

Abstract
Purpose:
Transposable elements are known to remodel gene structure and provide a known source of genetic variation. Retrotransposon gag-like-3 (RTL3) is a mammalian retrotransposon-derived transcript (MART) whose function in the skeletal tissue is unknown. This study aimed to elucidate the biological significance of RTL3 in chondrogenesis and type-II collagen (COL2A1) gene expression in chondrocytes.
Materials and Methods:
Expression of RTL3, SOX-9 and COL2A1 mRNAs was determined by TaqMan assays and the protein expression by immunoblotting. RTL3 and Sox-9 depletion in human chondrocytes was achieved using validated siRNAs. An RTL3 mutant (ΔRTL3) lacking the zinc finger domain was created using in vitro mutagenesis. Forced expression of RTL3, ΔRTL3 and SOX-9 was achieved using CMV promoter containing expression plasmids. CRISPR-Cas9 was utilized to delete Rtl3 and create a stable ATDC5Rlt3−/− cell line. Matrix deposition and Col2a1 quantification during chondrogenesis was determined by Alcian blue staining and Sircol™ Soluble Collagen Assay, respectively.
Results:
RTL3 is not ubiquitously expressed but showed strong expression in cartilage, chondrocytes and synoviocytes but not in muscle, brain or other tissues analyzed. Loss-of-function and gain-of-function studies demonstrated a critical role of RTL3 in the regulation of SOX-9 and COL2A1 expression and matrix synthesis during chondrogenesis. Both RTL3 and SOX-9 displayed co-regulated expression in chondrocytes. Gene regulatory activity of RTL3 requires the c-terminal CCHC zinc-finger binding domain.
Conclusions:
Our results identify a novel regulatory mechanism of COL2A1 expression in chondrocytes that may help to further understand the skeletal development and the pathogenesis of diseases with altered COL2A1 expression.
Keywords: ZCCHC5/RTL3, gene regulation, chondrocyte, chondrogenesis, extracellular matrix
Introduction
Transposable elements (TE) are mobile, repetitive genetic elements that comprise roughly 50% of the human genome1. The frequency and location of TE integrations influence genomic structure and evolution and affect gene and protein regulatory networks during development and in differentiated cell types2,3. Deleterious insertions and mutations disrupt proper gene function and contribute to the pathologies of inherited and sporadic human cancers and other diseases1,4.
The Sushi family of Ty3/gypsy long terminal repeat retrotransposons were first identified in teleost fish and Sushi-like neogenes were subsequently identified in mammals5,6. Mammalian retrotransposon-derived transcripts (MARTs) cannot transpose but have retained open reading frames, demonstrate high levels of evolutionary conservation and are subject to selective pressures, which suggests some have become neofunctionalized genes with new cellular functions7,8. Several cases support this theory and one of the MARTs, MART1, has been found associated with human melanoma and angiomyolipoma9,10.
Retrotransposon gag-like-3 (RTL3/ZCCHC5/MART3) is one of eleven Sushi-like neogenes identified in the human genome7 RTL3 encodes a 53 kDa protein with a nucleic acid binding domain (CX2CX4HX4C), gag-like region within the open reading frame and a ssDNA/RNA-binding homeobox-associated leucine zipper motif7,8,11 (Figure 1). At present, there is a scarcity of knowledge regarding the tissue expression and biological significance of RTL3 in mammals.
Figure 1:
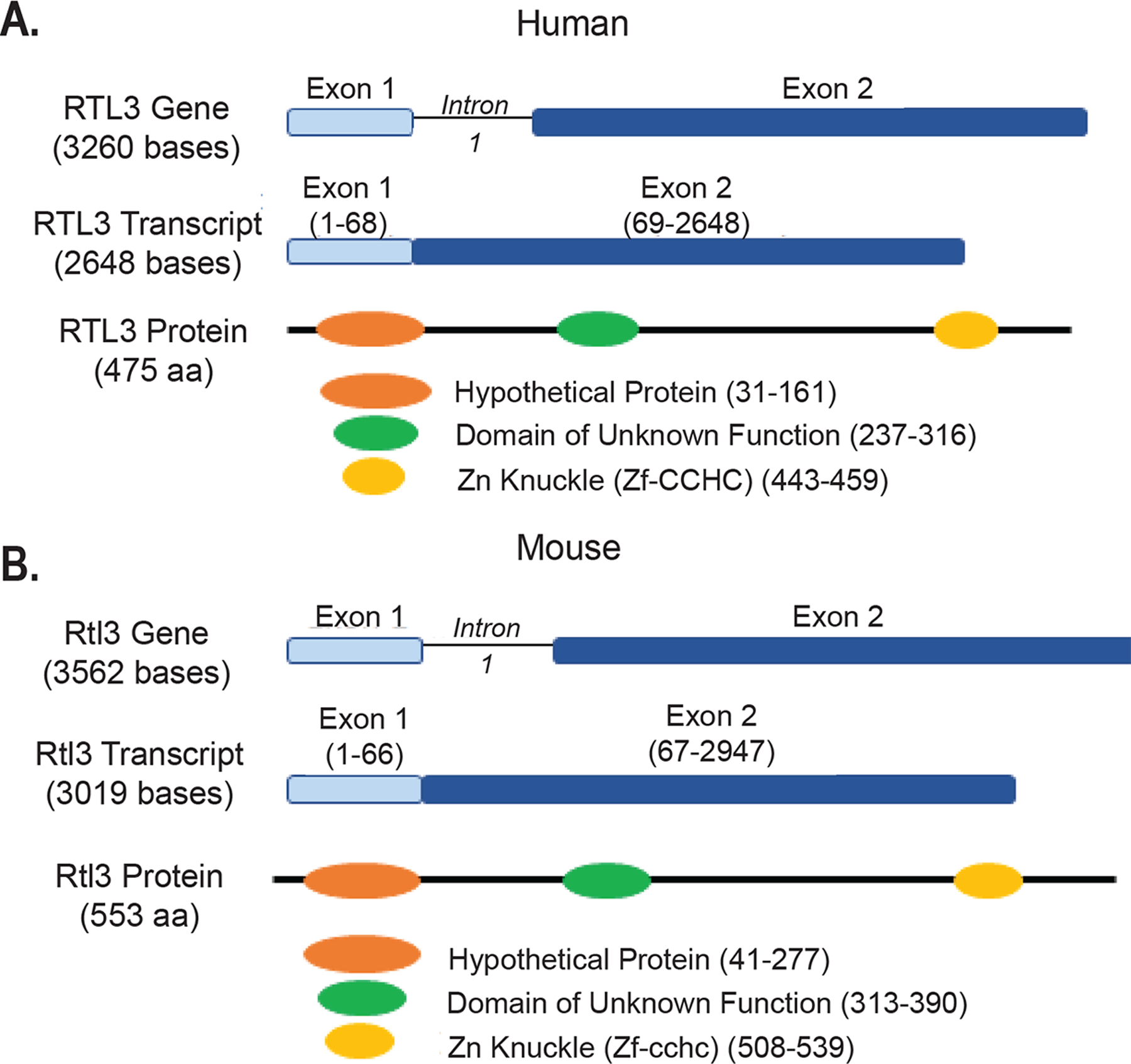
Genetic structure of RTL3 in human and mouse. The RTL3 gene encodes a 475 amino acid long peptide (A) in humans (NP_689907) and a 553 amino acid long peptide (B) in mouse (NP_955762).
To address this, we first determined RTL3 expression in human tissues and cell lines and found that RTL3 showed high expression in cartilage, chondrocytes, bone, and heart. Because of its abundant expression in cartilage and chondrocytes, we next assessed if the onset of the degenerative joint disease (DJD) Osteoarthritis (OA) affect RTL3 expression in human cartilage and discovered that the expression of RTL3 expression was significantly reduced in OA affected cartilage. Furthermore, in adult articular chondrocytes, siRNA-mediated knockdown of RTL3 or of SOX-9 results in the downregulation of the other and reduced Type-II Collagen (COL2A1) mRNA and protein expression. In cell types that normally do not express RTL3 and COL2A1 but express SOX-9, ectopic expression of RTL3 induced the expression of COL2A1. These data demonstrate that both RTL3 and SOX-9 are essential components of the COL2A1 gene expression regulatory network in chondrocytes. Importantly, we also demonstrate that the RTL3 zinc finger (CCHC) binding domain is required for the regulation of SOX-9 expression in chondrocytes. Finally, having established the role of RTL3 in SOX-9 and COL2A1 regulation, we assessed the effects of Rtl3 deletion on induced chondrogenesis in ATDC5 cells and demonstrate that Rtl3 depletion impairs matrix synthesis and delays chondrogenesis and also negatively impacts the expression of Sox-9 and Col2a1. Collectively, our data unearth the function of the neogene RTL3 and identify it as a key player in SOX-9-mediated regulation of COL2A1 expression.
Materials and Methods
Studies using discarded and de-identified human normal and OA cartilage were approved by the Institutional Review Boards (IRB) of Northeast Ohio Medical University (NEOMED) and Summa Health Systems, Barberton, Ohio as “non-human subject study under 45 CFR” prior to the initiation of the project. Normal (no history of rheumatic disease) cartilage was procured from National Disease Research Interchange (NDRI). All animal experiments were approved by the NEOMED IACUC prior to the initiation of the studies and were performed according to the approved protocols.
