

Abstract
In vitro cell-based toxicity testing methods generate large amounts of data informative for risk-based evaluations. To allow extrapolation of the quantitative outputs from cell-based tests to the equivalent exposure levels in humans, reverse toxicokinetic modeling is used to conduct in vitro-to-in vivo extrapolation (IVIVE) from in vitro effective concentrations to in vivo oral dose equivalents. IVIVE modeling approaches for individual chemicals are well-established; however, the potential implications of chemical-to-chemical interactions in mixture settings on IVIVE remain largely unexplored. We hypothesized that chemical coexposures could modulate both protein binding efficiency and hepatocyte clearance of the chemicals in a mixture, which would in turn affect the quantitative IVIVE toxicokinetic parameters. To test this hypothesis, we used 20 pesticides from the Agency for Toxic Substances and Disease Registry Substance Priority List, both individually and as equimolar mixtures, and investigated the concentration-dependent effects of chemical interactions on in vitro toxicokinetic parameters. Plasma protein binding efficiency was determined by using ultracentrifugation, and hepatocyte clearance was estimated in suspensions of cryopreserved primary human hepatocytes. We found that for single chemicals, the protein binding efficiencies were similar at different test concentrations. In a mixture, however, both protein binding efficiency and hepatocyte clearance were affected. When IVIVE was conducted using mixture-derived toxicokinetic data, more conservative estimates of activity-to-exposure ratios were produced as compared with using data from single chemical experiments. Because humans are exposed to mixtures of chemicals, this study is significant as it demonstrates the importance of incorporating mixture-derived parameters into IVIVE for in vitro bioactivity data in order to accurately prioritize risks and facilitate science-based decision-making.
Keywords: IVIVE, mixtures, in vitro, NAMs
Traditional chemical safety evaluations rely on testing for potential adverse effects of chemical in laboratory animals; these studies identify target organs and dose levels where the effects may be undetectable, information that is used to extrapolate to humans and other exposure levels. Animal-based testing approaches are highly informative for both cancer (Krewski et al., 2019) and noncancer (Olson et al., 2000) effects; however, these experiments are time- and resource-intensive and associated with a number of ethical and human relevance concerns, factors that make testing of a large number of environmental substances in animals impractical (Bell et al., 2018). Therefore, new approach methodologies (NAMs) are being developed for environmental and industrial chemicals, using in silico and in vitro models of various biological complexity (Krewski et al., 2020; Marx et al., 2020), followed by toxicokinetic modeling to extrapolate to in vivo human exposures through “in vitro-to-in vivo extrapolation” or IVIVE (Wambaugh et al., 2015; Wetmore et al., 2012). IVIVE calculations typically rely on the use of a steady-state model that incorporates 2 key experimental parameters: plasma protein binding and hepatocyte clearance. These values are then used to calculate steady-state plasma concentrations for a specific oral dose. Although the NAMs-derived data from in vitro-based tests are now available for thousands of chemicals, it is challenging to experimentally derive parameters that enable IVIVE on an equally massive scale (Wambaugh et al., 2015; Wetmore et al., 2013, 2014). Therefore, recent studies proposed computationally derived parameter estimation (Bell et al., 2020) to enable “high-throughput toxicokinetic” (HTTK) modeling (Pearce et al., 2017).
IVIVE is now widely recognized as a critical tool to facilitate chemical prioritization and decision making based on in vitro testing data (Bell et al., 2018; Yoon et al., 2012). Both in vitro tests and IVIVE analyses are most often performed on individual chemicals; however, real-life exposures are not limited to a single chemical, but rather are a mixture of chemicals (Drakvik et al., 2020). The application of NAMs to studies of mixtures is a nascent field (Chen et al., 2021; Escher et al., 2020; Fang et al., 2020; Hsieh et al., 2021), which calls for reevaluation of the suitability of current IVIVE approaches to mixture settings (Carpenter et al., 2002). For example, plasma protein binding determined through the use of a traditional rapid equilibrium dialysis (RED) method (Bohnert and Gan, 2013) is not suitable for testing many highly lipophilic environmental chemicals (Ferguson et al., 2019). In addition, biotransformation rate constants for polyaromatic hydrocarbons (PAHs) that were tested individually (Lee et al., 2014) were much greater than those obtained when these PAHs were tested as a mixture (OECD, 2018), suggesting that kinetics are different in a mixture setting compared with single chemical. Similarly, substance-specific differences in in vitro bioavailability were observed for the individual PAH tested as a mixture, or as components of complex petroleum substances (Luo et al., 2020).
Most studies of mixtures still consider individual chemical effects and then mathematically reconstruct the mixture IVIVE using a number of modeling approaches (van der Voet et al., 2020). Although indirect mixture modeling is common, most often there are no in vitro or in vivo toxicokinetic data available for the mixture as a whole, hence limiting the options for direct modeling of mixtures (Chang et al., 2021). Thus, there is a critical need to characterize in vitro toxicokinetic parameters of chemicals in a mixture setting. To address this need, we selected a subset of pesticides from the Superfund priority list (Agency for Toxic Substances and Disease Registry [ATSDR], 2019) and tested these compounds for plasma protein binding and hepatocyte clearance in both single and mixture settings. Next, we derived steady-state plasma concentrations for each chemical under single or mixture testing conditions. Finally, we combined these steady-state concentrations with publicly available in vitro bioactivity data and exposure estimates to derive oral equivalent doses (OEDs) and activity to exposure levels for each chemical individually or in a mixture. The results of this study are informative for cumulative risk assessment of chemical mixture exposures and show that IVIVE for mixtures needs experimental priors.
MATERIALS AND METHODS
Chemicals
Twenty pesticides and analytical internal standards (Table 1 and Supplementary Table 1) were purchased from Sigma Aldrich (St Louis, Missouri) or Chem Service (West Chester, Pennsylvania). Methanol (Cat No.: A456-500) and acetonitrile (Cat No.: A955-500) were purchased from Fischer Scientific (Hampton, New Hampshire). Pentane (Cat No.: 1.00882), diethyl ether (Cat No.: 309966), and distilled water with 0.1% formic acid (Cat No.: 1.59013) were acquired from Sigma Aldrich. Cell culture media used in these experiments was from the iCell Cardiomyocytes Media Kit (Catalog No.: R1151) and was purchased from FujiFilmCDI (Madison, Wisconsin), and was used as a representative example of in vitro media.
Table 1.
