cross-linked PG strands (51, 52). We speculate that this condition may be particularly toxic to mutants with long LTA polymers due to a resulting overactivity of PG hydrolases, as such mutants are already sensitized to the action of lytic enzymes. The susceptibility of ΔltaA and ΔugtP strains to beta-lactams provides further evidence that these mutants have cell wall alterations.
ΔugtP and ΔltaA mutants lyse when teichoic acid d-alanylation is blocked.
We found in previous work that ΔugtP cells become reliant on a tailoring modification of LTA called d-alanylation in which d-alanyl esters are attached to the C-2 hydroxyl groups of the phosphoglycerol repeats of LTA (53). This modification, although dispensable in wild-type strains, is important in resistance to cationic antimicrobial peptides and aminoglycosides (15, 19) and has been linked to regulation of autolysin function (54). We previously identified two structurally unrelated inhibitors, DBI-1 and amsacrine (amsa), that inhibit DltB, an essential component of the d-alanylation pathway (Fig. 1B and 2A) (53, 55). Using spot titer assays, we found here that ΔltaA strains, like ΔugtP strains, are susceptible to both DltB inhibitors (Fig. 2B; Fig. S2A). ΔltaA mutants are not as sensitive to the DltB inhibitors as ΔugtP mutants, reflecting an intermediate phenotype that is consistent with the intermediate length of LTA in ΔltaA mutants. This susceptibility provides additional evidence that the production of abnormal, long LTA causes defects that result in conditional essentiality of other cell envelope modifications.
FIG 2.

Mutants with long, abnormal LTA undergo lysis when treated with d-alanylation inhibitors. (A) Structures of the two DltB inhibitors used in this study. (B) Tenfold serial dilutions of RN4220 strains on 10 μg/ml DltB inhibitor or DMSO vehicle control. (C) TEM of RN4220 strains treated with DMSO or with 10 μg/ml amsa. Bars, 2 μm. (D) Growth curves of RN4220 strains treated with DMSO or 10 μg/ml amsa. The averages and standard deviations from 3 technical replicates are shown.
Terminal phenotypes can provide insight into processes that lead to cell death. We therefore used TEM to compare the phenotypes of wild-type, ΔltaA, and ΔugtP cells treated with the DltB inhibitor amsa. Fields of untreated and treated wild-type cells appeared similar by TEM, but there were large numbers of cell ghosts and fragments in images of the treated ΔltaA and ΔugtP mutants (Fig. 2C; Fig. S2B). Moreover, when we monitored biomass by optical density after treatment with inhibitor, we observed that the mutants stopped growing and then the optical density dropped (Fig. 2D; Fig. S2C and D). The TEM images of the untreated ΔugtP mutants also showed qualitatively more lysis than in wild-type cells. Taken together, our findings show that ΔltaA and ΔugtP cells have a greater propensity to lyse than wild-type cells, and they undergo catastrophic lysis when d-alanylation is inhibited. This suggests that d-alanylation may control the activity of cell wall hydrolases.
Reducing LTA length and abundance suppresses lethality of d-alanylation inhibitors.
Because suppressor analysis can provide clues to the underlying basis of a defect, we selected for suppressor mutations of amsa lethality in a ΔugtP strain. Whole-genome sequencing of one suppressor showed a mutation in ltaS that resulted in a leucine substitution at F93, and we found that LTA produced by this mutant was shorter and less abundant than in the parent strain (see below). We raised additional amsa-resistant mutants from multiple independent cultures in a few distinct ugtP-null strains, all of which could be complemented (Fig. S3A). Targeted sequencing of ltaS showed that mutations in this gene accounted for 75% of the 64 resistant colonies tested. Notably, only three of the mutations resulted in premature stop codons. In all other mutants, the reading frame of ltaS was maintained, suggesting that at least partial function of the encoded protein was retained.
To assess whether the activity of LtaS was affected by the ltaS mutations, we selected 14 strains that contained mutations spanning different regions of the encoded enzyme (Fig. 3A; Fig. S3B) and evaluated the abundance and length of their LTA. LtaS contains five predicted transmembrane (TM) helices and a C-terminal extracellular domain that contains the active site (4, 33). Three mutants contained 18-bp insertions that resulted in the insertion of six amino acids in the extracellular loop between TM1 and TM2. The other eleven mutants contained single amino acid substitutions in another extracellular loop, in the “helical linker” region, or in the extracellular domain. For the mutants for which LTA was detectable by Western blotting, we found that it was shorter than that in the ugtP parent strain and was typically much less abundant (Fig. 3B). For some mutants, LTA was undetectable. Targeted sequencing did not identify suppressor mutations known to restore viability to ltaS knockouts (16, 29, 30). Moreover, whole-genome sequencing of a subset of strains did not identify any shared mutations in other genes (Fig. S3C). Taken together, these results suggested that suppression of the lethality caused by loss of d-alanylation in the ΔugtP strain was a direct result of the reduced LTA length and abundance.
FIG 3.

The lethality of amsa to the ΔugtP mutant is suppressed by mutations in LtaS that decrease LTA length and/or abundance. (A) Schematic of LtaS depicting the location of the identified mutations. (B) Anti-LTA Western blot of exponential-phase-cell lysates from isolated ltaS mutants in ugtP-null backgrounds. While LTA abundance cannot be precisely compared between LTA of different lengths with the anti-LTA antibody, several mutations appear to drastically decrease LTA abundance. The wild type and ugtP::Tn strains are in the SEJ1 background, while the strain background of each isolated mutant is given in Fig. S3B. (C) Tenfold serial dilutions of strains on plates containing 10 μg/ml DltB inhibitor or DMSO. The ΔltaS strain contains a gdpP* hypomorphic suppressor mutation. gdpP::Tn is a transposon disruption of gdpP. ΔltaS strains are in the SEJ1 background, while gdpP::Tn strains are in the RN4220 background.
Consistent with this conclusion, we found that a ΔugtP ΔltaS double-deletion strain was not susceptible to amsa because it does not produce LTA (Fig. 3C). This strain contains a hypomorphic mutation in gdpP (gdpP*). GdpP is a cyclic-di-AMP phosphodiesterase, and this mutation results in increased cellular levels of cyclic di-AMP, a second messenger implicated in osmoregulation (56). The accumulation of cyclic di-AMP allows growth in the absence of ltaS (16). We therefore confirmed that a ΔugtP gdpP::Tn double mutant remained sensitive to amsa and produced LTA resembling that of a ΔugtP single mutant (Fig. 3C; Fig. S4A and B). Therefore, amsa resistance in the ΔugtP ΔltaS double mutant was due not to gdpP* but rather to the absence of LTA.