Human chondrocyte preparations and culture conditions
Chondrocytes were isolated from discarded and de-identified human cartilage of patients undergoing total knee arthroplasty (TKA) due to OA and culture confirmed by visual assessment of a rounded polygonal morphology and gene expression analyses (Supplementary Figure 1) as described in several of our publications12–16. Chondrocytes were cultured in monolayer at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s Medium (DMEM) with high glucose and F:12 (1:1), L-glutamine (HyClone, Cat # SH30022.02) and 100 IU/ml penicillin/100 μg/ml streptomycin (PS) (HyClone, Cat # SV30010) and medium was supplemented with 10% fetal calf serum (FCS) and cultured at 37°C and 5% CO2. Chondrocytes were utilized as primary cells without passage within 3–4 days of isolation. Human cell lines (HEK, U2OS, HTB-94) were purchased from ATCC and cultured in complete medium (DMEM 90%, FCS 10%, L-glutamine and 100 IU/ml penicillin/100 μg/ml streptomycin) at 37°C and 5% CO2.
Mouse primary chondrocyte isolation and culture conditions
Mouse chondrocytes were isolated from femoral condyle and tibial plateau cartilage of three-week-old mice. Chondrocytes were prepared via sequential enzymatic digestion as previously described17,18 and cultured in monolayer in supplemented DMEM media as described above.
Chondrocyte treatment with IL-1β
Prior to experimental treatment of chondrocytes, dosage and duration kinetic experiments were carried out to determine that best conditions (Supplementary Figure 2). Chondrocytes in monolayer were treated with 5 ng/ml IL-1β (R& D Systems, Cat# 201-LB-025) for 24 hrs in at 37°C and 5% CO2 before harvesting for RNA isolation as described below.
Human tissue and cell line qPCR
Human tissue RNAs for heart, brain, spleen, skeletal muscle and peripheral blood lymphocytes (H-PBL) were purchased from TakaRa (Mountain View, CA 94043). Genomic DNA-free total RNA from cartilage, primary chondrocytes and cell lines was isolated using Qiagen miRNeasy kit (Cat #217004). cDNA was synthesized using 1 μg total RNA following recommended protocols for the High Capacity cDNA synthesis kit (Applied Biosystems, Cat #4368814). Serial dilutions were used to obtain a standard curve for copy number quantification. Expression of RTL3 in tissues and cell lines was assessed by TaqMan assay with TaqMan master mix (IDT, Cat #1055770) on a StepOne Plus (Applied Biosystems) machine. Tissue and cell line expression was further verified by PCR using the primers designed with PrimerBlast and purchased from IDT (Coralville, Iowa): F: 5’CACAATAGCCCAAGAGCCCA3’ and R: 5’ACATTGCTCCAGTAAGGGGC3’. The predicted amplicon size was 581 base pairs and the specificity of the amplicon was verified via restriction digestion with the BamHI enzyme (New England Biolabs, Cat # R0136S) using the recommended buffer system (Supplementary Figure 4). Cartilage gene expression was determined using GeneQuery arrays and the data analyses were conducted following the manufacturer recommended protocols (ScienCell Research Laboratories, Cat #GK064).
CRISPR-Cas9 deletion of Rtl3 in ATDC5 cell line
ATDC5 cells were cultured in supplemented DMEM media as described above for primary chondrocyte cultures. A mouse Rtl3 gene knockout kit was purchased from OriGene (Cat #KN519666) to perform CRISPR-Cas9 deletion in ATDC cells. In brief, Turbofectin (OriGene, Cat# TF81005) was used to transfect gRNA and donor DNA into ATDC5 cells in DMEM media. Expression of GFP was used to visualize transfected cells and qPCR was used to estimate transfection efficiency. Post transfection, cells were serially split and grown in monoculture in DMEM supplemented with increasing doses (0.5–5nmol) of the selective antibiotic Puromycin (Sigma-Aldrich Cat # P8833) with one week intervals until the stable knockout cell line was established.
Chondrogenic differentiation and assessment in ATDC5 cell lines
Equivalent cell numbers for ATDC5Rlt3−/− and wild type ATDC5 cells, determined using hemacytometer, were chondrogenically differentiated in duplicate cultures in medium containing DMEM and F12 (1:1) supplemented with PS, 10% FCS and 1% ITS+1 liquid media supplement (Sigma-Aldrich, Cat #12521)19. Differentiation was completed over 21 days with daily media changes. Every 3 days matrix synthesis in one set of cultures was determined by Alcian Blue staining following published protocols20 while cells from the other set were harvested at the same time for gene expression analyses as described below. Images were taken on a Nikon Eclipse Ti microscope equipped with NIS Element imaging software (Nikon). Semi-quantification of Alcian Blue staining was completed using both percent area of proteoglycan staining with ImageJ software (NIH) as well as absorbance via overnight incubation in 8M guanidine hydrochloride solution with absorbance read at 600 nm on a 96-well plate reader (Synergy H1)20,21.
Total RNA isolation and gene expression analyses using TaqMan assays
Total RNA from cultured chondrocytes and other cell types was isolated using a modified Trizol-Chloroform method described earlier22. RNA was quantified using a Nanodrop 1000 Spectrophotometer (Thermo Fisher) and the integrity of the RNA preparation was determined using the TapeStation 4200 (Agilent Technologies) with High Sensitivity RNA ScreenTape analysis kit (Agilent, Cat #5067–5579). cDNA was synthesized using the High Capacity kit as described above. qPCR reactions were conducted using TaqMan assays with TaqMan master mix (IDT, Cat #1055770) on a StepOne Plus (Applied Biosystems) machine. Target mRNA levels were determined as fold change differences following the delta-delta Ct method with β-Actin expression level employed as the endogenous control for normalization.
Quantification of Soluble Collagen
Soluble collagen deposited by equivalent numbers of ATDC5Rlt3−/− and wild type ATDC5 cells was quantified using the Sircol™ Soluble Collagen Assay (BioColor, Cat #S1000) following the recommended protocol. Absorption at 555nM was read on a BioTek Synergy H1 96-well plate reader.
Generation of RTL3 Zinc finger CCHC deletion mutant
The RTL3 Zinc finger CCHC domain deletion mutant (ΔpRTL3) was generated by PCR based deletion strategy. Briefly, HindIII restriction enzyme site present in the RTL3 ORF (at position 748–752) was used in the forward primer (5’-CAGaagcttCCTGAGTTCCTGGTTCAGCTG-3’) and reverse primer (5’-GAgcggccgcGTTTCAGCTGGTGG-3’) encompassing positions 1242–1257 in the ORF was designed with Not-I restriction enzyme site to delete the Zinc finger domain. PCR was done with Vent DNA polymerase and the resulting PCR product was cloned into RC223801 plasmid (obtained from Origene having wild type RTL3) digested with HindIII and Not-I. The sequence of the clone was verified by DNA sequencing. Protein size of wild type RTL3 is 475 aa and that of zinc-finger deletion mutant is 418 aa.
siRNA-mediated depletion and forced expression of human RTL3
To deplete RTL3 expression, human chondrocytes were transfected with 100 nmoles of either scrambled control or RTL3-targeting siRNAs (OnTargetPlus, SMARTPOOL, Dharmacon, #Cat no L-016899-02-0005) using recommended protocols for X-tremeGENE siRNA Transfection Reagent (Sigma-Aldrich, Cat # 04476093001). For over expression experiments, human chondrocytes and HEK cells were transfected with pcDNA control plasmid, RTL3 over expression plasmid (Origene, RC223801) or ΔpRTL3 plasmid following recommended protocols for X-tremeGENE HP DNA Transfection Reagent (SigmaAldrich, Cat #06366236001). After transfection, cells were allowed to recover for 48 hours and either harvested for RNA isolation for gene expression analyses or cell lysate preparation for immunoblotting.
siRNA-mediated depletion and forced expression of human SOX-9
Human chondrocytes were transfected with 100 μM of either scrambled control or siRNAs targeting human SOX-9 (OnTargetPlus, SMARTPOOL, Dharmacon, #Cat no L-021507-00-005) using X-tremeGENE reagent as described above. For over expression experiments, human chondrocytes were transfected with pcDNA control plasmid or SOX-9 over expression plasmid (Origene, RC208944) using the recommended protocols for X-tremeGENE HP DNA Transfection Reagent (SigmaAldrich, Cat #06366236001). Cells were allowed to recover for 48 hours post-transfection prior to harvesting for RNA isolation for gene expression analyses or cell lysate preparation for immunoblotting.
Immunoblotting
Chondrocyte lysates, SDS-Page gels and immunoblotting were completed as previously described23. The primary antibody against RTL3 was purchased from Novus (NBP2–30851) and validated in the lab using RTL3 over expression plasmid (Supplementary Figure 3). The primary antibodies for β-Actin (sc-47778) and COL2A1 (sc-7764) were from Santa Cruz Biotechnology. A primary antibody against SOX-9 was purchased from Millipore (ab5535). Anti-rabbit (#7074) and anti-mouse (#7076) HRP-conjugated secondary antibodies were from Cell Signaling Technologies. Membrane development was done with Luminata Forte ECL reagent (Millipore, Cat # WBLUF0500) and visualized on a Pxi imaging system.
Immunofluorescence
Mouse primary chondrocytes were seeded (2 × 105) on cover slips in 24-well plates for 2 days after isolation and were fixed in 10% NBF before permeabilization with 0.3% Triton X-100 in PBS as described previously24. Treated cells were probed with DAPI and primary and secondary antibodies as indicated and then mounted with ProLong® Gold Antifade mounting medium (Thermofisher Scientific, Cat # P36930). Images were captured on an Olympus FV1000 confocal microscope with a 60X oil immersion lens.