Chemicals Used in This Study
| Chemical | CASRN | Analytical Assay | Ionization Mode | Mass Transitions (m/z) | CE(eV) |
|---|---|---|---|---|---|
| Test chemicals | |||||
| Aldrin | 309-00-2 | GC-MS/MS | EI | 263.0→191.0 | 40 |
| 293.0→186.0 | 40 | ||||
| DDD-p, p’ | 72-54-8 | GC-MS/MS | EI | 235.0→165.1 | 20 |
| 235.0→200.0 | 30 | ||||
| DDT-o, p’ | 789-02-6 | GC-MS/MS | EI | 235.0→165.0 | 30 |
| DDT-p, p’ | 50-29-3 | 235.0→199.0 | 18 | ||
| Dicofol | 115-32-2 | GC-MS/MS | EI | 249.9→215.1 | 10 |
| Dieldrin | 60-57-1 | GC-MS/MS | EI | 277.0→241.0 | 10 |
| 263.0→193.0 | 40 | ||||
| Endosulfan I | 115-29-7 | GC-MS/MS | EI | 240.9→205.9 | 15 |
| 240.9→203.9 | 20 | ||||
| Endrin | 72-20-8 | GC-MS/MS | EI | 279.0→243.0 | 10 |
| 281.0→211.0 | 30 | ||||
| Heptachlor epoxide B | 1024-57-3 | GC-MS/MS | EI | 352.9→281.9 | 20 |
| 352.9→262.8 | 15 | ||||
| Heptachlor | 76-44-8 | GC-MS/MS | EI | 272.0→236.8 | 25 |
| 337.0→266.0 | 20 | ||||
| Lindane | 58-89-9 | GC-MS/MS | EI | 218.8→145.0 | 20 |
| 218.8→183.0 | 5 | ||||
| Methoxychlor-o, p’ | 72-43-5 | GC-MS/MS | EI | 227.0→141.0 | 35 |
| 227.0→152.0 | 20 | ||||
| Parathion | 56-38-2 | GC-MS/MS | EI | 291.1→ 81.0 | 40 |
| 291.1→109.0 | 10 | ||||
| Trifluralin | 1582-09-8 | GC-MS/MS | EI | 306.0→264.0 | 7 |
| 306.0→160.0 | 25 | ||||
| 2,4-dinitrophenol | 51-28-5 | LC-MS/MS | ESI(−) | 183.0→137.0 | 5 |
| 183.0→123.0 | 5 | ||||
| Azinphos-methyl | 86-50-0 | LC-MS/MS | ESI(+) | 318.0→160.1 | 13 |
| 318.0→132.0 | 21 | ||||
| Chlorpyrifos | 2921-88-2 | LC-MS/MS | ESI(+) | 350.0→198.0 | 25 |
| 350.0→ 97.0 | 47 | ||||
| Diazinon | 333-41-5 | LC-MS/MS | ESI(+) | 305.0→169.1 | 31 |
| 305.0→153.1 | 29 | ||||
| Disulfoton | 298-04-4 | LC-MS/MS | ESI(+) | 275.0→ 89.2 | 11 |
| 275.0→ 61.2 | 33 | ||||
| Ethion | 563-12-2 | LC-MS/MS | ESI(+) | 384.8→199.2 | 15 |
| 384.8→142.9 | 39 | ||||
| Internal standards | |||||
| Atrazine | 1912-24-9 | GC-MS/MS | EI | 200.1→103.9 | 20 |
| 200.1→ 94.1 | 20 | ||||
| Benzo[a]anthracene | 56-55-3 | GC-MS/MS | EI | 228.0→226.2 | 30 |
| 228.0→202.1 | 30 | ||||
| Terbutryn | 886-50-0 | GC-MS/MS | EI | 241.0→185.0 | 15 |
| 241.0→170.0 | 20 | ||||
| Mifepristone | 84371-65-3 | LC-MS/MS | ESI(+) | 430.0→372.0 | 35 |
| Troglitazone | 97322-87-7 | LC-MS/MS | ESI(−) | 440.2→397.1 | 21 |
| 440.2→145.0 | 37 | ||||
See Supplementary Table 1 for vendor and catalog number information.
Selection of test compounds and preparation of a “designed” chemical mixture
The ATSDR maintains a priority list of hazardous substances/chemicals that are commonly detected at the U.S. National Priority List sites, also known as “Superfund” sites; these chemicals are known to be hazardous to human health (ATSDR, 2019). From the list of over 300 compounds, we selected 20 chemicals that are lipophilic and have IVIVE data in httk R package version 1.10.1 (Table 1). In addition, we created a molar-equivalent, “designed” mixture consisting of 20 chemicals (10 μM each) to investigate the impacts of chemical coexposure on quantitative IVIVE.
Determination of protein binding efficiency
Protein binding efficiency of 20 pesticides was determined using ultracentrifugation (Nakai et al., 2004). To calculate the unbound fractions of test compounds, 2 sets of samples were prepared in this experiment. First, 995 μl of the medium or human plasma was spiked with 5 μl of 0.2 or 2 mM test chemical, which resulted in the final test concentrations of 1 or 10 μM. Then, 300 μl of the reaction mixture was immediately transferred and extracted in an Eppendorf tube (as described in LC-MS/MS analyses and GC-MS/MS analyses sections), representing the initial concentration of the test compound. For the second set of samples, the reaction mixture was prepared using the identical procedures but further incubated at 37°C for 1 h. Next, a portion of 300 μl from the reaction mixture was transferred to a polycarbonate centrifuge tube (n = 3). Samples underwent ultracentrifugation with Optima MAX-XP Ultracentrifuge (Item No.: 393315; Beckman Coulter, Brea, California) at 90 000 rpm for 4.5 h at 4°C. One hundred microliters of the middle layer (clear portion) was transferred to a sample vial, representing the unbound concentration of the test compound. The collected samples were analyzed using liquid chromatography (LC) or gas chromatography (GC) followed by tandem mass spectrometry (MS/MS), as detailed below.
Determination of in vitro hepatocyte clearance
In vitro hepatocyte clearance was determined using a suspension culture of cryopreserved human hepatocytes (Lot No. HUE121, GIBCO, Frederick, Maryland) according to manufacturer’s protocol. In brief, the cryopreserved human hepatocytes were suspended in William’s E medium and adjusted to the cell concentration of 1 × 106 cells/ml. A portion of the cell working stock was heated at 95°C for 5 min to serve as negative control. Five hundred microliters of the chemical stock (20 μM) or designed mixture (20 chemicals, 20 μM each) was spiked in 500 μl of the cell working stock or inactivated cell control to a final chemical concentration of 10 µM and cell number of 5 × 105 cells/ml. One hundred microliters of the reaction mixture were removed subsequently at 0, 15, 30, 60, 90, and 120 min to individual 1.5 ml Eppendorf tubes for further sample extraction detailed below.