To verify that amsa resistance in the suppressor strains was indeed caused by the ltaS mutations, we constructed strains expressing inducible copies of three ltaS mutant alleles (resulting in the mutations Ins3, F93L, and L181S) or the wild-type allele. The mutations are found in extracellular loops of the transmembrane domain (Ins3 and F93L) or in the linker region between the transmembrane domain and the extracellular domain (L181S). We then deleted the chromosomal copy of ltaS and introduced marked deletions of ltaA or ugtP into each strain (Fig. 4A). RN4220 is a mutagenized model strain and has uncharacterized mutations that may influence reported phenotypes. However, we have verified that the LTA length patterns, beta-lactam sensitivity, and amsa susceptibility of the ΔltaA and ΔugtP mutants are consistent across multiple strain backgrounds. Given this, the advantages of using this transformable strain to confirm effects of ltaS mutations outweighed concerns about the uncharacterized mutations. The strains required inducer for growth, indicating that the mutant ltaS alleles, like the wild-type allele, performed an essential function (Fig. 4A). Strains expressing the ltaS mutants grew on amsa and DBI-1 regardless of whether ltaA or ugtP was present; however, strains expressing ltaSwt in a ΔltaA or ΔugtP background did not grow (Fig. 4A; Fig. S5A and B). Growth curves showed that amsa-treated cells expressing ltaSwt in the ΔugtP background underwent growth arrest followed by lysis, while cells expressing the ltaS mutants continued to grow (Fig. 4B). For two of the mutants, the ltaSF93L and ltaSL181S mutants, growth rates in amsa were comparable to those of the untreated controls, but the ltaSIns3 mutant was growth impaired. This mutant also reached only 20% of the optical density of the other mutants in overnight cultures. Despite differences in fitness, the reconstructed mutants confirmed that the selected ltaS mutations are necessary and sufficient to confer resistance to d-alanylation inhibitors in a ΔugtP background.
FIG 4.
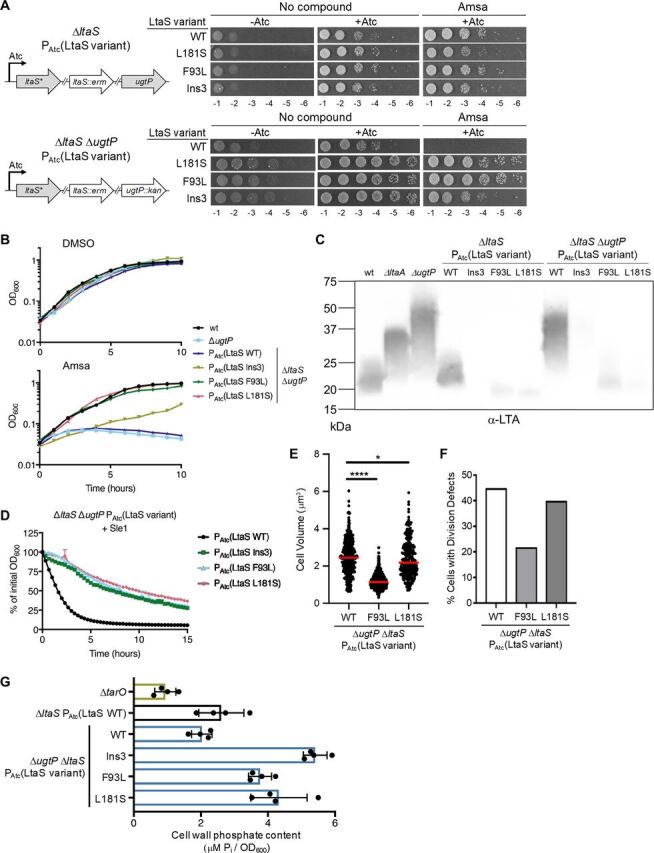
Mutations in ltaS that reduce LTA length and/or abundance correct defects of ΔugtP and increase WTA abundance. (A) RN4220 with an anhydrotetracyline (Atc)-inducible copy of selected ltaS mutants was transduced with an erythromycin (erm)-marked ltaS deletion and, optionally, a kanamycin (kan)-marked ugtP deletion. Rebuilt suppressor strains were spotted in a 10-fold dilution series on plates containing 10 μg/ml amsa or DMSO. Note that some residual growth is observed on plates without inducer due to growth of cells in the presence of inducer prior to plating. (B) Growth curves of RN4220 ΔugtP strains with ectopically expressed ltaS variants as the only source of LtaS. Strains were treated with 10 μg/ml amsa or DMSO. A representative growth curve from 3 replicates is shown for each strain. (C) Anti-LTA Western blot of exponential-phase lysates from rebuilt ltaS mutants. (D) RN4220 ΔugtP strains expressing ltaS alleles were suspended in phosphate-buffered saline, and the decrease in OD600 was tracked upon addition of Sle1. This phenotype is also observed with lysostaphin and when ltaS mutant alleles are expressed in the wild-type background (Fig. S7). The averages and standard deviations from 3 technical replicates are shown. (E) Cell volumes of reconstructed RN4220 ΔugtP strains expressing ltaS alleles were calculated from cells lacking visible septa. Expression of ltaSF93L (1.15 ± 0.31 μm3 [median ± median absolute deviation]) or ltaSL181S (2.17 ± 0.78 μm3) in the ΔugtP background reduces size compared to expression of ltaSwt (2.45 ± 0.73 μm3). The median is shown with a red bar. *, P < 0.05; ****, P < 0.0001. (F) RN4220 ΔugtP strains expressing ltaS alleles were classified based on cell cycle stage and presence of defects (see Fig. S1C for the classification scheme). The percentage of cells with division defects is decreased by expression of ltaSF93L (21.8%, n = 467) or ltaSL181S (39.9%, n = 308) compared to expression of ltaSwt (44.9%, n = 492). (G) Purified sacculi from overnight RN4220 cultures were hydrolyzed with HCl, and the released orthophosphate was quantified by an ammonium molybdate assay against a standard curve of KH2PO4. The ΔtarO strain is a mutant with a deletion of the first gene in the WTA biosynthesis pathway and does not produce WTA. The averages and standard deviations from 4 biological replicates are shown.
Access to a set of otherwise isogenic strains expressing different ltaS alleles allowed us to directly compare the impact of altering LTA length and abundance without confounding effects of other genetic differences. In the wild-type background, we found that LTA was undetectable in the ltaSIns3 strain; in strains with the other two ltaS mutant alleles, LTA was shorter and the signal was much less intense than in cells expressing ltaSwt (Fig. 4C). In the ΔugtP background, LTA was nearly undetectable in the ltaSIns3 mutant but appeared similar in length to that in the ltaSwt mutant. The other two mutants showed greatly reduced LTA signal intensity and appeared shorter than ltaSwt. Overall, these results confirmed that reducing LTA length or greatly reducing abundance in cells that produce long, abnormal LTA confers resistance to d-alanylation inhibitors. A question for future work is how d-alanylation protects cells from the lethality of long LTA polymers.