Promoter-reporter activity assays
Luciferase reporter assay was performed as described previously25. Briefly, human articular chondrocytes (2.5 × 105) in a 24 well plate were co-transfected with pcDNA or pGL3 control plasmids, RTL3 or SOX-9 expression plasmids, pCOL2A1-luciferase reporter plasmid (GeneCopoeia, Cat #HPRM22364-PG02) using X-tremeGENE HP DNA Transfection Reagent. Renilla luciferase reporter (pRL-TK, Promega) was co-transfected and used for normalization in all experiments. Chondrocytes were incubated with the required plasmid and the transfection reagent for 6 hours in serum-free media and then the medium was aspirated and replaced with medium containing 10% FCS and antibiotics. For siRNA-mediated depletion of RTL3 or SOX9, chondrocytes were first co-transfected with pcDNA or pGL3 control plasmids and pCOL2A1-luciferase reporter and incubated as described above. The cells were allowed to rest for 24 hours before transfection with either scramble control or siRNAs targeting either human RTL3 or SOX-9 as described earlier17 and were harvested 48 hours post-transfection, washed in cold PBS and luciferase activity was determined following the manufacturer’s recommended protocols for the Dual Glo® Luciferase Assay kit (Promega, Cat # E2920) on a Lumat LB 9507 machine (Berthold Technologies).
Quantification and statistical analyses
Statistical analyses were conducted with GraphPad Prism software. We calculated statistical significance using student t-tests or one-way analysis of variance (ANOVA) tests with Tukey’s post-hoc analyses. A p-value of ≤ 0.05 was regarded as significant.
Results
RTL3 is expressed in many, but not all, human tissues including cartilage and chondrocytes
Using TaqMan assay, we first determined the RTL3 mRNA expression in several human tissues and cell lines and found that RTL3 mRNA was not ubiquitously expressed. Expression of RTL3 was robust in long bone, cartilage, heart tissue, HTB94, U2OS, primary chondrocytes and synoviocytes but its expression was not detected in HEK, spleen and skeletal muscle (Figure 2A, Supplementary Figure 4). Because expression of RTL3 was strong in normal cartilage and chondrocytes, we next determined whether the expression of RTL3 is affected in OA-a disease which primarily targets articular cartilage in the joints. RTL3 mRNA (Figure 2B) and protein (Figure 2C) levels were found to be significantly reduced in OA cartilage (n=5 independent samples: 2M, 3F, Mean Age 61y) compared to cartilage from normal (no history of rheumatic disease) individuals (n=5 independent samples: 2M, 3F, Mean Age 44y). Furthermore, examination of catabolic gene expression in the same samples using a GeneQuery Array with 88 OA and cartilage related genes showed that several key catabolic genes (MMP-13, ADAMTS4 and ADAMTS5) associated with OA pathogenesis were significantly upregulated in OA cartilage compared to normal cartilage (Figure 2D). As inflammatory cytokines are an important factor in OA pathogenesis26–28, we next determined whether the inflammatory cytokine IL-1β can modulate its expression in human chondrocytes. Therefore, we treated human chondrocytes isolated from the damaged areas of OA cartilage (n=6 independent samples: 2M, 4F, Mean Age 64y) and normal cartilage (n=5 independent samples: 2M, 3F, Mean Age 44y) with IL-1β and determined the RTL3 expression by TaqMan assay. Our results demonstrated that the expression of RTL3 was suppressed in response to treatment with IL-1β in both cases but the expression level was most severely affected in chondrocytes isolated from the damaged OA cartilage (Figure 2E). As high degree of chondrocyte apoptosis is known to occur in OA29, we next determined whether inhibition of RTL3 expression plays a role in chondrocyte death in OA. siRNA-mediated depletion of RTL3 with a validated siRNA in human chondrocytes (n=6 independent samples: 2M, 4F, Mean Age 64y) resulted in significantly reduced RTL3 mRNA (Figure 2F) and protein levels (Figure 2G) compared to no treatment and siControl transfected chondrocytes without adversely affecting chondrocyte viability (Figure 2H), suggesting that inflammatory cytokine-mediated inhibition of RTL3 expression does not play a role in chondrocyte death in OA and that the low levels of RTL3 expression in OA cartilage were not due to the loss of chondrocytes.
Figure 2:
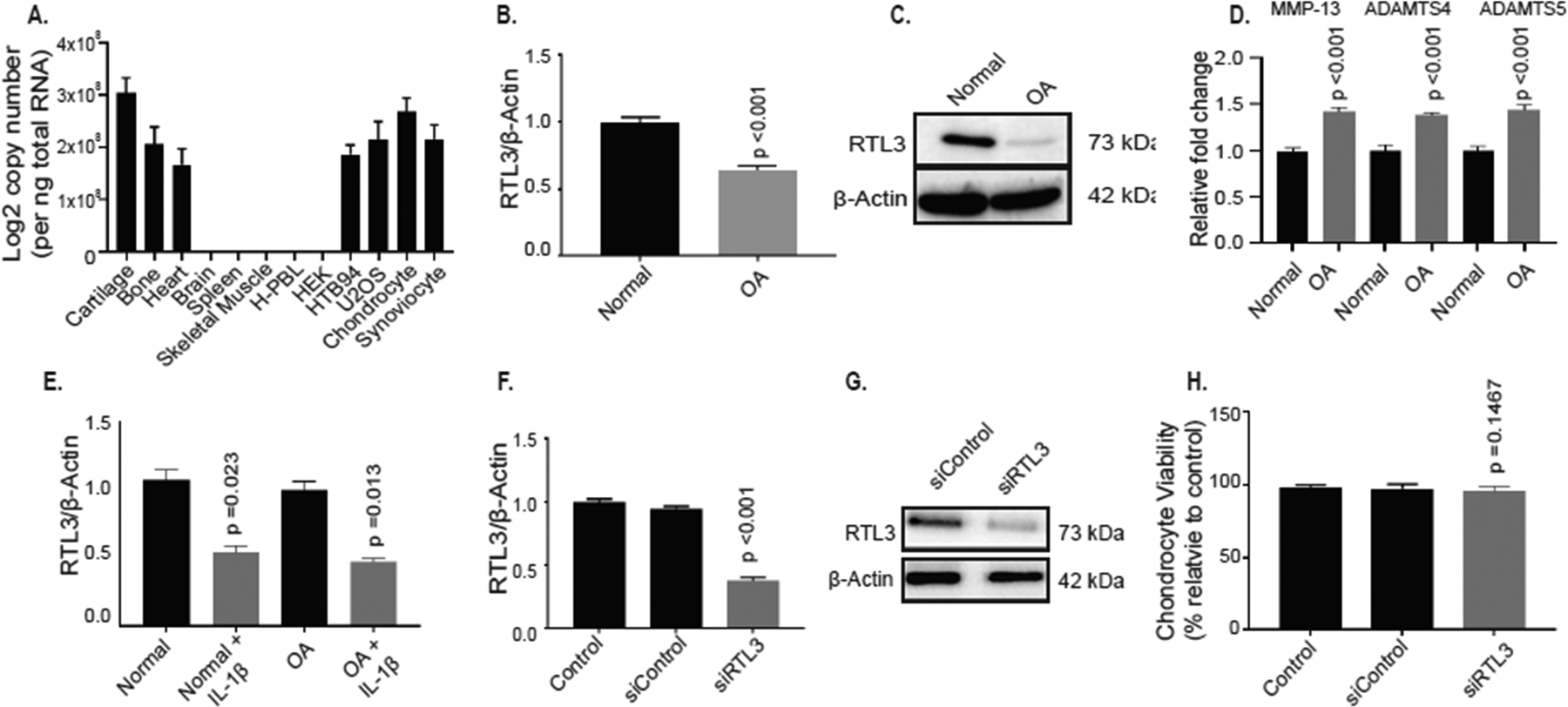
RTL3 is expressed in human tissues and cell lines and the expression is reduced in osteoarthritic cartilage and upon RTL3-depletion. (A) qPCR analysis showed that RTL3 mRNA was expressed in several human tissues and cell lines. Expression of RTL3 mRNA (B) and protein levels (C) were significantly lower in osteoarthritic cartilage compared to normal (no history of rheumatic disease) control. (D) GeneQuery qPCR assays detected significantly increased mRNA expression of MMP-13, ADAMTS4 and ADAMTS5 in OA cartilage compared to normal. Treatment with IL-1β significantly reduced RTL3 mRNA expression in both normal and OA chondrocytes (E). RTL3 depletion was successful in chondrocytes and resulted in significantly reduced mRNA (F) and protein levels (G) without adversely affecting cell viability (H).
RTL3 is primarily localized in the nucleus and its depletion negatively affects the expression of SOX-9 and COL2A1 and a COL2A1 promoter reporter activity in human chondrocytes
Having established a negative correlation between RTL3 inhibition and catabolic gene expression (Figure 2), we next determined the localization of RTL3 in chondrocytes and whether disruption in RTL3 expression affects the expression of key anabolic factor COL2A1 and the master transcription factor SOX-9 in terminally differentiated human chondrocytes using loss-of-function and gain-of-function approaches. The immunofluorescent localization of cellular compartmentalization of RTL3 in primary chondrocytes showed it to be chiefly present in the nucleus (Figure 3A). To determine the impact of downregulation of RTL3 expression on the expression of anabolic factors in chondrocytes, we knocked down its expression using validated siRNAs as above (Figure-2F–H). Concomitant with reduced RTL3 expression levels in chondrocytes with siRNA-mediated depletion of RTL3 expression was a significant reduction in the constitutive expression of SOX-9 and COL2A1 mRNAs (Figure 3B) and protein levels (Figure 3C). To determine if depletion of RTL3 affects the transcription of COL2A1, activity of a COL2A1 promoter reporter was determined. siRNA-mediated depletion of RTL3 in chondrocytes resulted in a significant reduction in COL2A1 promoter reporter activity suggesting that depletion of RTL3 affects COL2A1 expression at the transcriptional level (Figure 3D). As SOX-9 is known to regulate the expression of COL2A130,31 and was downregulated in chondrocytes with inhibition of RTL3 expression (Figure 3B), our data suggests that inhibition of COL2A1 expression in may be linked to the inhibition of SOX-9 expression.