LC-MS/MS analyses
Each sample (100 μl) was spiked with 10 μl of 10 μM internal standards, mixed with 200 μl of ice-cold acetonitrile, and then centrifuged at 10 000 × g for 5 min. The supernatant was dried under vacuum with a SpeedVac (Savant SPD1010, Thermo Scientific) and reconstituted with 100 μl of mobile phase prior to analyses. LC-MS/MS analysis was performed using 1290 Infinity II LC system and 6470 triple quadrupole mass spectrometer (both from Agilent Technologies, Santa Clara, California). Sample extract (10 μl) was chromatographed on a ZORBAX SSHD Eclipse Plus C18 column (3.0 × 50 mm, 1.8 μm, Cat No.: 959757-302; Agilent Technologies) with a guard column (2.1 × 5 mm, 1.8 μm, Cat No.: 821725-901; Agilent Technologies), and ionized with an electrospray ionization source. Analytical signal was acquired in positive or negative ion modes. For compounds analyzed in positive ion mode, chromatographic separation occurred following an LC gradient (time [A%]) at a flow rate of 400 μl/min: 0(98)-1(98)-3(20)-4(5)-5(98)-8(98), where mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in methanol. The analysis of 2,4-dinitrophenol followed the same LC gradient, except that the mobile phase A was water and mobile phase B was acetonitrile.
GC-MS/MS analyses
Sample extraction procedures for GC-MS/MS analysis were modified from (Moreno Frias et al., 2004). In brief, sample (100 μl) was spiked with 10 μl of 10 μM internal standards, mixed with 50 μl of methanol and 200 μl of pentane: diethyl ether (1:1, v/v), vortexed, and then centrifuged at 2500 rpm for 5 min. Supernatants were transferred to an amber vial for analyses. Detection of analytes was achieved using 7890B GC and 7010B GC/MS triple quadrupole mass spectrometer (both from Agilent Technologies). Samples (1 μl) were injected in splitless mode. Analytes were separated with a VF-5ms GC column (60 m × 250 μm × 0.25 μm, Agilent Technologies) and ionized with an electron ionization source. The column head pressure was set at 21.5 psi with a constant flow rate at 1.2 ml/min using helium gas. Initial column temperature was held at 70°C for 5 min, increased to 150°C at 50°C/min, ramped to 280°C at 4°C/min, and then held for 15 min. The total run time was 42.1 min. The injector temperature was set at 250°C. The ion source and transfer line temperatures were 300°C. Electron multiplier voltage was set at 1884 V. Nitrogen gas was used as the collision gas for all MS/MS experiments, and the pressure of collision gas was set at 16.8 psi. Ion transitions of test chemicals were adapted from (Chamkasem et al., 2013) and are listed in Table 1. Response ratio of analyte and internal standard was used to derive unbound fraction and in vitro hepatocyte clearance of test chemical. Fraction unbound (Fub), internal clearance (Clint), and steady-state blood concentration (Css) were determined as stated below.
Determination of Fub, Clint, and Css
The unbound fraction of chemical was calculated from the equation [1]:
| (1) |
where .
In vitro hepatocyte clearance (Clin vitro) of test compound was estimated by substrate depletion approach assuming a first-order kinetic for chemical elimination (Smith et al., 2012): Clin vitro = kV/N, where k = first-order elimination rate constant, V = incubation volume (1 ml), and N = number of cells in the incubation (5 × 105 cells). Clin vitro was further scaled up to the intrinsic hepatocyte clearance (Clint) according to the equation 2 (Wetmore, 2015):
| (2) |
where HPGL = hepatocytes per gram liver (137 × 106 cells/g) and Vl = volume of the whole liver (1820 g).
The Css was derived from equation 3 (Wetmore, 2015; Wilkinson and Shand, 1975):
| (3) |
where k0 = a unit input dose (1 mg/kg/day = 0.042 mg/kg/h); GFR = glomerular filtration rate (6.7 l/h); Fub = unbound fraction of tested compound at 10 μM; and Ql = liver blood flow (90 l/h).
Quantitative IVIVE
The in vitro point of departure values (95th percentile POD) were acquired from recent studies (Chen et al., 2020; Paul Friedman et al., 2020). The OEDs of these PODs were further derived according to equation 4:
| (4) |
Calculation of the activity-to-exposure ratios
The derived OEDs were compared with exposure estimates to obtain activity-to-exposure ratio (AER) using equation 5:
| (5) |
Exposure estimates were based on the estimated 95th percentile upper limit of aggregate exposure to U.S. population 20–65 years old acquired from EPA’s Comptox Dashboard (Williams et al., 2017).
RESULTS
In vitro toxicokinetic parameters of protein binding and hepatocyte clearance were evaluated in a set of 20 lipophilic pesticide active ingredients. The chemicals were tested on an individual basis and within equimolar mixtures of all compounds. These data were taken together in combination with reverse dosimetry modeling to derive human OEDs which were further compared with regulatory exposure estimates.
Protein Binding Assay
For studies in human plasma, protein binding values that are reported in httk R package version 1.10.1 (Pearce et al., 2017) were compared with the experimental data in our study. Free fraction values in plasma for httk, single, and mixture experiments ranged from 0% to 21.4%, 1.8% to 14.4%, and 1% to 9.8%, respectively. Free fraction values in httk were obtained through the use of RED and these studies reported that all chemicals tested herein should be highly bound to proteins in human plasma; however, the data obtained using ultracentrifugation show that the free fraction is typically higher than that derived using RED assay (Figure 1A, left panel). Additionally, we found poor concordance (Figure 1B) in the values for single compounds between httk RED and our ultra-centrifugation data, similar to a previous observation comparing RED and solid phase micro-extraction for PAHs (Ferguson et al., 2019). Also, there was no significant correlation of free fraction values from ultracentrifugation between single and mixture settings. Overall, free fraction values for the mixture setting were lower than those in a single chemical test setting.
Figure 1.

Comparison of protein binding using rapid equilibrium dialysis (RED, open circles) or ultracentrifugation (UCF) in a single (filled triangles) and mixture (black squares) setting. A, Plotted are free fractions (as % of tested, median ± 95% CI) of each chemical listed (the number in brackets indicates LogP of each compound). Data are shown for experiments with human plasma (left, chemicals tested at 10 µM) or cell culture media (middle and right, chemicals tested at 10 and 1 µM, respectively). B, Pair-wise correlation plots for % free values for each chemical tested under various conditions (as shown in a title of each plot). Pearson (r) correlations are listed in each graph together with a corresponding p-value. Correlations were considered statistically significant if p-value < .05. Gray-dotted line is a unit line. Values reported as 0% free were replaced with 0.1 for graphing purpose (see Supplementary Table 2 for individual data values).
Additionally, we investigated concentration-dependent effects on protein binding in cell culture media (Figure 1A, center and right panels). For the most part, compounds tested in single setting yielded free fraction values slightly higher than mixture setting in plasma when testing at 10 µM. However, this trend was less apparent when testing at 1 µM. Nonetheless, when tested at either 10 or 1 µM, there were highly significant correlations between single and mixture experiments. Moreover, free fraction values do not exhibit any concentration-dependent effect in media, and free fraction values were highly correlated in both single and mixture settings (r > 0.94, p < .0001) when the data at different concentrations were compared (data not shown).