Suppressors with low LTA retain the ability to produce large amounts of DAG.
We noted that most of the ltaS mutant genes with suppressor mutations maintained the reading frame, yet the LTA levels in the mutants were typically low. LtaS produces DAG in addition to LTA, with one molecule of DAG produced during each round of elongation (Fig. 1B). DAG is generally thought to be a by-product of LTA synthesis that serves mainly as a feedstock for glycolipid synthesis and for a recycling pathway that leads to more Ptd-Gro (57–59). To verify that the ltaS mutants retained the ability to produce appreciable amounts of DAG and to assess the correlation between DAG and LTA levels, we purified and reconstituted LtaSwt, LtaSIns3, LtaSF93L, and LtaSL181S in proteoliposomes and quantified DAG production (Fig. S6A). As reported previously, LtaSwt made abundant LTA polymer in this system (60), but LTA was low or undetectable in the three mutants (Fig. S6B). However, all the mutants retained the capability to produce considerable amounts of DAG, ranging from ∼20% of the levels in the LtaSwt strain for the LtaSIns3 strain to levels comparable to those of the LtaSwt strain for the LtaSL181S strain (Fig. S6C). Because these are technically challenging assays, we measured only endpoints, so relative DAG levels should be interpreted cautiously. Nonetheless, and with this caveat in mind, it appeared that the mutants produced more DAG than expected based on the LTA polymer detected, suggesting that DAG production may be partially uncoupled from LTA synthesis.
Reducing LTA length and abundance partially corrects cell size and division defects and protects against peptidoglycan hydrolases.
With our set of otherwise isogenic strains expressing ltaS variants, we next examined whether the ltaS suppressor mutations corrected other defects of ΔugtP. We found that the rebuilt strain expressing ltaSwt in the ΔugtP background was highly susceptible to both Sle1 and lysostaphin, but strains expressing ltaS variants were not (Fig. 4D; Fig. S7). We also quantified cell morphology by microscopy and found that cells expressing ltaSF93L or ltaSL181S in the ΔugtP background were 46% and 88% as large, respectively, as cells expressing ltaSwt (Fig. 4E). These mutants had correspondingly fewer cell division defects (Fig. 4F). Although ltaSF93L and ltaSL181S mutants produce LTA of similar lengths, the ltaSF93L mutation corrected size and division defects to a greater extent than the ltaSL181S mutation. We attribute this observation to the difference in LTA abundance produced by these two strains. The abundance of LTA in the ltaSF93L strain more closely matched the abundance of LTA in the wild-type strain, suggesting that the limited amount of LTA in the ltaSL181S strain may be insufficient to promote proper growth and division, as seen in ltaS depletion strains that grow abnormally large before lysing (5). Therefore, there may be both an ideal LTA length and abundance for optimal growth. The ltaSIns3 mutant could not be quantified due to severe clumping and morphological aberrations. Nevertheless, the results for the other mutants allow us to conclude that reducing LTA length and abundance partially corrects the cell size and division defects of ΔugtP mutant cells and also confers protection against PG hydrolases.
Suppressors with low LTA have high WTA levels.
Despite producing LTA at levels substantially below wild-type levels, the ltaS mutants we selected were viable and relatively healthy. Moreover, two grew similarly to the wild type (Fig. 4B). Because LTA and WTA have partially overlapping roles, we hypothesized that there may be compensatory changes in WTA abundance in the ltaS mutants that contribute to their fitness. To address this question, we treated purified sacculi with strong acid to release orthophosphate from WTA and quantified it. We found that sacculi from strains expressing the mutant ltaS alleles contained two to three times as much phosphate as sacculi from the strain expressing ltaSwt, consistent with a large increase in WTA (Fig. 4G). Polyacrylamide gel electrophoresis (PAGE) analysis of WTA polymers liberated from purified sacculi with base showed modest increases in WTA length in the strains expressing ltaS mutant alleles (Fig. S8). We conclude that S. aureus adapts to a reduction in LTA by increasing WTA. Previous work has shown that WTA positively regulates cell wall cross-linking (26). Moreover, WTA is known to decrease susceptibility to PG hydrolases (23, 25). Therefore, we surmise that increased WTA levels and the concomitant changes in cell wall structure lead to the decreased susceptibility of our ltaS mutant strains to cell wall hydrolases.
DISCUSSION
The principal goals of this investigation were to elucidate the roles of ugtP and LTA length in S. aureus cell growth and division and to identify mechanisms contributing to cell viability when LTA levels are low. First, by comparing multiple phenotypes of ΔltaA and ΔugtP mutants, we showed that cells producing long, abnormal LTA have cell growth and division defects and are also more susceptible to cell lysis than the wild type. Second, we showed that suppressor mutations in ltaS that reduced LTA length and abundance corrected these defects. Third, by comparing otherwise isogenic ltaS mutants that make either high or low levels of LTA, we showed that WTA levels correlated inversely with LTA levels. It is likely that the increased abundance of WTA partially compensated for low levels of LTA.
Previous work has shown that S. aureus ΔugtP mutants, which make long LTA, grow larger than normal (32). In B. subtilis, ugtP is also important in cell growth and division, although its absence led to decreased cell size in some studies (37, 38). B. subtilis UgtP is proposed to function as a nutrient sensor that, when present with sufficient UDP-glucose, slows Z-ring assembly so that cells grow to a larger size prior to division. Our results show that such a mechanism does not operate in S. aureus. First, ΔugtP mutant cells spend a longer time growing before initiating septal synthesis and consequently are larger rather than smaller than the wild type. Second, ΔltaA mutants are also larger than the wild type even though they express ugtP and produce intracellular Glc2DAG. Therefore, dysregulated cell growth in LTA pathway mutants upstream of the LtaS polymerase is not due to the absence of the proteins or their products but rather is due to the production of abnormally long LTA. Consistent with this conclusion, cell growth defects were reduced in a ΔugtP mutant background by mutations in ltaS that reduced LTA length.