Figure 3:
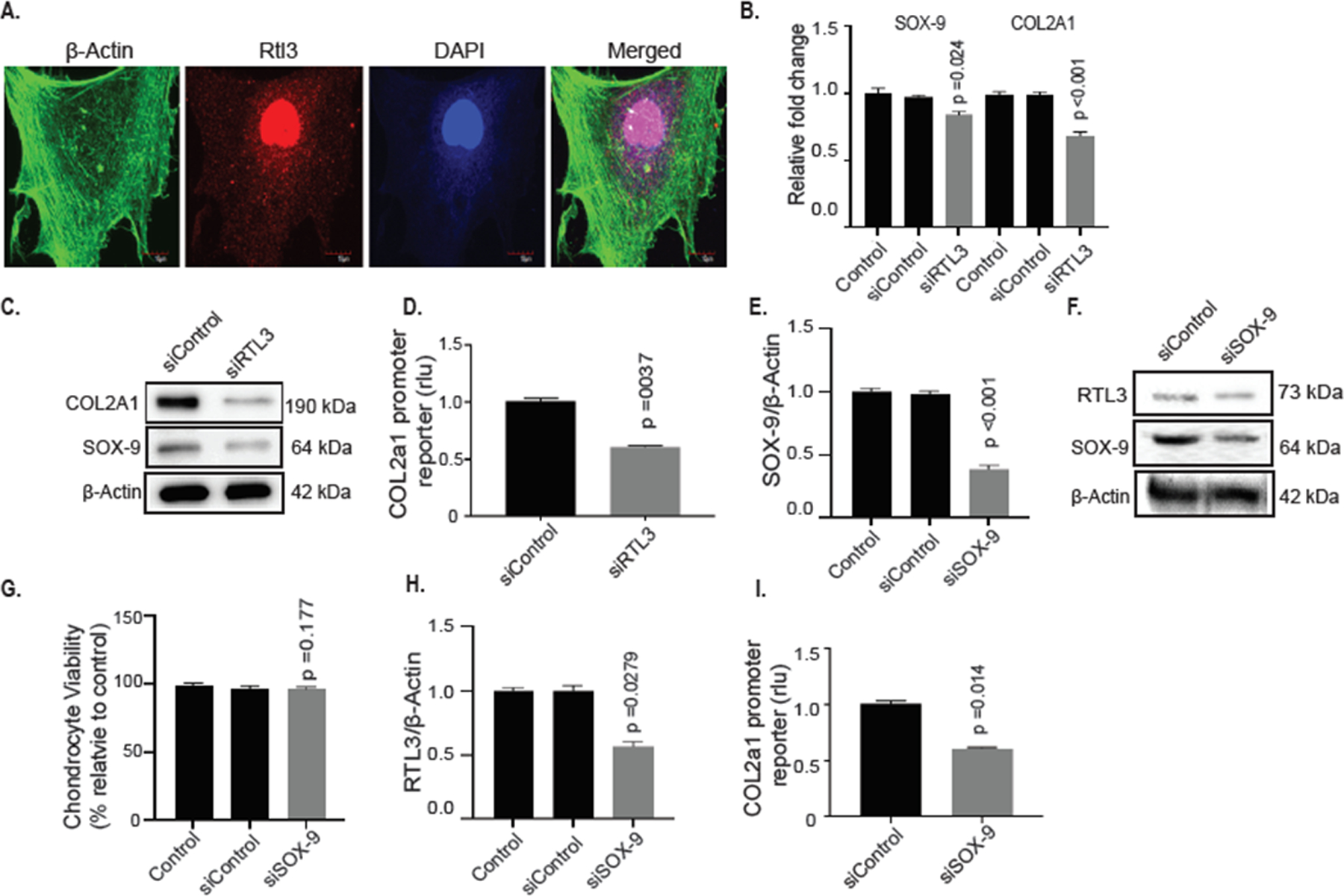
RTL3 co-localized to the nucleus in primary chondrocytes and depletion significantly reduced SOX-9 and COL2A1 mRNA and protein levels and siRNA-mediated depletion of SOX-9 negatively affects RTL3 expression in human chondrocytes. (A) Immunofluorescent staining with anti-RTL3 antibody (#NBP2–30741) followed by ActinGreen™ 488 ReadyProbes Reagent (#R37110) for 15 minutes and results demonstrated RTL3 nuclear co-localization. siRNA-mediated depletion of RTL3 significantly reduced SOX-9 and COL2A1 mRNA (B) and protein levels (C) and reduced the activity of the COL2A1 promoter reporter (D). Depletion of SOX-9 via a validated siRNA successfully reduced SOX-9 mRNA (E) and protein (F) levels in SOX-9-depleted chondrocytes without negatively affecting cell viability (G). siRNA-mediated depletion of SOX-9 significantly reduced RTL3 mRNA (H) and protein (F) levels and reduced the activity of the COL2A1 promoter reporter (I) levels of SOX-9 (E) and COL2A1 (F) as well as protein levels (G) were reduced as well as COL2A1 promoter reporter activity (H). Ectopic expression of RTL3 in HEK-293T cells increased COL2A1 promoter reporter activity in a dose-dependent manner (I) and increased SOX-9 (J) and COL2A1 (K) mRNA expression.
SOX9 depletion negatively affects RTL3 in chondrocytes
As SOX9 is known to regulate the expression of many genes in chondrocytes32, we next determined whether SOX-9 regulates the expression of RTL3 in chondrocytes. Using a validated siRNA, we successfully depleted SOX-9 mRNA expression (Figure 3E) and protein levels (Figure 3F) in primary chondrocytes (n=6 independent samples: 1M, 5F, Mean Age 68y) without significantly affecting chondrocytes viability (Figure 3G). Of importance is the finding that RTL3 mRNA expression was significantly reduced upon SOX-9 depletion compared to no treatment chondrocytes or those transfected with a scrambled control siRNA (Figure 3H). Furthermore, depletion of SOX-9 significantly reduced the activity of the COL2A1 promoter reporter in these samples (Figure 3I). As the promoter reporter used in these activities has a SOX-9 binding site, these data further validate successful SOX-9 depletion. Taken together, these results indicated that both RTL3 and SOX-9 positively regulates the expression of each other and that expression of both the genes is required for the robust expression of COL2A1 in chondrocytes, which is a novel finding.
Forced expression of RTL3 rescues SOX9 and COL2A1 expression in human chondrocytes
To further define the role of RTL3 in the regulation of COL2A1 expression, we next determined if ectopic expression of RTL3 would restore the COL2A1 gene expression in adult human chondrocytes with inhibited expression of COL2A1. For these experiments we used primary chondrocytes isolated from damaged OA cartilage (OARSI score ≥3) as they are known to be deficient in SOX-9 and COL2A1 gene expression33–36. Chondrocytes were transfected with a validated RTL3 expression plasmid and the results showed that the expressed RTL3 mRNA (Figure 4A) and protein levels (Figure 4B) were higher in the transfected chondrocytes compared to un-transfected and pcDNA-transfected chondrocytes and that cell viability was not affected (Figure 4C). Of importance is the finding that, concurrent with increased RTL3 expression in transfected OA chondrocytes, the constitutive expression of SOX-9 and COL2A1 mRNAs and proteins was also significantly enhanced compared to the levels detected in the control OA chondrocytes (Figure 4D, E, F). Furthermore, OA chondrocytes with forced expression of RTL3, which rescues the expression of SOX-9 (Figure-4D), and transfected with the COL2A1 promoter reporter vector showed increased activity of the reporter in a dose-dependent manner (Figure 4G). These data, together with the above findings, indicate that RTL3 plays an essential role in the regulation and maintenance of COL2A1 expression most likely via sustained expression of SOX-9 in differentiated human chondrocytes.