Hepatocyte Clearance Assay
Next, we compared the data for hepatocyte clearance between those in httk (Pearce et al., 2017) and our experiments (Figure 2). Metabolic clearance values derived in this study for single and mixture setting ranged from 0 to 43.2 and 0 to 5.5 µl/min/106 hepatocytes, respectively (Figure 2A). For a majority of chemicals in a mixture setting, we did not find any detectable hepatocyte clearance. In general, clearance values reported by httk were higher than either single or mixture setting values obtained in our study. Correlation analysis between httk and single setting testing did not show significant concordance, with only 2 compounds showing slightly greater clearance in our study as compared with httk data (Figure 2B). Additionally, correlation analysis between single and mixture setting displayed even less concordance, with only 2 chemicals demonstrating greater clearance in a mixture (Figure 2C).
Figure 2.

In vitro hepatocyte clearance of chemicals in httk (open circles), single (filled triangles), and mixture (black squares) setting. A, Plotted are in vitro hepatocyte clearances (median ± 95% CI) of each chemical listed. Data are shown for experiments using primary human hepatocytes. Each chemical was tested at 10 µM in single and mixture setting. Clearance values reported for each chemical in httk R package version 1.10.1 were tested at 10 µM. B, Pair-wise correlation plot for hepatocyte clearance values for each chemical tested in single setting and reported by httk. C, Pair-wise correlation plot for hepatocyte clearance values for each chemical tested in single and mixture setting. Pearson (r) correlations are listed in both graphs with corresponding p-values. Correlations were considered statistically significant if p-value < .05. Gray-dotted line is a unit line. Values reported as 0 clearance were replaced with 0.1 for graphing purposes (see Supplementary Table 3 for individual data values).
Steady-State Blood Concentrations
Data obtained through plasma protein binding and hepatocyte clearance assays were applied to calculate Css as detailed in Methods. Css average values determined from httk, single, and mixture setting ranged from 0.1 to 97.2 µM (Figure 3A). For most compounds where a comparison between httk and this study could be made, toxicokinetic data obtained from httk yielded the lowest Css values while data from mixture setting produced the highest Css values. Correlation analyses between httk and single setting (Figure 3B) and between mixture and single setting (Figure 3C) showed nonsignificant relationships. However, both analyses further highlight the trend of low Css values produced by httk and high Css values obtained through mixture setting.
Figure 3.

Variance of steady-state blood concentrations (Css) between httk (open circles), single (filled triangles), and mixture (black squares) setting. A, Plotted are Css (median ± 95% CI) of each chemical listed. Data are shown for Css values derived through single and mixture testing and reported in httk R package version 1.10.1. B, Pair-wise correlation plot for Css values for each chemical tested in single setting and reported in httk. C, Pair-wise correlation plot for Css values for each chemical tested in single and mixture setting. Pearson (r) correlations are listed in both graphs with corresponding p-values. Gray-dotted line is a unit line. Correlations were considered statistically significant if p-value < .05 (see Supplementary Table 4 for individual data values).
Mixture to Single Setting Ratios
In order to determine the impact of testing compounds in a mixture compared with individual setting, values obtained for protein binding, hepatocyte clearance, and Css were compared as ratios. The data from the mixture setting were divided by their corresponding values from the single setting. The resulting ratios were plotted (Figure 4) to determine whether mixture setting overall yielded higher, lower, or equal toxicokinetic parameters. Ratios for free fraction in plasma, media (10 µM), and media (1 µM) ranged from 0.18 to 2.65, 0.26 to 2.24, and 0.28 to 3.30, respectively. On average, unbound fractions were slightly lower in the mixture setting at 10 µM, regardless of whether the experiments were conducted in human plasma or cell culture media, and about the same at 1 µM. In all cases, the ratios were well within 1 order of magnitude.
Figure 4.
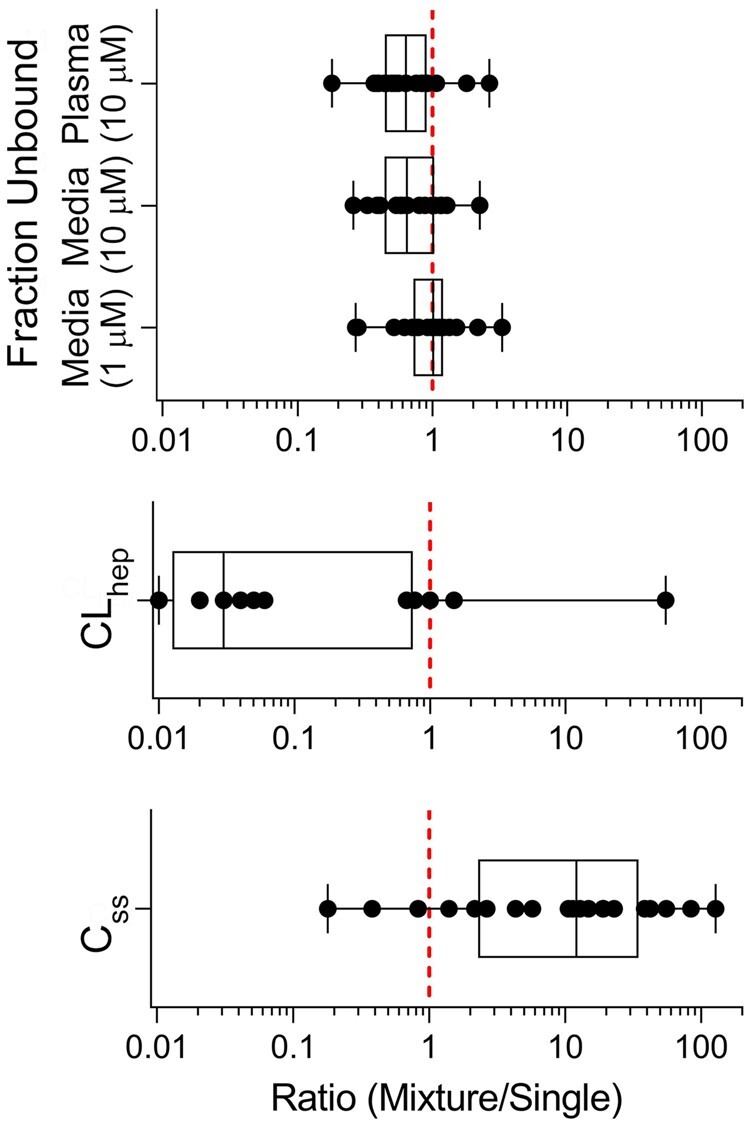
Comparison of ratios (mixture over single) for fraction unbound, hepatocyte clearance, and steady-state blood concentration (Css). Box plots are shown comparing ratio data values for each compound (black circle) corresponding to the listed parameter. Within each box, the black vertical lines inside the box denote median values; boxes extend from 25th to the 75th percentile of each group’s distribution of values; vertical extending lines denote minimum and maximum values. The vertical dotted line at 1 represents an equal ratio between mixture and single data values (see Supplementary Table 5 for individual data values).