Previous work has shown that cell division defects result when S. aureus cells grow larger than normal (6, 43–46). Consistent with these findings, we observed a remarkable correlation between cell size and frequency of cell division defects for the mutants studied here. For example, ΔltaA mutants were substantially smaller than ΔugtP mutants, and we found that ΔltaA mutants had 50% fewer cell division defects. Moreover, ltaS mutations that reduced cell size in a ΔugtP background had fewer cell division defects, with the magnitudes of the reduction in size and the reduction in division defects tracking closely. We therefore concluded that the cell division defects observed in ΔugtP and ΔltaA mutants are due to dysregulated coordination between cell growth and division.
We have shown that S. aureus WTA levels increase substantially when LTA levels are low. This finding is reminiscent of findings in Streptococcus pneumoniae showing that levels of WTA and LTA are inversely regulated (61). S. pneumoniae synthesizes both forms of teichoic acid through the same pathway, with the outcome being distinguished only by a final ligation step where the polymer precursor is transferred to PG or to a glycolipid (62). In S. pneumoniae, LTA synthesis predominates during exponential growth, but there is a switch to predominantly WTA synthesis as cells approach stationary phase. S. aureus WTA and LTA are synthesized by different pathways, with only the d-alanine tailoring modification as a shared feature. Nevertheless, there appears to be a mechanism to redistribute resources between the WTA and LTA pathways when one is lacking. Although LTA and WTA share some functions and act synergistically to maintain cell envelope integrity, their spatial localization is different. LTA is located between the membrane and the inner layer of PG, and WTA extends from the inner layer of PG beyond the outer layer. Consistent with this difference in localization, we have found that producing long, abundant LTA tends to promote cell lysis, whereas producing abundant WTA tends to limit cell lysis. Given this evidence that physiological roles of LTA and WTA in S. aureus are not identical, it would not be surprising to find that regulatory mechanisms exist to control the relative abundance of these polymers.
Expression of normal LTA is critical for S. aureus virulence. Previous work has shown that strains lacking ltaA or ugtP have attenuated pathogenicity (10, 11). While the physiological basis of this attenuation is not known, in light of our data on the susceptibility of ΔltaA and ΔugtP to PG hydrolases, it may involve an increased sensitivity to host lytic enzymes. We show here that the defects caused by expression of long, abnormal LTA also result in increased susceptibility of MRSA to beta-lactam antibiotics. We therefore anticipate that inhibitors of enzymes that act upstream of LtaS will have therapeutic potential, particularly if used in combination with an appropriate beta-lactam. Future work will focus on identifying such inhibitors and elucidating the mechanism for glycolipid-dependent control of LTA polymer length.
MATERIALS AND METHODS
General information.
Chemicals and reagents were purchased from Sigma-Aldrich unless otherwise indicated. Oligonucleotides were purchased from Integrated DNA Technologies. Detergents were purchased from Anatrace. Restriction enzymes were purchased from New England Biolabs. Staphylococcus aureus strains were grown with shaking at 30°C unless otherwise indicated in tryptic soy broth (TSB; Becton Dickinson Biosciences) or cation-adjusted Mueller-Hinton broth 2 (MHB2). Escherichia coli strains were grown at 37°C with shaking in LB-Miller broth (LB; Becton Dickinson Biosciences). Growth on solid medium used the appropriate broth plus 1.5% (wt/vol) agar (Becton Dickinson Biosciences). When required, antibiotics were used at the following concentrations: 50 μg/ml kanamycin, 50 μg/ml neomycin, 10 μg/ml erythromycin, 10 μg/ml chloramphenicol, 3 μg/ml tetracycline, and 100 μg/ml carbenicillin. Anhydrotetracycline (aTc) was used at 400 nM. S. aureus genomic DNA was isolated using a Wizard genomic DNA purification kit (Promega). Genomic DNA from S. aureus RN4220 or isolated amsacrine-resistant mutants was used as a template in PCRs to amplify S. aureus genes.
Strain construction.
To construct strains with aTc-inducible, integrated copies of genes via pTP63, plasmids were electroporated into S. aureus RN4220 containing the pTP44 plasmid. Transformants were selected on chloramphenicol at 30°C. Marked deletions, marked transposon insertions, and aTc-inducible genes were transduced via bacteriophage ϕ85 as described previously (63).
Analysis of LTA length by Western blotting.
Overnight cultures grown in TSB were diluted in fresh TSB and grown to an approximate optical density at 600 nm (OD600) of 0.8. A 0.5-ml portion of each culture (normalized by OD600) were harvested by centrifugation and suspended in 50 μl of a solution containing 50 mM Tris (pH 7.4), 150 mM NaCl, and 200 μg/ml lysostaphin (from Staphylococcus staphylolyticus; AMBI Products). The cells were incubated at 37°C for 10 min, diluted with one volume of 4× sodium dodecyl sulfate (SDS)-PAGE loading buffer, and boiled for 30 min. Samples were centrifuged for 10 min at 16,000 × g to pellet any insoluble material. The supernatant was diluted with 1 vol of water and treated with 0.5 μl of 20 mg/ml proteinase K (New England Biolabs) at 50°C for 2 h. Samples were separated on 4% to 20% Mini-Protean TGX acrylamide gels (Bio-Rad) with a running buffer consisting of 5 g/liter Tris base, 15 g/liter glycine, and 1 g/liter SDS and transferred to a polyvinylidene difluoride (PVDF) membrane. Western blotting was performed as described previously (60).
Transmission electron microscopy.
Overnight cultures grown in TSB were diluted in fresh TSB and grown to mid-log phase. Cells were treated with dimethyl sulfoxide (DMSO) or 16 μg/ml amsacrine for 4 h at 30°C, added to an equal volume of fixative solution (1.25% formaldehyde, 2.5% glutaraldehyde, and 0.03% picric acid in 100 mM sodium cacodylate [pH 7.4]), and pelleted for fixation. Samples were prepared for TEM by the Harvard Medical School Electron Microscopy Facility, and images were captured on a JEOL 1200EX instrument.
Microscopy.
Overnight cultures grown in TSB were diluted in fresh TSB and grown to an approximate OD600 of 0.5. To stain the cell membrane, FM 4-64 (Thermo Fisher Scientific) was added at a final concentration of 5 μg/ml to 1 ml of culture for 5 min at room temperature. Cells were pelleted at 4,000 × g for 1 min, washed with PBS (pH 7.4) (50 mM sodium phosphate [pH 7.4], 150 mM NaCl), and suspended in 100 μl of PBS (pH 7.4). A 1-μl aliquot of cells was spotted on top of a 2% agarose gel pad mounted on a glass slide. A 1.5-mm coverslip was placed over the cells and sealed with wax before imaging.