Figure 4:
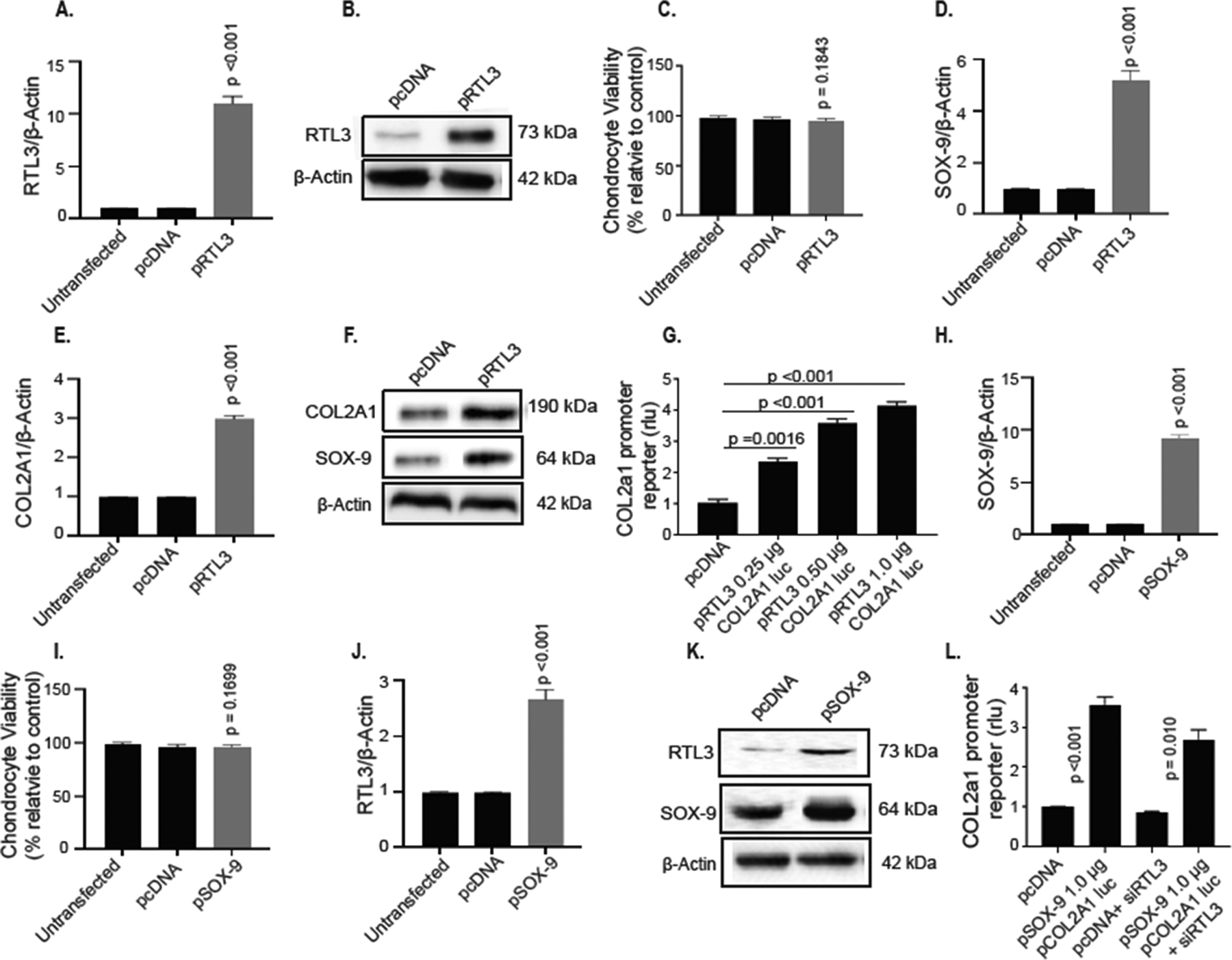
RTL3 over expression enhances SOX-9 and COL2A1 expression in human chondrocytes. Over expression of RTL3 enhanced RTL3 mRNA (A) and (B) protein without adverse effects to cell viability (C). RTL3 over expression rescued SOX-9 (D) and expression COL2A1 (E) mRNA expression and protein levels (F). RTL3 over expression also enhanced COL2A1 promoter reporter activity in a dose-dependent manner (G). Over expression of SOX-9 successfully enhanced SOX-9 mRNA (H) and protein (K) levels in transfected chondrocytes without adverse effects to cell viability (I). Surprisingly, expression of RTL3 mRNA (J) and protein (K) levels were also increased in pSOX-9 treated cells. When chondrocytes were transfected with pSOX-9 over expression plasmid as well as treated with siRNA-depletion of RTL3, the -COL2A1 promoter reporter activity failed to elicit the same robust activity (L).
RTL3 and SOX9 co-regulate each other
Data presented above showed that depletion of SOX-9 affect the expression of RTL3 (Figure-3H), therefore we next examined whether ectopic expression of SOX-9 would affect the RTL3 expression in human OA chondrocytes with inhibited expression of RTL3 (Figure-3G). For these studies, chondrocytes were isolated from damaged OA cartilage (OARSI score ≥3) and were transfected with a validated SOX-9 expression plasmid. Expression of SOX-9 mRNA (Figure 4H) and protein (Figure 4K) were significantly increased without negatively affecting cell viability (Figure 4I). Additionally, we found expression of RTL3 mRNA (Figure 4J) and protein (Figure 4K) to be significantly enhanced in pSOX-9 transfected chondrocytes compared to no treatment or pcDNA-transfected OA chondrocytes. As expected, when SOX-9 was over expressed in chondrocytes (n=6 independent samples) COL2A1 promoter reporter activity was increased. Having previously shown that RTL3 depletion negatively affects COL2A1 promoter reporter activity (Figure 3C), we next examined whether the increased transcription of COL2A1 in pSOX-9 treated chondrocytes would be affected if RTL3 was concomitantly depleted via COL2A1 promoter reporter assay. Interestingly, when SOX-9 was over expressed in chondrocytes (n=6 independent samples) combined with RTL3 depletion resulted in a reduction of COL2A1 promoter reporter activity significantly (Figure 4L). This lends further support to our assertion that the expression of both RTL3 and SOX9 is required for robust COL2A1 expression in chondrocytes.
In additional studies, we also determined whether forced expression of RTL3 can alter the expression of SOX-9 and/or COL2a1 in HEK-293-a cell line that does not express RTL3. We transfected the HEK-293 cells with the RTL3 expression plasmid used in experiments above for the induced expression of RTL3 in chondrocytes (Figure 4B) and observed an increase in the RTL3 mRNA expression without a decrease in viability in the transfected HEK-293 cells (Figure 5A, 5B). Next, we co-transfected the HEK-293 cells with the COL2A1 promoter reporter vector (Figures 3D, 4G) and the RTL3 expression plasmid. The data revealed that in the absence of expressed RTL3, COL2A1 promoter reporter activity was significantly lower compared to HEK-293 cells co-transfected with the RTL3 expression plasmid (Figure 5C). Further analyses using TaqMan assays also showed that in the HEK-293 cells with forced expression of RTL-3, the expression of SOX-9 and COL2A1 mRNA was significantly higher than the controls (Figure 5D, 5E) further supporting our hypothesis that the expression of both RTL3 and SOX-9 is needed for robust COL2A1 expression.
Figure 5:
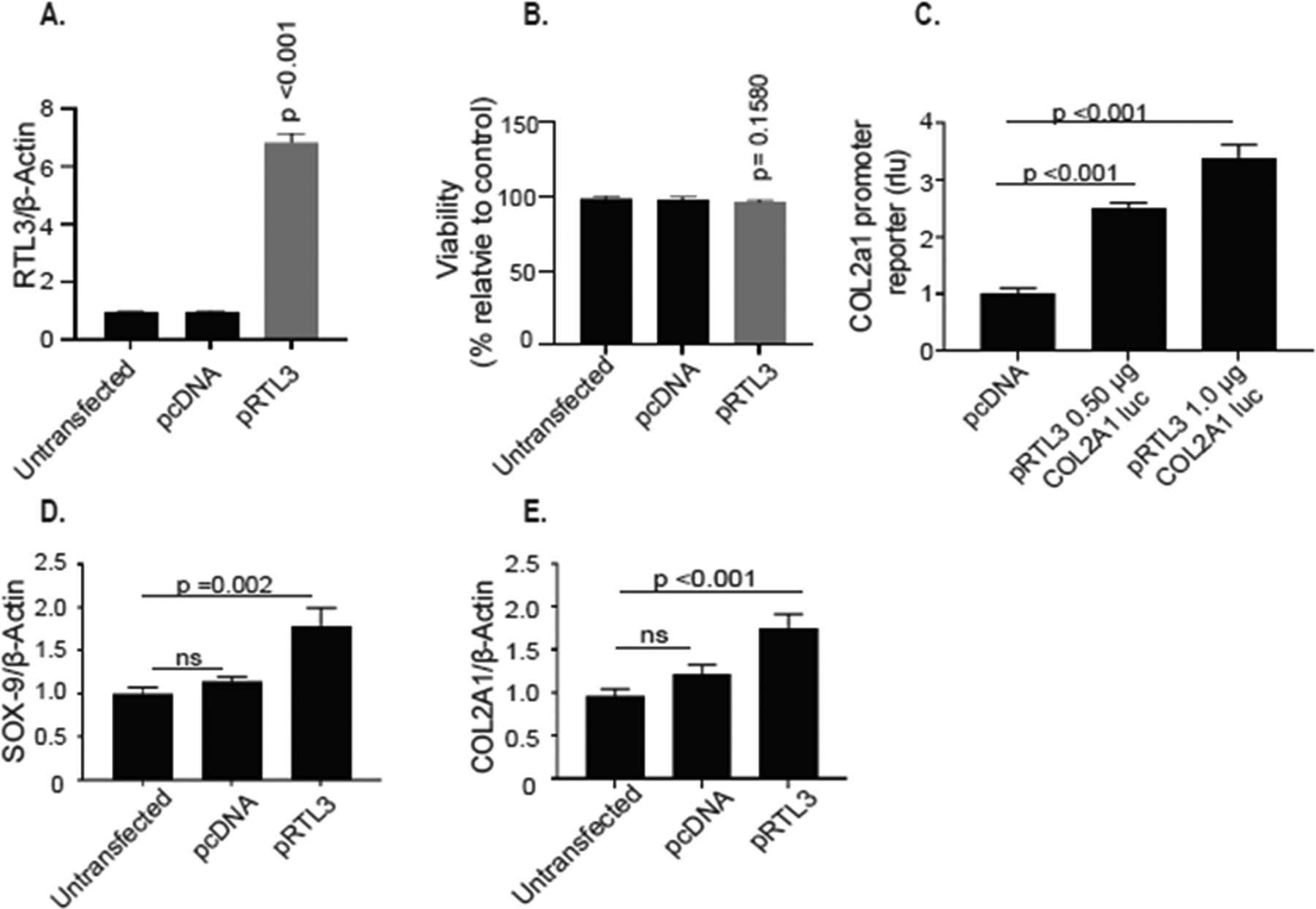
Over expression of RTL3 in HEK-293T cells, that do not express RTL3, increased ectopic SOX-9 and COL2A1 mRNA expression and increased activity of the COL2A1 promoter reporter. RTL3 was successfully over expressed in HEK-23T cells (A) without affecting cell viability (B). RTL3 over expression significantly increased activity of the COL2A1 promoter reporter (C) and mRNA expression of SOX-9 (D) and COL2A1 (E).