For hepatocyte clearance, the ratios ranged from 0.01 to 54.90. Overall, hepatocyte clearance values in the mixture setting were much lower compared to those in the single setting as evident by the median ratio value of 0.03. When the results from plasma protein binding and hepatocyte clearance are combined, Css ratios also ranged from 0.18 to 128.03, with the vast majority of the Css ratios above 1. These results indicate that testing in a mixture setting may result in Css that are up to 10 or 100-fold greater than those assumed from the data obtained for single chemicals.
Activity to Exposure Ratios
Using results of 2 recent large-scale in vitro studies of bioactivity of the test chemicals that have used these data to compare in vitro points of departure to estimates of human exposure, AERs (Figure 5) were calculated for each chemical based on converting bioactivity to an OED using Css values produced from httk, single, and mixture setting data as described in Methods. As a reference, we used the estimated 95th percentile upper limit of aggregate exposure to U.S. population 20–65 years old acquired from EPA’s Comptox Dashboard (Williams et al., 2017). Specifically, each AER was plotted based on httk, single, and mixture data and the bioactivity results from either Paul Friedman et al. (2020) (data were available for 10 of the 20 chemicals tested here) or Chen et al. (2020, data for 18–20 chemicals were available, depending on the cell type used in the experiments). We found that AERs calculated using httk-derived IVIVE data yielded the highest AER values, indicating the greatest margin of safety based on the in vitro results and the estimates of human exposures. Toxicokinetic parameters obtained in this study for the mixture setting produced the lowest AER values with both sets of bioactivity data. The median, upper, and lower quartiles of the AERs for mixture data using Paul Friedman et al. (2020) points of departure were 16 154, 78 026, and 5507, respectively. The median, upper, and lower quartiles of the AER distribution for mixture data using Chen et al. (2020) points of departure were 3283, 17 459, and 1089, respectively. The minimum AER derived from Paul Friedman et al. (2020) data was 4551 for azinphos-methyl, whereas the minimum AER from Chen et al. (2020) data was 127 for methoxychlor-o, p’.
Figure 5.
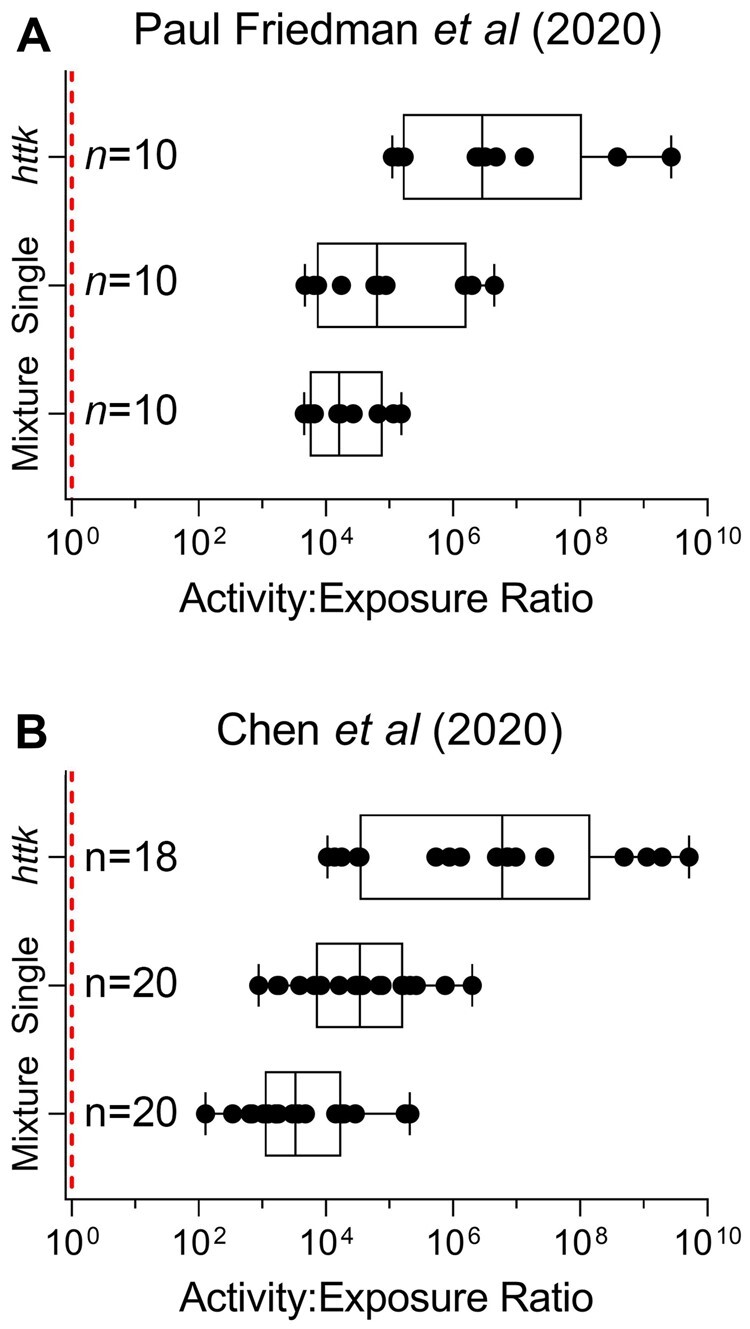
Distribution of activity to exposure ratios (AERs) derived from in vitro point of departures (PODs). A, Plotted are AERs for each chemical using PODs (95th percentile) reported by (Paul Friedman et al., 2020) derived from steady-state blood concentrations reported in httk or obtained in our study in single and mixture setting. For (A) an n = 10 due to an overlap of 10 chemicals between our study and (Paul Friedman et al., 2020). B, AERs for each chemical using PODs (95th percentile) reported by (Chen et al., 2020) derived in the same manner as (A). For (B), httk had an overlap of 18 chemicals with our study whereas single and mixture had n = 20. Vertical dotted line represents an AER of 1 (see Supplementary Table 6 for individual data values).
DISCUSSION
IVIVE is an essential tool for high-throughput screening (HTS) of environmental compounds with limited toxicity data. By combining in vitro kinetic and bioactivity data, IVIVE facilitates the translation of in vitro testing concentrations to in vivo oral doses (Paul Friedman et al., 2020; Rotroff et al., 2010; Wambaugh et al., 2015; Wetmore et al., 2012). The use of this well-established reverse toxicokinetic modeling approach enables regulators to prioritize chemicals through a risk-based interpretation of HTS data that incorporates not only hazard identification but also takes into consideration actual exposure estimates (Krewski et al., 2020; Thomas et al., 2013; Wambaugh et al., 2019).