The cells were imaged at 30°C as described previously (46). For each field of view, 3 images were taken: (i) phase-contrast, (ii) bright field, and (iii) fluorescence. The phase-contrast and bright-field images were acquired at a 100-ms camera exposure, while the fluorescence image was acquired at 500 ms. The bright-field images were used for cell segmentation for quantitative image analyses. Fluorescence images were used to detect division defects and sort cells, as depicted in Fig. S1C.
Image segmentation, cell volume quantification, and cellular phenotype classification were performed as described previously (46). Cell volumes were calculated from cells lacking visible septa. For each strain, 600 to 1,000 cells were quantified. A two-tailed Mann-Whitney U nonparametric test was used to calculate the P value for the differences in distribution of cell sizes among strains. Three hundred to 600 cells of each strain were assessed for cell division phenotypes.
Expression and purification of MBP-Sle1.
S. aureussle1 (SAOUHSC_00427) was cloned into the NdeI and BamHI sites of pMAL-c5X (New England Biolabs) with primers oTD22 and oTD23 and transformed into E. coli NEB Express cells. An overnight culture grown at 30°C was used to inoculate fresh medium, and the culture was grown at 37°C to an approximate OD600 of 0.5. Cultures were cooled on ice and induced with 0.3 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 16°C overnight. Cells were pelleted at 3,600 × g for 15 min at 4°C, suspended in lysis buffer (20 mM Tris [pH 7.2], 200 mM NaCl, 10% glycerol) plus 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 100 μg/ml lysozyme, and 100 μg/ml DNase, and lysed with 3 passages through an EmulsiFlex-C3 cell disruptor (Avestin). All subsequent steps were performed at 4°C. Insoluble material was pelleted at 119,000 × g for 35 min, and the supernatant was bound to amylose resin (New England Biolabs). The resin was washed with lysis buffer and eluted with lysis buffer plus 10 mM maltose. The elution was concentrated with a 30-kDa-molecular-weight-cutoff spin concentrator (EMD Millipore) and further purified on a Superdex 200 Increase 10/300 GL (GE Life Sciences) equilibrated in lysis buffer. Appropriate fractions were concentrated, flash frozen with liquid nitrogen, and stored at –80°C.
Whole-cell lytic assays.
Overnight cultures grown in TSB were diluted in fresh TSB and grown to an approximate OD600 of 3. A 1.5-ml portion of culture (normalized by OD600) was harvested by centrifugation, washed with PBS (pH 7.2), and suspended in 1.6 ml of PBS (pH 7.2). A 75-μl portion of cell suspension was added to 75 μl of 25 nM lysostaphin, 250 to 500 nM purified Sle1, or PBS (pH 7.2). Samples were incubated at 25°C with shaking in a SpectraMax Plus 384 microplate reader, and OD600 was monitored over time.
MIC determination.
Overnight cultures grown in TSB were diluted in fresh MHB2 (without antibiotics) and grown to an approximate OD600 of 0.8. Cultures were diluted with medium to an OD600 of 0.001, and 146 to 147 μl was added to each well of a 96-well plate. Three to four microliters of a compound dilution series was added, and cultures were grown with shaking at 37°C for 18 h. Each condition was tested in technical triplicates, and the MIC was determined as the lowest concentration that prevented growth.
Spot dilutions.
Overnight cultures of each strain grown in TSB were diluted in fresh TSB and grown to an approximate OD600 of 0.8. Cultures were normalized to an OD600 of 0.1, a 10-fold dilution series from 10−1 to 10−6 was created, and dilutions were spotted on TSB agar containing any desired compounds. Strains grown with anhydrotetracycline were washed once with TSB before diluting. Plates were incubated at 30°C and imaged the next day with a Nikon D3400 DSLR camera fitted with an AF Micro-Nikkor 60-mm f/2.8D lens.
Growth curves.
Overnight cultures of each strain grown in TSB were diluted in fresh TSB and grown to an approximate OD600 of 0.8. Cultures were diluted to an OD600 of 0.03, and amsacrine or DBI-1 (in DMSO) was added at a final concentration of 10 μg/ml. DMSO was added to untreated control cultures at a final concentration of 2%. Cultures were grown at 30°C in a 150-μl volume with shaking in a SpectraMax Plus 384 microplate reader (Molecular Devices), and OD600 was monitored over time.
Raising amsacrine-resistant mutants.
Amsacrine-resistant mutants were raised in the following strains: SEJ1 ugtP::Tn, HG003 ugtP::Tn, RN4220 ΔugtP, and SEJ1 ΔugtP::Kan. For mutants in the SEJ1 ugtP::Tn background, 50 μl of undiluted overnight cell culture was plated on TSB agar plus 6 μg/ml amsacrine at 30°C for 2 days. For mutants in the backgrounds SEJ1 ΔugtP::Kan, HG003 ugtP::Tn, and RN4220 ΔugtP, overnight cultures were diluted in TSB and grown at 30°C to an OD600 of 1.0. One milliliter of this culture was harvested, suspended in 100 μl fresh TSB, and plated on TSB agar plus 10 μg/ml amsacrine at 30°C for 2 days. Multiple independent cultures were used to increase the diversity of mutants. Whole-genome sequencing of select mutants was performed with an Illumina MiSeq as described previously (46).
Construction of strains with anhydrotetracycline-inducible ltaS alleles.
Genes encoding wild-type LtaS and Ins3, F93L, and L181S LtaS variants were cloned from the genomic DNA of RN4220 or their respective suppressor mutants into pTP63 with the primers iLtaS_1 and iLtaS_2 and electroporated into RN4220 bearing pTP44 for integration (45). Each resulting strain was transduced with ϕ85 lysate from a strain with an erythromycin-marked ltaS deletion. These strains were then optionally transduced with ϕ85 lysate from a strain with a kanamycin-marked ugtP or ltaA deletion.
Phosphate quantification from purified sacculi.
Sacculi containing covalently linked WTA were isolated in a manner similar to that described previously (64). Two milliliters of an overnight culture grown in TSB (normalized by OD600) was harvested, washed with buffer 1 (50 mM MES [pH 6.5]), and suspended in buffer 2 (50 mM MES [pH 6.5], 4% SDS). Cells were boiled for 1 h, and pellets were harvested at 16,000 × g for 10 min. The supernatant was discarded, and the pellets were washed twice with buffer 2, once with buffer 3 (50 mM MES [pH 6.5], 342 mM NaCl), and twice with buffer 1. Pellets were treated with 50 μg/ml DNase and 50 μg/ml RNase in buffer 1 at 37°C for 1 h. Pellets were harvested, washed with buffer 1, and suspended in a solution containing 20 mM Tris (pH 8), 0.5% SDS. Samples were treated with 20 μg/ml proteinase K at 50°C for 2 h with light shaking. After pellets were harvested by centrifugation, pellets were washed once with buffer 3 and then three times with water.