RTL3 zinc finger domain is necessary for its gene regulatory activity in chondrocytes
RTL3 contains a nucleic acid binding Zinc finger CCHC domain (Figure 1) and we hypothesized that this DNA-binding region may be essential for the regulation of SOX-9 expression. To test this, we created a CCHC domain deletion mutant and cloned it into an expression plasmid (ΔRTL3). Human normal chondrocytes transfected with the ΔRTL3 plasmid were validated to ensure that the mutant RTL3 mRNA and protein was expressed in transfected chondrocytes (Figure 6A, 6B). Chondrocytes expressing the mutant RTL3 mRNA and protein appeared healthy and showed no negative effect on the viability (Figure 6C). Of importance is our finding that the expression of ΔRTL3 failed to increase the expression of the endogenous SOX-9 or COL2A1 in the transfected chondrocytes as opposed to the chondrocytes with the forced expression of the wild type RTL3 (Figure 6D, E). Additionally, there was no increase in the activity of the COL2A1 promoter reporter in the chondrocytes transfected with the ΔRTL3 plasmid lacking the CCHC domain (Figure 6F). Taken together, these data indicate that the RTL3 zinc finger binding domain is essential for its regulatory activity of SOX-9 expression (which also regulate the expression of COL2A1), and possibly of other genes, in human chondrocytes.
Figure 6:
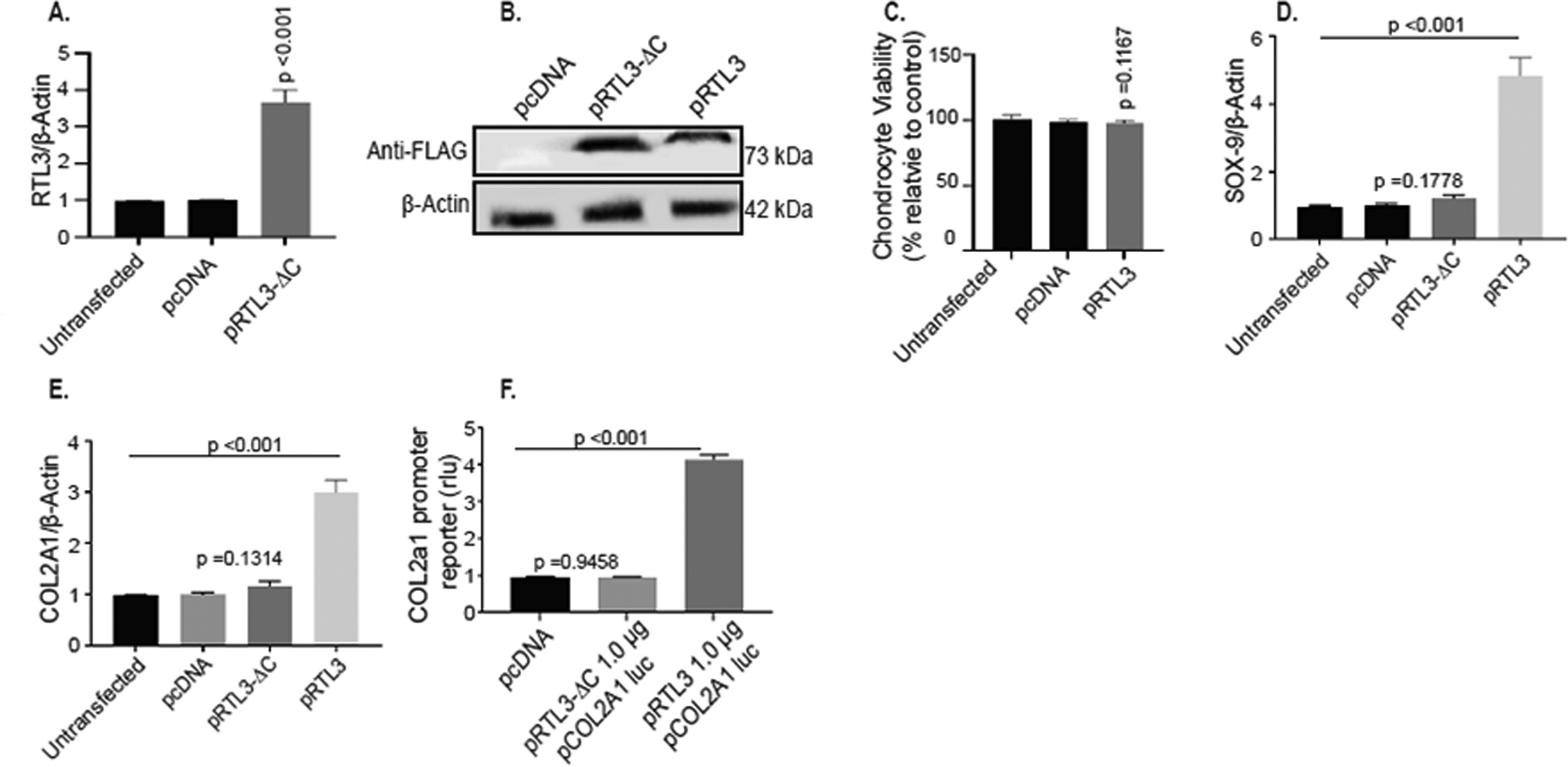
RTL3 modulates SOX-9 expression through interactions with the zinc finger CCHC binding domain. (A) mRNA expression of RTL3 was increased upon transfection with the ΔRTL3 plasmid. (B) Immunoblotting with anti-RTL3 Novus antibody confirmed production of the truncated ΔRTL3 protein. Transfection with ΔRTL3 did not affect cell viability (C) and did not induce chondrocyte mRNA expression of SOX-9 (D) or of COL2A1 (E) nor increase the COL2A1 promoter reporter activity (F).
Genomic deletion of Rtl3 gene retarded the chondrogenic differentiation of ATDC5 cells
Our above data showed that the modulation of RTL3 expression in chondrocytes affect the expression of SOX-9. As SOX-9 plays an essential role in chondrocyte lineage commitment, promotes cell survival, and transcriptionally activates the genes for many cartilage-specific structural components and regulatory factors during chondrogenesis, we next determined whether depletion of RTL3 will affect the expression of SOX9 and the progress of induced chondrogenesis in ATDC5 cells31. To test this, we generated a stable Rtl3 knockout ATDC5cell line via CRISPR-Cas9 editing (ATDC5Rlt3−/−). Chondrogenic differentiation of wild type and ATDC5Rlt3−/− cells was performed for 21 days and we confirmed the Rtl3 gene deletion and absence of Rtl3 mRNA expression during the length of the studies by TaqMan assay (Figure 7A). Both the wild type and the ATDC5Rlt3−/− cells undergoing induced chondrogenesis deposited the matrix, determined by Alcian Blue staining, indicating successful induction (Figure 7B). Matrix deposition was semi-quantified by percent area analyses with ImageJ (Figure 7C). Of importance is the finding that the wild type ATDC5Rtl3+/+ cells showed robust chondrogenesis and matrix deposition while ATDC5Rlt3−/− chondrocytes demonstrated a noticeable reduction in matrix deposition (Figure 7B, 7C) and impaired chondrogenic differentiation at each time point analyzed as indicated by the reduced expression of Sox9 and Col2a1 genes in the ATDC5Rlt3−/− chondrocytes (Figures 7D, 7E). Inhibition of Col2a1 expression also resulted in a noticeable reduction in Col2a1 synthesis and protein deposition in the matrix in the ATDC5Rlt3−/− chondrocytes cultures as determined by quantification of pepsin soluble Col2a1 protein in the synthesized matrix (Figure 7F). These data suggest an important role of Rtl3 in the regulation of the extracellular matrix (ECM) components expression during chondrogenesis.
Figure 7:
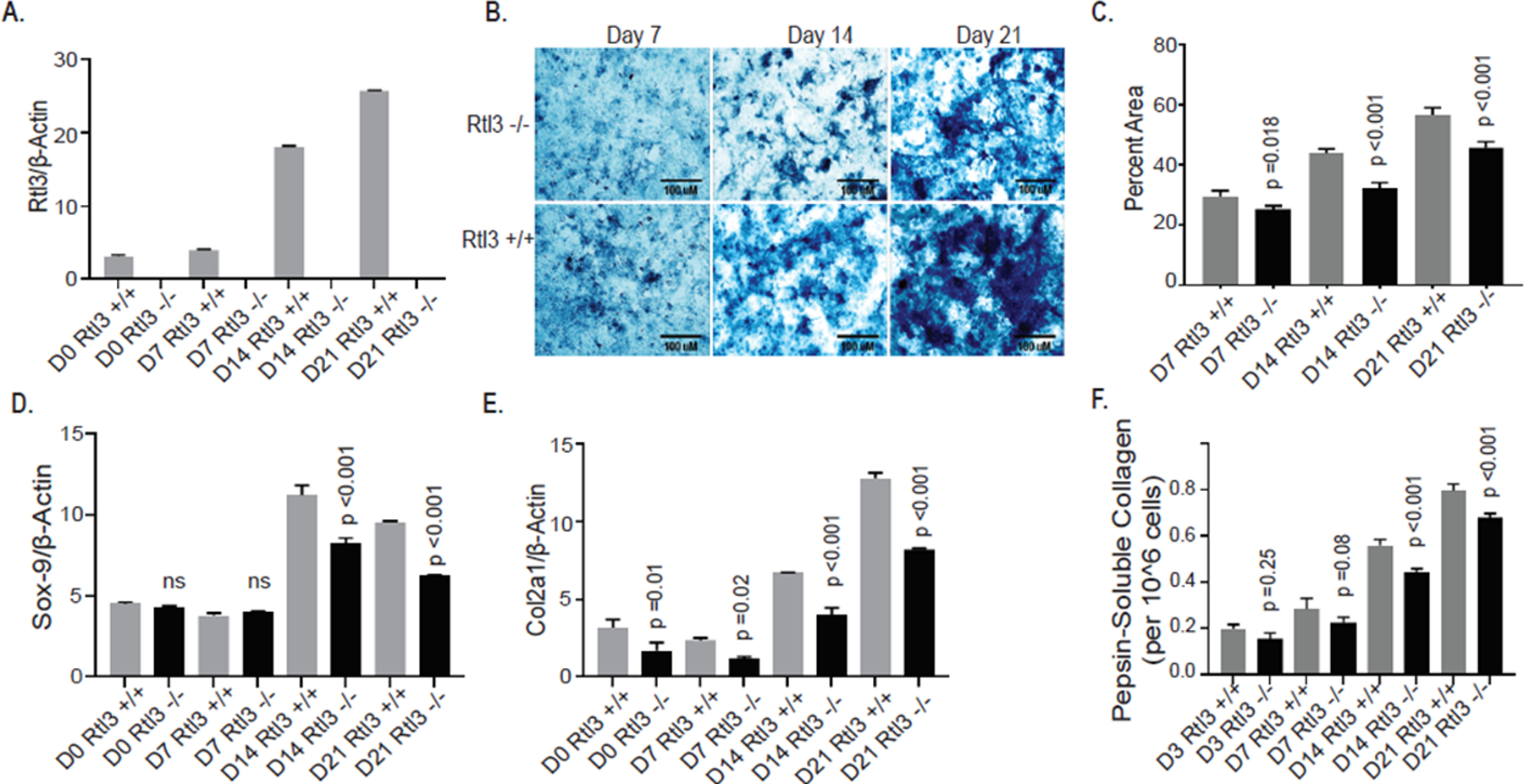
CRISPR-Cas9 mediated deletion of Rtl3 impairs chondrogenesis and delays matrix synthesis in ATDC5 cells. (A) ATDC5Rlt3−/− cells did not express Rtl3 mRNA at any chondrogenic time point. (B) Reduced matrix deposition was visualized in wild type and ATDC5Rlt3−/− cells with Alcian Blue stain (scale bar 100 μm) and (C) semi-quantified by percent area analyses (ImageJ). (D) Sox-9 mRNA levels were significantly reduced in ATDC5Rlt3−/− cells assessed by qPCR. ATDC5Rlt3−/− cells demonstrated a significant reduction in Col2a1 mRNA (E) and (F) protein levels compared to wild type ATDC5 cells.