In this study, in vitro kinetic assays were performed on 20 lipophilic environmental chemicals chosen as a subset from the ATSDR’s Substance Priority List (ATSDR, 2019). The in vitro kinetic assays were applied to derive 2 most informative parameters of IVIVE—plasma protein binding and hepatocyte clearance. All chemicals were tested on an individual basis, as well as in an equimolar mixture in order to investigate mixture effects on in vitro kinetics. The results from the in vitro assays were combined with IVIVE to estimate each chemical’s concentration in blood at steady state for specific human exposure situations. Next, reverse dosimetry was incorporated to calculate oral doses that would produce a Css equivalent to the 95th percentile point of departure value of each chemical’s in vitro bioactivity data. Last, OEDs were compared with exposure estimates to attain AERs under various testing conditions.
Currently, IVIVE has been applied to thousands of chemicals to yield large data sets that are highly informative for decision-making (Wambaugh et al., 2015, 2019; Wetmore, 2015). Although a number of studies and analyses have used in vitro bioactivity to assess mixture effects from an exposure or cumulative risk point of view, they have all still used IVIVE kinetic data and models based on single chemicals, given the lack of IVIVE data for chemical mixtures (Chang et al., 2021; Lichtenstein et al., 2020; van der Voet et al., 2020). Similarly, available computational approaches to characterize in vitro bioavailability are currently limited to individual chemicals (Armitage et al., 2014; Fischer et al., 2017; Stadnicka-Michalak et al., 2021). Therefore, our study is novel as it reported and compared IVIVE parameters for a large number of environmental chemicals that were tested in single and mixture settings. Additionally, compared with IVIVE data reported in httk, our study used ultracentrifugation to estimate plasma protein binding instead of RED assay. As previously mentioned, RED has been shown to not be a suitable assay to calculate plasma protein binding of lipophilic environmental compounds (Ferguson et al., 2019). From our chemical set, almost half of all compounds were reported to have 0% unbound fraction in human plasma according to httk; however, by using ultracentrifugation we were able to obtain unbound fraction values >0%, albeit still relatively low (<10%). Taken together, our study demonstrates not only the impact of more accurate estimates of plasma protein binding, but also the effects on kinetics by chemical mixtures compared with traditional single chemical analysis. Knowing that most chemical exposures occur as mixtures rather than individual compounds (Hernández and Tsatsakis, 2017) we believe this study is highly informative for cumulative risk assessment of environmental chemical exposures.
Our testing approach showed that a vast majority of the 20 pesticides screened in our study did not show any clear impacts on plasma protein binding capabilities when tested as a mixture compared with individual compound analysis. In both testing conditions, all compounds appeared to be highly bound to proteins suggesting that the amount of albumin present in plasma is not a limiting factor in binding efficiency. However, hepatocyte metabolic clearance data showed a pronounced effect when tested as a mixture. In general, almost all chemicals had no detectable clearance under mixture setting. This observation potentially indicates that hepatocytes were saturated with chemicals under mixture setting and thus, could not metabolically clear them as efficiently compared with exposure to a single chemical. The same effect has been noted in previous studies conducted on hepatocyte clearance of environmental compounds (Lee et al., 2014). It is not surprising that a major reduction in hepatocyte clearance under mixture setting yielded Css much higher compared with single chemical data. When compared with our study, httk data had the lowest Css that are likely due to very low unbound fractions derived using RED and higher hepatocyte clearance values. Using our mixture data to estimate OEDs that would result in bioactive concentrations appeared to be the most conservative approach compared with using our single chemical testing data or IVIVE data from httk. When OEDs obtained from mixture data were compared with upper exposure estimates (95th percentile) for the U.S. general population the data revealed that the margin for safety from chemical exposure is not nearly as high compared with using data from single chemical screening. This conservative approach takes into consideration simultaneous exposure to multiple chemicals which we believe to be more relevant to actual human exposure scenarios.
We note a few limitations in our study. First, our designed mixture of 20 compounds was created as an equimolar mixture with each chemical present at a final concentration of 10 µM. Chemicals present in the environment occur at varying concentrations (Carpenter et al., 2002; Hsieh et al., 2021; Martin et al., 2013), and that 10 µM across all 20 compounds may be rather high for general population environmental exposure, or may be potentially cytotoxic. Still, we reason that in the general population, this saturation effect may be less pronounced than we have observed. In addition, we note that it is unlikely that the mixture as tested would have had an adverse effect on the hepatocytes in the study of clearance because exposure was limited to 4 h and most of these substances were without effect on cytotoxicity in HepaRG cells or human hepatocytes as reported in ToxCast (Supplementary Table 7). Also, in order to facilitate direct comparison between single and mixture setting, we decided to use 10 µM as the set concentration based on typical testing concentrations for IVIVE studies (Rotroff et al., 2010; Wetmore et al., 2012). Alternative designs can be used, such as 0.5 µM of the 20 chemicals for a cumulative concentration of the mixture at 10 µM. Nonetheless, our results suggest the importance of considering reduced hepatocyte clearance in real-life mixture settings, because AERs based on single-chemical data may not be conservative. Even though such AERs may still be adequate for prioritization, there is less confidence in their use in screening-level decision-making. Additionally, other existing limitations of IVIVE as it is currently practiced (eg, steady-state, hepatic-only metabolism, etc.) are well known; still, several studies comparing IVIVE-based oral equivalents with in vivo data have suggested that errors are about an order of magnitude (Honda et al., 2019; Wambaugh et al., 2018, 2019). We also acknowledge that physicochemical properties such as water solubility play a key role in determining the exact concentrations these lipophilic compounds may exist in vitro and in the environment. For instance, available computational models for in vitro bioavailability have utilized a range of chemical and system property information, including various partition coefficients, temperature and dimensions of the test system, and presence of cosolvents (Armitage et al., 2014; Fischer et al., 2017; Punt et al., 2021).
Second, protein precipitation with organic solvents may release some of the bound chemical, which may cause overestimation for the unbound fraction. Nevertheless, the method we used to determine free fraction in human plasma has been previously shown to yield free fraction values similar to those measured in vivo (Nakai et al., 2004). Third, all the environmental compounds in our study were limited to only pesticides. Despite there being various classes of pesticides within our study, we understand there are various other chemical classes in the environment such as PAHs, polybrominated biphenyls, etc. Follow up studies using untargeted analyses would be highly beneficial to characterize which chemical compounds are commonly found together in the environment.