Purified sacculi were suspended in 1 M HCl. A dilution series of K2HPO4 in 1 M HCl was also prepared. Samples were treated at 80°C for 16 h and cooled to room temperature. Any insoluble material remaining was pelleted by centrifugation, and the supernatant was retained. An ammonium molybdate reagent was prepared by mixing, in order, equal volumes of 2 M H2SO4, 2.5% (wt/vol) ammonium molybdate, and 10% (wt/vol) ascorbic acid. One volume of ammonium molybdate reagent was added to each sample, and samples were incubated at 37°C for 1 h. Orthophosphate was quantified by absorbance at 820 nm with the K2HPO4 standard curve.
Polyacrylamide gel electrophoresis of WTA polymers.
Sacculi containing covalently linked WTA were isolated as described above but treated with 100 mM NaOH at room temperature for 16 h. Three volumes of loading buffer (50% glycerol, 100 mM Tris-Tricine, 0.02% bromophenol blue) were added to each sample.
High-resolution 20- by 20-cm polyacrylamide gels were prepared as described previously (64), but with a stacking gel consisting of 3% acrylamide (3% T–3.3% C, where T is total acrylamide and C is the percentage of T consisting of bisacrylamide), 900 mM Tris (pH 8.5), 0.1% ammonium persulfate, and 0.01% tetramethylethylenediamine. Gels were run at 4°C in a Protean II xi Cell electrophoresis system (Bio-Rad) at 40 mA/gel with a running buffer consisting of 100 mM Tris-Tricine (pH 8.2) until the bromophenol blue loading dye was near the bottom of the gel. Gels were washed with water, stained with 1 mg/ml aqueous alcian blue for 30 min, destained with water and 40% ethanol–5% acetic acid, and rehydrated with water. Silver staining was performed with a Silver Stain Plus kit (Bio-Rad) without the fixation step. Images were taken with a Nikon D3400 DSLR camera fitted with an AF Micro-Nikkor 60-mm f/2.8D lens and converted to an 8-bit image using ImageJ.
Purification of LtaS mutants and proteoliposome analysis of DAG production.
Mutant LtaS constructs were cloned from genomic DNA isolated from the original suppressor mutants and assembled into pET28b with the primers LtaS_F and LtaS_R as previously described (60). LtaS constructs were expressed, purified, and reconstituted into proteoliposomes as previously described (60). Proteoliposomes were added to 9 volumes of a solution containing 20 mM succinate (pH 6.0), 50 mM NaCl, and 5% DMSO. Reaction mixtures were incubated in the presence or absence of 1 mM MnCl2. LTA was detected by Western blotting as previously described (60). To measure DAG production, reactions proceeded for 4 h at 30°C, and then DAG was extracted and assayed according to the instructions provided by the Cell BioLabs DAG assay kit. Reactions were performed in duplicate and plotted using GraphPad Prism. Absolute activity was calculated by subtracting activity values calculated from reactions that did not use MnCl2 from the values from reactions that did use MnCl2. Activity was compared between mutants by setting the activity in reactions with proteoliposomes containing wild-type LtaS to 100%.
Data availability.
Whole-genome sequencing data (accession number PRJNA612838) can be found in the NCBI BioProject database.
ACKNOWLEDGMENTS
Some strains were generated with the help of Lincoln Pasquina and Wonsik Lee, and we thank them for their generous assistance.
This project was funded by the National Institutes of Health (P01-AI083214 to S.W. and R01-AI139011 to S.W. and R.L.) and the National Science Foundation (DGE1144152 to A.R.H. and to T.D.).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Rajagopal M, Walker S. 2017. Envelope structures of Gram-positive bacteria. Curr Top Microbiol Immunol 404:1–44. doi: 10.1007/82_2015_5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santa MariaJP, Jr, Sadaka A, Moussa SH, Brown S, Zhang YJ, Rubin EJ, Gilmore MS, Walker S. 2014. Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc Natl Acad Sci U S A 111:12510–12515. doi: 10.1073/pnas.1404099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oku Y, Kurokawa K, Matsuo M, Yamada S, Lee B-L, Sekimizu K. 2009. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J Bacteriol 191:141–151. doi: 10.1128/JB.01221-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schirner K, Marles-Wright J, Lewis RJ, Errington J. 2009. Distinct and essential morphogenic functions for wall‐ and lipo‐teichoic acids in Bacillus subtilis. EMBO J 28:830–842. doi: 10.1038/emboj.2009.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gründling A, Schneewind O. 2007. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc Natl Acad Sci U S A 104:8478–8483. doi: 10.1073/pnas.0701821104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell J, Singh AK, Santa MariaJP, Jr, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. 2011. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weidenmaier C, Kokai-Kun JF, Kristian SA, Chanturiya T, Kalbacher H, Gross M, Nicholson G, Neumeister B, Mond JJ, Peschel A. 2004. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat Med 10:243–245. doi: 10.1038/nm991. [DOI] [PubMed] [Google Scholar]
- 8.Weidenmaier C, Peschel A, Kempf VA, Lucindo N, Yeaman MR, Bayer AS. 2005. DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 73:8033–8038. doi: 10.1128/IAI.73.12.8033-8038.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wanner S, Schade J, Keinhörster D, Weller N, George SE, Kull L, Bauer J, Grau T, Winstel V, Stoy H, Kretschmer D, Kolata J, Wolz C, Bröker BM, Weidenmaier C. 2017. Wall teichoic acids mediate increased virulence in Staphylococcus aureus. Nat Microbiol 2:16257. doi: 10.1038/nmicrobiol.2016.257. [DOI] [PubMed] [Google Scholar]
- 10.Gründling A, Schneewind O. 2007. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J Bacteriol 189:2521–2530. doi: 10.1128/JB.01683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheen TR, Ebrahimi CM, Hiemstra IH, Barlow SB, Peschel A, Doran KS. 2010. Penetration of the blood-brain barrier by Staphylococcus aureus: contribution of membrane anchored lipoteichoic acid. J Mol Med (Berl) 88:633–639. doi: 10.1007/s00109-010-0630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins LV, Kristian SA, Weidenmaier C, Faigle M, van Kessel KPM, van Strijp JAG, Götz F, Neumeister B, Peschel A. 2002. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis 186:214–219. doi: 10.1086/341454. [DOI] [PubMed] [Google Scholar]
- 13.Simanski M, Gläser R, Köten B, Meyer-Hoffert U, Wanner S, Weidenmaier C, Peschel A, Harder J. 2013. Staphylococcus aureus subverts cutaneous defense by D-alanylation of teichoic acids. Exp Dermatol 22:294–296. doi: 10.1111/exd.12114. [DOI] [PubMed] [Google Scholar]