Discussion
Transposable elements (TEs) are known to influence mammalian development by acting as host gene enhancers, conveying new function to protein coding genes via insertional mutagenesis or through domestication, where TE-derived elements execute vital host functions3,37,38. Several domesticated Sushi-like neogenes have already been shown to effect human health and perform essential roles in mouse placental development9,10,39,40. However, nothing has been published or postulated regarding the expression of RTL3 in mammalian tissues or the role of this gene in mammalian biology. Here, we provide the first evidence that RTL3 is a neofunctionalized gene with important roles in both chondrogenesis and COL2A1 regulation during chondrogenesis and in adult articular chondrocytes.
We first assessed the expression pattern of RTL3 in human tissues and cell lines. RTL3 was not found to be ubiquitously expressed but was highly expressed in cartilage and bone tissue and the primary synoviocytes and chondrocytes that comprise the joint capsule. Chondrocytes are the resident cell type present in cartilage. During development chondrocytes lay the foundation for endochondral ossification and skeletal growth and, in adult tissues, enable smooth joint articulation and cushion underlying bone from compressive and tensile stressors41,42. They are also responsible for the production, secretion and maintenance of an extracellular matrix (ECM) rich in fibrous type-II collagen (COL2A1) supplemented with lubricating glycosaminoglycans and compression resistant proteoglycans41.
In OA, joint tissues are damaged due to age, mechanical injury or inflammation and chondrocytes are not able to properly repair the damage43–45. Instead, chondrocytes proliferate, undergo hypertrophy and alter the matrix composition via decreasing expression of key anabolic genes such as SOX-9 and COL2A1. Since expression of RTL3 was high in chondrocytes and that its knockdown affected the COL2A1 expression, we next assessed if RTL3 expression was altered in OA cartilage, where expression of COL2A1 is known to be reduced, and found a significant decrease in RTL3 mRNA and protein levels in the damaged OA cartilage. This is a novel finding and has not been reported previously.
Having established that RTL3 is down-regulated in the damaged OA cartilage, we used loss-of-function and gain-of-function approaches to determine whether RTL3 was directly involved in COL2A1 regulation in human chondrocytes. siRNA-mediated depletion of RTL3 resulted in a significant reduction in COL2A1 mRNA and protein expression and also significantly reduced the activity of a COL2A1 promoter reporter vector in vitro. Gain-of-function approach demonstrated that the forced expression of RTL3 enhanced the COL2A1 expression in the transfected chondrocytes and also enhanced the activity of the COL2A1 promoter reporter in a dose- dependent manner. Additionally, forced expression of RTL3 was able to enhance COL2A1 promoter reporter activity in HEK-293T cells, which do not endogenously produce RTL3. Together, these results demonstrate a new role for a transposable element-derived gene in the regulation of COL2A1 expression in chondrocytes during health and disease. Future studies on the expression of RTL3 during development and aging is likely to shed new light on the pathogenesis of aging-associated diseases in which altered expression of COL2A1 plays a role.
Having observed a reduction in SOX-9 expression upon siRNA-mediated depletion of RTL3 expression in chondrocytes, we wished to determine if RTL3 modulate COL2A1 expression via suppression of its known regulator SOX-9-a member of the SOX family of transcription factors and is essential for chondrocyte specification and differentiation during development and is a known regulator of COL2A1 expression30,31,46. Expression of SOX-9 is known to decrease with OA progression resulting in chondrocyte phenotypic instability and dedifferentiation and their role in matrix degradation, decreased joint function and OA progression47–49. The altered expression of SOX-9 with disease progression has been attributed to alterations in the epigenetic status and increased methylation of the SOX-9 promoter and/or alterations in the expression of miRNAs targeting SOX-945,50–52.In mice, the conditional deletion of Sox9 in chondrocytes results in a phenotype resembling the semi-lethal human skeletal disorder Campomelic Dysplasia32,53. Interestingly, siRNA-mediated depletion of SOX-9 expression significantly reduced the mRNA expression of not only SOX-9 but also of RTL3 indicating that SOX-9 also plays a role in the regulation of RTL3 expression in chondrocytes. Over expression of SOX-9, was able to increase the RTL3 mRNA and protein expression and when these findings are taken with other data presented above suggests that SOX9 and RTL3 coregulate each other by a yet unknown mechanism. These are novel findings and unraveling the details of this mechanism will shed new light on the skeletal development and associated diseases pathologies.
As Rtl3 protein contains a nucleic acid binding domain, and we next examined whether RTL3 may be affecting SOX-9 through the zinc finger CCHC domain. Interestingly, forced expression of a mutant plasmid lacking the CCHC domain failed to increase the mRNA expression of either SOX-9 or COL2A1 and the activity of the COL2A1 promoter reporter was not increased either, suggesting the RTL3 zinc finger binding domain is essential for the induction of SOX-9 which is a known regulator of the COL2A1 expression in chondrocytes. The fine details of this mechanism in vivo warrants further studies.
Our above data presents a composite picture in which both RTL3 and SOX9 co-regulate each other and that the absence of either affects the regulation of COL2A1 in chondrocytes. As TEs have also been implicated in development3,40, we determined the role of Rtl3 in chondrogenic differentiation and matrix deposition in an ATDC5 cell line. Derived from mouse teratocarcinoma cells, ATDC5 is a well-characterized line utilized for the study of induced chondrocyte differentiation and matrix deposition19,54. The Rtl3-depleted chondrocytes (ATDC5Rlt3−/−) were capable of successful chondrogenic differentiation, indicated by Col2a1 expression, albeit at a lower level, suggesting that Rtl3 is not required for the induction of chondrogenic differentiation. However, matrix synthesis and deposition, as visualized with Alcian Blue stain and Col2a1 expression levels, was impaired in ATDC5Rlt3−/− chondrocytes versus induced wild type chondrocytes at all the timepoints analyzed. The delay and reduction in Sox-9 expression and subsequent Col2a1 synthesis and activity in Rtl3 deficient chondrocytes suggests that these cells may deposit an ECM less resistant to tensile forces and which may be more prone to biomechanical erosion since collagen provides tensile strength41. Analysis and biological significance of the deposition of a matrix deficient in Col2a1 component in an in vivo model lacking Rtl3 is being investigated in ongoing studies.
In conclusion, the present study is the first to demonstrate that a neofunctionalized gene RTL3 plays a key role during chondrogenesis, as deletion of Rtl3 impaired chondrogenesis and reduced the matrix synthesis in a model of induced chondrogenesis in vitro, and that RTL3 inhibition in differentiated human chondrocytes downregulated the expression of SOX-9 and COL2A1. Our findings are the first to demonstrate that both RTL3 and SOX9 co-regulate each other and also indicate that RTL3 does not directly regulate COL2A1 expression, but instead RTL3 interacts with and enhances the expression of SOX-9 via its zinc-finger binding domain to regulate COL2A1 expression. This mechanism of COL2A1 regulation is novel and has not been previously reported in chondrocytes.
Supplementary Material
Funding:
This work was supported in part by USPHS/National Institutes of Health grants RO1-AT-007373 and RO1-AR-067056 and funds from Northeast Ohio Medical University to TMH.
Footnotes
Disclosure of Interest: The authors report no conflict of interest.
Data Availability Statement: All data supporting the findings of this study are available within the article and its Supplementary Information Files and from the corresponding author on reasonable request.