Overall, this case study used 20 chemicals commonly found in the environment to demonstrate how chemical coexposures affect in vitro pharmacokinetics of plasma protein binding and hepatocyte metabolic clearance which directly impact IVIVE. The current paradigm shift of toxicity testing from traditional animal models to more reliance on HTS assays (Krewski et al., 2014, 2020) requires an equal investment in the proper characterization of pharmacokinetics of not only single chemicals, but mixtures as well. Data generated from mixture analyses provide a more comprehensive guideline for cumulative risk assessment compared with individual chemical testing. Thus, the impact of mixtures may require more attention as chemical coexposures are expected to impact both bioavailability in vitro as well as in vitro toxicokinetic parameters used to conduct IVIVE.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENT
The authors wish to show gratitude to Dr Tracy Clement at Texas A&M University for facilitating access to some of the equipment used in these studies.
FUNDING
National Institute of Environmental Health Sciences (P42 ES027704).
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Armitage J. M., Wania F., Arnot J. A. (2014). Application of mass balance models and the chemical activity concept to facilitate the use of in vitro toxicity data for risk assessment. Environ. Sci. Technol. 48, 9770–9779. [DOI] [PubMed] [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). (2019). ATSDR’s Substance Priority List. Available at: http://www.atsdr.cdc.gov/SPL/index.html. Accessed April 8, 2021.
- Bell S., Abedini J., Ceger P., Chang X., Cook B., Karmaus A. L., Lea I., Mansouri K., Phillips J., McAfee E., et al. (2020). An integrated chemical environment with tools for chemical safety testing. Toxicol. In Vitro 67, 104916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S. M., Chang X., Wambaugh J. F., Allen D. G., Bartels M., Brouwer K. L. R., Casey W. M., Choksi N., Ferguson S. S., Fraczkiewicz G., et al. (2018). In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicol. In Vitro 47, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert T., Gan L. S. (2013). Plasma protein binding: From discovery to development. J. Pharm. Sci. 102, 2953–2994. [DOI] [PubMed] [Google Scholar]
- Carpenter D. O., Arcaro K., Spink D. C. (2002). Understanding the human health effects of chemical mixtures. Environ. Health Perspect. 110(Suppl. 1), 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamkasem N., Ollis L. W., Harmon T., Lee S., Mercer G. (2013). Analysis of 136 pesticides in avocado using a modified QuEChERS method with LC-MS/MS and GC-MS/MS. J. Agric. Food Chem. 61, 2315–2329. [DOI] [PubMed] [Google Scholar]
- Chang X., Abedini J., Bell S., Lee K. M. (2021). Exploring in vitro to in vivo extrapolation for exposure and health impacts of e-cigarette flavor mixtures. Toxicol. In Vitro 72, 105090. [DOI] [PubMed] [Google Scholar]
- Chen Z., Liu Y., Wright F. A., Chiu W. A., Rusyn I. (2020). Rapid hazard characterization of environmental chemicals using a compendium of human cell lines from different organs. ALTEX 37, 623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Lloyd D., Zhou Y. H., Chiu W. A., Wright F. A., Rusyn I. (2021). Risk characterization of environmental samples using in vitro bioactivity and polycyclic aromatic hydrocarbon concentrations data. Toxicol. Sci. 179, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakvik E., Altenburger R., Aoki Y., Backhaus T., Bahadori T., Barouki R., Brack W., Cronin M. T. D., Demeneix B., Hougaard Bennekou S., et al. (2020). Statement on advancing the assessment of chemical mixtures and their risks for human health and the environment. Environ. Int. 134, 105267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher B. I., Stapleton H. M., Schymanski E. L. (2020). Tracking complex mixtures of chemicals in our changing environment. Science 367, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W., Peng Y., Yan L., Xia P., Zhang X. (2020). A tiered approach for screening and assessment of environmental mixtures by omics and in vitro assays. Environ. Sci. Technol. 54, 7430–7439. [DOI] [PubMed] [Google Scholar]
- Ferguson K. C., Luo Y. S., Rusyn I., Chiu W. A. (2019). Comparative analysis of rapid equilibrium dialysis (RED) and solid phase micro-extraction (SPME) methods for in vitro-in vivo extrapolation of environmental chemicals. Toxicol. In Vitro 60, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F. C., Henneberger L., Konig M., Bittermann K., Linden L., Goss K. U., Escher B. I. (2017). Modeling exposure in the Tox21 in vitro bioassays. Chem. Res. Toxicol. 30, 1197–1208. [DOI] [PubMed] [Google Scholar]
- Hernández A. F., Tsatsakis A. M. (2017). Human exposure to chemical mixtures: Challenges for the integration of toxicology with epidemiology data in risk assessment. Food Chem. Toxicol. 103, 188–193. [DOI] [PubMed] [Google Scholar]
- Honda G. S., Pearce R. G., Pham L. L., Setzer R. W., Wetmore B. A., Sipes N. S., Gilbert J., Franz B., Thomas R. S., Wambaugh J. F. (2019). Using the concordance of in vitro and in vivo data to evaluate extrapolation assumptions. PLoS One 14, e0217564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh N. H., Chen Z., Rusyn I., Chiu W. A. (2021). Risk characterization and probabilistic concentration-response modeling of complex environmental mixtures using new approach methodologies (NAMs) data from organotypic in vitro human stem cell assays. Environ. Health Perspect. 129, 17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewski D., Andersen M. E., Tyshenko M. G., Krishnan K., Hartung T., Boekelheide K., Wambaugh J. F., Jones D., Whelan M., Thomas R., et al. (2020). Toxicity testing in the 21st century: Progress in the past decade and future perspectives. Arch. Toxicol. 94, 1–58. [DOI] [PubMed] [Google Scholar]
- Krewski D., Rice J. M., Bird M., Milton B., Collins B., Lajoie P., Billard M., Grosse Y., Cogliano V. J., Caldwell J. C., et al. (2019). Concordance between sites of tumor development in humans and in experimental animals for 111 agents that are carcinogenic to humans. J. Toxicol. Environ. Health B Crit. Rev. 22, 203–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewski D., Westphal M., Andersen M. E., Paoli G. M., Chiu W. A., Al-Zoughool M., Croteau M. C., Burgoon L. D., Cote I. (2014). A framework for the next generation of risk science. Environ. Health Perspect. 122, 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. S., Lee D. H., Delafoulhouze M., Otton S. V., Moore M. M., Kennedy C. J., Gobas F. A. (2014). In vitro biotransformation rates in fish liver S9: Effect of dosing techniques. Environ. Toxicol. Chem. 33, 1885–1893. [DOI] [PubMed] [Google Scholar]