- 14.Bunk S, Sigel S, Metzdorf D, Sharif O, Triantafilou K, Triantafilou M, Hartung T, Knapp S, von Aulock S. 2010. Internalization and coreceptor expression are critical for TLR2-mediated recognition of lipoteichoic acid in human peripheral blood. J Immunol 185:3708–3717. doi: 10.4049/jimmunol.0901660. [DOI] [PubMed] [Google Scholar]
- 15.Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev 67:686–723. doi: 10.1128/mmbr.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corrigan RM, Abbott JC, Burhenne H, Kaever V, Gründling A. 2011. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 7:e1002217. doi: 10.1371/journal.ppat.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, McGuinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T. 2013. Discovery of wall teichoic acid inhibitors as potential anti-MRSA beta-lactam combination agents. Chem Biol 20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuster CF, Wiedemann DM, Kirsebom FCM, Santiago M, Walker S, Gründling A. 2020. High-throughput transposon sequencing highlights the cell wall as an important barrier for osmotic stress in methicillin resistant Staphylococcus aureus and underlines a tailored response to different osmotic stressors. Mol Microbiol 113:699–717. doi: 10.1111/mmi.14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gale RT, Götz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 20.Kohler T, Weidenmaier C, Peschel A. 2009. Wall teichoic acid protects Staphylococcus aureus against antimicrobial fatty acids from human skin. J Bacteriol 191:4482–4484. doi: 10.1128/JB.00221-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown S, Xia G, Luhachack LG, Campbell J, Meredith TC, Chen C, Winstel V, Gekeler C, Irazoqui JE, Peschel A, Walker S. 2012. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc Natl Acad Sci U S A 109:18909–18914. doi: 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, Yang SJ. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One 9:e107426. doi: 10.1371/journal.pone.0107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankel MB, Schneewind O. 2012. Determinants of murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J Biol Chem 287:10460–10471. doi: 10.1074/jbc.M111.336404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fedtke I, Mader D, Kohler T, Moll H, Nicholson G, Biswas R, Henseler K, Götz F, Zähringer U, Peschel A. 2007. A Staphylococcus aureus ypfP mutant with strongly reduced lipoteichoic acid (LTA) content: LTA governs bacterial surface properties and autolysin activity. Mol Microbiol 65:1078–1091. doi: 10.1111/j.1365-2958.2007.05854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sieradzki K, Tomasz A. 2003. Alterations of cell wall structure and metabolism accompany reduced susceptibility to vancomycin in an isogenic series of clinical isolates of Staphylococcus aureus. J Bacteriol 185:7103–7110. doi: 10.1128/jb.185.24.7103-7110.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atilano ML, Pereira PM, Yates J, Reed P, Veiga H, Pinho MG, Filipe SR. 2010. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc Natl Acad Sci U S A 107:18991–18996. doi: 10.1073/pnas.1004304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiwari KB, Gatto C, Walker S, Wilkinson BJ. 2018. Exposure of Staphylococcus aureus to targocil blocks translocation of the major autolysin Atl across the membrane, resulting in a significant decrease in autolysis. Antimicrob Agents Chemother 62:e00323-18. doi: 10.1128/AAC.00323-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol 188:4183–4189. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bæk KT, Bowman L, Millership C, Dupont Søgaard M, Kaever V, Siljamäki P, Savijoki K, Varmanen P, Nyman TA, Gründling A, Frees D. 2016. The cell wall polymer lipoteichoic acid becomes nonessential in Staphylococcus aureus cells lacking the ClpX chaperone. mBio 7:e01228-16. doi: 10.1128/mBio.01228-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karinou E, Schuster CF, Pazos M, Vollmer W, Gründling A. 2018. Inactivation of the monofunctional peptidoglycan glycosyltransferase SgtB allows Staphylococcus aureus to survive in the absence of lipoteichoic acid. J Bacteriol 201:e00574-18. doi: 10.1128/JB.00574-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jorasch P, Warnecke DC, Lindner B, Zähringer U, Heinz E. 2000. Novel processive and nonprocessive glycosyltransferases from Staphylococcus aureus and Arabidopsis thaliana synthesize glycoglycerolipids, glycophospholipids, glycosphingolipids and glycosylsterols. Eur J Biochem 267:3770–3783. doi: 10.1046/j.1432-1327.2000.01414.x. [DOI] [PubMed] [Google Scholar]
- 32.Kiriukhin MY, Debabov DV, Shinabarger DL, Neuhaus FC. 2001. Biosynthesis of the glycolipid anchor in lipoteichoic acid of Staphylococcus aureus RN4220: role of YpfP, the diglucosyldiacylglycerol synthase. J Bacteriol 183:3506–3514. doi: 10.1128/JB.183.11.3506-3514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu D, Wörmann ME, Zhang X, Schneewind O, Gründling A, Freemont PS. 2009. Structure-based mechanism of lipoteichoic acid synthesis by Staphylococcus aureus LtaS. Proc Natl Acad Sci U S A 106:1584–1589. doi: 10.1073/pnas.0809020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duckworth M, Archibald AR, Baddiley J. 1975. Lipoteichoic acid and lipoteichoic acid carrier in Staphylococcus aureus H. FEBS Lett 53:176–179. doi: 10.1016/0014-5793(75)80013-5. [DOI] [PubMed] [Google Scholar]
- 35.Taron DJ, ChildsWC, III, Neuhaus FC. 1983. Biosynthesis of D-alanyl-lipoteichoic acid: role of diglyceride kinase in the synthesis of phosphatidylglycerol for chain elongation. J Bacteriol 154:1110–1116. doi: 10.1128/JB.154.3.1110-1116.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jerga A, Lu YJ, Schujman GE, de Mendoza D, Rock CO. 2007. Identification of a soluble diacylglycerol kinase required for lipoteichoic acid production in Bacillus subtilis. J Biol Chem 282:21738–21745. doi: 10.1074/jbc.M703536200. [DOI] [PubMed] [Google Scholar]