References:
- 1.Kazazian HH Jr., Moran JV. Mobile DNA in Health and Disease. N Engl J Med. 2017. July 27;377(4):361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandt J, Schrauth S, Veith AM, et al. Transposable elements as a source of genetic innovation: expression and evolution of a family of retrotransposon-derived neogenes in mammals. Gene. 2005. January 17;345(1):101–11. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Perez JL, Widmann TJ, Adams IR. The impact of transposable elements on mammalian development. Development. 2016. November 15;143(22):4101–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancks DC, Kazazian HH, Jr. Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulter R, Butler M. A retrotransposon family from the pufferfish (fugu) Fugu rubripes. Gene. 1998. July 30;215(2):241–9. [DOI] [PubMed] [Google Scholar]
- 6.Volff J, Korting C, Schartl M. Ty3/Gypsy retrotransposon fossils in mammalian genomes: did they evolve into new cellular functions? Mol Biol Evol. 2001. February;18(2):266–70. [DOI] [PubMed] [Google Scholar]
- 7.Alzohairy AM, Gyulai G, Jansen RK, et al. Transposable elements domesticated and neofunctionalized by eukaryotic genomes. Plasmid. 2013. January;69(1):1–15. [DOI] [PubMed] [Google Scholar]
- 8.Volff JN, Brosius J. Modern genomes with retro-look: retrotransposed elements, retroposition and the origin of new genes. Genome Dyn. 2007;3:175–190. [DOI] [PubMed] [Google Scholar]
- 9.Curry BJ, Myers K, Hersey P. MART-1 is expressed less frequently on circulating melanoma cells in patients who develop distant compared with locoregional metastases. J Clin Oncol. 1999. August;17(8):2562–71. [DOI] [PubMed] [Google Scholar]
- 10.Jungbluth AA, Iversen K, Coplan K, et al. Expression of melanocyte-associated markers gp-100 and Melan-A/MART-1 in angiomyolipomas. An immunohistochemical and rt-PCR analysis. Virchows Arch. 1999. May;434(5):429–35. [DOI] [PubMed] [Google Scholar]
- 11.Henke C, Strissel PL, Schubert MT, et al. Selective expression of sense and antisense transcripts of the sushi-ichi-related retrotransposon--derived family during mouse placentogenesis. Retrovirology. 2015. February 3;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed S, Rahman A, Hasnain A, et al. Green tea polyphenol epigallocatechin-3-gallate inhibits the IL-1 beta-induced activity and expression of cyclooxygenase-2 and nitric oxide synthase-2 in human chondrocytes. Free Radic Biol Med. 2002. October 15;33(8):1097–105. [DOI] [PubMed] [Google Scholar]
- 13.Akhtar N, Haqqi TM. MicroRNA-199a* regulates the expression of cyclooxygenase-2 in human chondrocytes. Ann Rheum Dis. 2012. June;71(6):1073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansari MY, Haqqi TM. Interleukin-1beta induced Stress Granules Sequester COX-2 mRNA and Regulates its Stability and Translation in Human OA Chondrocytes. Sci Rep. 2016. June 8;6:27611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green JA, Ansari MY, Ball HC, et al. tRNA-derived fragments (tRFs) regulate post-transcriptional gene expression via AGO-dependent mechanism in IL-1beta stimulated chondrocytes. Osteoarthritis Cartilage. 2020. August;28(8):1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haseeb A, Ansari MY, Haqqi TM. Harpagoside suppresses IL-6 expression in primary human osteoarthritis chondrocytes. J Orthop Res. 2017. February;35(2):311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ansari MY, Khan NM, Ahmad N, et al. Genetic Inactivation of ZCCHC6 Suppresses Interleukin-6 Expression and Reduces the Severity of Experimental Osteoarthritis in Mice. Arthritis Rheumatol. 2019. Apr;71(4):583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gosset M, Berenbaum F, Thirion S, et al. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–60. [DOI] [PubMed] [Google Scholar]
- 19.Newton PT, Staines KA, Spevak L, et al. Chondrogenic ATDC5 cells: an optimised model for rapid and physiological matrix mineralisation. Int J Mol Med. 2012. November;30(5):1187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terry DE, Chopra RK, Ovenden J, et al. Differential use of Alcian blue and toluidine blue dyes for the quantification and isolation of anionic glycoconjugates from cell cultures: application to proteoglycans and a high-molecular-weight glycoprotein synthesized by articular chondrocytes. Anal Biochem. 2000. October 15;285(2):211–9. [DOI] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012. Jul;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad N, Ansari MY, Bano S, et al. Imperatorin suppresses IL-1beta-induced iNOS expression via inhibiting ERK-MAPK/AP1 signaling in primary human OA chondrocytes. Int Immunopharmacol. 2020. August;85:106612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansari MY, Khan NM, Ahmad I, et al. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage. 2018. August;26(8):1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ansari MY, Ahmad N, Haqqi TM. Butein Activates Autophagy Through AMPK/TSC2/ULK1/mTOR Pathway to Inhibit IL-6 Expression in IL-1beta Stimulated Human Chondrocytes. Cell Physiol Biochem. 2018;49(3):932–946. [DOI] [PubMed] [Google Scholar]
- 25.Rasheed Z, Akhtar N, Anbazhagan AN, et al. Polyphenol-rich pomegranate fruit extract (POMx) suppresses PMACI-induced expression of pro-inflammatory cytokines by inhibiting the activation of MAP Kinases and NF-kappaB in human KU812 cells. J Inflamm (Lond). 2009. January 8;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haseeb A, Haqqi TM. Immunopathogenesis of osteoarthritis. Clin Immunol. 2013. March;146(3):185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed Pharmacother. 2020. July 3;129:110452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahmati M, Mobasheri A, Mozafari M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone. 2016. April;85:81–90. [DOI] [PubMed] [Google Scholar]
- 29.Zamli Z, Sharif M. Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int J Rheum Dis. 2011. May;14(2):159–66. [DOI] [PubMed] [Google Scholar]
- 30.de Crombrugghe B, Lefebvre V, Behringer RR, et al. Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol. 2000. September;19(5):389–94. [DOI] [PubMed] [Google Scholar]
- 31.Lefebvre V, Dvir-Ginzberg M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect Tissue Res. 2017. January;58(1):2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CF, Samsa WE, Zhou G, et al. Transcriptional control of chondrocyte specification and differentiation. Semin Cell Dev Biol. 2017. February;62:34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldring MB. Articular cartilage degradation in osteoarthritis. HSS J. 2012. February;8(1):7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makki MS, Haqqi TM. Histone Deacetylase Inhibitor Vorinostat (SAHA) Suppresses IL-1beta-Induced Matrix Metallopeptidase-13 Expression by Inhibiting IL-6 in Osteoarthritis Chondrocyte. Am J Pathol. 2016. October;186(10):2701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poole AR, Kobayashi M, Yasuda T, et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann Rheum Dis. 2002. November;61 Suppl 2:ii78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong L, Huang X, Karperien M, et al. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int J Mol Sci. 2016. July 14;17(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evsikov AV, Marin de Evsikova C. Friend or Foe: Epigenetic Regulation of Retrotransposons in Mammalian Oogenesis and Early Development. Yale J Biol Med. 2016. December;89(4):487–497. [PMC free article] [PubMed] [Google Scholar]
- 38.Gerdes P, Richardson SR, Mager DL, et al. Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol. 2016. May 9;17:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono R, Nakamura K, Inoue K, et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat Genet. 2006. January;38(1):101–6. [DOI] [PubMed] [Google Scholar]
- 40.Sekita Y, Wagatsuma H, Nakamura K, et al. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet. 2008. February;40(2):243–8. [DOI] [PubMed] [Google Scholar]
- 41.Akkiraju H, Nohe A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J Dev Biol. 2015. December;3(4):177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson DL, Kersh ME, Walsh NC, et al. Mechanical properties of normal and osteoarthritic human articular cartilage. J Mech Behav Biomed Mater. 2016. August;61:96–109. [DOI] [PubMed] [Google Scholar]
- 43.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016. July;12(7):412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martel-Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers. 2016. October 13;2:16072. [DOI] [PubMed] [Google Scholar]
- 45.Sherwood J Osteoarthritis year in review 2018: biology. Osteoarthritis Cartilage. 2019. March;27(3):365–370. [DOI] [PubMed] [Google Scholar]
- 46.Ng LJ, Wheatley S, Muscat GE, et al. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997. March 1;183(1):108–21. [DOI] [PubMed] [Google Scholar]
- 47.Ashraf S, Cha BH, Kim JS, et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage. 2016. February;24(2):196–205. [DOI] [PubMed] [Google Scholar]
- 48.Singh P, Marcu KB, Goldring MB, et al. Phenotypic instability of chondrocytes in osteoarthritis: on a path to hypertrophy. Ann N Y Acad Sci. 2019. April;1442(1):17–34. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Q, Ji Q, Wang X, et al. SOX9 is a regulator of ADAMTSs-induced cartilage degeneration at the early stage of human osteoarthritis. Osteoarthritis Cartilage. 2015. December;23(12):2259–2268. [DOI] [PubMed] [Google Scholar]
- 50.Guerit D, Philipot D, Chuchana P, et al. Sox9-regulated miRNA-574–3p inhibits chondrogenic differentiation of mesenchymal stem cells. PLoS One. 2013;8(4):e62582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim KI, Park YS, Im GI. Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J Bone Miner Res. 2013. May;28(5):1050–60. [DOI] [PubMed] [Google Scholar]
- 52.Mak IW, Singh S, Turcotte R, et al. The epigenetic regulation of SOX9 by miR-145 in human chondrosarcoma. J Cell Biochem. 2015. January;116(1):37–44. [DOI] [PubMed] [Google Scholar]
- 53.Kist R, Schrewe H, Balling R, et al. Conditional inactivation of Sox9: a mouse model for campomelic dysplasia. Genesis. 2002. February;32(2):121–3. [DOI] [PubMed] [Google Scholar]
- 54.Yao Y, Wang Y. ATDC5: an excellent in vitro model cell line for skeletal development. J Cell Biochem. 2013. June;114(6):1223–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.