- Lichtenstein D., Luckert C., Alarcan J., de Sousa G., Gioutlakis M., Katsanou E. S., Konstantinidou P., Machera K., Milani E. S., Peijnenburg A., et al. (2020). An adverse outcome pathway-based approach to assess steatotic mixture effects of hepatotoxic pesticides in vitro. Food Chem. Toxicol. 139, 111283. [DOI] [PubMed] [Google Scholar]
- Luo Y. S., Ferguson K. C., Rusyn I., Chiu W. A. (2020). In vitro bioavailability of the hydrocarbon fractions of dimethyl sulfoxide extracts of petroleum substances. Toxicol. Sci. 174, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin O. V., Martin S., Kortenkamp A. (2013). Dispelling urban myths about default uncertainty factors in chemical risk assessment–sufficient protection against mixture effects? Environ. Health 12, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx U., Akabane T., Andersson T. B., Baker E., Beilmann M., Beken S., Brendler-Schwaab S., Cirit M., David R., Dehne E. M., et al. (2020). Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX 37, 365–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Frias M., Jimenez Torres M., Garrido Frenich A., Martinez Vidal J. L., Olea-Serrano F., Olea N. (2004). Determination of organochlorine compounds in human biological samples by GC-MS/MS. Biomed. Chromatogr. 18, 102–111. [DOI] [PubMed] [Google Scholar]
- Nakai D., Kumamoto K., Sakikawa C., Kosaka T., Tokui T. (2004). Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J. Pharm. Sci. 93, 847–854. [DOI] [PubMed] [Google Scholar]
- OECD. (2018). Guidance Document on the Determination of in vitro Intrinsic Clearance Using Cryopreserved Hepatocytes (RTHEP) or Liver S9 sub-Cellular Fractions (RT-S9) from Rainbow Trout and Extrapolation to In Vivo Intrinsic Clearance. Environment Directorate, Organisation for Economic Co-operation and Development, Paris, France. [Google Scholar]
- Olson H., Betton G., Robinson D., Thomas K., Monro A., Kolaja G., Lilly P., Sanders J., Sipes G., Bracken W., et al. (2000). Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67. [DOI] [PubMed] [Google Scholar]
- Paul Friedman K., Gagne M., Loo L. H., Karamertzanis P., Netzeva T., Sobanski T., Franzosa J. A., Richard A. M., Lougee R. R., Gissi A., et al. (2020). Utility of in vitro bioactivity as a lower bound estimate of in vivo adverse effect levels and in risk-based prioritization. Toxicol. Sci. 173, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce R. G., Setzer R. W., Strope C. L., Wambaugh J. F., Sipes N. S. (2017). httk: R package for high-throughput toxicokinetics. J. Stat. Softw. 79, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punt A., Pinckaers N., Peijnenburg A., Louisse J. (2021). Development of a web-based toolbox to support quantitative in-vitro-to-in-vivo extrapolations (QIVIVE) within nonanimal testing strategies. Chem. Res. Toxicol. 34, 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotroff D. M., Wetmore B. A., Dix D. J., Ferguson S. S., Clewell H. J., Houck K. A., LeCluyse E. L., Andersen M. E., Judson R. S., Smith C. M., et al. (2010). Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci. 117, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. M., Nolan C. K., Edwards M. A., Hatfield J. B., Stewart T. W., Ferguson S. S., Lecluyse E. L., Sahi J. (2012). A comprehensive evaluation of metabolic activity and intrinsic clearance in suspensions and monolayer cultures of cryopreserved primary human hepatocytes. J. Pharm. Sci. 101, 3989–4002. [DOI] [PubMed] [Google Scholar]
- Stadnicka-Michalak J., Bramaz N., Schonenberger R., Schirmer K. (2021). Predicting exposure concentrations of chemicals with a wide range of volatility and hydrophobicity in different multi-well plate set-ups. Sci. Rep. 11, 4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. S., Philbert M. A., Auerbach S. S., Wetmore B. A., Devito M. J., Cote I., Rowlands J. C., Whelan M. P., Hays S. M., Andersen M. E., et al. (2013). Incorporating new technologies into toxicity testing and risk assessment: Moving from 21st century vision to a data-driven framework. Toxicol. Sci. 136, 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Voet H., Kruisselbrink J. W., de Boer W. J., van Lenthe M. S., van den Heuvel J., Crepet A., Kennedy M. C., Zilliacus J., Beronius A., Tebby C., et al. (2020). The MCRA toolbox of models and data to support chemical mixture risk assessment. Food Chem. Toxicol. 138, 111185. [DOI] [PubMed] [Google Scholar]
- Wambaugh J. F., Hughes M. F., Ring C. L., MacMillan D. K., Ford J., Fennell T. R., Black S. R., Snyder R. W., Sipes N. S., Wetmore B. A., et al. (2018). Evaluating in vitro-in vivo extrapolation of toxicokinetics. Toxicol. Sci. 163, 152–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambaugh J. F., Wetmore B. A., Pearce R., Strope C., Goldsmith R., Sluka J. P., Sedykh A., Tropsha A., Bosgra S., Shah I., et al. (2015). Toxicokinetic triage for environmental chemicals. Toxicol. Sci. 147, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambaugh J. F., Wetmore B. A., Ring C. L., Nicolas C. I., Pearce R. G., Honda G. S., Dinallo R., Angus D., Gilbert J., Sierra T., et al. (2019). Assessing toxicokinetic uncertainty and variability in risk prioritization. Toxicol. Sci. 172, 235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore B. A. (2015). Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 332, 94–101. [DOI] [PubMed] [Google Scholar]
- Wetmore B. A., Allen B., Clewell H. J. 3rd, Parker T., Wambaugh J. F., Almond L. M., Sochaski M. A., Thomas R. S. (2014). Incorporating population variability and susceptible subpopulations into dosimetry for high-throughput toxicity testing. Toxicol. Sci. 142, 210–224. [DOI] [PubMed] [Google Scholar]
- Wetmore B. A., Wambaugh J. F., Ferguson S. S., Li L., Clewell H. J., Judson R. S., Freeman K., Bao W., Sochaski M. A., Chu T.-M., et al. (2013). Relative impact of incorporating pharmacokinetics on predicting in vivo hazard and mode of action from high-throughput in vitro toxicity assays. Toxicol. Sci. 132, 327–346. [DOI] [PubMed] [Google Scholar]
- Wetmore B. A., Wambaugh J. F., Ferguson S. S., Sochaski M. A., Rotroff D. M., Freeman K., Clewell H. J. 3rd, Dix D. J., Andersen M. E., Houck K. A., et al. (2012). Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci. 125, 157–174. [DOI] [PubMed] [Google Scholar]
- Wilkinson G. R., Shand D. G. (1975). Commentary: A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 18, 377–390. [DOI] [PubMed] [Google Scholar]
- Williams A. J., Grulke C. M., Edwards J., McEachran A. D., Mansouri K., Baker N. C., Patlewicz G., Shah I., Wambaugh J. F., Judson R. S., et al. (2017). The CompTox Chemistry Dashboard: A community data resource for environmental chemistry. J. Cheminform. 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M., Campbell J. L., Andersen M. E., Clewell H. J. (2012). Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit. Rev. Toxicol. 42, 633–652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.