- 37.Price KD, Roels S, Losick R. 1997. A Bacillus subtilis gene encoding a protein similar to nucleotide sugar transferases influences cell shape and viability. J Bacteriol 179:4959–4961. doi: 10.1128/jb.179.15.4959-4961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weart RB, Lee AH, Chien AC, Haeusser DP, Hill NS, Levin PA. 2007. A metabolic sensor governing cell size in bacteria. Cell 130:335–347. doi: 10.1016/j.cell.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazarevic V, Soldo B, Medico N, Pooley H, Bron S, Karamata D. 2005. Bacillus subtilis alpha-phosphoglucomutase is required for normal cell morphology and biofilm formation. Appl Environ Microbiol 71:39–45. doi: 10.1128/AEM.71.1.39-45.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuoka S, Chiba M, Tanimura Y, Hashimoto M, Hara H, Matsumoto K. 2011. Abnormal morphology of Bacillus subtilis ugtP mutant cells lacking glucolipids. Genes Genet Syst 86:295–304. doi: 10.1266/ggs.86.295. [DOI] [PubMed] [Google Scholar]
- 41.Chien AC, Zareh SK, Wang YM, Levin PA. 2012. Changes in the oligomerization potential of the division inhibitor UgtP co-ordinate Bacillus subtilis cell size with nutrient availability. Mol Microbiol 86:594–610. doi: 10.1111/mmi.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuoka S, Seki T, Matsumoto K, Hara H. 2016. Suppression of abnormal morphology and extracytoplasmic function sigma activity in Bacillus subtilis ugtP mutant cells by expression of heterologous glucolipid synthases from Acholeplasma laidlawii. Biosci Biotechnol Biochem 80:2325–2333. doi: 10.1080/09168451.2016.1217147. [DOI] [PubMed] [Google Scholar]
- 43.Pinho MG, Errington J. 2003. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol Microbiol 50:871–881. doi: 10.1046/j.1365-2958.2003.03719.x. [DOI] [PubMed] [Google Scholar]
- 44.Jorge AM, Hoiczyk E, Gomes JP, Pinho MG. 2011. EzrA contributes to the regulation of cell size in Staphylococcus aureus. PLoS One 6:e27542. doi: 10.1371/journal.pone.0027542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang T, Wang X, Lim HC, Bernhardt TG, Rudner DZ. 2017. The nucleoid occlusion factor Noc controls DNA replication initiation in Staphylococcus aureus. PLoS Genet 13:e1006908. doi: 10.1371/journal.pgen.1006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Do T, Schaefer K, Santiago AG, Coe KA, Fernandes PB, Kahne D, Pinho MG, Walker S. 2020. Staphylococcus aureus cell growth and division are regulated by an amidase that trims peptides from uncrosslinked peptidoglycan. Nat Microbiol 5:291–303. doi: 10.1038/s41564-019-0632-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler CA, Schuhardt VT. 1964. Lysostaphin: a new bacteriolytic agent for the staphlyococcus. Proc Natl Acad Sci U S A 51:414–421. doi: 10.1073/pnas.51.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson JM, Hardman JK, Sloan GL. 1979. Relationship between lysostaphin endopeptidase production and cell wall composition in Staphylococcus staphylolyticus. J Bacteriol 137:1158–1164. doi: 10.1128/JB.137.3.1158-1164.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M. 2005. Identification and molecular characterization of an N-acetylmuramyl-L-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol Microbiol 58:1087–1101. doi: 10.1111/j.1365-2958.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- 50.Meredith TC, Wang H, Beaulieu P, Gründling A, Roemer T. 2012. Harnessing the power of transposon mutagenesis for antibacterial target identification and evaluation. Mob Genet Elements 2:171–178. doi: 10.4161/mge.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waxman DJ, Yu W, Strominger JL. 1980. Linear, uncross-linked peptidoglycan secreted by penicillin-treated Bacillus subtilis. J Biol Chem 255:11577–11587. [PubMed] [Google Scholar]
- 52.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasquina L, Santa MariaJP, Jr, Wood BM, Moussa SH, Matano LM, Santiago M, Martin SE, Lee W, Meredith TC, Walker S. 2016. A synthetic lethal approach for compound and target identification in Staphylococcus aureus. Nat Chem Biol 12:40–45. doi: 10.1038/nchembio.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peschel A, Vuong C, Otto M, Götz F. 2000. The D-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob Agents Chemother 44:2845–2847. doi: 10.1128/aac.44.10.2845-2847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matano LM, Morris HG, Wood BM, Meredith TC, Walker S. 2016. Accelerating the discovery of antibacterial compounds using pathway-directed whole cell screening. Bioorg Med Chem 24:6307–6314. doi: 10.1016/j.bmc.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fahmi T, Port GC, Cho KH. 2017. c-di-AMP: an essential molecule in the signaling pathways that regulate the viability and virulence of Gram-positive bacteria. Genes (Basel) 8:197. doi: 10.3390/genes8080197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsuoka S, Hashimoto M, Kamiya Y, Miyazawa T, Ishikawa K, Hara H, Matsumoto K. 2011. The Bacillus subtilis essential gene dgkB is dispensable in mutants with defective lipoteichoic acid synthesis. Genes Genet Syst 86:365–376. doi: 10.1266/ggs.86.365. [DOI] [PubMed] [Google Scholar]
- 58.Parsons JB, Rock CO. 2013. Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res 52:249–276. doi: 10.1016/j.plipres.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuhn S, Slavetinsky CJ, Peschel A. 2015. Synthesis and function of phospholipids in Staphylococcus aureus. Int J Med Microbiol 305:196–202. doi: 10.1016/j.ijmm.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Vickery CR, Wood BM, Morris HG, Losick R, Walker S. 2018. Reconstitution of Staphylococcus aureus lipoteichoic acid synthase activity identifies Congo red as a selective inhibitor. J Am Chem Soc 140:876–879. doi: 10.1021/jacs.7b11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flores-Kim J, Dobihal GS, Fenton A, Rudner DZ, Bernhardt TG. 2019. A switch in surface polymer biogenesis triggers growth-phase-dependent and antibiotic-induced bacteriolysis. Elife 8:e44912. doi: 10.7554/eLife.44912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heß N, Waldow F, Kohler TP, Rohde M, Kreikemeyer B, Gómez-Mejia A, Hain T, Schwudke D, Vollmer W, Hammerschmidt S, Gisch N. 2017. Lipoteichoic acid deficiency permits normal growth but impairs virulence of Streptococcus pneumoniae. Nat Commun 8:2093. doi: 10.1038/s41467-017-01720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee W, Do T, Zhang G, Kahne D, Meredith TC, Walker S. 2018. Antibiotic combinations that enable one-step, targeted mutagenesis of chromosomal genes. ACS Infect Dis 4:1007–1018. doi: 10.1021/acsinfecdis.8b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meredith TC, Swoboda JG, Walker S. 2008. Late-stage polyribitol phosphate wall teichoic acid biosynthesis in Staphylococcus aureus. J Bacteriol 190:3046–3056. doi: 10.1128/JB.01880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Whole-genome sequencing data (accession number PRJNA612838) can be found in the NCBI BioProject database.